
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic identification of fragment ions of DNA/RNA building blocks: the case of pyrimidine†
Kuntal
Chatterjee
 and
Otto
Dopfer
*
and
Otto
Dopfer
*
Institut für Optik und Atomare Physik, TU Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 7th July 2020
Abstract
Pyrimidine (Pym, 1,3-diazine, 1,3-diazabenzene) is an important N-heterocyclic building block of nucleobases. Understanding the structures of its fragment and precursor ions provides insight into its prebiotic and abiotic synthetic route. The long-standing controversial debate about the structures of the primary fragment ions of the Pym+ cation (C4H4N2+, m/z 80) resulting from loss of HCN, C3H3N+ (m/z 53), is closed herein with the aid of a combined approach utilizing infrared photodissociation (IRPD) spectroscopy in the CH and NH stretch ranges (νCH/NH) and density functional theory (DFT) calculations. IRPD spectra of cold Ar/N2-tagged fragment ions reveal that the C3H3N+ population is dominated by cis-/trans-HCCHNCH+ ions (∼90%) along with a minor contribution of the most stable H2CCCNH+ and cis-/trans-HCCHCNH+ isomers (∼10%). We also spectroscopically confirm that the secondary fragment resulting from further loss of HCN, C2H2+ (m/z 26), is the acetylene cation (HCCH+). The spectroscopic characterization of the identified C3H3N+ isomers and their hydrogen-bonded dimers with Ar and N2 provides insight into the acidity of their CH and NH groups. Finally, the vibrational properties of Pym+ in the 3 μm range are probed by IRPD of Pym+-(N2)1–2 clusters, which shows a high π-binding affinity of Pym+ toward a nonpolar hydrophobic ligand. Its νCH spectrum confirms the different acidity of the three nonequivalent CH groups.
1. Introduction
Nucleobases, the molecular building blocks of the fundamental genetic materials DNA and RNA, are primarily composed of two different aromatic nitrogen heterocycles (N-heterocylces), namely pyrimidine (1,3-diazine, 1,3-diazabenzene, Pym, C4H4N2) and purine. The pyrimidine nucleobases are uracil, thymine, and cytosine, whereas adenine and guanine are purine bases. The ubiquity of these few selected nucleobases involved in the evolution of life on Earth shows a clear example of restricted natural selectivity upon which life as we know it developed.1–4 Apart from nucleobases, Pym is also a building block of other natural and artificial bioactive molecules (e.g., vitamin B1 or thiamine, barbituric acid).5,6The early Earth was exposed to strong solar irradiation (particularly UV), because of the absence of the protecting ozone layer. Therefore, only photochemically stable molecules survived during that time. Interestingly, pyridine and diazine molecules with their short first excited state (nπ*) lifetimes fall in such a photochemically robust category.3,7–12 The canonical DNA/RNA bases comprised of these N-heterocycles also exhibit such short-lived excited states because of ultrafast internal conversion to the ground electronic state, and their high photostability may explain their abundance in genetic materials.3,4,13–19
Because of their abundance on Earth, these nucleobases and their building blocks such as Pym have been intensively searched for in extraterrestrial media to understand their possible delivery to Earth.20–23 Although such investigations have failed so far, the presence of these aromatic N-heterocycles in the interstellar medium has received widespread acceptance.24–32 For example, the 6.2 μm feature in the unidentified infrared emission spectrum was suggested to originate from polycyclic aromatic hydrocarbons with at least one N atom in the aromatic skeleton.23 The detection of Pym- and purine-based compounds in carbonaceous meteorites further strengthens the possibility of the extraterrestrial presence of N-heterocycles.33–36 Because of their biological importance, prebiotic synthetic routes for these nucleobases have been well explored.37,38 It has been proposed that such N-containing macromolecules may be synthesized through complex reactions between abundant N-bearing species such as NH3 or HCN and carbonaceous precursors in interstellar ice grains under the influence of UV photons and cosmic ray analogs (e.g., protons and electrons).39–43 Recent laboratory experiments under interstellar-like conditions demonstrate the formation of uracil and cytosine upon UV irradiation of H2O:Pym and H2O:NH3:Pym ices, respectively.44,45 These experiments triggered theoretical investigations to gain further insight into the involved chemical conversion scheme.46 Similar studies of CH3OH:Pym, H2O:CH3OH:Pym, and CH4:Pym ices reveal the generation of thymine, along with numerous amino pyrimidines and their hydroxyl derivatives.40 Related photon processing of H2O:NH3:purine ices results in the formation of adenine, guanine, and other substituted purine derivatives.41,47 Interestingly, apart from photon irradiation of simple H2O:Pym ices leading to thymine formation, all other photon processing results in the production of RNA nucleobases and thus supports the hypothesis of RNA being the first type of genetic material used by early life.48
The Pym nucleobases inherit certain properties like their geometry and core and valence electronic structure from the parent Pym moiety,49,50 which renders this N-heterocycle a subject of demanding attention. Understanding the abiotic origin of such genetic material is of fundamental interest for prebiotic chemistry and astrobiology. Several prudent experimental techniques, such as gas-phase photon or electron ionization (EI) and mass spectrometry, have been employed along with computations to explore abiotic routes of both Pym formation and fragmentation.51–66 These studies have established two main fragment ions of the Pym+ cation, C3H3N+ (m/z 53) and C2H2+ (m/z 26), through the elimination of one and two HCN molecules, respectively.55–57,61,64,65 Interestingly, there is a long-standing debate about the molecular structure of the C3H3N+ fragment. An early mass-spectrometric study using isotope labeling suggested the kinetically favoured HCCHNCH+ structure (4/5) because this fragmentation channel can eliminate HCN containing C4 (e.g., H4C4N3), without further structural arrangement.60 Subsequently, the acrylonitrile cation (H2CCHCN+, 6) was considered,56,66 which however was later discarded because of the dissimilarity of its collision-induced dissociation (CID) spectrum and enthalpy of formation (ΔfH) from those of the C3H3N+ ion originating from Pym+.56,58,64 Instead, the most stable H2CCCNH+ isomer of C3H3N+ (1) with a CCCN backbone was suggested as the major contributing structure of this daughter ion, because it is more stable than 6 and other C3H3N+ isomers, such as HCCCHNH+ (11/12) and HCCHCNH+ (2/3).64 However, the generation of 1 from Pym+ demands a large activation barrier, as revealed from a detailed computational study of all feasible C3H3N+ isomers.67 A photoionization–fragmentation study suggested either 6 or H2CNCCH+ (7).63 On the other hand, a theoretical study showed effective radical site formation on the N atom upon ionization of Pym that disintegrates into 6 or H2CCNCH+ (8) by eliminating HCN.65 An electron-ionization mass spectrometric study concluded the kinetically favorable HCCHNCH+ isomer (4/5) to be the most probable C3H3N+ structure.51 A recent mass-spectrometric and computational investigation reported the formation of Pym+ by the sequential barrierless addition of two HCN molecules to the acetylene cation (HCCH+, HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH+), and the suggested intermediate C3H3N+ ions have again been 4/5.68 In the reverse direction, the C3H3N+ ion dissociates via HCN elimination into C2H2+ (m/z 26), which was identified as the acetylene cation (HCCH+) by comparing their ΔfH values.51,57,59,69 Quantum chemical investigations demonstrate an efficient production scheme of the HCCH+ cation from 4/5 by HCN elimination.54,58,61,62,67
CH+), and the suggested intermediate C3H3N+ ions have again been 4/5.68 In the reverse direction, the C3H3N+ ion dissociates via HCN elimination into C2H2+ (m/z 26), which was identified as the acetylene cation (HCCH+) by comparing their ΔfH values.51,57,59,69 Quantum chemical investigations demonstrate an efficient production scheme of the HCCH+ cation from 4/5 by HCN elimination.54,58,61,62,67
So far, the identification of the m/z 53 and 26 fragment ions of Pym+ has been solely based on mass spectrometry, thermochemistry, and quantum chemistry. Unambiguous spectroscopic confirmation of the structures suggested for these two important fragment ions of Pym+ are lacking. To this end, this work aims at the infrared (IR) spectroscopic identification of these two major Pym+ fragments. For this purpose, we employ an EI technique to produce Pym+ which dissociates in the source mainly into C3H3N+ and C2H2+. We generate the Ar/N2-tagged clusters of both Pym+ and its fragment ions, whose structures are then probed by IR photodissociation (IRPD) spectroscopy. These fingerprint spectra are analyzed with the aid of dispersion-corrected density functional theory (DFT) calculations to unambiguously identify the contributing structural isomers and competing ligand binding sites. This combined spectroscopic/computational approach has previously been applied to various aromatic and nonaromatic cluster ions in our laboratory to unravel their structure and bonding.30–32,70–77
2. Experimental methods
IRPD spectra of mass-selected Pym+-(N2)n clusters (n = 1–2) and the Ar/N2-tagged clusters of Pym+ fragments, i.e. C3H3N+-Ln (n = 0, n = 1 for Ar, n = 1–3 for N2) and C2H2+-L (L = Ar/N2), are acquired between 2900 and 3600 cm−1 in a tandem quadrupole mass spectrometer coupled to an EI source and an octupole ion guide.78,79 Briefly, the investigated ions and their clusters are produced in a pulsed supersonic plasma expansion utilizing electron and chemical ionization close to the nozzle orifice. Electrons are emitted from two tungsten filaments close to the nozzle orifice. The expanding gas mixture is produced by seeding the vapor of Pym (Sigma-Aldrich, >98%, used without further purification) heated to 333 K in N2 or Ar carrier gas (10 bar). The desired parent cluster ions are mass-selected in the first quadrupole and irradiated in the adjacent octupole ion guide with a tunable IR laser pulse (νIR) emitted from an optical parametric oscillator laser pumped by a nanosecond Q-switched Nd:YAG laser operating at 10 Hz. The IR laser pulses are characterized by a bandwidth of 1 cm−1 and energies of 1–5 mJ. Resonant vibrational excitation leads to the elimination of all weakly bound ligands. The C3H3N+ monomer spectrum is recorded in the HCN loss channel. The resulting daughter ions are mass-selected by the second quadrupole and monitored with a Daly detector as a function of νIR to derive the IRPD spectrum of the parent cluster. The photofragmentation spectra are linearly normalized for energy fluctuations of the laser pulses recorded with a pyroelectric detector. To obtain the IRPD yield, the background contribution arising from metastable decay (laser-off signal) is subtracted from the laser-on signal by triggering the ion source at twice the laser repetition rate. The observed widths of the vibrational transitions are mainly due to the unresolved rotational structure, lifetime broadening, sequence hot bands involving low-frequency inter- and intramolecular modes (νCH/NH + νx ← νx), and possible overlapping contributions from various structural isomers. Low-energy collision-induced dissociation (CID) spectra at ∼10 eV collision energy in the laboratory frame are recorded by introducing 10−5 mbar N2 into the octopole to confirm the composition of the ions and their clusters. IRPD spectra of Pym+-Arn clusters can not be recorded with this setup because of the mass overlap of Pym+ with the much more abundant isobaric Ar2 ion (m/z 80). A typical mass spectrum of the EI ion supersonic expansion source using N2 carrier gas is quite similar to the published standard EI spectrum of isolated Pym (apart from signals arising from N2 and H2O impurity),80 with dominant peaks at m/z 80 (Pym+), 53 (C3H3N+), and 28 (C2H2+), strongly suggesting that that the ionization and fragmentation processes are rather similar under both conditions (Fig. S1 in the ESI†). This scenario is supported by a previous energy-resolved ionization study of Pym,56 which concludes that Pym+ cations generated in higher electronic states undergo rapid internal conversion into the lowest electronic state before statistical dissociation occurs on the ground state potential. The CID spectra of mass-selected Pym+ and C3H3N+ ions show the exclusive loss of HCN (Fig. S2, ESI†).3. Computational methods
