
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe unique catalytic role of water in aromatic C–H activation at neutral pH: a combined NMR and DFT study of polyphenolic compounds†
Aneela
Fayaz
a,
Michael G.
Siskos
*b,
Panayiotis C.
Varras
 b,
M. Iqbal
Choudhary
acd,
Atia-tul-Wahab
c and
Gerothanassis P.
Ioannis
*abc
b,
M. Iqbal
Choudhary
acd,
Atia-tul-Wahab
c and
Gerothanassis P.
Ioannis
*abc
aH.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan
bSection of Organic Chemistry and Biochemistry, Department of Chemistry, University of Ioannina, Ioannina, GR-45110, Greece. E-mail: igeroth@uoi.gr; msiskos@uoi.gr
cPanjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan
dDepartment of Biochemistry, Faculty of Science, King Abdul-Aziz University, Jeddah-21589, Saudi Arabia
First published on 9th July 2020
Abstract
Direct activation of aromatic C–H bonds in polyphenolic compounds in a single step, without the use of late transition metals, is demonstrated with the use of D2O and common phosphate buffer at neutral pD and near ambient temperatures. Detailed variable temperature and pD 1H NMR studies were carried out to investigate, for the first time, the Gibbs activation energy (ΔG‡), the activation enthalpy (ΔH‡), and activation entropy (TΔS‡) of H/D exchange reactions of the natural product catechin and the model compounds resorcinol and phloroglucinol. NMR and DFT calculations support a catalytic cycle comprising two water molecules in a keto–enol tautomeric process. The reduction of ΔG‡ values due to the catalytic role of two molecules of water by a factor of 20–30 kcal mol−1 and the resulting acceleration of the H/D exchange rate by a factor of 1020–1030 should be compared with a minor reduction in ΔG‡ of 0.4 to 4.5 kcal mol−1 due to the effect of an additional electron donating oxygen group and the deprotonation of OH groups. It can therefore be concluded that although the H/D exchange process can be accelerated by a small amount of an acid or a base to break a C–H bond, water as a catalyst plays the major role. This approach opens a new vistas for the combined use of NMR and DFT studies as tools to understand the molecular basis of the catalytic role of water.
Introduction
The growing interest in H/D-exchange reactions at carbon centers1,2 is due to intensive basic research on C–H bond activation,3–5 in mechanistic investigations on catalysts and reaction pathways,6–8 and the increasing demand for isotopically labeled compounds as reference materials in mass spectrometry and for pharmacokinetic and metabolic studies.9,10 The methods for H/D exchange may be broadly divided into two classes, pH-dependent H/D exchange and metal-catalyzed H/D exchange. Acid-catalyzed methods, through an electrophilic aromatic substitution reaction mechanism, have been utilized with strong deuterated Brønsted acids or alternatively Lewis acids, in combination with a deuterium source.11–13 Base-catalyzed H/D-exchange reactions can also provide a convenient method for the exchange of acidic hydrogen atoms by means of keto–enol equilibria in a variety of carbonyl compounds.14–16A central component of the methodological developments of H/D and H/T exchange was the study of metal-catalyzed activations, thus resulting in exchange reactions under milder conditions in sensitive classes of compounds.17–21 Since the seminal research studies in the late 1960s on H/D exchange by homogeneous catalysis, many efficient methods have been developed which allow a high degree of deuteration in both aromatic and aliphatic substrates.22–25 An important technical advantage was the development of heterogeneous catalysis due to the possibility to remove the catalyst by simple filtration at the end of the reaction. However, side reactions such as the formation of products of dehalogenation, hydrogenation, and hydrolysis, as well as epimerization and racemization have been frequently encountered.26
Traditionally most organic catalytic reactions have been conducted in aprotic or non-polar organic solvents. These notions, arising mainly from reactivity and solubility considerations, have been modified with the discovery of the effect of water as a solvent in a variety of organic reactions.27 Water can have a significant catalytic effect because it can act as both a proton donor and a proton acceptor and, thus, mediates intra- and inter-molecular proton transfer reactions. In particular, a great importance has been attributed to the explicit role of one or more water molecules shuttling the proton between various reaction sites. Nevertheless, despite the considerable efforts directed into understanding the physicochemical basis of the rate acceleration in homogeneous and heterogeneous water reactions,27–35 the nature of the interactions that influence Gibbs activation energy and, thus, reactivity was not fully understood.36
Herein, we describe the investigation of the unique catalytic role of H2O in aromatic C–H activation of a natural product catechin (a) and the model compounds resorcinol (b) and phloroglucinol (c) (Fig. 1) through keto–enol tautomerization at neutral pH and near ambient temperatures with the use of variable temperature and pD NMR studies and DFT calculations.
