
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nuclear spin-induced optical rotation of functional groups in hydrocarbons†
Petr
Štěpánek
 *
*
NMR Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, FI-90014, Oulu, Finland. E-mail: petr.stepanek@oulu.fi
First published on 15th July 2020
Abstract
Nuclear spin-induced optical rotation (NSOR) is a nuclear magneto-optic effect manifesting as a rotation of the plane of polarization of linearly polarized light induced by nuclear magnetic moments within a molecule. NSOR probes molecular optical properties through localized nuclear interactions and has potential to be developed into a new spectroscopic tool. However, so far the connection between the molecular structure and NSOR response has not been systematically investigated. To obtain insight into this relation and to assess its viability as a foundation for a new spectroscopic method, NSOR of 1H and 13C nuclei in a set of hydrocarbon molecules with various structural motifs is theoretically investigated using density functional theory calculations. The results reveal that NSOR intensities are correlated with several structural features of the molecules, such as the position of the nucleus in the carbon chain, isomerism and presence of nearby unsaturated groups. Specific patterns connecting NSOR to the local chemical environment of the nucleus can be observed. It is also shown that this effect can be to a good approximation modelled as a sum of individual contributions from nearby chemical groups, allowing for a rapid estimation of its values. The demonstrated systematic dependence of the NSOR signal on the molecular structure is a desirable feature for theoretical and experimental development of new spectroscopic methods based on this phenomenon.
1 Introduction
Modern spectroscopic methods provide invaluable insight into the structure of molecules and materials. A prominent example of this is nuclear magnetic resonance (NMR), a technique known for its versatility in the investigation of molecular structure,1 quantifying microscopic dynamic phenomena,2 and non-invasive imaging.3 The strength of NMR comes from its ability to distinguish between individual atoms in a molecule based on their local chemical surroundings.Recently, a branch of spectroscopy related to NMR, known as nuclear magneto-optic spectroscopy (NMOS), has been explored both experimentally4–11 and theoretically.10–29 In contrast to classical NMR, which detects the signal through electromagnetic induction from the precessing nuclear magnetic moments, NMOS measures how these magnetic moments change the polarization state of the light passing through the sample. The interactions between the nuclei and the light, which are mediated by the electron cloud of the molecule, have shown promise for opening new types of spectroscopic measurements, such as observation of dipolar couplings in isotropic liquids26 or mapping the properties of excited states.23 So far, five NMOS effects have been described: nuclear spin-induced optical rotation (NSOR),4–18 nuclear spin-induced circular dichroism (NSCD),19–23 nuclear spin-induced Cotton–Mouton effect (NSCM),24,25 nuclear spin-induced Cotton–Mouton effect in an external magnetic field (NSCM-B),26,27 and nuclear quadrupole-induced Cotton–Mouton effect (NQCM).28,29 Out of these, only NSOR, a circular birefringence caused by nuclear magnetic moments oriented along the light beam, has been experimentally measured so far.4–11
The NMOS field is still in its infancy with broad room for development both experimentally and theoretically. So far, several general observations have been reported, such as enhancement of NSOR due to a nearby electronic resonance14 or conditions necessary for appearance of the NSCD signal23 and its correlation with the global electronic structure.22 A simple semi-empirical model for estimation of ratios of NSOR signals in a single molecule has also been proposed.17 However, despite multiple studies on the subject the general connection between the molecular structure motifs and the corresponding NMOS response has not been established for any of these effects and the interpretation of experiments still largely relies on full rigorous quantum-chemical calculations.10–13
Motivated by the lack of such insight, this paper investigates the existence of systematic trends or characteristic features that connect chemical moieties in the molecular structure with NMOS response. In particular, the study focuses on NSOR since it is the most experimentally developed NMOS effect and is likely to be one of the first to find an application in chemical structure determination in the near future.
In contrast to NMR, in which signals of nuclei of one isotope have the same strength, NSOR intensity, and even sign, depends on the chemical environment of the nucleus, an effect known as optical chemical shift.10,14,17 The goal of the presented report is to quantify this variation with respect to molecular structure.
The study is based on computational evaluation of NSOR for a set of hydrocarbon molecules including alkanes, alkenes, alkynes and dienes. The hydrocarbons were chosen as the model set because they provide multiple isomers with recurring structural motifs and thus can produce a sufficiently large data set for assessing common features. They are also likely to be one of the first groups of molecules to be investigated in a systematic manner by high-resolution NSOR.
The analysis of the results reveals that NSOR signals of individual nuclei correlate with the local chemical structure, such as position of the nucleus in the chain, isomerism and presence of nearby unsaturated bonds. In addition, it is also found that NSOR signals can be approximately reconstructed from individual contributions of neighbouring atom groups. The validity of this approximation is tested by comparing the NSOR values obtained from the contribution model with full quantum-chemical calculations for several hydrocarbons not present in the original set. An overall good agreement is found, supporting the existence of common spectral markers of NSOR for nuclei in specific moieties, and their additivity.
2 Methods
The set of model molecules consisted of all structural isomers of linear and branched hydrocarbons which are possible to create from up to six carbon atoms. Because some structural features are less represented in this group than others, a number of larger hydrocarbons were added to the set, chosen so as to increase the number of data points corresponding to less abundant structural motifs. The list of all molecules is given in ESI† in Tables S1–S6. The studied systems included alkanes, alkenes, alkynes and dienes (isolated, conjugated and cumulated).Each molecule was represented by a single conformer, built as an extended chain to minimize intramolecular interactions between different parts of the chain. The geometries were then optimized in Turbomole 7.4.130,31 at the RI-B3LYP/def-TZVP level.32–34 The presence of a local geometry minimum was verified by calculation of harmonic vibrational frequencies at the same level of theory. For comparison, several highly branched molecules were also optimized with the addition of DFT-D3 dispersion correction by Grimme.35
The NSOR parameters were calculated in DALTON36,37 using the quadratic response function formalism.38 The angle of optical rotation per unit length, nuclear concentration and spin polarization can be expressed using the quadratic response function 〈〈μα;μβ,hhfK,γ〉〉ω,0 as19
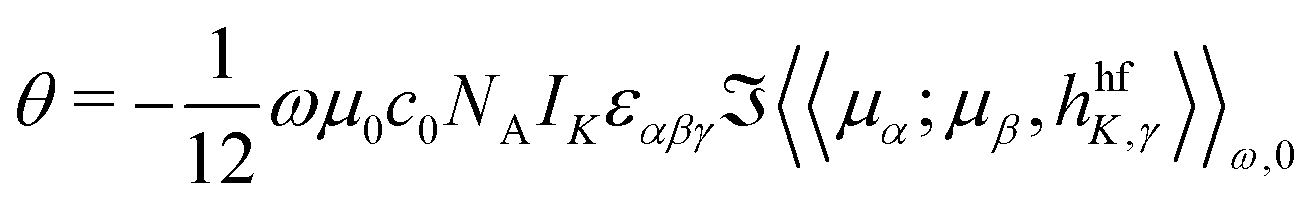 | (1) |
![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/b_i_char_e0b3.gif) and hyperfine interaction ĥhfK, the latter corresponding in non-relativistic theory of closed-shell systems to paramagnetic nuclear spin–electron orbit operator ĥPSOK:
and hyperfine interaction ĥhfK, the latter corresponding in non-relativistic theory of closed-shell systems to paramagnetic nuclear spin–electron orbit operator ĥPSOK: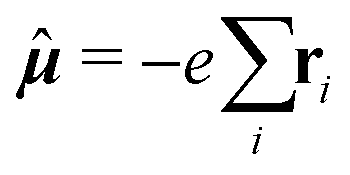 | (2) |
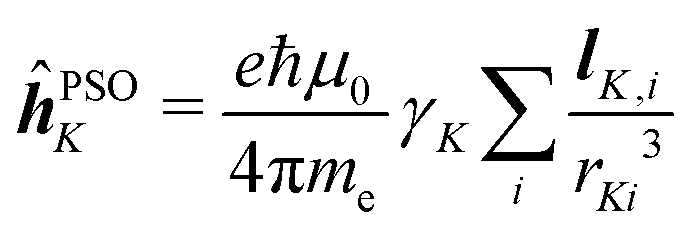 | (3) |
Calculations of the response functions were performed using the BHandHLYP33,39 density functional with the co2 basis set, in vacuum. The BHandHLYP functional has been shown to provide NSOR close to experimental and correlated ab initio coupled cluster singles and doubles (CCSD) results for organic molecules.10,11 The basis set co2 is a completeness-optimized basis set40 developed and tested for NSOR.16 Additional benchmark tests against larger completeness-optimized basis sets16,41 have shown that it provides a sufficient quality with modest computational effort (see Fig. S1 in ESI†). To assess the effect of solvent, several NSOR calculations were performed with the inclusion of an implicit solvent using the integral equation formalism of the polarizable continuum model (IEF-PCM) with solvent parameters for n-heptane.42
All calculations43 were carried out for a laser wavelength of 405 nm, which is a commonly available wavelength in commercial laser diodes. The corresponding energy is also far enough from the lowest excitation energy for all studied molecules, avoiding accidental near-resonance effects from close lying excited states, which may significantly affect the intensity of NSOR signals.14 Although such amplification of NSOR may be desirable on its own as a spectroscopic feature, accidental excitations would complicate assessment of the common trends in the present study. The NSOR values for chemically equivalent nuclei were averaged.