Various structural isomers of the C3H3N+ ion and conceivable ligand binding sites of Ar and N2 to Pym+, C3H3N+, and C2H2+ are explored at the B3LYP-D3/aug-cc-pVTZ level of DFT theory to analyze their experimental IR spectra.81 This dispersion-corrected functional accounts well for the electrostatic, induction, and dispersion forces of the investigated clusters.30–32,74–77 Fully relaxed potential energy surface calculations are performed during the search for stationary points, and their nature as minima or transition states are verified by harmonic frequency analysis. Harmonic intramolecular vibrational frequencies are scaled with 0.96221, obtained by fitting the νCH/OH frequencies of Pym and H2O to their experimental values in a parallel study of H+Pym-(H2O)n. Computed scaled IR stick spectra are convoluted with Gaussian line profiles with FWHM = 10 cm−1 to facilitate convenient comparison with the experimental spectra. All relative energies (Ee) and equilibrium dissociation energies (De) are corrected for harmonic zero-point vibrational energy to derive E0 and D0 values. Gibbs free energies (G) are reported for T = 298.15 K. Cartesian coordinates and energies of all relevant structures are listed in the ESI.† The atomic charge distribution and second-order perturbation energies (E(2)) of the donor–acceptor orbitals involved in the hydrogen bond (H-bond) interaction are calculated using natural bond orbital (NBO) analysis.82 Only doublet electronic states are considered.674. Results and discussion
The IRPD spectra of Pym+-(N2)n=1–2, C3H3N+, C3H3N+-L, and C2H2+-L (L = Ar/N2) recorded between 2900 and 3550 cm−1 are summarized in Fig. 1. The positions, widths, and vibrational and isomer assignments of the experimental transitions (labeled A–F) are listed in Tables 1–3. The investigated spectral range covering the NH and CH stretch fundamentals (νNH/CH) is diagnostic for identifying the contributing isomers along with their competing ligand binding sites. The free and bound XH stretch modes (X = N/C) are classified as νfXH and νbXH, respectively. The IRPD spectra of Pym+-(N2)n=1–2 reveal the vibrational properties of Pym+ in the 3 μm range, yielding all νCH frequencies of Pym+. The broad IR spectrum of the C3H3N+ monomer becomes narrower after tagging with an inert gas (Ar/N2) through the substantial reduction of both the internal energy of the ions and the lowest dissociation threshold. At first, we discuss the structural, vibrational, and electronic parameters of Pym, Pym+, and Pym+-(N2)n and compare the effects of ionization and cluster formation. Subsequently, similar analyses are performed for C3H3N+ and C2H2+ and their Ar and N2 clusters. | ||
| Fig. 1 IRPD spectra of Pym+-(N2)n with n = 1–2, C3H3N+-Ln (n = 0–1), and C2H2+-L clusters (L = Ar/N2) recorded between 2900 and 3550 cm−1 covering the CH and NH stretch range. The positions, widths, and vibrational and isomer assignments of the transitions observed (A–F) are listed in Tables 1–3. The IRPD spectra of the clusters are monitored in the monomer ion fragment channel, while the spectrum of bare C3H3N+ is recorded in the HCN loss channel. | ||
| Exp. (cm−1) | Calc.a (cm−1) | Vibration | Isomer | |
|---|---|---|---|---|
| a Vibrational symmetry species and IR intensities (in km mol−1) are listed in parentheses. b Ref. 9, 88 and 91. c Extrapolated from the Pym+-(N2)n data. | ||||
| Pym | 3074b | 3076 (9, a1) | ν C5H (ν13) | Pym |
| 3050b | 3043 (13, a1) | ν C2H (ν20a) | Pym | |
| 3039b | 3032 (18, b2) | ν C4H/C6H(as) (ν7b) | Pym | |
| 3002b | 3029 (14, a1) | ν C4H/C6H(s) (ν2) | Pym | |
| Pym+ | 3124 ± 2c | 3117 (20, a1) | ν C2H (ν13) | Pym+ |
| 3112 ± 2c | 3103 (15, a1) | ν C5H (ν20a) | Pym+ | |
| 3045 ± 2c | 3031 (49, b2) | ν C4H/C6H(as) (ν7b) | Pym+ | |
| 3045 ± 2c | 3031 (13, a1) | ν C4H/C6H(s) (ν2) | Pym+ | |
| Pym+-N2 | A1 3125 (20) | 3119 (18, a′), 3117 (18, a′) | ν fC2H | Pym+-N2(π), Pym+-N2(C4) |
| A2 3111 (16) | 3103 (14, a′), 3103 (15, a′) | ν fC5H | Pym+-N2(π), Pym+-N2(C4) | |
| B 3046 (15) | 3032 (45, a′′), 3032 (29, a′) | ν fC4H/C6H(as), νfC6H | Pym+-N2(π), Pym+-N2(C4) | |
| 3032 (12, a′) | ν fC4H/C6H(s) | Pym+-N2(π) | ||
| C 3001 (20) | 3008 (200, a′) | ν bC4H | Pym+-N2(C4) | |
| Pym+-(N2)2 | A1 3128 (8) | 3120 (17, a1), 3119 (17) | ν fC2H | Pym+-(N2)2(π/π), Pym+-N2(C4/π) |
| A2 3112 (10) | 3103 (13, a1), 3103 (14) | ν fC5H | Pym+-(N2)2(π/π), Pym+-N2(C4/π) | |
| B 3048 (16) | 3033 (41, b2), 3033 (27) | ν fC4H/C6H(as), νfC6H | Pym+-(N2)2(π/π), Pym+-N2(C4/π) | |
| 3033 (11, a1) | ν fC4H/C6H(s) | Pym+-(N2)2(π/π) | ||
| C 3003 (14) | 3011 (187) | ν bC4H | Pym+-N2(C4/π) | |
| 3011 (79, a1), 3010 (301, b2) | ν bC4H/C6H(s/a) | Pym+-N2(C4/C6) | ||
| Exp. (cm−1) | Calc.a (cm−1) | Vibration | Isomer | |
|---|---|---|---|---|
| a Vibrational symmetry species and IR intensities (in km mol−1) are listed in parentheses. | ||||
| C3H3N+ | D 3160 (200) | 3229 (334, a′) | ν fCH | 5 |
| 3155 (46, a′) | ν fCH | 5 | ||
| 3040 (39, a′) | ν fCH | 5 | ||
| 3231 (333, a′) | ν fCH | 4 | ||
| 3149 (62, a′) | ν fCH | 4 | ||
| 3000 (30, a′) | ν fCH | 4 | ||
| C3H3N+-Ar | E 3356 (61) | 3362 (2370, a1) | ν bNH | 1-Ar |
| 3347 (2119, a′) | ν bNH | 3-Ar | ||
| 3347 (2131, a′) | ν bNH | 2-Ar | ||
| D1 3232 (16) | 3235 (313, a′) | ν fCH | 5-Ar(π) | |
| D2 3170 (26) | 3151 (785, a′) | ν bCH | 4-Ar | |
| 3148 (768, a′) | ν bCH | 5-Ar(H) | ||
| 3150 (53, a′) | ν fCH | 4-Ar | ||
| 3156 (47, a′) | ν fCH | 5-Ar(H) | ||
| 3157 (43, a′) | ν fCH | 5-Ar(π) | ||
| C3H3N+-N2 | D3 3170 (40) | 3150 (61, a′) | ν fCH | 4-N2 |
| 3156 (43, a′) | ν fCH | 5-N2 | ||
| E 3118 (60) | 3144 (3357, a1) | ν bNH | 1-N2 | |
| 3133 (652, a′) | ν bNH/νfCH(s) | 3-N2 | ||
| 3131 (2348, a′) | ν bNH/νfCH(a) | 3-N2 | ||
| 3132 (2415, a′) | ν bNH | 2-N2 | ||
| 3128 (626, a′) | ν fCH | 2-N2 | ||
| D2 3090 (55) | 3055 (1076, a′) | ν bCH | 4-N2 | |
| 3053 (959, a′) | ν bCH | 5-N2 | ||
| C3H3N+-(N2)2 | D3 3171 (13) | ν fCH | 4-(N2)2 | |
| ν fCH | 5-(N2)2 | |||
| E 3132 (30) | ν bNH | 1-(N2)2 | ||
| ν bNH/νfCH(s) | 3-(N2)2 | |||
| ν bNH/νfCH(a) | 3-(N2)2 | |||
| ν bNH | 2-(N2)2 | |||
| ν fCH | 2-(N2)2 | |||
| D2 3110 (25) | ν bCH | 4-(N2)2 | ||
| ν bCH | 5-(N2)2 | |||
| C3H3N+-(N2)3 | D3 3172 (14) | ν fCH | 4-(N2)3 | |
| ν fCH | 5-(N2)3 | |||
| E 3141 (16) | ν bNH | 1-(N2)3 | ||
| ν bNH/νfCH(s) | 3-(N2)3 | |||
| ν bNH/νfCH(a) | 3-(N2)3 | |||
| ν bNH | 2-(N2)3 | |||
| D2 3131 (24) | ν fCH | 2-(N2)3 | ||
| ν bCH | 4-(N2)3 | |||
| ν bCH | 5-(N2)3 | |||
| Exp. (cm−1) | Calc.a (cm−1) | Vibration | Isomer | |
|---|---|---|---|---|
| a Vibrational symmetry species and IR intensities (in km mol−1) are listed in parentheses. b Ref. 80 and 98. | ||||
| C2H2+ | ∼3234b | 3225 (0, σg) | ν 1 | HCCH+ |
| 3136b | 3123 (443, σu) | ν 3 | HCCH+ | |
| C2H2+-Ar | F1 3234 (15)b | 3258 (2, a′) | ν 1 | HCCH+-Ar(π) |
| F2 3153 (77) | 3160 (373, a′) | ν 3 | HCCH+-Ar(π) | |
| C2H2+-N2 | F2 3165 (30) | 3166 (358, a′) | ν 3 | HCCH+-N2(π) |
| F3 3035 (68) | 2962 (1063, σ) | ν bCH | HCCH+-N2(H) | |
4.1 Pym, Pym+, and Pym+-(N2)n=1–2 clusters
Pym has a planar equilibrium structure with C2v symmetry in its 1A1 ground electronic state (Fig. 2), as revealed from electron diffraction and microwave, IR, and Raman spectroscopy.9,83–88 Our calculated geometric and vibrational parameters agree well with available experimental data,83–87 demonstrating the reliability of our chosen computational approach (Table 1 and Table S1, ESI†). Information about the electronic and vibrational properties of the ground electronic state of the Pym+ cation comes from photoelectron and photoionisation spectroscopy, and calculations.49,59,89–93 Ionization of Pym into its 2B2 cation ground electronic state (C2v) occurs by removal of an electron from the highest occupied in-plane b2 orbital of Pym, which is a linear combination of the nonbonding lone pair orbitals of the N atoms and the bonding σ orbitals of the aromatic skeleton (Fig. S3, ESI†). In line with the shape of this HOMO, the N1–C2 (N3–C2) and N1–C6 (N3–C4) bonds elongate (by 11 and 18 mÅ), while the C4–C5 (C5–C6) bonds contract (by 6 mÅ). As expected, in contrast to the significant deformation of the heavy atom skeleton, the C–H bonds are much less effected by ionization, with changes of −1, +2, and −4 mÅ for C5–H, C4/6-H, and C2–H, respectively. As a result of these modest geometry changes, the previous photoionization spectra do not reveal any experimental information about the νCH frequencies of Pym+ due to vanishing Franck–Condon factors.91–93 Our computations predict that ionization slightly contracts the C–H bonds leading to an increase in average νCH frequency (ΔνCH = +74, +27, −1, +2 cm−1 for νC2H, νC5H, νC4H/C6H(a), νC4H/C6H(s)) and a slight increase in IR intensity (Fig. 3). The predicted adiabatic ionization energy is comparable to the previously measured value (IE = 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 568 vs. 75261 cm−1), and also our computed frequencies agree well with available measured ones (Table S2, ESI†).91–93 The positive charge resides mainly on the peripheral hydrogens (Fig. S4, ESI†). All efforts to record an IRPD spectrum of bare Pym+ in the HCN loss channel failed because of (i) high metastable decay background, which exceeds by far the achieved IRPD yield and/or (ii) the high energy required for dissociation (∼200–300 kJ mol−1).51,57–59,68
568 vs. 75261 cm−1), and also our computed frequencies agree well with available measured ones (Table S2, ESI†).91–93 The positive charge resides mainly on the peripheral hydrogens (Fig. S4, ESI†). All efforts to record an IRPD spectrum of bare Pym+ in the HCN loss channel failed because of (i) high metastable decay background, which exceeds by far the achieved IRPD yield and/or (ii) the high energy required for dissociation (∼200–300 kJ mol−1).51,57–59,68
 | ||
| Fig. 2 Optimized structures of Pym, Pym+, and most stable isomers of Pym+-(N2)1–2 calculated at the B3LYP-D3/aug-cc-pVTZ level. Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parentheses correspond to relative energies in cm−1 (E0). For Pym, the atomic numbering according to IUPAC is given as well. | ||
 | ||
| Fig. 3 Comparison of experimental IRPD spectra of Pym+-(N2)1–2 with linear IR absorption spectra of Pym, Pym+, and most stable Pym+-(N2)1–2 isomers calculated at the B3LYP-D3/aug-cc-pVTZ level (Fig. 2 and Table 1). | ||
Cluster formation of Pym+ with N2 occurs mainly through two different ligand binding motifs, namely π-bonding to the aromatic ring and linear CH⋯N2 ionic H-bonding (Fig. 2). The latter class leads to three different planar isomers (Cs or C2v) because of the three nonequivalent CH groups. The highest D0 value of the Pym+-N2(π) dimer (D0 = 868 cm−1, Cs) results mostly from the strong charge-quadrupole and charge-induced dipole interaction between Pym+ and N2, along with minor dispersion forces involving the highly polarizable aromatic π electrons. The π-attachment negligibly affects the ring structure, and thus the νCH frequencies probed in this work remain nearly unchanged from those of bare Pym+ (ΔνCH ≤ 2 cm−1, Table 1 and Fig. 3). The slight frequency increase arises from the small noncooperative effect induced by π-bonded N2 and agrees with the minor computed reduction in the C–H bond lengths (ΔrCH ≤ 0.2 mÅ). Among the three nonequivalent H-bonded isomers, the CH⋯N2 H-bond is the strongest in the Pym+-N2(C4) dimer (D0 = 784 cm−1), which is rationalized by the highest acidity of the corresponding CH group, as is evidenced from its longest C–H bond and highest atomic charge (0.275 e) in bare Pym+ (Fig. 2 and Fig. S4, ESI†). This relatively strong CH⋯N2 H-bond lowers the bound νbC4H stretch frequency of the C4H donor (by 23 cm−1) and enhances its IR intensity by a factor 7 to 200 km mol−1 (Fig. 3), while the properties of the three remaining free CH groups remain nearly unaffected (ΔνfCH ≤1 cm−1). Similar results are also predicted for the other two less stable H-bonded isomers, Pym+-N2(C2) and Pym+-N2(C5), with D0 = 775 and 690 cm−1 (Fig. 2 and 3). For each of the two isomers, the frequency of the intense νbCH mode is calculated near 3100 cm−1 (νbC2H/C5H = 3100/3097 cm−1, −ΔνbC2H/C5H = 17/6 cm−1) because of its higher intrinsic frequency, resulting from its inherently weaker acidic nature (νC2H/C5H = 3117/3103 cm−1 for Pym+).