 | ||
| Fig. 1 Chemical structures of catechin (a), resorcinol (b), and phloroglucinol (c). | ||
Results and discussion
NMR studies
It has been suggested that the mechanism of H/D exchange reaction of aromatic C–H protons in acid or basic conditions of polyphenolic compounds37–41 follows a two-step process,41 as shown in Fig. 2. In the first step, the solvent D+ is transferred to the aromatic carbon atom with the formation of an intermediate σ-complex carbocation. In the second step, the hydrogen atom linked to the same carbon is eliminated as H+ from the σ-complex. Furthermore, it was found that oxygen substituents on the aromatic ring are expected to stabilize the σ-complex carbocation intermediate42 and, thus, to increase the exchange rate. | ||
| Fig. 2 Suggested mechanism of the H/D exchange process of aromatic C–H protons of flavonoids.41 | ||
Due to the mesomeric effects of electron-donating substitution groups, such as hydroxyl groups, the aromatic rings of flavonoids have certain electronic density at specific sites. For instance, the substitution pattern of ring A can facilitate the electrophilic aromatic substitution reactions involving hydrogen exchange with deuterium at a particular carbon atom. In a basic solution, a path for exchange via a ketone–enolate intermediate has been suggested,43,44 which also results in the formation of σ-complexes.
The H/D exchange reaction of catechin (a), resorcinol (b), and phloroglucinol (c) (2.5 mM) was investigated using variable temperature and pD 1D 1H NMR spectroscopy. The NMR experiments were performed with the use of phosphate buffer (25 mM) in order to maintain a constant ionic strength and minimize temperature variation of the pD of solution due to the pKa values of the compounds investigated45–47 (Table S1 in ESI†). Fig. 3 illustrates the expanded region