All NSOR signals throughout the text are reported normalized to unit spin polarization, concentration of nuclei and sample thickness in units of μrad mol−1 dm3 cm−1. The units are written in the text as μrad for short and normalization is always implied.
All NSOR signals were categorized based on the chemical identity of the nucleus according to two structural criteria: (a) atom type, as determined by its position in the chain, i.e., how many other carbon atoms it is bound to, and (b) distance from the nearest unsaturated bond. An example of the labeling of nuclei is given in Fig. 1.
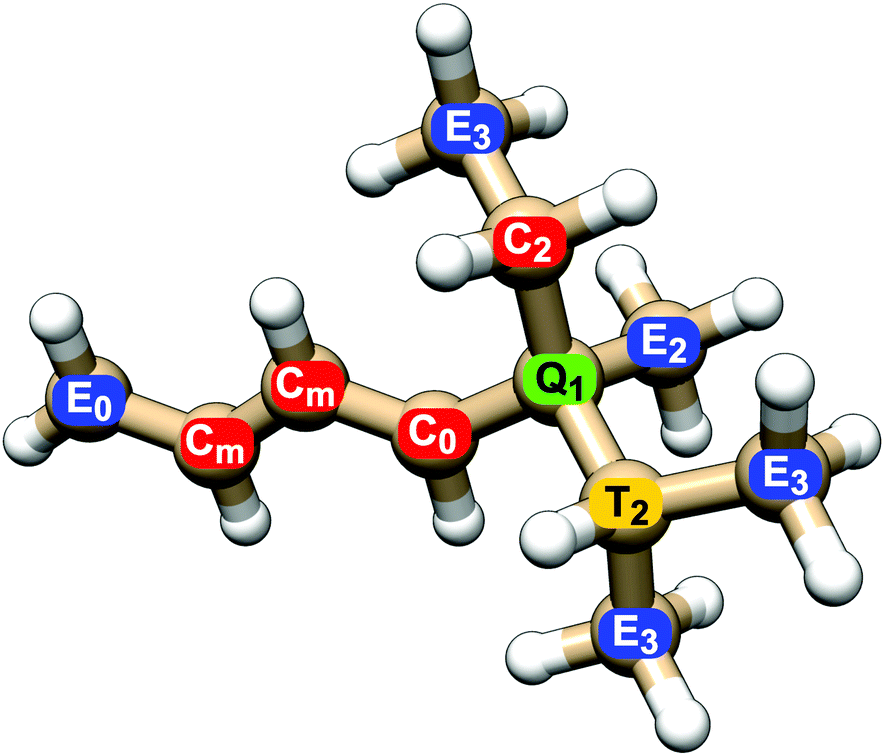 | ||
| Fig. 1 Explanation of the labelling system of nuclei. Colours and capital letters distinguish carbon nuclei at the end of the chain (E), inside the chain bound to two carbon atoms (C), nuclei at a branching point sharing three carbon–carbon bonds (T) and quaternary carbon with four carbon neighbours (Q). The subscripts indicate the distance from the nearest unsaturated bond, counted as number of carbon–carbon bonds, with 0 zero being assigned to atoms sharing the bond and m to the atoms in the middle of a larger unsaturated system. | ||
The first criterion for assigning the nuclei is their position in the hydrocarbon chain. For carbon nuclei we can distinguish four possibilities: nucleus in the group at the end of the chain (E, end), nucleus in the middle of a linear chain (C, chain), atom at a T-branch (T, tertiary), and quaternary carbon (Q). This notation reflects the number of attached carbon atoms, from 1 in primary carbons (E) to 4 in quaternary carbons (Q).
The second criterion for categorization of nuclei is their distance from the nearest unsaturated bond. The nuclei of carbons that share the unsaturated bond are labelled 0, their direct neighbours are labelled 1, the next ones in the chain 2, etc. (Fig. 1). In the case of molecules with several unsaturated bonds, the nucleus is numbered according to the closest one. This choice is based on the expected locality of NSOR due to the presence of the locally acting spin–orbit operator, as was already shown for NSOR14 and closely related NSCD property.23 The assumption that closer perturbation will have larger impact than more remote one is first verified on systems with only one unsaturated bond before applying it to more complex systems. In the case of alkanes only the first criterion is used to categorize the atoms as there are no unsaturated bonds.
A subtype of nuclei, labelled m instead of a number, is introduced for dienes. It describes nuclei sandwiched between two double bonds. The corresponding atom type Cm occurs in the following typical chains: (a) in isolated dienes it is a carbon nucleus neighboured on both sides by double bonds, in the chain of the form C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–Cm–CC; (b) in conjugated dienes it is the nucleus in the middle of the conjugated chain, i.e. CCm–CmC; (c) in cumulated dienes it corresponds to the nucleus of the atom sharing both double bonds, in structure CCmC.
C–Cm–CC; (b) in conjugated dienes it is the nucleus in the middle of the conjugated chain, i.e. CCm–CmC; (c) in cumulated dienes it corresponds to the nucleus of the atom sharing both double bonds, in structure CCmC.
The above described labelling is used for carbon atoms. The hydrogen nuclei share for simplicity the atom type with the carbons they are attached to. For example, hydrogen atoms bound to carbon atom E0 in Fig. 1 are also labelled E0, even though they do not participate in the double bond themselves. In other words, all nuclei in a given CHn group share the label of the carbon nucleus.
The molecular models were rendered using UCSF Chimera.44
3 Results
As noted above, the following results were obtained from a limited number of model molecules, each represented by a single conformer in vacuum, and with different number of data points for each type of nucleus. Therefore, the results should be understood as a qualitative description of the essential features of NSOR with respect to the general molecular framework. The quantitative prediction of experimental data for each molecule would have to include other effects, such as conformational flexibility and influence of the environment. So far, the available high-resolution experimental data for atom-specific NSOR signals6 do not have a sufficiently high signal-to-noise ratio for quantitative comparison.It should also be noted that since NSOR originates from the alignment of the light beam and nuclear magnetic moments, it is naturally modulated by the precession of the magnetization and is detected at the Larmor frequency corresponding to the NMR shift of the nucleus. This allows one to experimentally separate the NSOR signals according to their chemical shift in cases where NSOR intensities alone might lead to ambiguous results.
3.1 Simple hydrocarbons
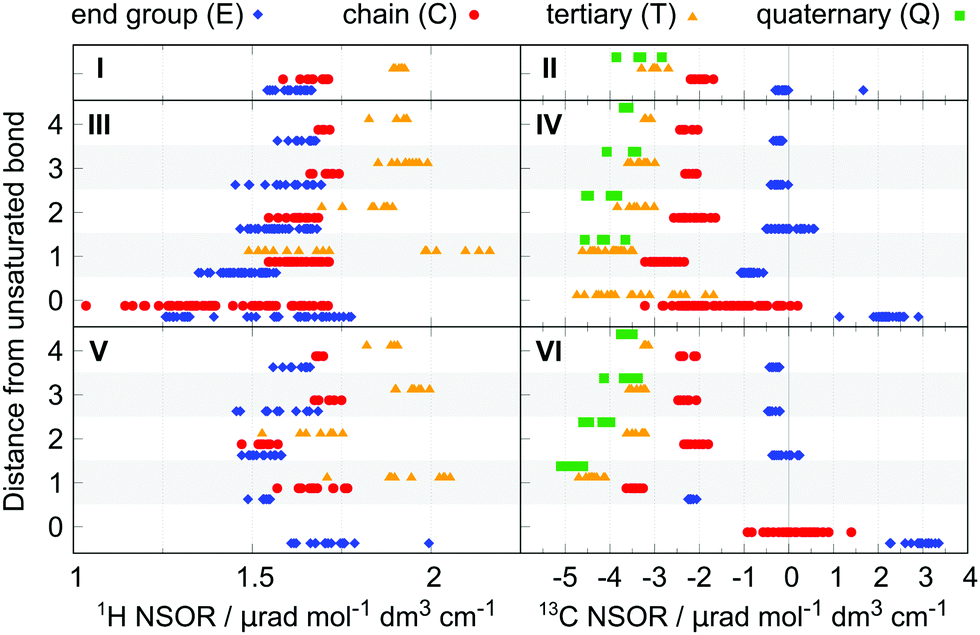 | ||
| Fig. 2 Comparison of 1H (left) and 13C (right) NSOR signals for alkanes (top row, panels I and II), alkenes (middle row, panels III and IV) and alkynes (bottom row, panels V and VI). On the x-axis is the optical rotation normalized for unit path length, concentration of nuclei and unit spin polarization, and on the y-axis is the distance of the atom from the unsaturated bond. The chart distinguishes nuclei corresponding to atoms at the end of the chain (E, blue diamonds), inside the chain (C, red circles), carbons with three carbon neighbours (T, orange triangles) and quaternary carbons (Q, green squares). | ||
In the case of 1H, there is an overlap between the E and C groups, while the hydrogen nuclei T bound to tertiary carbons are offset at higher NSOR values. The total spread of signals is rather limited, ranging between 1.5 and 2 μrad. The NSOR overall gets stronger with increasing branching at the site of the carbon it is attached to, as seen in increasing intensity from E to T.