The experimental Pym+-N2 spectrum in Fig. 3 features a clear doublet A1 and A2 at 3125 and 3111 cm−1, along with an intense band B at 3046 cm−1 and a weak transition C at 3001 cm−1. The relative intensities and positions of the first three bands show good correspondence with the spectrum computed for the most stable Pym+-N2(π) isomer (3119, 3103, 3032 cm−1), with deviations of less than 15 cm−1 (which is of the order of the widths of the bands). The relative intensities of the bands A1, A2, and B bands quantitatively match the predicted pattern, indicating a dominant contribution of the most stable π-bonded isomer. Similar agreement is not observed for the two H-bound Pym+-N2(C5) and Pym+-N2(C2) local minima. Particularly, their predicted intense νbC2H/C5H modes near 3100 cm−1 are not consistent with the intensity observed for band A2, suggesting no (or at most very low) population of these two isomers. This is further corroborated by their smaller binding energies (D0 = 775 and 690 cm−1). Therefore, we will not consider these two structures for further cluster growth. The remaining small band C at 3001 cm−1 agrees well with the νbC4H mode of Pym+-N2(C4) calculated at 3008 cm−1, the unique spectroscopic signature of this most stable H-bonded isomer. Its other predicted modes overlap with the CH stretch bands A1, A2, and B predominantly assigned to the π-bonded global minimum. The substantial discrepancy between the experimental and calculated intensity ratios of the CH stretch bands demonstrates the significantly smaller population of Pym+-N2(C4) compared to Pym+-N2(π), which is in line with their thermochemical data (D0 = 784 vs. 868 cm−1). Taking the relative intensity of bands C and B (1:6) along with the computed IR oscillator strengths (4:1) yields a crude estimate for the abundance ratio of 1:24 (or 4%) for the C4 and π isomers.
Attachment of a second N2 ligand to Pym+-N2(π) on the opposite side of the aromatic ring leads to the most stable Pym+-(N2)2(π/π) trimer (C2v) with D0 = 1724 cm−1 (Fig. 2). The two N2 ligands are equivalent and their bonding is only slightly weaker than in the dimer, because effective shielding of the aromatic ring causes only small nonadditive noncooperative effects. Similar to the n = 1 case, the second π-addition has a negligible influence on the C–H bond properties of Pym+. The νCH modes and their IR intensity remain almost unchanged (ΔνCH ≤ 3 cm−1). We consider in Fig. 2 two further isomers resulting from attachment of the second N2 ligand to the much less abundant Pym+-N2(C4) dimer, namely Pym+-(N2)2(C4/π) and Pym+-(N2)2(C4/C6). The π-bonded N2 in the first isomer does not perturb the Pym+-N2(C4) dimer (ΔνCH ≤ 3 cm−1). On the other hand, in Pym+-(N2)2(C4/C6) the C6–H bond slightly elongates upon H-bonding (ΔrCH = 1.3 mÅ). The corresponding symmetric and antisymmetric νbC4H/C6H modes are calculated at 3011 and 3010 cm−1, respectively, with the latter being more intense (ICH = 79 vs. 301 km mol−1). This trimer with two CH⋯N2 H-bonds is slightly less stable than Pym+-(N2)2(C4/π) by ΔD0 = 98 cm−1, resulting from the higher π-affinity of the N2 ligand (as compared to H-bonding).
The appearance of the n = 2 spectrum is very similar to the dimer spectrum with respect to both frequency and relative intensity of the four bands A–C, with frequency shifts of less than 3 cm−1 upon attachment of the second N2 ligand (Fig. 3). However, the doublet structure now appears as two clearly separated peaks at 3128 (A1) and 3112 (A2) cm−1, due to colder clusters and smaller rotational constants leading to narrower transitions for the larger cluster. The intense band B at 3048 cm−1 and the weak transition C at 3003 cm−1 remain almost unshifted. Similar to the dimer case, the predicted νCH modes of the Pym+-(N2)2(π/π) trimer at 3120, 3103, and 3033 cm−1 nicely agree with the three dominant bands A1, A2, and B, respectively. The calculation satisfactorily reproduces the slight experimental blue shifts of 1–3 cm−1 (Table 1), which originate from the noncooperativity induced by the π-bound N2 ligand. Thus, the most stable Pym+-(N2)2(π/π) isomer dominates the experimental spectrum. In analogy to the dimer spectrum, band C (3003 cm−1) in the trimer spectrum is explained by the νbCH mode(s) of both the (C4/π) and (C4/C6) isomers. From the relative intensities of bands B and C, the contribution of these two local minima is estimated to be below 5% of the abundance of the most stable calculated Pym+-(N2)2(π/π) global minimum. From the π-bonded isomers of Pym+-(N2)n, we can reliably and accurately estimate the four CH stretch frequencies of bare Pym+ as 3124 ± 2 (νC2H), 3112 ± 2 (νC5H), 3045 ± 2 (νC4H/6H(s)), and 3045 ± 2 cm−1 (νC4H/6H(a)) by taking into account the predicted small complexation shifts. The IRPD signal of Pym+-(N2)2 is observed exclusively in the Pym+ fragment channel. This result is consistent with the total binding energy of D0 = 1724 cm−1, which is much smaller than the energy of a single photon absorbed in the CH stretch range (νIR > 3000 cm−1).
4.2 C3H3N+ monomer (m/z = 53)
In our EI source, Pym+ mainly disintegrates into the C3H3N+ fragment (m/z 53), which further dissociates into the smaller C2H2+ fragment (m/z 26). This is demonstrated by the mass spectrum of the EI source, as well as CID spectra of the mass-selected Pym+ and C3H3N+ radical cations, both of which show exclusive loss of HCN (Fig. S1 and S2, ESI†). Mass spectrometric studies on isotopically-labelled Pym indicate that H4C4N3 is the by far dominant neutral fragment.60 We consider all conceivable isomers and IR spectra of the C3H3N+ ion in Fig. 4 and Fig. S5 (1–13) (ESI†), which were suggested as the most probable isomers by previous computational and thermochemical studies.58,61,62,64,67,68,94 These show a considerable structural variety and spread in energy. For example, a systematic global optimization search yielded 17 isomers within 200 kJ mol−1.67 Consistent with the previous reports,58,61,62,64,67,94 the H2CCCNH+ isomer (1, H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCN+H, C2v, 2B2) with a linear CCCN backbone is calculated to be by far the most stable C3H3N+ structure, with an energy gap of ∼50 kJ mol−1 to the other isomers. This ion features a free NH group and its characteristic νfNH mode calculated at 3545 cm−1 dominates its IR spectrum in the CH and NH stretch ranges (INH = 1022 km mol−1). The next stable C3H3N+ monomers are the cis/trans isomers of HCCHCNH+ (3/2, HCC(H)–C+NH, Cs, 2A′) at E0 = 53/49 kJ mol−1, which also have a CCCN backbone and a terminal NH group with similar NH stretch properties (νfNH = 3538 cm−1). Further isomers with a CCCN backbone and terminal NH group are rather unstable (11/12, cis-/trans-HCCCHNH+, H–CC–C+(H)NH, E0 > 130 kJ mol−1, Cs, 2A′, Fig. S5, ESI†), and were not considered in all previous systematic calculations,58,61,62,67,94 with one exception.64 Acrylonitrile (6, H2CCHCN+, H2CC+(H)–CN, Cs, 2A′′, E0 = 74 kJ mol−1), the initially proposed candidate of the elusive C3H3N+ structure,56 also exhibits a bent CCCN backbone but lacks a NH group. Its characteristic CH stretch bands appear as three weak transitions between 3000 and 3140 cm−1 (ICH ∼ 30 km mol−1). Two further high-energy nonplanar structures with a cyclic CCC ring and a NH group are also computed, namely HCCHCNH+(r) (9, C1, 2A) and HCCCHNH+(b) (13, Cs, 2A′) at 83 and 299 kJ mol−1. The IR spectrum of a cyclic CCC structure with a NH2 group (10, CCHCHNH2+, Cs, 2A′) at 103 kJ mol−1 is characterized by two NH stretch modes at 3335 and 3395 cm−1, respectively. Two isomers with a (nearly) linear CCNC backbone, H2CNCCH+ (7, H2CN+–CCH, C2v, 2B2) and H2CCNCH+ (8, H2CC+–NCH, Cs, 2A′), are calculated at relatively low energy (E0 = 61 and 67 kJ mol−1). Each of them exhibits a characteristic intense νfCH mode at 3246 (ICH = 187 km mol−1) and 3138 cm−1 (ICH = 269 km mol−1), respectively. We further calculate the cis and trans forms of the kinetically favored HCCHNCH+ isomer (5 and 4, HCC(H)–NC+H, E0 = 107 and 102 kJ mol−1, Cs, 2A′′).58,60,67,68 Unlike all other isomers, which demand a significant activation barrier for their formation, 4 and 5 exhibit a suitable CCNC backbone that facilitates fragmentation of the Pym+ ring via simple neutral HCN elimination.58,60,67,68 Their intense νfCH modes are predicted around 3230 cm−1 (ICH ∼ 330 km mol−1).