of the 1H NMR spectra of catechin (a) recorded at different time intervals. The assignment of the resonances was confirmed using 2D 1H–13C HSQC and HMBC experiments. The H2′, H5′ and H6′ resonances of ring B, which are well separated from the other aromatic signals, do not show H/D exchange and, thus, were used as an internal integration reference. In contrast, H/D exchange at C(8)–H and C(6)–H of ring A was clearly observed even at pD = 6.92 and 7.93 and at 298 K. The pD values of the solutes in D2O were estimated using the following relationship:48
| pD = pH (reading) + 0.4 | (1) |
 | ||
| Fig. 3 Selected 1D 1H NMR (400 MHz) spectral range illustrating C(6)–H and C(8)–H H/D exchange of 2.5 mM catechin (a) in D2O, T = 298 K and 25 mM phosphate buffer at pD = 6.92 (a), pD = 7.93 (b), and pD = 8.88 (c). The spectra were recorded at time intervals of 6.6 min. Total experimental time 8 h, and 48 min for (a–c), respectively. | ||
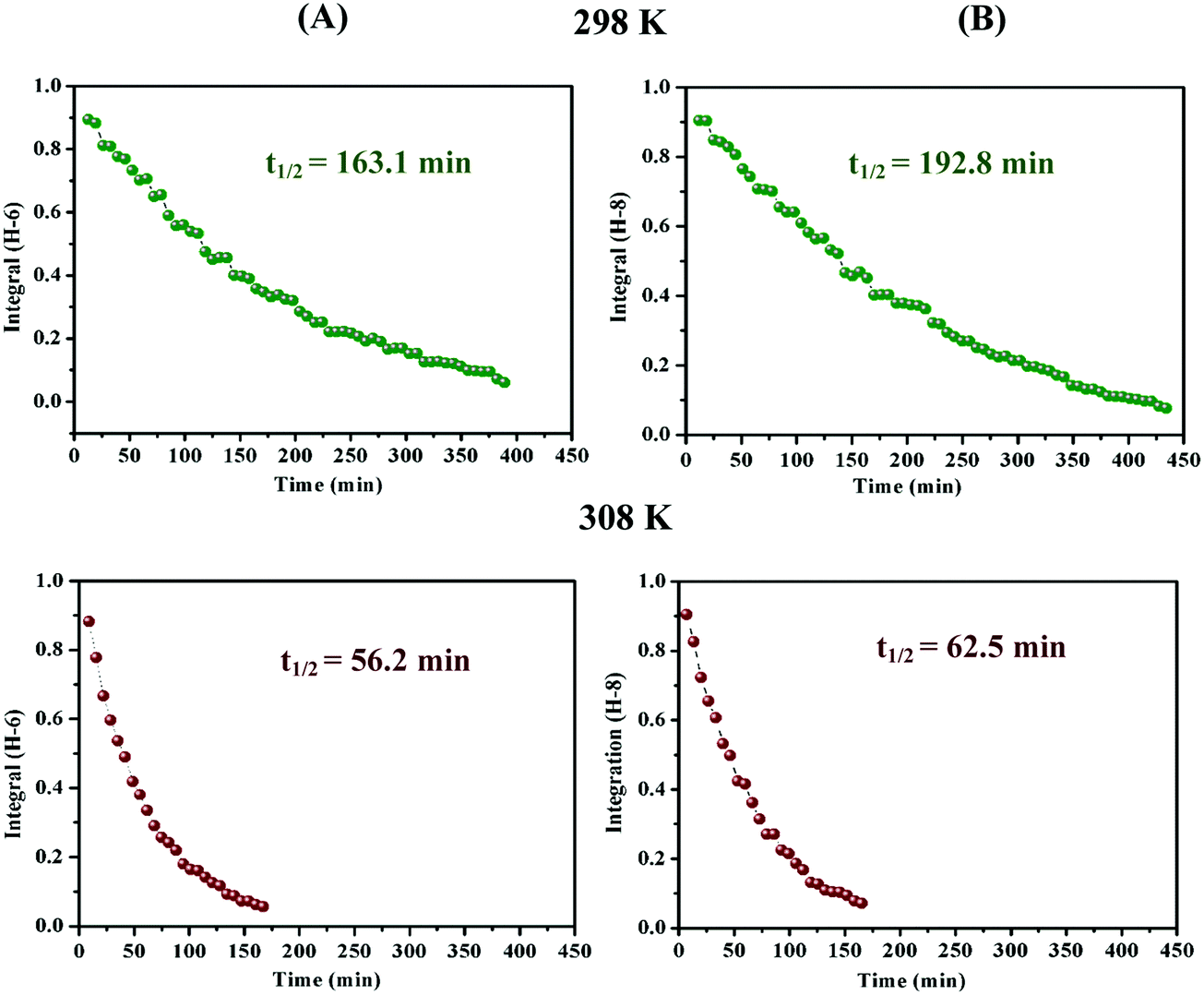 | ||
| Fig. 4 C-6 (A) and C-8 (B) H/D exchange kinetic curves of 2.5 mM catechin (a) in D2O, phosphate buffer solution (25 mM), pD = 6.93 at 298 K and 308 K. | ||
 | ||
| Fig. 5 Composite C(2)–H, C(4)–H and C(6)–H H/D exchange kinetic curves of 2.5 mM phloroglucinol (c) in D2O, phosphate buffer solution (25 mM): (A) pD = 6.94 at 298 K (a) and 303 K (b), (B) pD = 7.94 at 298 K (a) and 303 K (b), (C) D = 8.94 at 293 K (a) and 298 K (b). | ||
 | ||
| Fig. 6 Selected 1D 1H NMR (400 MHz) spectral range illustrating the C(2)–H and C(4,6)–H H/D exchange of 2.5 mM resorcinol (b) in D2O, T = 333 K, and 25 mM phosphate buffer solution, pD = 9.91. The spectra were recorded at time intervals of 0 h (a), 10 h (b), 20 h (c), and 30 h (d). | ||
According to the linear form of the Eyring equation:
 | (2) |
 plot. Eqn (2) is a straight line with a negative slope,