Signals of 13C nuclei form three distinct clusters corresponding to separate groups for atom types E and C, and the overlapping group of T and Q. The values are distributed over a broader range compared to 1H, from around 0 μrad for atom type E down to −4 μrad for T and Q. The data point at 1.67 μrad corresponds to methane and due to the unique structure of this molecule is not counted as a typical feature of alkanes. In general, a similar trend can be observed as in 1H where the strength of NSOR signals increases with the number of carbon neighbours, although in this case with reversed sign and larger relative magnitude.
In summary, NSOR of each atom type in alkanes, as described by their position in the carbon skeleton, falls into a rather narrow range of values for both 1H and 13C nuclei and its magnitude is positively correlated with the number of neighbouring carbon atoms. The NSOR intensity can thus distinguish between primary, secondary, and to a limited degree tertiary and quaternary CHx groups in alkanes.
At first sight it is apparent that the signals now cover a much wider range. In the case of 1H the NSOR values of C0 and E0-type nuclei at the double bond are not anymore clustered into separate groups. Instead, they are spread rather evenly, mostly towards negative with respect to alkanes, and overlap significantly. As we will discuss in detail below, this wide spread of values reflects cis/trans isomerism around the double bond. Next to the double bond the signals of atom types E1 and C1 start to cluster, with E1 having an overall weaker signal than either C1 or T1 and also being weaker than E in alkanes. NSOR of atom type C1 appears in roughly the same region as in alkanes and the signals of nuclei T1 are spread over a very wide range, providing the strongest signals at around 2.2 μrad, but also appearing at much lower values, overlapping with the whole range of C1 and part of E1. This spread is partially explained by cis/trans isomerism as NSOR of nuclei of this type in cis-alkenes appears around 1.6 μrad while trans-alkenes and alk-1-enes show higher NSOR values (see Fig. S2 in ESI†). However, this does not explain the wide separation fully, which is linked to other structural details not apparent from this set of data points. Further from the double bond, the NSOR strength of the E2 atom type slightly increases and T2 signals aggregate around 1.8 μrad. Interestingly, with increasing distance from the unsaturated bond the signals of all atom types drift towards roughly the same intervals as in alkanes. The double bond thus appears to affect the NSOR of nearby nuclei the most, and this effect falls off with the distance.
In the case of 13C the atoms participating in the double bond show a wide distribution, but over quite characteristic regions. Similarly as in the case of 1H, this wide spread can be linked to isomerism, and will be discussed below. The E0-type nuclei have strongly positive NSOR around 2.3 μrad, C0-type occupy a wide range between 0 and −3 μrad and T0 nuclei also show a large spread, but shifted to more negative values between −1.5 and −5 μrad. The outlier E0 signal at 1.13 μrad belongs to ethene. The nuclei neighbouring the double bond form more compact groups with T1 and Q1 around −4 μrad, C1 around −2.8 μrad, and E1 switching sign to slightly negative values around −0.9 μrad. In position 2 the signals of E2 nuclei drift towards zero, C2 signals towards −2 μrad, T2 signals move closer to −3 μrad, and Q2-type remain the most negative at around −4 μrad. At larger distances signals of all atom types monotonically shift towards the range of values characteristic for alkanes, similarly as in the case of 1H.
We can see from these results that NSOR can distinguish, e.g., the terminal CH2 group, which gives a positive signal for the E0 nucleus, from the C0 in –CHCH– inside the chain. The spectroscopic value of NSOR can be further enhanced when we take into account the Larmor frequency of the nuclei: the signals in position 0 will appear at chemical shifts characteristic for unsaturated bonds, while the NSOR of other nuclei will be modulated with frequencies typical for saturated hydrocarbons. This allows one, for example, to identify the 13C E1 signal as it is the only NSOR signal in the range around −1 μrad with chemical shift outside of the alkene region.
Based on the observations we can argue that the double bond acts as a local perturbation of NSOR with respect to an alkane chain and affects most strongly nuclei up to roughly two to three bond-lengths away. Nuclei further from the double bond see rapidly diminishing influence. The local nature of NSOR has been observed before as signal enhancement of nuclei located in the chromophore when the energy of the incident light approaches its electronic resonance14 and the present results suggest that the locality of NSOR manifests also at energies far from excitations. This behaviour will be observed in the forthcoming cases as well.
For 1H nuclei the E0 is located around 1.7 μrad, at slightly higher values than E in alkanes and covers the upper range of E0 of alkenes. The outlier signal at 1.99 μrad is ethyne. Next to the triple bond the NSOR of C1 and T1 is spread out at around the same mean value as in alkanes while E1 nuclei give slightly smaller NSOR. The intensities thus keep the general trend T > C > E. Interestingly, in position 2 the NSOR of C2 and T2 appears to be slightly shifted towards smaller values by about 0.1 and 0.2 μrad, respectively, compared to alkanes and alkenes. At distances further from the triple bond the NSOR converges again towards values of alkanes for all three atom types.
The NSOR of 13C qualitatively resembles that of alkenes, but with overall slightly stronger intensities. For atoms participating in the triple bond the E0 nuclei give NSOR around 3 μrad, compared to 2.3 μrad in alkenes. On the other hand, the signals of C0 are the exception and are overall weaker and spread quite evenly around zero. Next to the triple bond the NSOR of different types of nuclei forms distinct compact groups with all signals negative and following the progression E1 > C1 > T1 > Q1. The most notable increase in intensity compared to alkenes is for E1. As in previous cases the influence of the unsaturated bond can be observed to a lesser degree still in Q2 and T2, but drops off with distance.
From the point of view of spectroscopic application the carbon nuclei E0 and C0 at the triple bond are the most characteristic, being well separated in their magnitude. The other NSOR signals, which have chemical shifts in the region of saturated hydrocarbons, can be assigned for E≥2 nuclei with NSOR around zero, and for T/Q atom types at strongly negative NSOR values <−4 μrad. We note in passing that the T and Q could be further experimentally distinguished by their different NMR relaxation rates. The regions around −2 μrad and −3.5 μrad are ambiguous since NSOR values of E1/Cn≥2 and C1/Tn≥2/Qn≥3 overlap there, respectively.
Comparing the overall features of the alkynes, alkenes and alkanes, we can see that the NSOR of carbon nuclei E0, E1, C1, T1 and Q1 increases in intensity as the system becomes more unsaturated. On the other hand, the NSOR intensity of C0 atom type decreases towards zero. Additionally, the signals in alkenes are much more widely spread compared to the other two cases. This is partially due to the cis/trans isomerism and possibly also because of the sensitivity of NSOR of nuclei near the double bond to conformations and possible intramolecular interactions (see Fig. S3 in ESI†). Alkynes might be less sensitive to this effect, in part because the rest of the chain points straight away from the triple bond and thus decreases the possibility of close interactions, while alkenes can create more folded structures due to the bond angle of sp2 carbon atoms.
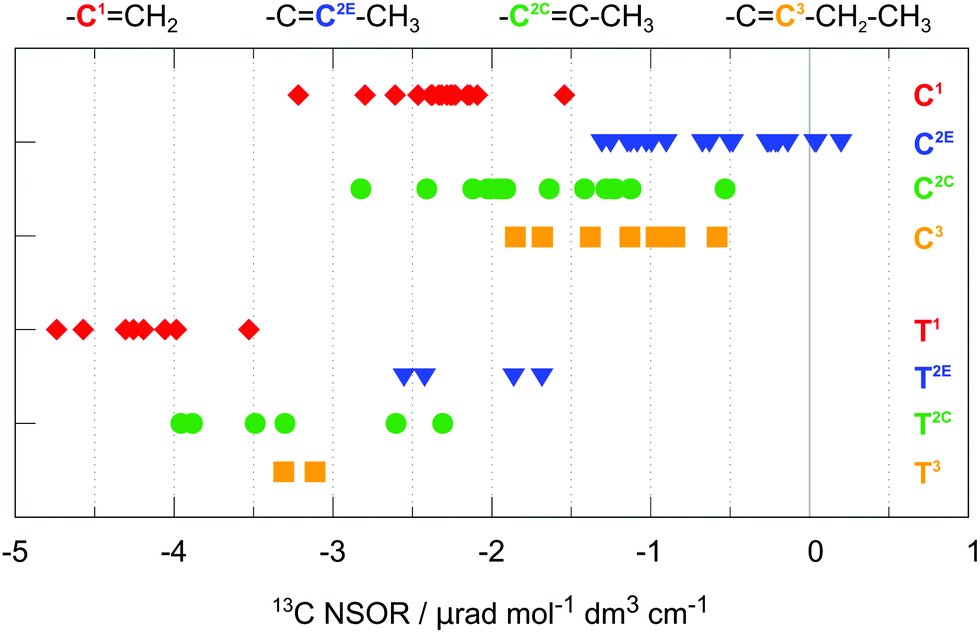 | ||
| Fig. 3 Effect of position of the double bond in the alkene chain on 13C NSOR of atom types C and T. The x-axis represents the normalized optical rotation, and the y-axis distinguishes different atom positions. | ||
Thus, there are two possible general patterns: in the case of alk-1-enes, one of the two NSOR signals with chemical shifts corresponding to the unsaturated bond will be strongly positive (E0) and one strongly negative (C0, second carbon from the end, C1 in Fig. 3). In the case of alk-2-enes, or in general, alk-n-enes, where the double bond is inside the chain, the positive E0 signal will be absent and instead two negative, less intense C0 signals will be observed (C2C, C2E or C3 in Fig. 3). A similar argument can be made for the nuclei of type T0, depending on whether they are located next to the terminus of the molecule or inside the chain.