CCN+H, C2v, 2B2) with a linear CCCN backbone is calculated to be by far the most stable C3H3N+ structure, with an energy gap of ∼50 kJ mol−1 to the other isomers. This ion features a free NH group and its characteristic νfNH mode calculated at 3545 cm−1 dominates its IR spectrum in the CH and NH stretch ranges (INH = 1022 km mol−1). The next stable C3H3N+ monomers are the cis/trans isomers of HCCHCNH+ (3/2, HCC(H)–C+NH, Cs, 2A′) at E0 = 53/49 kJ mol−1, which also have a CCCN backbone and a terminal NH group with similar NH stretch properties (νfNH = 3538 cm−1). Further isomers with a CCCN backbone and terminal NH group are rather unstable (11/12, cis-/trans-HCCCHNH+, H–CC–C+(H)NH, E0 > 130 kJ mol−1, Cs, 2A′, Fig. S5, ESI†), and were not considered in all previous systematic calculations,58,61,62,67,94 with one exception.64 Acrylonitrile (6, H2CCHCN+, H2CC+(H)–CN, Cs, 2A′′, E0 = 74 kJ mol−1), the initially proposed candidate of the elusive C3H3N+ structure,56 also exhibits a bent CCCN backbone but lacks a NH group. Its characteristic CH stretch bands appear as three weak transitions between 3000 and 3140 cm−1 (ICH ∼ 30 km mol−1). Two further high-energy nonplanar structures with a cyclic CCC ring and a NH group are also computed, namely HCCHCNH+(r) (9, C1, 2A) and HCCCHNH+(b) (13, Cs, 2A′) at 83 and 299 kJ mol−1. The IR spectrum of a cyclic CCC structure with a NH2 group (10, CCHCHNH2+, Cs, 2A′) at 103 kJ mol−1 is characterized by two NH stretch modes at 3335 and 3395 cm−1, respectively. Two isomers with a (nearly) linear CCNC backbone, H2CNCCH+ (7, H2CN+–CCH, C2v, 2B2) and H2CCNCH+ (8, H2CC+–NCH, Cs, 2A′), are calculated at relatively low energy (E0 = 61 and 67 kJ mol−1). Each of them exhibits a characteristic intense νfCH mode at 3246 (ICH = 187 km mol−1) and 3138 cm−1 (ICH = 269 km mol−1), respectively. We further calculate the cis and trans forms of the kinetically favored HCCHNCH+ isomer (5 and 4, HCC(H)–NC+H, E0 = 107 and 102 kJ mol−1, Cs, 2A′′).58,60,67,68 Unlike all other isomers, which demand a significant activation barrier for their formation, 4 and 5 exhibit a suitable CCNC backbone that facilitates fragmentation of the Pym+ ring via simple neutral HCN elimination.58,60,67,68 Their intense νfCH modes are predicted around 3230 cm−1 (ICH ∼ 330 km mol−1).
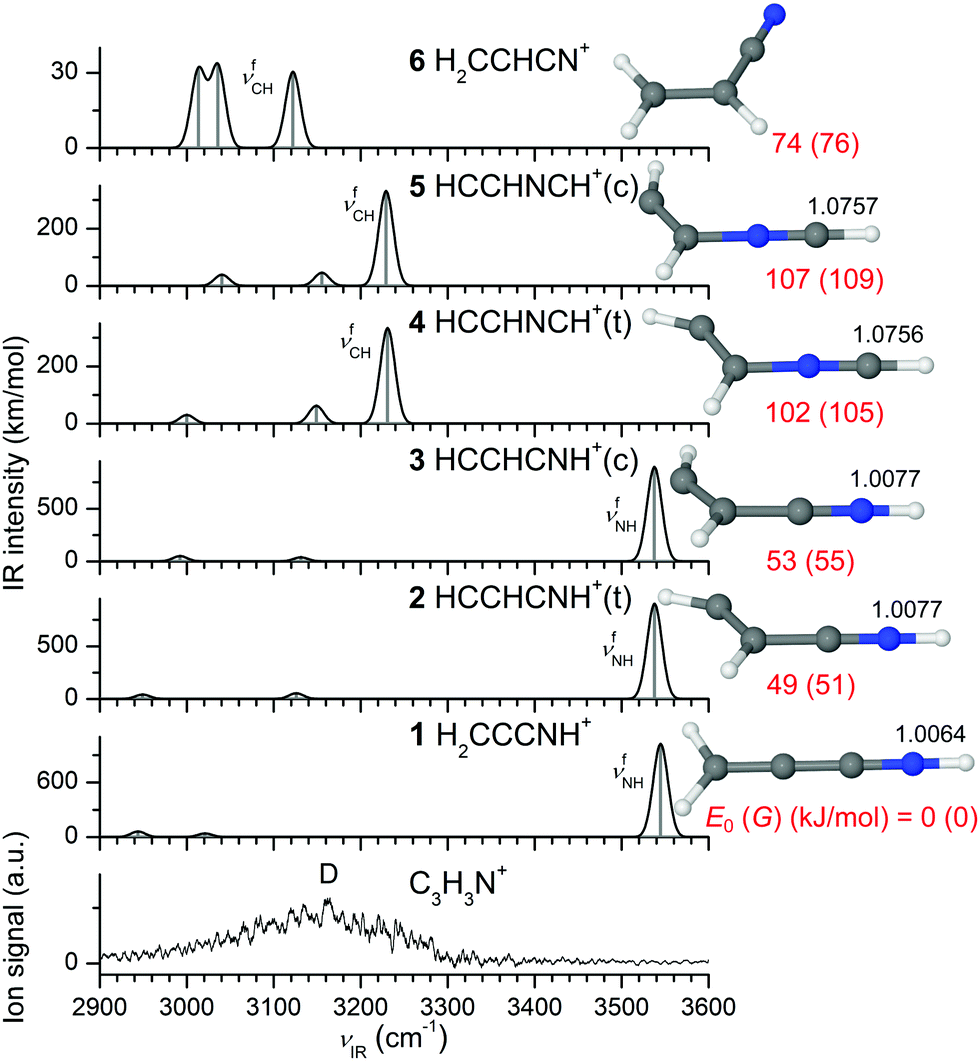 | ||
| Fig. 4 Comparison of experimental IRPD spectrum of C3H3N+ recorded in the HCN loss channel with linear IR absorption spectra of various C3H3N+ isomers along with the corresponding optimized structures calculated at the B3LYP-D3/aug-cc-pVTZ level. The relative energies (E0), free energies (G) in parentheses and bond lengths are given in kJ mol−1 and Å, respectively. A comparison with spectra computed for other C3H3N+ isomers is available in Fig. S5 (ESI†). | ||
The IRPD spectrum of bare C3H3N+ recorded in the C2H2+ fragment ion channel (HCN loss, Fig. 4) is dominated by a broad transition (D) ranging from 2950 to 3290 cm−1 and peaking near 3160 cm−1 (FWHM ∼ 200 cm−1). The signal to noise ratio is limited because of the large background arising from metastable decay of hot ions, which keep high internal energy after Pym+ → C3H3N+ fragmentation due to insufficient cooling in the supersonic expansion after HCN elimination. The broad and unstructured spectrum results from the substantial internal energy of the bare C3H3N+ ion, which becomes much narrower upon tagging with an inert Ar/N2 ligand (Fig. 1) leading to the observation of much colder ions. The unfocussed IR laser beam allows only for single-photon absorption processes, so that only ions with energy around 3000 cm−1 below the dissociation threshold can be detected upon IRPD. Because HCN loss requires typically much higher energies for all C3H3N+ ions considered,58,61,62,67,94 only ions with very high internal energy are probed in the recorded IRPD spectrum, leading to the observation of hot bands and sequence transitions giving rise to the broad band contour. The calculated free νfNH frequencies of all isomers with a NH group (>3300 cm−1 for 1–3 and 9–13) appear to be too high to explain the observed hot band transitions contributing to band D, suggesting their absence and/or inefficient photofragmentation resulting from high dissociation thresholds. However, the intense νfCH modes predicted at 3230 cm−1 for the isomers 4 and 5 are within the experimental absorption range of band D centered around 3160 cm−1. The difference in frequency (∼70 cm−1) can readily by rationalized by anharmonic coupling typical for hot band transitions, which arises from the elevated internal energy of the C3H3N+ fragments populating vibrational excited states with high quantum numbers. Such anharmonic coupling (diagonal and off-diagonal) results in red shifts of the transitions compared to the fundamentals predicted by the calculations. Thus, isomers 4 and 5 are plausible candidates to explain the experimental C3H3N+ spectrum despite their relatively high E0 values (>100 kJ mol−1). Similarly, the intense νfCH modes of 7 and 8 predicted at 3246 and 3138 cm−1 match the measured IRPD spectrum. The computed νfCH modes of 6 also lie within the observed spectral range, which suggests that acrylonitrile+ could also contribute to the monomer spectrum, although their fundamentals are calculated close to the red edge of transition D and their IR oscillator strengths are weaker by one order of magnitude than those of the other potential candidates. Thus, from the warm C3H3N+ spectrum it is rather difficult to unambiguously determine the dominant contribution of the observed isomers that do not have a NH group. The approach of inert gas tagging discussed in the next section reduces the temperature of the probed ions, and thus increases the spectral resolution of the IRPD spectrum and reduces the effective dissociation threshold, which is required for a more reliable and more definitive identification of the contributing isomers.
4.3 C3H3N+-L (L = Ar/N2) dimers
Various Ar and N2 ligand binding motifs of the C3H3N+ monomers identified as potential candidates in Section 4.2 are displayed in Fig. 5 and 6 (along with their IR spectra), while others are presented in Fig. S6 and S7 (ESI†). The isomers with a NH group (1–3, 9–13) strongly prefer the formation of a NH⋯L H-bond of L to the acidic NH group because of the high partial charge on the NH proton (e.g., q = 0.498 e for 1, Fig. S4, ESI†). In contrast, in the 4-L and 5-L dimers, the ligand forms a CH⋯L ionic H-bond to the strongly positively charged proton of the terminal CH group adjacent to N (e.g., q = 0.718 e for 4). For 5-Ar, a π-bound structure is also calculated, in which the Ar ligand interacts weakly with both the CN π-cloud and the adjacent CH group (as evidenced from the NBO calculations, Fig. S8, ESI†). At this binding site, the Ar ligand has no significant influence on the monomer geometry. Thus, its predicted intense νCH frequency at 3235 cm−1 remains comparable to that of bare 5. A similar π-bonding motif is also obtained for 6-Ar. However, attempts to optimize such a π-bound structure with N2 fail because of the strong preference of N2 for H-bonding to acidic CH/NH protons over π-bonding. The XH⋯L ionic H-bonds (X = N/C, L = Ar/N2) significantly perturb the XH proton donor groups and leads to an elongation of the X–H bond along with a reduction of the corresponding νbXH frequency. For the most stable H-bonded 1-Ar/N2 clusters with a linear NH⋯L H-bond (C2v), the red shift is predicted as −ΔνNH = 183/401 cm−1. The higher proton affinity of N2 as compared to Ar (PA = 494 vs. 369 kJ mol−1)95 causes a larger red shift resulting from the stronger and shorter NH⋯L H-bond (D0 = 1950 vs. 928 cm−1, RNH⋯L = 1.849 vs. 2.259 Å for L = N2 and Ar). This view is further corroborated by the higher E(2) energy of the NH⋯N2 H-bond as compared to the NH⋯Ar bond (E(2) = 88.2 vs. 35.1 kJ mol−1, Fig. S8, ESI†). The NH⋯L bonds in 2/3-L are similar to those in 1-L, because of their similar N–H bond properties. Similar results are also obtained for the CH⋯L H-bonds in 4/5-L(H) with L = Ar/N2. For these C3H3N+ isomers, the Ar ligand again perturbs the H-bonded CH group less strongly than N2, which results in a smaller D0 value for Ar as compared to N2 (e.g., D0 = 815 vs. 1500 cm−1 for 5) and a longer H-bond (RCH⋯L = 2.436 vs. 2.047 Å). As a result, the red shift of the Ar-bound νbCH mode is smaller than for N2 (−ΔνbCH = 81 vs. 176 cm−1). The E(2) values also support this trend (E(2) = 26.2 vs. 52.0 kJ mol−1). Analogous results are obtained for the H-bonded 4-L dimers with L = Ar/N2. However, in both cases, 4/5-L, the CH proton donor group is less strongly perturbed by H-bonding than the more acidic NH groups in isomers with NH⋯L H-bonds. The relatively low-energy isomers 7/8-Ar (E0 ∼ 70 kJ mol−1) have their intense νbCH modes at 3211 and 3081 cm−1, respectively, and these frequencies are further red shifted down to 3157 and 3015 cm−1 for the more strongly bonded N2 clusters. As usual, the N2-bound CH stretch modes show higher IR intensity because of its larger H-bond affinity than the Ar-bound νbCH modes reflecting their higher D0 value. In general, charge transfer from C3H3N+ to the ligands L is small, as expected for the weak intermolecular H-bonds (e.g., Δq ≤ 0.06 e for 1/2/4-N2, Fig. S4, ESI†).