plot. Eqn (2) is a straight line with a negative slope,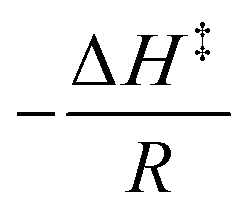 , and a y-intercept
, and a y-intercept  . The Gibbs energy of activation, ΔG‡ (kcal mol−1), is given by
. The Gibbs energy of activation, ΔG‡ (kcal mol−1), is given by| ΔG‡ = ΔH‡ − TΔS‡ | (3) |
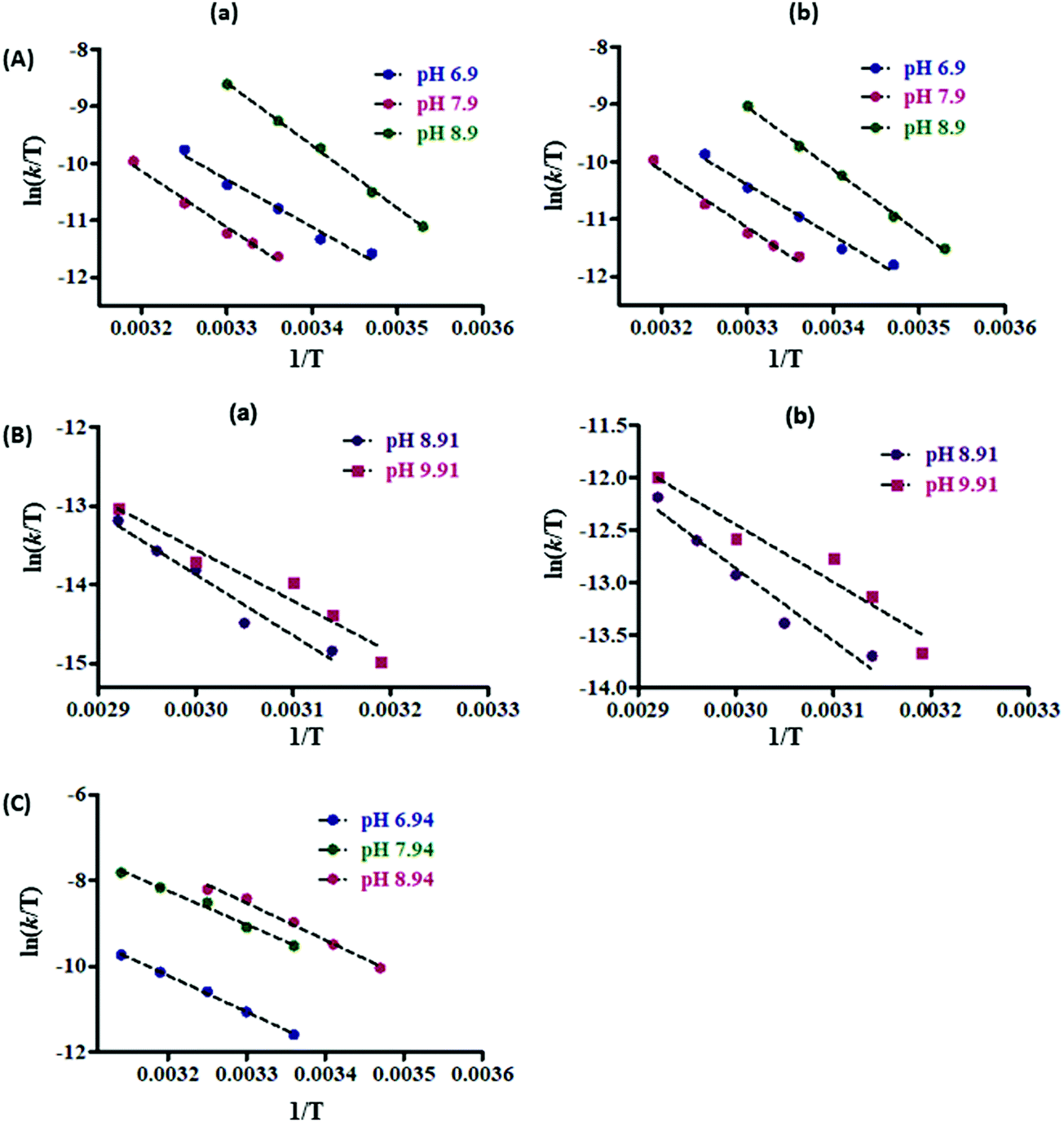 | ||
| Fig. 7 Eyring plots of (A) C(6)–H (a) and C(8)–H (b) protons of 2.5 mM catechin (a); (B) C(2)–H (a), and C(4,6)–H (b) protons of 2.5 mM resorcinol (b), and (C) C(2,4,6)–H protons of 2.5 mM phloroglucinol (c) in D2O, phosphate buffer solution (25 mM) at various pD values. | ||
| Compound | pD | ΔH‡ (kcal mol−1) | −TΔS‡ (kcal mol−1) | ΔG‡ (kcal mol−1) | ΔH‡ (kcal mol−1) | −TΔS‡ (kcal mol−1) | ΔG‡ (kcal mol−1) |
|---|---|---|---|---|---|---|---|
| Catechin | C(8)–H | C(6)–H | |||||
| 6.90 | 17.25 ± 1.31 | 3.24 ± 0.39 | 20.50 | 16.00 ± 1.32 | 4.42 ± 0.60 | 20.42 | |
| 7.90 | 18.43 ± 1.63 | 2.52 ± 0.33 | 20.96 | 18.03 ± 1.56 | 2.90 ± 0.37 | 20.94 | |
| 8.90 | 20.38 ± 0.65 | −0.64 ± 0.02 | 19.74 | 20.93 ± 0.66 | −1.43 ± 0.06 | 19.50 | |
| Resorcinol | C(2)–H | C(4,6)–H | |||||
| 8.91 | 15.14 ± 1.92 | 8.74 ± 2.94 | 23.88 | 13.72 ± 1.79 | 9.41 ± 3.47 | 23.13 | |
| 9.91 | 12.91 ± 1.67 | 10.56 ± 4.81 | 23.47 | 11.48 ± 1.64 | 11.20 ± 5.10 | 22.68 | |
| Phloroglucinol | C(2,4,6)–H | ||||||
| 6.94 | 17.46 ± 0.30 | 3.50 ± 0.09 | 20.96 | ||||
| 7.94 | 16.05 ± 0.78 | 3.69 ± 0.26 | 19.74 | ||||
| 8.94 | 16.55 ± 1.15 | 2.86 ± 0.29 | 19.41 | ||||
Literature pKa values45–47 for the molecules investigated in the present work are shown in Table S1 in ESI.