This observation is another demonstration of the influence of local electronic structure on NSOR. The atoms not only feel the presence of the nearby double bonds, but also the surrounding chain structure in their vicinity. The former effect appears to be the dominant one, though.
cis/trans isomerism. Another structural motif in alkenes is cis/trans isomerism. This has been briefly discussed in relation with 1H T1, which shows some dependence on the isomerism of the molecules in which it is present. Here we look in more detail at the nuclei directly connected next to the double bond. For this purpose, the nuclei were categorized into four groups, according to the orientation of that particular nucleus with respect to the carbon chain(s) continuing on the opposite side of the double bond. When a carbon chain is attached in the cis or trans position, the nucleus is correspondingly labelled as cis and trans, respectively. If both cis and trans positions are occupied with carbon substituents, the nucleus is assigned to group cis + trans. Conversely, when there are no carbon chains attached at the other side of the double bond, the nucleus is labelled unsubstituted. We note that unsubstituted implies that the nucleus is located in a group neighbouring the end of the molecule.
The situation for 1H and 13C is summarized in Fig. 4 in panels I and II, respectively. The plot distinguishes between the atoms that are part of the end group of the molecule (type E0 for 1H and E1 for 13C) or inside the chain (type C0 or C1, respectively). The atom type T does not show any significant trend in this respect in the available data and is not reported. The cis/trans isomerism is marked in colour and shape of the symbols and by offset on the y-axis.
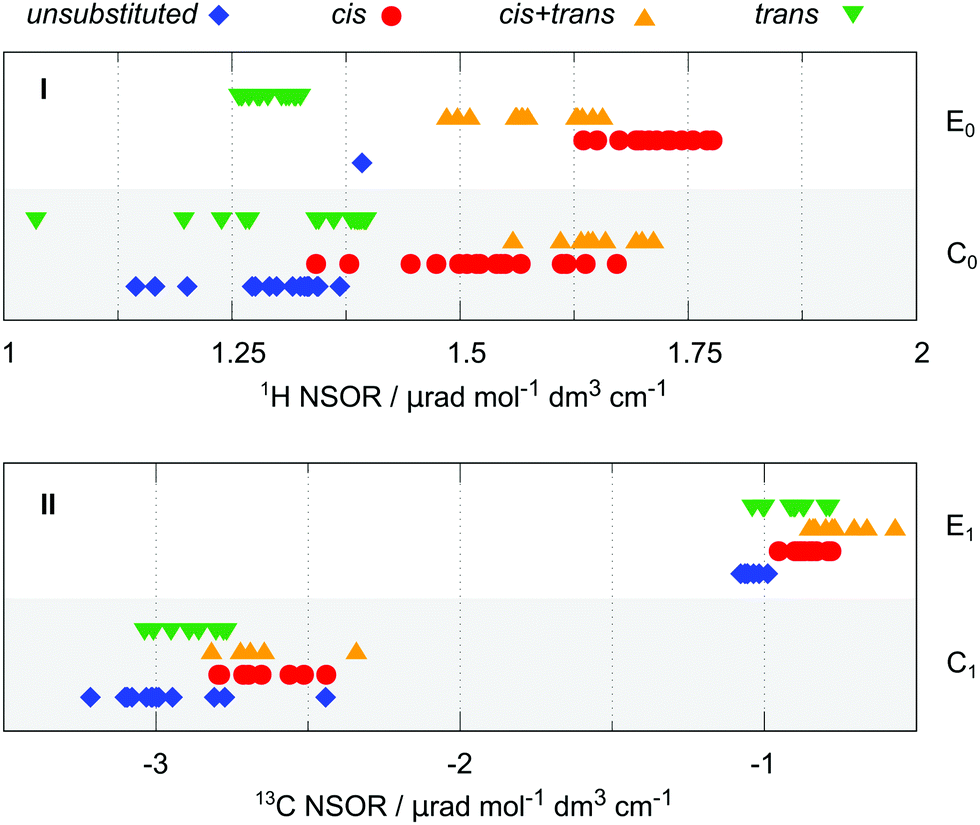 | ||
| Fig. 4 Effect of cis/trans isomerism in alkenes for 1H (top panel I) and 13C (bottom panel II) NSOR signals. On the x-axis is the normalized optical rotation, and the y-axis distinguishes different atom positions. | ||
The 1H NSOR shows a separation of different signals according to their local isomerism, with the signals of E0 nuclei decreasing in intensity in the order cis > cis + trans > trans. The single point in the unsubstituted group corresponds to ethene. Although there is an overlap between cis + trans and cis groups, the trans one is set apart at much lower intensity, thus being of a potential spectroscopic value.
The hydrogen nuclei C0 cover a slightly wider region of intensities. The groups of signals of nuclei in cis and cis + trans positions are close together at higher intensities than the trans and unsubstituted groups, similarly as in the case of E0. Interestingly, the cis + trans group provides in this case stronger average signal than cis and gives now the strongest NSOR, relative to the total spread of the signals. Because of the overall wider range of the values of the cis group, it now also overlaps with trans. Nevertheless, the region below 1.25 μrad is exclusively occupied by trans and unsubstituted groups, while cis and cis + trans are the only species present above 1.5 μrad.
In the case of 13C the trend of intensities for different groups of atom type E1 follows roughly the pattern cis + trans > cis ≈ trans > unsubstituted. Interestingly, this pattern differs from that of the 1H E1 nuclei, where cis is the group with the strongest signals. The C1 nuclei show the trend cis + trans ≈ cis > trans > unsubstituted. Although there is a slight progression of intensities between the groups, their separation is quite small, with significant overlap between them. Therefore, at least at this level of approximation, the 13C signals might not be specific enough for unambiguous determination of the molecular structure. This issue is exacerbated by the presence of notable outliers in the case of C1.
In summary the 1H signals are more specific than the 13C signals in distinguishing cis/trans isomerism. Although the different groups are not completely separated, there is a rather strong distinction between cis and cis + trans on the one hand and trans and unsubstituted on the other hand, promising a potential spectroscopic feature of NSOR for structural elucidation. Given the constraints of this study it is not clear whether such distinction would be unambiguous to resolve all four cases under real experimental conditions. However, the results suggest at least a possibility to distinguish the limiting cases near the ends of this NSOR range.
3.2 Dienes
We will now discuss dienes as examples of common, more complicated hydrocarbon systems. We can distinguish three groups of dienes, depending on the mutual distance of the double bonds: isolated dienes, where the double bonds are separated by two or more single bonds; conjugated dienes, containing alternating single and double bonds; and cumulated dienes, with double bonds directly next to each other.The NSOR for all three types is plotted in Fig. 5 together with the essential results for alkenes for comparison. The nuclei at distances up to 2 bonds from the nearest double bond are shown, as they are the most influenced and thus of the largest interest. (For extended figure showing also the further nuclei see ESI,† Fig. S4.)