 | ||
| Fig. 5 Comparison of experimental IRPD spectrum of C3H3N+-Ar with linear IR absorption spectra of various C3H3N+-Ar isomers along with the corresponding optimized structures calculated at the B3LYP-D3/aug-cc-pVTZ level (Table 2). Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parentheses correspond to relative energies in kJ mol−1 (E0). A comparison with spectra computed for other C3H3N+-Ar isomers is available in Fig. S6 (ESI†). | ||
 | ||
| Fig. 6 Comparison of experimental IRPD spectra of C3H3N+-(N2)1–3 recorded in the C3H3N+ channel with linear IR absorption spectra of various C3H3N+-N2 isomers along with the corresponding optimized structures calculated at the B3LYP-D3/aug-cc-pVTZ level (Table 2). Binding energies (D0) and bond lengths are given in cm−1 and Å, respectively. Numbers in parentheses correspond to relative energies in kJ mol−1 (E0). A comparison with spectra computed for other C3H3N+-N2 isomers is available in Fig. S7 (ESI†). | ||
Unlike the broad and unresolved C3H3N+ monomer spectrum, the C3H3N+-L dimer spectra with both L = Ar and N2 exhibit several discernible narrow features because of colder ions. The C3H3N+-Ar spectrum displays three main bands E, D1, and D2 at 3344, 3232, and 3170 cm−1 and is compared in Fig. 5 and Fig. S6 (ESI†) to spectra computed for potential cluster structures. The satellite bands to the blue of the transitions E and D2 are characteristic of proton donor stretch vibrations involved in H-bonding,30,79,96,97 while the rather symmetric band D1 is indicative of a free XH stretch oscillator. Although band E agrees with the νbNH mode predicted for the most stable 1–3-Ar(H) dimers (3362 and 3347 cm−1), their strong calculated IR intensities (>2000 km mol−1) are not reproduced by the measured spectrum. This discrepancy indicates at most a minor contribution of these C3H3N+ isomers to the measured spectrum despite their superior stability. The appearance of this band in the spectrum of the Ar-tagged ion reinforces the above hypothesis of inefficient photofragmentation of the bare 1–3 ions that leads to the absence of their νfNH bands in the monomer spectrum (Section 4.2). This observation illustrates the importance of tagging in order to reduce the fragmentation threshold and to detect all present C3H3N+ isomers (independent of their dissociation threshold). Band D2 is assigned to the intense νbCH modes of the 4/5-Ar(H) dimers calculated near 3150 cm−1 and band D1 matches the free intense νfCH mode predicted for the 5-Ar(π) dimer (3235 cm−1). The sequence hot band transitions of band D2 contribute to the intensity of band D1. The remaining free νfCH modes of 5-Ar(π) are not visible in the experimental spectrum due to their weak IR activity. Also, the spectra predicted for the cyclic isomers (9/10-Ar) and 8-Ar agree poorly with the experimental spectrum indicating their absence (or negligible population, Fig. S6, ESI†). Similarly, the intense νbCH mode of H2CCNCH+-Ar at 3081 cm−1 disagrees with the measured spectrum. Although the characteristic bound νbCH mode of 7-Ar at 3211 cm−1 occurs close to transition D1 at 3232 cm−1, its symmetric band contour typical for a free XH stretch band argues against this interpretation. Furthermore, the predicted νbNH modes of 11/12-Ar agree to some extent with the band D2. However, because the predicted 11/12-N2 spectra fail to explain the measured C3H3N+-N2 spectrum, we also exclude these isomers from the observed C3H3N+ ion population (Fig. S7, ESI†). There is no indication for the presence of 6-Ar, possibly due to its low Ar binding energy and the low computed IR cross sections. Thus, from the IRPD spectrum of C3H3N+-Ar, we infer a predominant population of 4/5, along with a minor contribution of 1–3. Using the observed intensity ratios of D2 and E (4:1) and the calculated IR cross sections (2:5), we can extract a crude estimate of 10:1 for the isomer population of 4/5 and 1–3 (assuming similar efficiencies for Ar tagging because of their similar Ar binding energies).
The IRPD spectrum of the N2-tagged dimers shown in Fig. 6 confirms the conclusions drawn from the spectrum of the Ar-tagged C3H3N+ ions, namely the predominant population of 4/5 and the minor abundance of 1–3. The C3H3N+-N2 spectrum also exhibits three main transitions at 3170 (D3), 3118 (E), and 3090 (D2) cm−1, which differ substantially from the C3H3N+-Ar spectrum, because of the stronger H-bonds of N2 causing larger red shifts in the proton-donor stretch modes (Fig. 1). Band E with its satellite bands to the blue is assigned to the νbNH mode of the most stable 1–3-N2 dimer predicted at 3131–3144 cm−1. Its somewhat enhanced IR intensity compared to the Ar spectrum originates from its increased oscillator strength (e.g., INH = 3354 vs. 2370 km mol−1 for 1-N2) resulting from the stronger NH⋯N2 H-bond, and the additional contribution of the sequence hot band transitions from the adjacent D2 band. The measured difference of the bands E (238 cm−1 for 1-L) agrees well with the difference in the predicted νbNH bands for the Ar and N2 clusters (218 cm−1) supporting this assignment. The blueshaded transition D2 is attributed to the νbCH mode of the 4/5-N2 isomers calculated near 3055 cm−1, whereas the symmetric D3 transition is attributed to their free νfCH mode predicted near 3155 cm−1. Again, good agreement between the difference of the bands D (80 cm−1) and the predicted νbNH bands for the Ar and N2 clusters (96 cm−1 for 1-N2) strengthens this assignment. Although the C3H3N+-N2 spectra computed for the two cyclic (9/13-N2) and 7-N2 isomers fit the experimental spectrum, we exclude them because the spectra of their Ar-tagged clusters do not match with the experiment (Fig. S6 and S7, ESI†). Similarly, we can exclude 8/10/11/12-N2 because of their mismatch with the recorded N2 spectrum. In 6-N2, the N2 ligand prefers H-bonding to the acidic CH group (while a π-bonded structure to the CN group like in 6-Ar(π) does not converge to a local minimum), leading to a strong enhancement of the corresponding νbCH mode, which is however still absent in the IRPD spectrum of C3H3N+-N2. From this observation, we estimate an upper limit for the abundance of 6 as 30% from the signal-to-noise ratio (∼45) and the IR cross section ratio (∼15). To this end, the broad spectrum of the C3H3N+ monomer is disentangled by tagging with Ar and N2, and both cold IRPD spectra can simultaneously be explained only by the presence of the 4/5 (major) and 1–3 (minor) isomers.
4.4 C3H3N+-(N2)n>1 clusters
The IRPD spectra recorded for C3H3N+-(N2)2–3 in Fig. 6 further confirm the assignment of the C3H3N+ core ions. Efforts to record similar spectra for C3H3N+-Arn≥2 failed due to inefficient cluster production, probably due to their low binding energy and insufficient cooling of the C3H3N+ ions produced by fragmentation of Pym+. No calculations are performed for the N2 clusters with n ≥ 2. With increasing number of N2 ligands the internal energy of the clusters decreases, which produces higher-resolution spectra with narrower bands. As a result, the shading of the proton-donor bands D2 and E becomes less pronounced and band D3 becomes better resolved. All three transitions D3, E, and D2 observed for n = 1 show small monotonic blue shifts as n increases to 3, which is typical for interior ion solvation.30,79,96 This noncooperative threebody effect is more pronounced for proton-donor stretch modes involved in H-bonds and relatively small for free XH stretch oscillators. In addition, the monotonic incremental blueshifts become smaller as the number of ligands increase. As a result, bands D2 and E assigned to bound νbCH of 4/5-(N2)n and bound νbNH of 1–3-(N2)n show larger incremental shifts (+20/+11 and +14/+9 cm−1 for n = 2/3) than D3 assigned to free νfCH of 4/5-(N2)n (+1/+1 cm−1). Because the appearance of the C3H3N+-(N2)n spectra is similar for n = 1–3, the vibrational and isomer assignments are the same. The fact that the IRPD spectrum of C3H3N+-(N2)3 is detected in the C3H3N+ channel implies that the total binding energy of the three N2 ligands is below the IR photon energy (D0 < νIR = 2900–3600 cm−1) for all isomers, in line with the computed binding energies for the most strongly bound first N2 ligands.4.5 C2H2+-L dimers (L = Ar/N2)
The C3H3N+ ion (m/z 53) further dissociates into the lighter C2H2+ ion (m/z 26) by HCN elimination (Fig. S2, ESI†). We probe the structure of the C2H2+ product ion by IRPD of its Ar/N2-tagged dimer (Fig. 7), which confirms it as the acetylene cation HCCH+ (H–CC–H+). Out of the two possible νCH normal modes of linear HCCH+ (D∞h) in its 2Πu ground electronic state, the symmetric one computed at ν1 = 3225 cm−1 is IR forbidden, while the lower-frequency antisymmetric mode at ν3 = 3124 cm−1 has high IR activity (ICH = 443 km mol−1). Tagging with the inert gas ligand reduces the symmetry which makes the dark ν1 mode weakly IR allowed. The ligand may either form a π-bond to the CC group, HCCH+-L(π), or a linear CH⋯L ionic H-bond to one of available CH groups, HCCH+-L(H).
 | ||
| Fig. 7 Comparison of experimental IRPD spectra of C2H2+-Ar/N2 with linear IR absorption spectra of various HCCH+-Ar/N2 isomers along with the corresponding optimized structures calculated at the B3LYP-D3/aug-cc-pVTZ level (Table 3). Binding energies (D0), bond lengths and bond angles are provided in cm−1, Å and degrees, respectively. Numbers in parentheses correspond to relative energies in cm−1 (E0). | ||
In the more stable HCCH+-L(π) global minima, the ligand is slightly displaced toward one of the CH groups, which becomes weakly distorted (Fig. 7 and Table 3). The structure computed for HCCH+-Ar(π) agrees with previous high level CCSD(T) calculations.98,99 The magnitude of this distortion is stronger for N2 than for Ar because of its higher binding energy (D0 = 2720 vs. 2338 cm−1).72,100 The predicted ν1/3 modes shift slightly to the blue by +30/+41 (N2) and +28/+36 cm−1 (Ar) due to modest charge transfer from HCCH+ to L leading to C–H bond contractions (ΔrCH ≤ −5.2 mÅ). IR activation of ν1 upon π-bonding is rather minor (ICH = 2/6 km mol−1 for Ar/N2). In the less stable linear HCCH+-L(H) local minima (D0 = 910 vs. 1635 cm−1 and RCH⋯L = 2.341 vs. 2.026 Å for Ar and N2) with C∞v symmetry, the CH proton donor group experiences a substantial elongation by H-bonding. As a result, unlike the π-bound minima, the CH stretch local modes become largely decoupled. While the free ν1 (νfCH) modes at 3202 and 3195 cm−1 for Ar and N2 become weakly IR active (I1 = 19 and 57 km mol−1), the strongly allowed H-bonded ν3 (νbCH) modes at 3057 and 2962 cm−1 are substantially redshifted and further enhanced in IR intensity (I3 = 791 vs. 1063 km mol−1).