† Catechin (a) exists in the uncharged (non-deprotonated) state at pD = 6.9; at pD = 7.9, ∼20% of the molecules exist in the mono-deprotonated form, and at pD = 8.9, nearly 60% of the molecules exist in the mono-deprotonated form. At pD = 7.9, the Gibbs activation enthalpy of catechin (a) is slightly larger. At pD = 8.9, the exchange rate of catechin (a) increases significantly, and the Gibbs activation energy is reduced by 0.76 and 0.92 kcal mol−1 for C-8 and C-6 protons, respectively. This demonstrates that the deprotonation of the OH group can result in the reduction of the ΔG‡ values. Phloroglucinol (c) at pD = 6.94 exists mainly (>90%) in the uncharged state; at pD = 7.94, 40% of the molecules exist in the mono-deprotonated state which results in the reduction of the ΔG‡ values by ∼1.2 kcal mol−1. At pD = 8.94, one of the OH groups is 90% and a second one is 40% in the deprotonated states. Despite the subtle changes in the ionization state, the ΔG‡ value reduces by only ∼1.55 kcal mol−1 which is slightly larger compared with the results obtained for catechin (a). Resorcinol (b) at pD = 8.91 exists essentially in the uncharged state; at pD = 9.91, 70% of the molecules exist in the mono-deprotonated state but the reduction of the ΔG‡ value for both C-2 proton and the C-4 and C-6 protons is only 0.4 to 0.5 kcal mol−1. This demonstrates that the acceleration of the H/D exchange rate and, thus, the reduction in ΔG‡ values of catechin (a) and phloroglucinol (c) upon deprotonation of an OH group are not only due to the synergistic effect of the two –OH groups in meta position, but also due to the presence of an additional electron donating oxygen group.
Several experiments were also performed at constant concentration (2.5 mM) of resorcinol and catechin with variable concentration of the phosphate buffer (c = 5 mM, 25 mM and 50 mM at pD = 7.5 and C = 5 mM, 25 mM and 100 mM at pD = 9.6). The maximum reduction of the ΔG‡ values at high buffer concentration was found to be ≤1.5 kcal mol−1. This demonstrates a minor catalytic effect of the phosphate buffer.