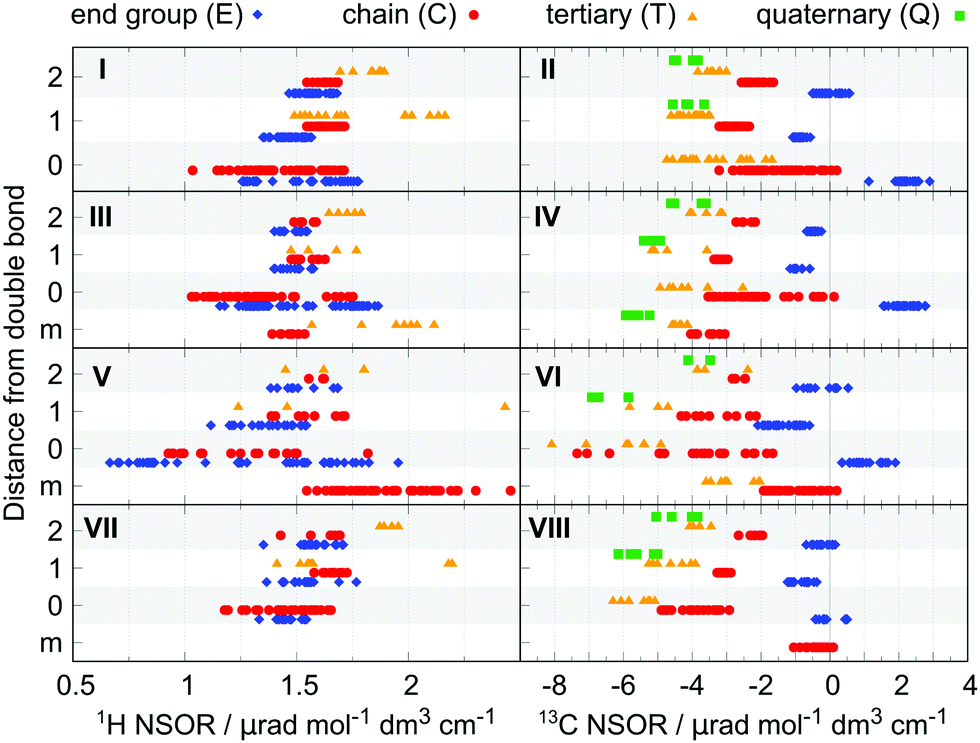 | ||
| Fig. 5 Comparison of 1H (left) and 13C (right) NSOR signals for alkenes (top row, panels I and II), isolated dienes (second row, panels III and IV), conjugated dienes (third row, panels V and VI) and cumulated dienes (bottom row, panels VII and VIII). On the x-axis is the normalized optical rotation, and on the y-axis is the distance of the atom from the nearest unsaturated bond. The chart distinguishes nuclei corresponding to atoms at the end of the chain (E, blue diamonds), inside the chain (C, red circles), carbons with three carbon neighbours (T, orange triangles) and quaternary carbons (Q, green squares). Signals labelled m correspond to nuclei between two double bonds (see the Methods section for details). | ||
For 1H the signals of atom types E0 and C0 spread over the intervals of about 1.25–2.0 μrad and 1.0–1.75 μrad, respectively, and become more compact with increasing distance from the double bond. Both the spread of values and their mean position reflect those of alkenes. This similarity can be observed also in the signals of 13C. A small deviation is present in Q1 and T1, which are pushed slightly towards negative. This can be explained by the influence of the second double bond, to which the Q is more sensitive than C at larger distances, as seen in Q2 compared to C2 in alkenes.
Of special interest are the signals of nuclei which neighbour simultaneously both double bonds. They can thus be considered a special case of atom types in position 1. In the case of 1H the NSOR signals of Cm are slightly more negative compared to the C1 in alkenes, indicating some additional influence of the second double bond. On the other hand, NSOR of Tm is spread over a similar region as T1. However, we should note the wide distribution of the values, which makes this conclusion more tentative.
For 13C nuclei the signals of Cm form quite a compact group and shift towards negative values compared to C1. This indicates that the presence of a second adjacent double bond to some degree strengthens the influence of the first one. Moreover, the shift from C in alkanes to Cm is roughly twice as large as the difference between C in alkanes and C1 in alkenes (−1.52 μrad compared to −0.83 μrad), suggesting that the effect of the two double bonds is approximately additive. The strengthening of the NSOR by the second double bond is also seen to a limited degree for Tm, which is shifted by −1.33 μrad from the alkane value compared to the shift of −0.91 μrad for T1, and to an even larger extent in Qm values compared to Q1 in alkenes (shift of −2.35 μrad and −0.79 μrad, respectively). This suggests that both double bonds contribute to the NSOR perturbation, a feature that will be explored later in the construction of an approximate model for this property.
Overall, the features of the isolated dienes reflect those of alkenes for their common atom types, suggesting the local nature of NSOR in molecules with non-interacting double bonds. In addition, the cis/trans isomerism also influences the NSOR in a similar manner as in alkenes (see Fig. S5 in ESI†).
In the case of 1H the spread is mostly towards weaker NSOR. The signals of atom types E0 completely overlap with the range of C0 and reach very low values around 0.7 μrad. Next to the double bond the nuclei E1 and C1 are spread less, but still over a wider interval than in alkenes. They also start to differ in their mean values with C1 being stronger than E1. In position 2 the signals approach the corresponding alkene values. The signals of T1 and T2 are found in a wider region than in alkenes, with no appreciable shift of the mean value, taking into account the limited number of points.
In 13C the NSOR values for atom type C0 and T0 are shifted towards negative compared to alkenes, and cover a broader range. In particular, T0 shows the strongest negative NSOR among the studied molecules, up to −8 μrad. On the other hand, the signals of E0 are weaker than in alkenes and with comparable spread. Nuclei E1 and C1 neighboring the conjugated system both give signals distributed over wider regions than in alkenes, but with similar mean values while the NSOR of Q1 and T1 is quite significantly shifted towards negative. In position 2 all four atom types approach values similar to those in alkenes.
The nuclei of type m, corresponding to the atoms in the middle of the conjugated chain, do not have a directly comparable atom type in alkenes. In the case of 1H the Cm nuclei exhibit significantly stronger NSOR compared to other types, reaching up to almost 2.5 μrad, which is one of the largest observed values for 1H among the tested molecules. For 13C the NSOR of Cm is at slightly negative values, approximately covering the upper range of C0 in alkenes. Likewise, tertiary carbons Tm also cover the upper range of T0 in alkenes. This might suggest some similarity between the NSOR of nuclei inside conjugated systems and those in double bonds of alk-n-enes, which occupy this region.
The overall feature of conjugated dienes is that both 1H and 13C signals cover a wide interval of values. In fact, the strongest and the weakest NSOR for 1H and the most negative NSOR for 13C can be found in this set of molecules. This suggests that NSOR of conjugated systems might be more sensitive to minor structural changes. Similarly as in the case of alkenes, some of the variability of the NSOR can be explained by the cis/trans isomerism (see Fig. S5 in ESI†). Further studies on extended conjugated systems, aromatic and anti-aromatic molecules might provide some additional insight.
The 1H NSOR is rather similar to that of alkenes for all atom types, having similar mean values and spread with slightly more compact width for the nuclei in position 0. In the case of 13C the NSOR of nuclei of type C0 and T0 is shifted towards negative values compared to alkenes. On the other hand, the signals of E0-type nuclei are spread closely around zero. Interestingly, this can be seen as a trend for E0 signals which decrease in intensity from alkanes/isolated dienes through conjugated dienes to cumulated ones as the double bond system gets more compact. In position 1 the values for C1 and E1 are already close to the NSOR of alkenes, while T1 and Q1 are slightly more negative. In position 2 all four atom types approach values close to alkenes.
Concerning the atom type Cm, it shows a rather weak signal between 0 and −1 μrad. It is worth noting that similarly as for E0, the NSOR values of this atom type steadily decrease going from isolated dienes to conjugated to cumulated, as the double bonds approach one another.
3.3 Effect of solvent and geometry
As has been shown, NSOR is sensitive to the environment surrounding the molecule.10–13 To gain insight into this effect for the present case, NSOR calculations including the implicit solvent model (PCM) were performed for a subset of molecules, with two representatives from each group of investigated hydrocarbons. The list of selected structures is given in Table S7 in ESI.†n-Heptane was used as solvent in the PCM, as it is a typical hydrocarbon and thus should provide a reasonable description if we consider the molecule to be surrounded by the bulk of its own neat liquid phase or other similar, nonpolar solvent.The results are shown in Fig. 6. As we can see, the inclusion of the implicit solvent model rather systematically increases the NSOR values by about 25% for all atom types, with only small deviations. As the change is approximately proportional to the signal intensity, this effect enhances the distinction between the different atom types, but does not change the overall trends.