The measured C2H2+-Ar spectrum reported in Fig. 7 features a strong transition F2 centered at 3153 cm−1 and a very weak band F1 at 3234 cm−1. Although recorded at lower resolution, this spectrum perfectly reproduces the previously published high-resolution spectrum of C2H2+-Ar assigned to π-bound HCCH+-Ar(π),98 as illustrated in Fig. S9 (ESI†). In the previous studies,98,99 C2H2+-Ar was prepared in a supersonic expansion of acetylene so that it is unambiguous that the C2H2+ ion in the Ar cluster is indeed HCCH+. This is further confirmed by the analysis of the fully rotationally resolved IR spectrum of the ν3 band.98 In agreement with the past result, band F2 is assigned to ν3 of HCCH+-Ar(π) predicted at 3160 cm−1 (and it displays the weakly-resolved Q-branches associated with this vibrational transition, Fig. S9, ESI†), while the weak band F1 corresponds to ν1 computed at 3258 cm−1. The C2H2+-Ar spectrum lacks any feature near 3050 cm−1, which indicates the absence of the less stable HCCH+-Ar(H) isomer (ν1 = 3057 cm−1). The C2H2+-N2 spectrum is reported herein for the first time. Its strong transition F2 at 3165 cm−1 matches the ν3 mode of HCCH+-N2(π) calculated at 3166 cm−1. Unlike the Ar case, the two orders of magnitude weaker ν1 mode calculated at 3260 cm−1 is not observed and below the detection limit. Instead, the C2H2+-N2 spectrum exhibits a relatively broad transition F3 peaking near 3035 cm−1, which is assigned to the ν3 mode (νbCH) of the less stable H-bonded HCCH+-N2 dimer predicted at 2962 cm−1. Apparently, the B3LYP-D3 calculations somewhat overestimate the shift for this transition. Our anharmonic calculations reveal that the difference does not result from anharmonicity but from an overestimated force constant. A similar effect is also observed in our unpublished spectra of larger C2H2+-Arn clusters, in which Ar ligands start to form H-bonds to HCCH+ for cluster sizes n ≥ 3. Analysis of the observed band intensities along with computed IR cross sections yields a population of ∼10% for the H-bonded local minimum, in line with its smaller binding energy (D0 = 1635 vs. 2720 cm−1). We can safely exclude the H2CC+ ion both from energetics101,102 and its IR spectrum (Fig. S10, ESI†).
4.6 Further discussion
The 3 μm spectra of Pym+-(N2)n=1=2 provide the first experimental information about the properties of the C–H bonds of the radical cation of the fundamental Pym molecule, a basic heterocyclic building block of nucleobases. The N2 ligands prefer π-bonding at the aromatic ring to H-bonding with the weakly acidic CH protons. This binding motif hardly influences the C–H bond parameters, with very minor changes due to noncooperative effects. Thus, the νCH frequencies of Pym+-N2(π) and Pym+-(N2)2(π/π) provide an accurate estimate of the four IR active νCH frequencies of bare Pym+ as 3124 ± 2, 3112 ± 2, 3045 ± 2, and 3045 ± 2 cm−1. These are substantially higher than those measured for neutral Pym (3074, 3050, 3039, and 3002 cm−1),9,88,91 indicating that the C–H bonds become on average stronger and less acidic upon ionization. Among the CH groups in Pym+, the acidity increases in the order C2H < C5H < C4H, in line with the C–H bond lengths and the H-bond energy with N2 (and also HCN).68 This is the reason, why a minor population of the observed Pym+-(N2)2 cluster cations have a C4H⋯N2 ionic H-bond.Previous information about the interaction of Pym+ with nonpolar and polar ligands comes from photoelectron and photoionization spectra of Pym-Ar1/2 and Pym-N2 clusters91,103 and mass spectrometric studies of Pym+-(H2O)n clusters.104 It was found that Pym+-Ar1/2 clusters have a (π) and (ππ) configurations, similar to the structures found here for the most stable structures of Pym-(N2)1/2. No conclusion was presented for the structure of Pym+-N2.91 The most stable structures computed for Pym+-(H2O)1–3 clusters have linear or bifurcated CH⋯O ionic H-bonds with computed and measured binding energies of ∼10 kcal mol−1 (∼3500 cm−1),104 indicating that the interaction of Pym+ with dipolar ligands is not only much stronger than with N2 (because of the additional charge-dipole forces) but also via a different binding motif. Results for the related imidazole+-N2 or pyrrole+-N2 cations show that N2 strongly prefers H-bonding to an acidic NH group of heterocyclic cations (if available) over π or CH bonding.105,106
All previous studies about the primary C3H3N+ fragment of Pym+ have been based on mass spectrometry, thermochemistry, and quantum chemistry, and no definitive conclusion has been derived about its structure. All probable isomers of the primary C3H3N+ fragment of Pym+ produced through elimination of HCN are considered herein to decipher its so far controversial structure by spectroscopy. By comparing the IRPD spectra of the bare hot monomer and its much colder Ar/N2-tagged clusters, the major contributions of HCCHNCH+(c/t) (4/5) and the minor population of H2CCCNH+ and HCCHCNH+(c/t) (1–3) are clearly established. In contrast, all other conceivable isomers do not agree with the measured IR spectra and are thus concluded to be below the detection limit. The minor fragment ions, 1–3, are in fact the most stable C3H3N+ isomers (E0 = 0, 49, 53 kJ mol−1), and thus their production may be expected for thermodynamic reasons. On the other hand, the predominant fragment ions, 4/5, are much higher in relative energy (E0 = 102 and 107 kJ mol−1) but their production is strongly favored for kinetic reasons. They can be formed by simple HCN elimination without any other structural rearrangement. The production of the other isomers at lower energies is strongly hindered by high activation barriers en route from the parent Pym+ monomer to the product ions. Although some of the conclusions drawn here have previously been put forward based on mass spectrometry and quantum chemistry, the approach of IR spectroscopy used herein provides for the first time an unequivocal determination of the structures of the detected C3H3N+ fragment ions, along with a crude estimate of the branching ratios. Of particular importance has been the tagging approach with weakly bonded inert ligands. First, tagging reduces the temperature leading to higher resolution IR spectra with resolved vibrational structure, which is sufficient to identify all present isomers by their fingerprints. A further effect of tagging is the reduction of the effective dissociation energy, which guarantees that all isomers can be detected by single-photon IRPD, independent of their monomer dissociation energy. This has been vital in the detection of the most stable but minor 1–3 ions. Second, recording IRPD spectra of fragment ions with different tags (here Ar and N2) further increases the selectivity of this spectroscopic approach because H-bonding of the tag modifies the IR spectrum depending on its binding affinity, and the identification of the fragment ion should be independent of the tag leading to a multiple test for cases with similar spectra for a single tag. In this way, we can exclude here the isomers 7, 9, and 11–13, because their IRPD spectra fit the IR spectra computed for either the Ar-tagged or N2-tagged clusters but not both of them. Finally, the IR spectra reported for the 4/5 and 1–3 isomers correspond to the first (IR) spectroscopic detection of these radical cations and thus provide a useful probe of the acidity of their CH and NH groups and ability to form H-bonds with (nonpolar) ligands.
The C3H3N+ ion further disintegrates by elimination of a second HCN molecule into C2H2+ (m/z 26), which was previously established as the acetylene cation (HCCH+) from thermochemical analysis. We successfully tag this product ion with Ar and N2, and the resulting IRPD spectra indeed confirm this fragment as HCCH+ for the first time by spectroscopic means. The IR spectrum of the alternative high-energy H2CC+ ion does not match. The C2H2+-Ar spectrum reproduces the HCCH+-Ar spectrum recorded previously, in which the cluster was prepared in an EI plasma expansion of HCCH. The Ar ligand binds to HCCH+ solely through π-bonding. In contrast, the HCCH+-N2 spectrum is presented for the first time and reveals also a H-bound local minimum in addition to the π-bound structure, thus directly probing the acidity of the acetylenic CH groups. The higher H-bond affinity of N2 is explained by its higher proton affinity compared to Ar (PA = 494 vs. 369 kJ mol−1)95 leading to a stronger CH⋯L ionic H-bond (D0 = 1635 vs. 910 cm−1). However, the population of HCCH+-N2(H) is only ∼10% of the HCCH+-N2(π) global minimum, in line with the computed binding energies.
5. Conclusions
We apply herein a combined IR spectroscopic and quantum chemical approach to characterize the structures and bonding of tagged clusters of Pym+ and its two most prominent fragment ions, C3H3N+ and C2H2+. The most salient results may be summarized as follows.The 3 μm spectra of Pym+-(N2)n yield the first experimental information about the frequency and thus acidity of the CH groups of this fundamental building block for nucleobases in its radical cation ground electronic state. In general, removal of the b2 electron of Pym results in an overall contraction of the C–H bonds, as illustrated by the increase in CH stretch frequencies upon ionization used as a sensitive probe of the C–H bond strength. The C4H group is the most acidic one and serves as preferred proton donor in CH⋯N2 ionic H-bonding. As a result of the reduced acidity of the CH groups, the inert N2 ligands strongly prefer π-bonding to the aromatic ring over H-bonding to the CH groups.
The controversial structures and branching ratios of the primary elusive C3H3N+ fragments (m/z 53) of Pym+ produced by elimination of HCN upon EI are unambiguously identified by spectroscopic means as HCCHNCH+(c/t) (4/5, major, kinetically favoured) and H2CCCNH+/HCCHCNH+ (1–3, minor, thermodynamically favoured) by the tagging approach. All other conceivable isomers are below the detection limit. It is demonstrated that the IRPD approach of clusters with different tags is a generally applicable approach for fragment identification, providing high resolution IR spectra with high sensitivity and selectivity. The minor 1–3 fragment ions are the most stable C3H3N+ isomers and thus expected from thermochemical arguments. The predominant 4/5 fragment ions are high in energy (E0 ∼ 100 kJ mol−1) but strongly favored by kinetic arguments because they can be formed by simple HCN elimination involving no or low barriers. Clearly, the spectroscopic approach used herein is superior compared to all previous efforts using mass spectrometry, quantum chemistry, and thermochemistry, and provides for the first time an unequivocal determination of the structures of the detected C3H3N+ fragment ions, along with a crude estimate of their branching ratio. As a further result, the first IR spectra reported for the 4/5 and 1–3 radical cations provide a direct probe of the acidity of their C–H and N–H bonds and their ability to form H-bonds with neutral ligands. Finally, the secondary C2H2+ fragment ion (m/z 26) resulting from further HCN loss of C3H3N+ is clearly identified as HCCH+ ion by spectroscopic means, confirming previous less certain mass spectrometric and thermochemical analysis. While Ar exclusively binds to the CC triple bond, N2 shows some weaker tendency to form a CH⋯N2 ionic H-bond, thereby probing the acidity of the CH groups of the HCCH+ radical in its cation ground electronic state. In conclusion, this spectroscopic study provides fundamental characterization of the Pym+ precursor and fragment ions of this important biomolecular building block and thus, in the reverse direction, paves the way for understanding the formation of large biomolecules from their smaller building blocks. In general, our spectroscopic results represent a qualitative step forward in the understanding of fragmentation of Pym+ and thus provide valuable input into astronomical models simulating the generation of precursor molecules of nucleobases in the interstellar medium. In that sense, it will be further intriguing to spectroscopically investigate similar backbone structures of other biomolecules that play a pivotal role to understand their prebiotic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Deutsche Forschungsgemeinschaft (DFG, project DO 729/3-3).References
- A. Abo-Riziq, L. Grace, E. Nir, M. Kabelac, P. Hobza and M. S. de Vries, Photochemical Selectivity in Guanine–cytosine Base-pair Structures, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 20–23 CrossRef CAS PubMed.
- A. Broo, A Theoretical Investigation of the Physical Reason for the Very Different Luminescence Properties of the Two Isomers Adenine and 2-Aminopurine, J. Phys. Chem. A, 1998, 102, 526–531 CrossRef CAS.
- M. S. de Vries and P. Hobza, Gas-Phase Spectroscopy of Biomolecular Building Blocks, Annu. Rev. Phys. Chem., 2007, 58, 585–612 CrossRef CAS PubMed.
- J.-P. Schermann, Spectroscopy and Modeling of Biomolecular Building Blocks, Elsevier, Amsterdam, 2008 Search PubMed.
- T. P. Selvam, C. R. James, P. V. Dniandev and S. K. Valzita, A Mini Review of Pyrimidine and Fused Pyrimidine Marketed Drugs, Res. Pharm., 2015, 2, 01–09 Search PubMed.
- V. Sharma, N. Chitranshi and A. K. Agarwal, Significance and Biological Importance of Pyrimidine in the Microbial World, Int. J. Med. Chem., 2014, 202784 Search PubMed.
- A. K. Jameson and E. C. Lin, Non-radiative decay from photoselected vibrational levels of pyrimidine vapor Franck–Condon effects on 1pπ* → 3ππ* intersystem crossing, Chem. Phys. Lett., 1981, 79, 326–330 CrossRef CAS.
- G. A. Pino, G. Feraud, M. Broquier, G. Grégoire, S. Soorkia, C. Dedonder and C. Jouvet, Non-radiative processes in protonated diazines, pyrimidine bases and an aromatic azine, Phys. Chem. Chem. Phys., 2016, 18, 20126–20134 RSC.