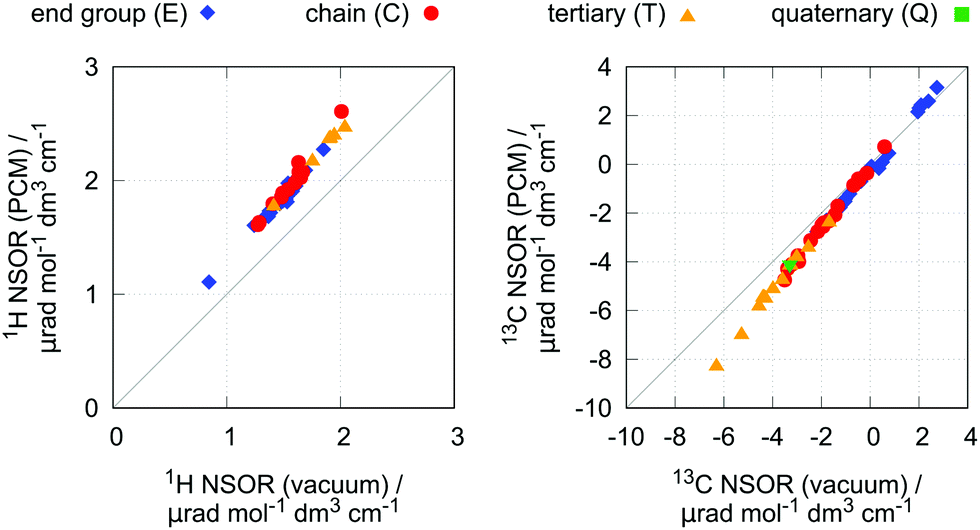 | ||
| Fig. 6 Effect of the implicit solvent model on 1H NSOR (left) and 13C NSOR (right). The x-axis represents NSOR calculated for molecules in vacuum, and the y-axis shows NSOR calculated with the inclusion of the polarizable continuum model. Each point represents one chemically distinct nucleus. | ||
The increase of NSOR intensity can be compared to the changes in optical properties induced by the solvent. The excitation energies calculated in vacuum and with the PCM show very small differences (see ESI,† Fig. S6), and the increase in NSOR is thus not due to the presence of closer electronic resonances.14 On the other hand, electric transition dipole moments, which formally appear in the sum-over-states expression of NSOR, are overall larger, correlating with the change in NSOR.
We note that this description gives only a partial insight, since NSOR of different nuclei can also be influenced by specific localized solvent–solute intermolecular interactions, which are not present in the PCM. Thus, further changes in NSOR, likely more sensitive to the chemical nature of the nucleus, can be expected by including an explicit solvent. Such interactions might be prominent particularly in the case of more polar molecules than hydrocarbons.
Another aspect which might influence NSOR is geometry of the molecular structure. The effect of small variations was investigated by including the dispersion DFT-D3 term35 in the geometry optimization of eight structures selected from the most branched molecules, where the influence of dispersion on the geometry is expected to be the most pronounced (see Table S7 in ESI†). It is found that this geometry perturbation does not change NSOR significantly (see Fig. S7 in ESI†). It should be noted, however, that the dispersion might have secondary effects on NSOR by affecting the relative populations of conformers.
The molecular conformation affects the NSOR intensity noticeably (see Fig. S3 in ESI†). The present results suggest that the saturated parts of molecules are less sensitive to such changes, and their NSOR reflects primarily the structural features determined by the molecular skeleton. The effect of conformation on the unsaturated parts of the molecule appears more pronounced. Quantitative assessment of the role of conformational freedom and its impact on NSOR and its trends is beyond the scope of the present work. Given that the differences in NSOR between conformers are comparable in magnitude to the effect of the solvent, even modelled implicitly, such evaluation would most likely require an accurate model including explicit solvent molecules.
3.4 Trends in NSOR of hydrocarbons
Let's summarize the main observations about NSOR in hydrocarbons so far:(a) signals of alkanes are all present in narrow regions for each of the atom types E, C, T and Q
(b) an unsaturated bond locally perturbs the NSOR of nearby nuclei, shifting the values from that of alkanes; this change is largest for atoms participating in the bond and for those adjacent to it; NSOR of nuclei further apart approaches values characteristic for alkanes
(c) there is a monotonic progression in NSOR intensity for 13C nuclei in position 0 and 1 from alkanes to alkenes and alkynes, and for E0 and Cm in dienes from isolated to cumulated, suggesting dependence of NSOR on the electron richness of the system
(d) for nuclei not involved in the unsaturated bond, the signal strength follows the pattern Q ≈ T > C > E for 13C and T > C ≥ E for 1H
(e) generally, 13C-NSOR is more sensitive to the backbone structure of the molecule, while 1H is more sensitive to isomerism.
In addition, in systems with two isolated double bonds, their influence on NSOR of adjacent nuclei appears to be approximately additive as seen for Cm, Tm and Qm in isolated dienes. This, however, is not observed in conjugated and cumulated dienes – if the unsaturated bonds form more extended systems, the new complex chromophore has its own specific NSOR properties.
Based on the results from the isolated dienes we may tentatively propose that NSOR can be approximated for non-interacting chromophores as an additive property. The central hypothesis of the model is that each chromophore contributes to the NSOR of nearby nuclei separately. Each contribution is specific for the atom type of the nucleus being influenced (e.g., E), the type of the chromophore (e.g., a double bond) and the distance between the chromophore and the nucleus. Because the NSOR values of nuclei far from the chromophore approach those of alkanes, we can take the alkane values as a baseline for each atom type and add the corrections, which are most significant near the chromophore and fall off towards zero with distance. Thus, the corrections can be estimated for each atom type AT at the distance N from the chromophore C as
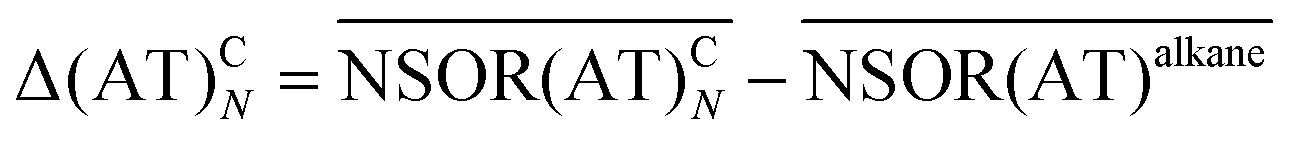 | (4) |
| Chromophore | Atom type of Cx | |||
|---|---|---|---|---|
| E | C | T | Q | |
| Alkane | −0.16 | −2.00 | −3.04 | −3.34 |
| CH2Cx– (alk-1-ene) |
— | −0.34 | −1.16 | — |
| –CCx–CH3 (alk-2-ene) |
— | 1.42 | 0.91 | — |
| –CxC–CH3 (alk-2-ene) |
— | 0.32 | −0.17 | — |
| –CCx– (general alkene) |
2.42 | 0.58 | −0.36 | — |
| –CC–Cx– |
−0.72 | −0.83 | −0.91 | −0.79 |
| –CC–C–Cx– |
0.12 | −0.15 | −0.32 | −0.72 |
| –CC–C–C–Cx– |
−0.07 | −0.16 | −0.29 | −0.24 |
–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Cx– Cx– |
3.06 | 2.19 | — | — |
| –CC–Cx– |
−2.02 | −1.44 | −1.34 | −1.51 |
| –CC-C-Cx– |
0.08 | −0.11 | −0.35 | −0.94 |
| –CC–C–C–Cx– |
−0.20 | −0.31 | −0.38 | −0.28 |
| –CCxC– |
— | 1.64 | — | — |
| –CCCx– |
0.11 | −1.88 | −2.59 | — |
| –CCC–Cx– |
−0.67 | −1.13 | −1.70 | −2.21 |
| –CCC–C–Cx– |
−0.13 | −0.21 | −0.78 | −1.04 |
| –CCC–C–C–Cx– |
−0.11 | −0.16 | −0.22 | −0.58 |
| Chromophore | Atom type of Hx | ||
|---|---|---|---|
| E | C | T | |
| Alkanes | 1.61 | 1.68 | 1.91 |
| –CCHxcis |
0.11 | −0.15 | — |
| –CCHxtrans |
−0.32 | −0.35 | — |
| –CCHxcis + trans |
−0.03 | −0.03 | — |
| –CCHxunsubstituted |
— | −0.39 | — |
| –CC–CHx |
−0.13 | −0.05 | −0.14 |
| –CC–C–CHx |
−0.04 | −0.05 | −0.07 |
| –CC–C–C–CHx |
−0.01 | 0.03 | 0.02 |
| –CCHx |
0.09 | 0.00 | — |
| –CC–CHx |
−0.08 | −0.15 | 0.02 |
| –CC–C–CHx |
−0.08 | 0.03 | −0.23 |
| –CC–C–C–CHx |
−0.03 | 0.00 | 0.04 |
| –CCCHx |
−0.15 | −0.21 | — |
| –CCC–CHx |
−0.07 | −0.03 | −0.27 |
| –CCC–C–CHx |
−0.03 | −0.06 | 0.00 |
| –CCC–C–C–CHx |
−0.01 | −0.03 | 0.00 |
Using these data we can estimate the NSOR for a system composed of several chromophores as follows:
1. Identify the atom type (E, C, T or Q) for the nucleus of interest. Assign the nucleus the baseline NSOR value corresponding to its atom type in alkanes.
2. Find chromophores near the nucleus, at least up to two bonds away.
3. Add all relevant corrections corresponding to the atom type, chromophore, and the distance between them. Alkane substituents do not contribute.