- K. K. Innes, I. G. Ross and W. R. Moomaw, Electronic states of azabenzenes and azanaphthalenes: A revised and extended critical review, J. Mol. Spectrosc., 1988, 132, 492–544 CrossRef CAS.
- I. Yamazaki, T. Murao, K. Yoshihara, M. Fujita, K. Sushida and H. Baba, Picosecond fluorescence decays from vibrational levels in the S1(n,π*) state of pyridine vapor, Chem. Phys. Lett., 1982, 92, 421–424 CrossRef CAS.
- D. B. McDonald, G. R. Fleming and S. A. Rice, Intermediate case radiationless decay: the excited state dynamics of pyrazine, Chem. Phys., 1981, 60, 335–345 CrossRef CAS.
- E. Nir, K. Kleinermanns and M. S. de Vries, Pairing of isolated nucleic-acid bases in the absence of the DNA backbone, Nature, 2000, 408, 949–951 CrossRef CAS PubMed.
- H. Kang, K. T. Lee, B. Jung, Y. J. Ko and S. K. Kim, Intrinsic Lifetimes of the Excited State of DNA and RNA Bases, J. Am. Chem. Soc., 2002, 124, 12958–12959 CrossRef CAS PubMed.
- K. Kleinermanns, D. Nachtigallová and M. S. de Vries, Excited state dynamics of DNA bases, Int. Rev. Phys. Chem., 2013, 32, 308–342 Search PubMed.
- P. R. Callis, Electronic States and Luminescence of Nucleic Acid Systems, Annu. Rev. Phys. Chem., 1983, 34, 329–357 CrossRef CAS.
- A. Pinchuk, Optical constants and dielectric function of DNA's nucleotides in UV range, J. Quant. Spectrosc. Radiat. Transfer, 2004, 85, 211–215 CrossRef CAS.
- A. Zalar, D. Tepfer, S. V. Hoffmann, J. M. Kenney and S. Leach, Directed exospermia: I. Biological modes of resistance to UV light are implied through absorption spectroscopy of DNA and potential UV screens, Int. J. Astrobiol., 2007, 6, 229–240 CrossRef CAS.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Ultrafast Excited-State Dynamics in Nucleic Acids, Chem. Rev., 2004, 104, 1977–2020 CrossRef PubMed.
- C. Canuel, M. Elhanine, M. Mons, F. Piuzzi, B. Tardivel and I. Dimicoli, Time-resolved photoelectron and photoion fragmentation spectroscopy study of 9-methyladenine and its hydrates: a contribution to the understanding of the ultrafast radiationless decay of excited DNA bases, Phys. Chem. Chem. Phys., 2006, 8, 3978–3987 RSC.
- S. B. Charnley, Y.-J. Kuan, H.-C. Huang, O. Botta, H. M. Butner, N. Cox, D. Despois, P. Ehrenfreund, Z. Kisiel, Y.-Y. Lee, A. J. Markwick, Z. Peeters and S. D. Rodgers, Astronomical searches for nitrogen heterocycles, Adv. Space Res., 2005, 36, 137–145 CrossRef CAS.
- Y.-J. Kuan, C.-H. Yan, S. B. Charnley, Z. Kisiel, P. Ehrenfreund and H.-C. Huang, A search for interstellar pyrimidine, Mon. Not. R. Astron. Soc., 2003, 345, 650–656 CrossRef CAS.
- M. N. Simon and M. Simon, Search for Interstellar Acrylonitrile, Pyrimidine, and Pyridine, Astrophys. J., 1973, 184, 757–762 CrossRef CAS.
- D. M. Hudgins, J. C. W. Bauschlicher and L. J. Allamandola, Variations in the Peak Position of the 6.2 μm Interstellar Emission Feature: A Tracer of N in the Interstellar Polycyclic Aromatic Hydrocarbon Population, Astrophys. J., 2005, 632, 316–332 CrossRef CAS.
- L. J. Allamandola, A. G. G. M. Tielens and J. R. Barker, Interstellar Polycyclic Aromatic-Hydrocarbons – The Infrared-Emission Bands, The Excitation Emission Mechanism, and The Astrophysical Implications, Astrophys. J., Suppl. Ser., 1989, 71, 733–775 CrossRef CAS PubMed.
- F. Galliano, S. C. Madden, A. G. G. M. Tielens, E. Peeters and A. P. Jones, Variations of the Mid-IR Aromatic Features inside and among Galaxies, Astrophys. J., 2008, 679, 310–345 CrossRef CAS.
- J. L. Puget and A. Léger, A New Component of the Interstellar Matter: Small Grains and Large Aromatic Molecules, Annu. Rev. Astron. Astrophys., 1989, 27, 161–198 CrossRef CAS.
- A. G. G. M. Tielens, Interstellar Polycyclic Aromatic Hydrocarbon Molecules, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- B. A. McGuire, A. M. Burkhardt, S. Kalenskii, C. N. Shingledecker, A. J. Remijan, E. Herbst and M. C. McCarthy, Detection of the Aromatic Molecule Benzonitrile (c-C6H5CN) in the Interstellar Medium, Science, 2018, 359, 202–205 CrossRef CAS PubMed.
- C. Joblin and J. Cernicharo, Detecting the building blocks of aromatics, Science, 2018, 359, 156–157 CrossRef CAS PubMed.
- K. Chatterjee and O. Dopfer, Infrared Signatures of Protonated Benzonitrile, Astrophys. J., 2018, 865, 114 CrossRef.
- K. Chatterjee and O. Dopfer, Switching of binding site from nonpolar to polar ligands toward cationic benzonitrile revealed by infrared spectroscopy, J. Chem. Phys., 2018, 149, 174315 CrossRef PubMed.
- K. Chatterjee and O. Dopfer, Intracluster Proton Transfer in Protonated Benzonitrile–(H2O)n≤6 Nanoclusters: Hydrated Hydronium Core for n ≥ 2, Phys. Chem. Chem. Phys., 2019, 21, 25226–25246 RSC.
- R. Hayatsu, Orgueil Meteorite: Organic Nitrogen Contents, Science, 1964, 146, 1291–1293 CrossRef CAS PubMed.
- C. E. Folsome, J. Lawless, M. Romiez and C. Ponnamperuma, Heterocyclic Compounds indigenous to the Murchison Meteorite, Nature, 1971, 232, 108–109 CrossRef CAS PubMed.
- P. G. Stoks and A. W. Schwartz, Uracil in carbonaceous meteorites, Nature, 1979, 282, 709–710 CrossRef CAS.
- M. P. Callahan, K. E. Smith, H. J. Cleaves, J. Ruzicka, J. C. Stern, D. P. Glavin, C. H. House and J. P. Dworkin, Carbonaceous meteorites contain a wide range of extraterrestrial nucleobases, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13995–13998 CrossRef CAS.
- R. Shapiro, Prebiotic cytosine synthesis: A critical analysis and implications for the origin of life, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4396–4401 CrossRef CAS.
- A. W. Schwartz and G. J. F. Chittenden, Synthesis of uracil and thymine under simulated prebiotic conditions, BioSystems, 1977, 9, 87–92 CrossRef CAS.
- B. Basile, A. Lazcano and J. Oró, Prebiotic syntheses of purines and pyrimidines, Adv. Space Res., 1984, 4, 125–131 CrossRef CAS.
- C. K. Materese, M. Nuevo, P. P. Bera, T. J. Lee and S. A. Sandford, Thymine and Other Prebiotic Molecules Produced from the Ultraviolet Photo-Irradiation of Pyrimidine in Simple Astrophysical Ice Analogs, Astrobiology, 2013, 13, 948–962 CrossRef CAS PubMed.
- C. K. Materese, M. Nuevo, B. L. McDowell, C. E. Buffo and S. A. Sandford, The Photochemistry of Purine in Ice Analogs Relevant to Dense Interstellar Clouds, Astrophys. J., 2018, 864, 44 CrossRef.
- Y. Oba, Y. Takano, H. Naraoka, N. Watanabe and A. Kouchi, Nucleobase synthesis in interstellar ices, Nat. Commun., 2019, 10, 4413 CrossRef PubMed.
- R. Glaser, B. Hodgen, D. Farrelly and E. McKee, Adenine Synthesis in Interstellar Space: Mechanisms of Prebiotic Pyrimidine-Ring Formation of Monocyclic HCN-Pentamers, Astrobiology, 2007, 7, 455–470 CrossRef CAS PubMed.
- M. Nuevo, S. N. Milam and S. A. Sandford, Nucleobases and Prebiotic Molecules in Organic Residues Produced from the Ultraviolet Photo-Irradiation of Pyrimidine in NH3 and H2O + NH3 Ices, Astrobiology, 2012, 12, 295–314 CrossRef CAS PubMed.
- M. Nuevo, S. N. Milam, S. A. Sandford, J. E. Elsila and J. P. Dworkin, Formation of Uracil from the Ultraviolet Photo-Irradiation of Pyrimidine in Pure H2O Ices, Astrobiology, 2009, 9, 683–695 CrossRef CAS PubMed.
- P. P. Bera, M. Nuevo, S. N. Milam, S. A. Sandford and T. J. Lee, Mechanism for the abiotic synthesis of uracil via UV-induced oxidation of pyrimidine in pure H2O ices under astrophysical conditions, J. Chem. Phys., 2010, 133, 104303 CrossRef PubMed.
- C. K. Materese, M. Nuevo and S. A. Sandford, The Formation of Nucleobases from the Ultraviolet Photoirradiation of Purine in Simple Astrophysical Ice Analogues, Astrobiology, 2017, 17, 761–770 CrossRef CAS.
- M. P. Robertson and G. F. Joyce, The Origins of the RNA World, Cold Spring Harbor Perspect. Biol., 2012, 4, a003608 Search PubMed.
- Z. Yang, P. Duffy and F. Wang, Inheritance and correlation of nucleic acid pyrimidine bases, Int. J. Quantum Chem., 2013, 113, 2312–2318 CAS.
- F. Wang and Q. Zhu, Inner-shell chemical shift of DNA/RNA bases and inheritance from their parent purine and pyrimidine, J. Synchrotron Radiat., 2008, 15, 624–631 CrossRef CAS PubMed.
- I. Linert, M. Dampc, B. Mielewska and M. Zubek, Cross sections for ionization and ionic fragmentation of pyrimidine molecules by electron collisions, Eur. Phys. J. D, 2012, 66, 20 CrossRef.
- H. Abdoul-Carime and L. Sanche, Fragmentation of short single DNA strands by 1–30 eV electrons: dependence on base identity and sequence, Int. J. Radiat. Biol., 2002, 78, 89–99 CrossRef CAS PubMed.
- J. E. Elsila, M. R. Hammond, M. P. Bernstein, S. A. Sandford and R. N. Zare, UV photolysis of quinoline in interstellar ice analogs, Meteorit. Planet. Sci., 2006, 41, 785–796 CrossRef CAS.
- H. Ichikawa and M. Ogata, Molecular orbital approach to the interpretation of organic mass spectra. III. Determination of intermediate ion structure in the mass spectrometry of heteroaromatic compounds, J. Am. Chem. Soc., 1973, 95, 806–811 CrossRef CAS.
- J. M. Rice, G. O. Dudek and M. Barber, Mass Spectra of Nucleic Acid Derivatives. Pyrimidines, J. Am. Chem. Soc., 1965, 87, 4569–4576 CrossRef CAS PubMed.
- R. Buff and J. Dannacher, The least endothermic fragmentation pathways of the diazine cations, Int. J. Mass Spectrom. Ion Processes, 1984, 62, 1–15 CrossRef CAS.
- M. Schwell, H.-W. Jochims, H. Baumgärtel and S. Leach, VUV photophysics and dissociative photoionization of pyrimidine, purine, imidazole and benzimidazole in the 7–18 eV photon energy range, Chem. Phys., 2008, 353, 145–162 CrossRef CAS.
- H. K. Ervasti, K. J. Jobst, P. Gerbaux, P. C. Burgers, P. J. A. Ruttink and J. K. Terlouw, The reaction of the acrylonitrile ion CH2CH–CN+ with HCN: Proton-transport catalysis vs. formation of ionized pyrimidine, Chem. Phys. Lett., 2009, 482, 211–216 CrossRef CAS.
- O. Plekan, M. Coreno, V. Feyer, A. Moise, R. Richter, M. D. Simone, R. Sankari and K. C. Prince, Electronic state resolved PEPICO spectroscopy of pyrimidine, Phys. Scr., 2008, 78, 058105 CrossRef.
- F. Milani-nejad and H. D. Stidham, Spectra of C2ν deuterium substituted pyrimidines, Spectrochim. Acta, Part A, 1975, 31, 1433–1453 CrossRef.
- K. J. Jobst, S. A. Hasan and J. K. Terlouw, Does the Ion–molecule Reaction between HCCH+ and HCN Lead to CH2CH–CN+? A Computational and Experimental Study of the Reverse Process, Chem. Phys. Lett., 2008, 450, 243–247 CrossRef CAS.
- N. Takagi, K. Fukuzawa, Y. Osamura and H. F. Schaefer, Ion-molecule Reactions Producing HC3NH+ in Interstellar Space: Forbiddenness of the Reaction between Cyclic C3H3+ and the N Atom, Astrophys. J., 1999, 525, 791–798 CrossRef CAS.
- J. G. Philis, The Multiphoton Ionization Spectrum of Jet-cooled Pyrimidine in the 3p Rydberg and 3B1 (π,n) States, J. Mol. Spectrosc., 2005, 232, 26–29 CrossRef CAS.
- D. J. Lavorato, T. K. Dargel, W. Koch, G. A. McGibbon, H. Schwarz and J. K. Terlouw, Pyrimidine-ylidenes Produced using Neutralization–reionization Mass Spectrometry and Probed by Density Functional Methods, Int. J. Mass Spectrom., 2001, 210–211, 43–57 CrossRef.
- L. D. Fondren, J. McLain, D. M. Jackson, N. G. Adams and L. M. Babcock, Studies of reactions of a series of ions with nitrogen containing heterocyclic molecules using a selected ion flow tube, Int. J. Mass Spectrom., 2007, 265, 60–67 CrossRef CAS.
- S. Petrie, C. G. Freeman and M. J. McEwan, The ion–molecule chemistry of acrylonitrile: astrochemical implications, Mon. Not. R. Astron. Soc., 1992, 257, 438–444 CrossRef CAS.
- P. P. Bera, T. J. Lee and H. F. Schaefer, Are Isomers of the Vinyl Cyanide Ion Missing Links for Interstellar Pyrimidine Formation?, J. Chem. Phys., 2009, 131, 074303 CrossRef PubMed.
- A. M. Hamid, P. P. Bera, T. J. Lee, S. G. Aziz, A. O. Alyoubi and M. S. El-Shall, Evidence for the Formation of Pyrimidine Cations from the Sequential Reactions of Hydrogen Cyanide with the Acetylene Radical Cation, J. Phys. Chem. Lett., 2014, 5, 3392–3398 CrossRef CAS PubMed.
- G. Vall-llosera, M. Coreno, P. Erman, M. A. Huels, K. Jakubowska, A. Kivimäki, E. Rachlew and M. Stankiewicz, VUV photoionisation of free azabenzenes: Pyridine, pyrazine, pyrimidine, pyridazine and s-triazine, Int. J. Mass Spectrom., 2008, 275, 55–63 CrossRef CAS.
- N. Solcà and O. Dopfer, Protonated Benzene: IR Spectrum and Structure of C6H7+, Angew. Chem., Int. Ed., 2002, 41, 3628–3631 CrossRef.
- A. Patzer, S. Chakraborty, N. Solcà and O. Dopfer, IR Spectrum and Structure of the Phenyl Cation, Angew. Chem., Int. Ed., 2010, 49, 10145–10148 CrossRef CAS PubMed.
- H.-S. Andrei, N. Solcà and O. Dopfer, IR Spectrum of the Ethyl Cation: Evidence for the Nonclassical Structure, Angew. Chem., Int. Ed., 2008, 47, 395–397 CrossRef CAS PubMed.
- A. Patzer, M. Schütz, T. Möller and O. Dopfer, Infrared Spectrum and Structure of the Adamantane Cation: Direct Evidence for Jahn–Teller Distortion, Angew. Chem., Int. Ed., 2012, 51, 4925–4929 CrossRef CAS PubMed.
- K. Chatterjee, O. Dopfer, K. Chatterjee and O. Dopfer, Microhydration of PAH+ Cations: Evolution of Hydration Network in Naphthalene+-(H2O)n clusters (n ≤ 5), Chem. Sci., 2018, 9, 2301–2318 RSC.
- K. Chatterjee and O. Dopfer, Protonation of Naphthalene–(Water)n Nanoclusters: Intracluster Proton Transfer to Hydration Shell Revealed by Infrared Photodissociation Spectroscopy, J. Phys. Chem. A, 2020, 124, 1134–1151 CrossRef CAS PubMed.
- K. Chatterjee and O. Dopfer, Microhydration Structures of Protonated Oxazole, J. Phys. Chem. A, 2019, 123, 7637–7650 CrossRef CAS PubMed.
- K. Chatterjee, Y. Matsumoto and O. Dopfer, Aromatic Charge Resonance Interaction Probed by Infrared Spectroscopy, Angew. Chem., Int. Ed., 2019, 58, 3351–3355 CrossRef CAS PubMed.
- O. Dopfer, Spectroscopic and Theoretical studies of CH3+-Rgn clusters (Rg = He, Ne, Ar): From weak intermolecular forces to chemical reaction mechanisms, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- O. Dopfer, IR Spectroscopy of Microsolvated Aromatic Cluster Ions: Ionization-Induced Switch in Aromatic Molecule–Solvent Recognition, Z. Phys. Chem., 2005, 219, 125 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standards and Technology, Gaithersburg MD, 2017, p. 20899, http://webbook.nist.gov Search PubMed.
- M. J. Frisch, et al., Gaussian 09, version D.01, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry, University of Wisconsin, Madison, 2013 Search PubMed.
- L. Fernholt and C. Romming, Molecular-structure of Gaseous Pyrimidine, Acta Chem. Scand., 1978, 32, 271–273 CrossRef.
- S. Albert and M. Quack, High resolution rovibrational spectroscopy of pyrimidine: Analysis of the B1 modes ν10b and ν4 and B2 mode ν6b, J. Mol. Spectrosc., 2007, 243, 280–291 CrossRef CAS.
- S. Breda, I. D. Reva, L. Lapinski, M. J. Nowak and R. Fausto, Infrared spectra of pyrazine, pyrimidine and pyridazine in solid argon, J. Mol. Struct., 2006, 786, 193–206 CrossRef CAS.
- R. C. Lord, A. L. Marston and F. A. Miller, Infra-red and Raman spectra ot the diazines, Spectrochim. Acta, 1957, 9, 113–125 CrossRef CAS.
- Z. N. Heim, B. K. Amberger, B. J. Esselman, J. F. Stanton, R. C. Woods and R. J. McMahon, Molecular Structure Determination: Equilibrium Structure of Pyrimidine (m-C4H4N2) from Rotational Spectroscopy (rSEe) and High-level ab initio Calculation (re) Agree within the Uncertainty of Experimental Measurement, J. Chem. Phys., 2020, 152, 104303 CrossRef PubMed.
- L. Bokobza-Sebagh and J. Zarembowitch, Apport de la coordination des hétérocycles à la connaisance de leurs spectres i.r. Raman—II. spectres de la pyrimidine, Spectrochim. Acta, Part A, 1976, 32, 797–805 CrossRef.
- R. Gleiter, E. Heilbronner and V. Hornung, Photoelectron Spectra of Azabenzenes and Azanaphthalenes: I. Pyridine, diazines, s-triazine and s-tetrazine, Helv. Chim. Acta, 1972, 55, 255–274 CrossRef CAS.
- A. W. Potts, D. M. P. Holland, A. B. Trofimov, J. Schirmer, L. Karlsson and K. Siegbahn, An experimental and theoretical study of the valence shell photoelectron spectra of purine and pyrimidine molecules, J. Phys. B: At., Mol. Opt. Phys., 2003, 36, 3129–3143 CrossRef CAS.
- S. Sato, K. Omiya and K. Kimura, Cation vibrational spectra of pyrimidine and its van der Waals complexes with Ar and N2 by ZEKE photoelectron spectroscopy, J. Electron Spectrosc. Relat. Phenom., 1998, 97, 121–129 CrossRef CAS.
- J. H. Kim, J. H. Lee, H. Hwang, H. L. Kim and C. H. Kwon, Determination of precise pyrimidine cationic structure by vacuum ultraviolet mass-analyzed threshold ionization spectroscopy, Phys. Chem. Chem. Phys., 2014, 16, 1590–1596 RSC.
- M. Riese and J. Grotemeyer, Mass-analyzed threshold ionization spectroscopy of pyrimidine: determining the geometry in the first excited and the ionic ground states, Anal. Bioanal. Chem., 2006, 386, 59–68 CrossRef CAS PubMed.
- K. Elkner, R. C. Binning Jr and D. E. Bacelo, Structures and G3X energies of C3H3N+ intermediates, Int. J. Quantum Chem., 2006, 106, 3043–3047 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- N. Solcà and O. Dopfer, Microsolvation of the Phenol Cation (Ph+) in Nonpolar Environments: Infrared Spectra of Ph+–Ln (L = He, Ne, Ar, N2, CH4), J. Phys. Chem. A, 2001, 105, 5637–5645 CrossRef.
- R. V. Olkhov and O. Dopfer, Spectroscopic and ab initio studies of ionic hydrogen bonds: the O–H stretch vibration of SiOH+–X dimers (X = He, Ne, Ar, N2), Chem. Phys. Lett., 1999, 314, 215–222 CrossRef CAS.
- O. Dopfer, R. V. Olkhov, M. Mladenović and P. Botschwina, Intermolecular Interaction in an Open-shell π-bound Cationic Complex: IR Spectrum and Coupled Cluster Calculations for C2H2+-Ar, J. Chem. Phys., 2004, 121, 1744–1753 CrossRef CAS PubMed.
- R. A. Relph, J. C. Bopp, J. R. Roscioli and M. A. Johnson, Structural Characterization of (C2H2)1–6+ Cluster Ions by Vibrational Predissociation Spectroscopy, J. Chem. Phys., 2009, 131, 114305 CrossRef PubMed.
- O. Dopfer, H.-S. Andrei and N. Solcà, IR Spectra of C2H5+-N2 Isomers: Evidence for Dative Chemical Bonding in the Isolated Ethanediazonium Ion, J. Phys. Chem. A, 2011, 115, 11466–11477 CrossRef CAS PubMed.
- J. Baker, On the Stability of the Vinylidene Radical Cation, Chem. Phys. Lett., 1989, 159, 447–451 CrossRef CAS.
- S. Boyé-Péronne, D. Gauyacq and J. Liévin, Vinylidene-acetylene cation isomerization investigated by large scale ab initio calculations, J. Chem. Phys., 2006, 124, 214305 CrossRef.
- H. Abe, Y. Ohyanagi, M. Ichijo, N. Mikami and M. Ito, Vibrational predissociation and nonradiative process of electronically excited van der Waals complexes of pyrimidine, J. Phys. Chem., 1985, 89, 3512–3521 CrossRef CAS.
- A. M. Hamid, P. Sharma, M. Samy El-Shall, R. Hilal, S. Elroby, S. G. Aziz and A. O. Alyoubi, Hydration of the Pyrimidine Radical Cation and Stepwise Solvation of Protonated Pyrimidine with Water, Methanol, and Acetonitrile, J. Chem. Phys., 2013, 139, 084304 CrossRef PubMed.
- H.-S. Andrei, N. Solcà and O. Dopfer, Interaction of Ionic Biomolecular Building Blocks with Nonpolar Solvents: Acidity of the Imidazole Cation (Im+) Probed by IR Spectra of Im+–Ln Complexes (L = Ar, N2; n ≤ 3), J. Phys. Chem. A, 2005, 109, 3598–3607 CrossRef CAS PubMed.
- M. Schütz, Y. Matsumoto, A. Bouchet, M. Öztürk and O. Dopfer, Microsolvation of the Pyrrole Cation (Py+) with Nonpolar and Polar Ligands: Infrared Spectra of Py+–Ln with L = Ar, N2, and H2O (n ≤ 3), Phys. Chem. Chem. Phys., 2017, 19, 3970–3986 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02919j |
| This journal is © the Owner Societies 2020 |