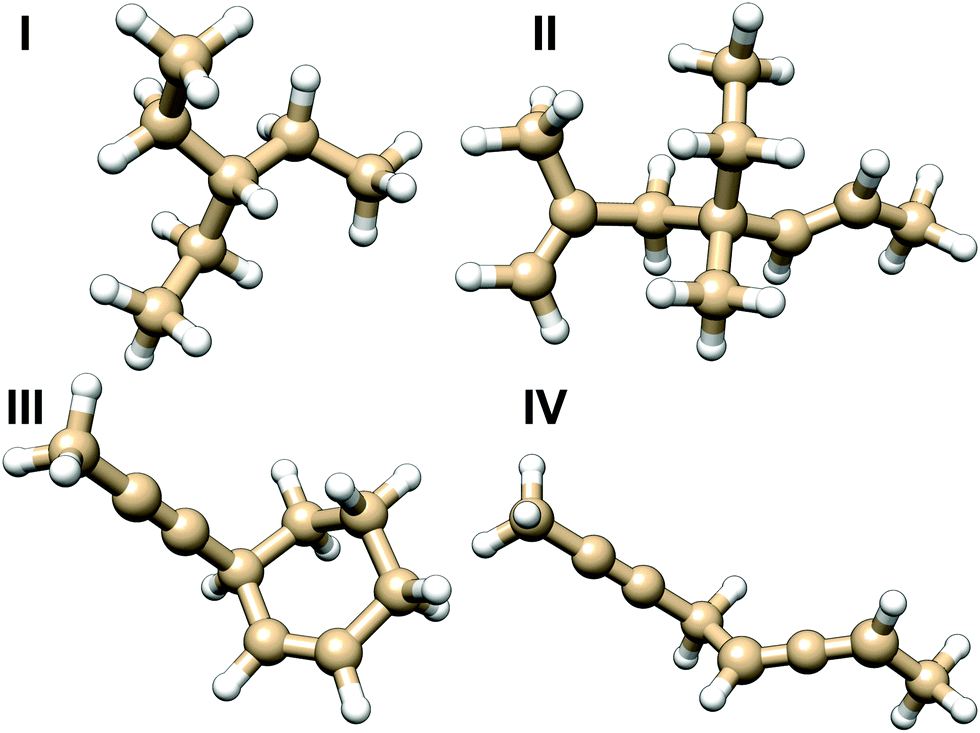 | ||
| Fig. 7 Molecules used for testing the additive model of NSOR. The results for different variations of molecule I are in Table 3, and for molecules II, III and IV in Fig. 8. | ||
The molecule I was chosen as the first test case to benchmark the model. Several structures were built by replacing some of the ethyl substituents attached to the central carbon with different side chains. The created structures are shown in Table 3 together with comparison of NSOR obtained from full calculation and from the additive model. Overall a reasonable agreement is found, with the largest relative difference between the model and the full calculation being about 21%. This is a rather good result considering the simplicity of the model with respect to full quantum-chemical computation. The error increases with the number of attached unsaturated chromophores, which is expected as the molecules in the training set contained only a single chromophore each.
| Molecule | Substituent | Contribution | Model | Full | Δ r % | ||||
|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S1 | S2 | S3 | ||||
| I 1 | –CH2–CH3 | –CH2–CH3 | –CH2–CH3 | — | — | — | −3.04 | −3.34 | 9 |
| I 2 | –CH2–CH3 | –CH2–CH3 | –CHCH2 |
— | — | −0.91 | −3.96 | −3.95 | 0 |
| I 3 | –CH2–CH3 | –CHCH2 |
–CHCH2 |
— | −0.91 | −0.91 | −4.87 | −4.80 | 2 |
| I 4 | –CHCH2 |
–CHCH2 |
–CHCH2 |
−0.91 | −0.91 | −0.91 | −5.79 | −5.02 | 15 |
| I 5 | –CHCH2 |
–CHCH2 |
–CCH |
−0.91 | −0.91 | −1.34 | −6.21 | −5.12 | 21 |
| I 6 | –CH2–CH3 | –CH2–CH3 | –CCH |
— | — | −1.34 | −4.38 | −4.46 | 2 |
| I 7 | –CH2–CH3 | –CHCH2 |
–CCH |
— | −0.91 | −1.34 | −5.29 | −5.14 | 3 |
| I 8 | –CH2–CH3 | –CCH |
–CCH |
— | −1.34 | −1.34 | −5.72 | −5.03 | 14 |
| I 9 | –CH2–CH3 | –CHCH2 |
–CHCCH2 |
— | −0.91 | −1.70 | −5.66 | −5.41 | 5 |
| I 10 | –CH2–CH3 | –CHCCH2 |
–CHCCH2 |
— | −1.70 | −1.70 | −6.45 | −5.76 | 12 |
| I 11 | –CHCCH2 |
–CHCCH2 |
–CHCCH2 |
−1.70 | −1.70 | −1.70 | −8.16 | −7.23 | 13 |
The molecules II, III and IV feature a wider range of atom types at different distances from the chromophores. Their NSOR values obtained from full calculation and from the contribution model are shown in Fig. 8. The nuclei are numbered in this case with two indices, representing the distance from the first and the second chromophore, indicated for each molecule in the figure by red and orange, respectively. For each atom type the values obtained from full calculation are shown as black bars, and from the approximate model in green. The error bars show estimate of the precision of the values in the model, calculated for each nucleus as 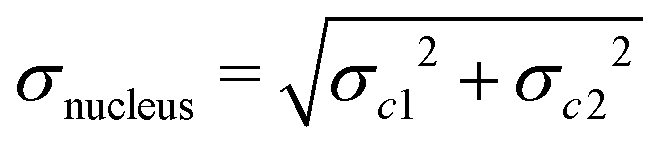 , where σc1, σc2 are the standard sample deviations of NSOR of the chromophores calculated for the corresponding atom type and distance. The coloured stacked bars show the baseline NSOR of alkane in blue and contributions from the two unsaturated groups in red and orange. Missing values are due to the particular carbons having no hydrogens attached, or more chemically inequivalent hydrogens being attached to the same carbon.
, where σc1, σc2 are the standard sample deviations of NSOR of the chromophores calculated for the corresponding atom type and distance. The coloured stacked bars show the baseline NSOR of alkane in blue and contributions from the two unsaturated groups in red and orange. Missing values are due to the particular carbons having no hydrogens attached, or more chemically inequivalent hydrogens being attached to the same carbon.
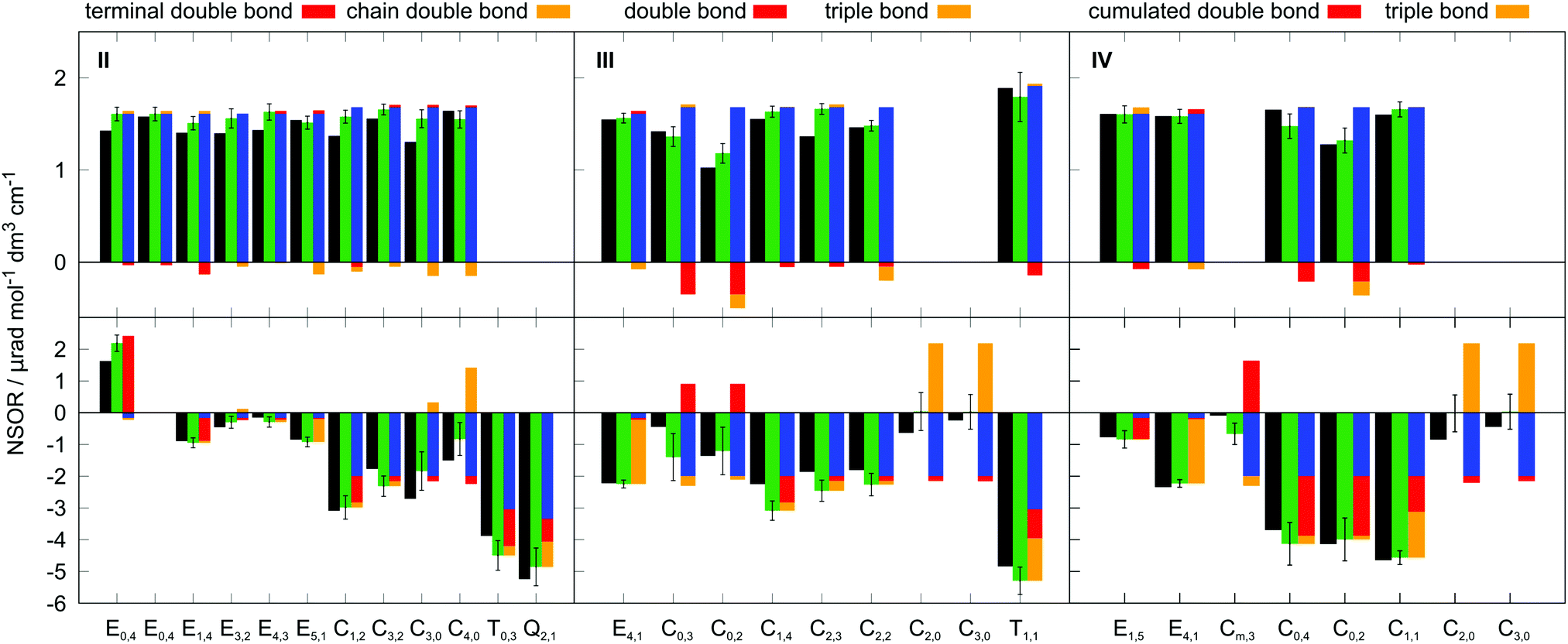 | ||
| Fig. 8 Results of calculations of 1H (top) and 13C (bottom) NSOR for the test molecules II (left), III (middle) and IV (right). The black bars show NSOR calculated directly for the whole molecule, and the green bars correspond to the estimates from the additive model, together with the standard deviation. The stacked bars show the contributions from which the green bar was constructed: the blue bars represent the baseline values of alkanes, and the red and orange are contributions from the chromophores labeled above each panel. The subscripts in nucleus labels indicate the distance from the chromophores in order or red contribution, orange contribution. | ||
Overall a good agreement is found for all three molecules. Similarly as in all other cases, the 13C NSOR covers a wider range of values compared to 1H, which is dominated by the alkane contribution and is only slightly changed by the influence of the chromophores. On the other hand, 13C nuclei close to the unsaturated systems see significant shifts from the alkane baseline. In general, the corrections shift the NSOR towards the values obtained from the full calculations for majority of cases. The most notable exceptions are the C3,0 and C4,0 nuclei in molecule II. This disagreement can be attributed to the discussed wide spread of NSOR values of C0 in alkenes, which can shift towards both positive and negative with respect to alkanes. In this particular case the influence of the double bond is in the opposite direction than the approximate average contribution for C0 used in the model. This is also reflected in the large error bar for these nuclei. Nevertheless, the model provides a reasonable agreement with the full quantum-chemical calculation and can be used for pre-screening estimation of NSOR in molecules of potential experimental interest before expensive response function-based computations.
4 Conclusion
Nuclear spin-induced optical rotation (NSOR) of 1H and 13C nuclei was investigated by density functional theory computations on a set of hydrocarbon molecules with various structural motifs. The results were discussed with respect to the local chemical environment of the nuclei. It is shown that NSOR is sensitive in a systematic manner to the position of the nucleus in the carbon backbone, isomerism, presence of a particular chromophore in the molecule and the distance of the nucleus from it. Different combinations of these features give rise to a wide range of NSOR responses and characteristic spectroscopic features for molecular structure elucidation. Further patterns might be revealed by extending the study beyond hydrocarbons to molecules containing functional groups with heteroatoms.In addition, it has been demonstrated that NSOR can be to a good approximation modelled as an additive property, where the signal of each nucleus can be constructed by adding contributions from the neighbouring chemical groups. This allows one to quickly estimate NSOR with a reasonable accuracy without a need for extensive quantum-chemical calculations.
The results provide a proof of concept that the NSOR effect correlates with the underlying molecular structure and therefore could be experimentally exploited as a basis for a viable spectroscopic method.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author is grateful for the financial support from Academy of Finland (Grant 316180). The support from the Kvantum institute (University of Oulu) and grants of computer capacity from the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533) are also acknowledged. The author thanks Sarah Mailhiot for reviewing the manuscript for clarity.Notes and references
- J. Keeler, Understanding NMR Spectroscopy, John Wiley & Sons, 2010 Search PubMed.
- P. T. Callaghan, Translational Dynamics and Magnetic Resonance, Oxford University Press, 2011 Search PubMed.
- E. Haake, R. Brown, M. Thompson and R. Venkatesan, Magnetic Resonance Imaging. Physical Principles and Sequence Design, John Wiley & Sons, Inc., 1999 Search PubMed.
- I. M. Savukov, S. Lee and M. V. Romalis, Nature, 2006, 442, 1021–1024 CrossRef CAS PubMed.
- D. Pagliero, W. Dong, D. Sakellariou and C. A. Meriles, J. Chem. Phys., 2010, 133, 154505 CrossRef PubMed.
- D. Pagliero and C. A. Meriles, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19510–19515 CrossRef CAS PubMed.
- I. M. Savukov, H. Chen, T. Karaulanov and C. Hilty, J. Magn. Reson., 2013, 232, 31–38 CrossRef CAS PubMed.
- Y. Zhu, Y. Gao, S. Rodocker, I. Savukov and C. Hilty, J. Phys. Chem. Lett., 2018, 9, 3323–3327 CrossRef CAS PubMed.
- I. Savukov, D. Filin, Y. Zhu, R. Castro and C. Hilty, Eur. Phys. J. D, 2019, 73, 156 CrossRef.
- J. Shi, S. Ikäläinen, J. Vaara and M. V. Romalis, J. Phys. Chem. Lett., 2013, 4, 437–441 CrossRef CAS PubMed.
- P. Štěpánek and A. M. Kantola, J. Phys. Chem. Lett., 2019, 10, 5458–5462 CrossRef PubMed.
- S. Ikäläinen, P. Lantto and J. Vaara, J. Chem. Theory Comput., 2012, 8, 91–98 CrossRef PubMed.
- T. S. Pennanen, S. Ikäläinen, P. Lantto and J. Vaara, J. Chem. Phys., 2012, 136, 184502 CrossRef PubMed.
- S. Ikäläinen, M. V. Romalis, P. Lantto and J. Vaara, Phys. Rev. Lett., 2010, 105, 153001 CrossRef PubMed.
- G. Yao, M. He, D. Chen, T. He and F. Liu, Chem. Phys., 2011, 387, 39–47 CrossRef CAS.
- J. Vähäkangas, P. Lantto and J. Vaara, J. Phys. Chem. C, 2014, 118, 23996–24005 CrossRef.
- F. Chen, G. Yao, T. He, D. Chen and F. Liu, Chem. Phys., 2014, 453, 57–61 CrossRef.
- Y. Zhu, C. Hilty and I. Savukov, eMagRes, 2019, 8, 205–214 CAS.
- J. Vaara, A. Rizzo, J. Kauczor, P. Norman and S. Coriani, J. Chem. Phys., 2014, 140, 134103 CrossRef PubMed.
- M. Straka, P. Štěpánek, S. Coriani and J. Vaara, Chem. Commun., 2014, 50, 15228–15231 RSC.
- F. Chen, G. Yao, Z. Zhang, F. Liu and D. Chen, ChemPhysChem, 2015, 16, 1954–1959 CrossRef CAS PubMed.
- P. Štěpánek, S. Coriani, D. Sundholm, V. A. Ovchinnikov and J. Vaara, Sci. Rep., 2017, 7, 46617 CrossRef PubMed.
- P. Štěpánek and S. Coriani, Phys. Chem. Chem. Phys., 2019, 21, 18082–18091 RSC.
- T. Lu, M. He, D. Chen, T. He and F. Liu, Chem. Phys. Lett., 2009, 479, 14–19 CrossRef CAS.
- L. Fu and J. Vaara, J. Chem. Phys., 2013, 138, 204110 CrossRef PubMed.
- L. Fu and J. Vaara, ChemPhysChem, 2014, 15, 2337–2350 CrossRef CAS PubMed.
- G. Yao, M. He, D. Chen, T. He and F. Liu, ChemPhysChem, 2012, 13, 1325–1331 CrossRef CAS PubMed.
- L. Fu and J. Vaara, J. Chem. Phys., 2014, 140, 024103 CrossRef PubMed.
- L. Fu, A. Rizzo and J. Vaara, J. Chem. Phys., 2013, 139, 181102 CrossRef PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- TURBOMOLE V7.4 2019, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Phys. Chem., 1994, 100, 5829 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- DALTON, a molecular electronic structure program, Release DALTON2017 (2017), see http://daltonprogram.org.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekstrom, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernandez, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hattig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. K. H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS.
- P. Sałek, O. Vahtras, T. Helgaker and H. Ågren, J. Chem. Phys., 2002, 117, 9630 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- P. Manninen and J. Vaara, J. Comput. Chem., 2006, 27, 434–445 CrossRef CAS PubMed.
- J. Vähäkangas, S. Ikäläinen, P. Lantto and J. Vaara, Phys. Chem. Chem. Phys., 2013, 15, 4634–4641 RSC.
- L. Frediani, H. Ågren, L. Ferrighi and K. Ruud, J. Chem. Phys., 2005, 123, 144117 CrossRef PubMed.
- P. Štěpánek, Dataset: Nuclear spin-induced optical rotation of functional groups in hydrocarbons, http://urn.fi/urn:nbn:fi:att:ff3a6b18-e02d-4df1-b7a3-2b4295df4055, 2020.
- E. F. Pettersen, T. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and R. E. Ferrin, J. Comput. Chem., 2004, 13, 1605–1612 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: List of molecules, basis set benchmark, additional figures discussing the effects of molecular conformation, isomerism and solvent on the NSOR signal. See DOI: 10.1039/d0cp02856h |
| This journal is © the Owner Societies 2020 |