DFT mechanistic studies
Our experimental NMR results that H/D substitution reactions can be performed at neutral pD and at ambient temperatures clearly demonstrate that water can play a major catalytic role. An alternative reaction mechanism, therefore, via a keto–enol tautomerization was investigated with quantum chemical methods in which a water-assisted proton transfer takes place from the phenol OH group to the α-carbon.The catalytic effect of water molecules in resorcinol (b) was investigated: (i) at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level of theory,50 by incorporating also the semi-empirical Grimme's dispersion correction GD3BJ,51 and (ii) at the M0-62X/6-31+G(d)52 and ωB97XD/6-31+G(d)53 level with van der Waals functionals. The following cases were investigated: (i) in vacuum, (ii) in IEF-PCM (water) without explicit water molecules, and (iii) in IEF-PCM (water) in the presence of one, two, and four explicit molecules of water (Table 2 and Tables S3, S4 in ESI†). In all cases the keto–enol species are in thermodynamic equilibrium with overwhelming preference of the phenol-type tautomer.
| Functional | Complex | Group | ΔH‡, kcal mol−1 | −TΔS‡, kcal mol−1 | ΔG‡, kcal mol−1 |
|---|---|---|---|---|---|
| B3LYP/6-31+G(d) (IEF-PCM water) | Resorcinol (in–out conformer) | C(2)–H | 61.7 | 0.3 | 62.0 |
| C(4,6)–H | 61.0 | 0.4 | 61.4 | ||
| B3LYP/6-31+G(d) (IEF-PCM water) | Resorcinol + 2H2O (in–out conformer) | C(2)–H | 24.8 | 3.7 | 28.5 |
| C(4,6)–H | 26.0 | 5.0 | 31.0 | ||
| B3LYP-GD3BJ/6-31+G(d) (IEF-PCM water) | Resorcinol + 2H2O (in–out conformer) | C(2)–H | 22.1 | 3.5 | 25.6 |
| C(4,6)–H | 22.3 | 3.4 | 25.7 | ||
| B3LYP/6-31+G(d) (IEF-PCM water) | Resorcinol + 4H2O (in–out conformer) | C(2)–H | 21.4 | 7.5 | 28.9 |
| C(4,6)–H | 27.2 | 2.5 | 29.7 | ||
| B3LYP-GD3BJ/6-31+G(d) (IEF-PCM water) | Resorcinol + 4H2O (in–out conformer) | C(2)–H | 22.5 | 4.8 | 27.3 |
| C(4,6)–H | 26.6 | 2.0 | 28.6 | ||
| B3LYP/6-31+G(d) (IEF-PCM water) | Resorcinol + 4H2O (in–in conformer) | C(2)–H | 23.8 | 2.5 | 26.3 |
| C(4,6)–H | — | — | — | ||
| B3LYP-GD3BJ/6-31+G(d) (IEF-PCM water) | Resorcinol + 4H2O (in–in conformer) | C(2)–H | 22.1 | 2.1 | 24.2 |
| C(4,6)–H | — | — | — | ||
| B3LYP/6-31+G(d) (IEF-PCM water) | Phloroglucinol + 2H2O | C(2,4,6)–H | 26.8 | 2.1 | 28.9 |
| B3LYP-GD3BJ/6-31+G(d) (IEF-PCM water) | Phloroglucinol + 2H2O | C(2,4,6)–H | 19.6 | 3.6 | 23.2 |
| B3LYP/6-31+G(d) (IEF-PCM water) | Catechin + 2H2O | C(6)–H | 23.5 | 4.3 | 27.8 |
| C(8)–H | 23.1 | 2.4 | 25.5 |
For the ground state of resorcinol (b), three orientations of the two –OH groups in meta position were investigated (Fig. S3 in ESI†). The phenolic OH groups were found to invariably lie in the plane of the aromatic ring. The homodrome configuration54 (in–out) of the two –OH groups was found to be of lower energy. The resulting activation energies of this conformer in vacuum were found to be ΔG‡ (C(2)–H) = 62.5 kcal mol−1 and 50.9 kcal mol−1, and ΔG‡ (C(4,6)–H) = 61.6 kcal mol−1 and 50.1 kcal mol−1, with optimization of the structures at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level, respectively. These values were significantly reduced in the water continuum model (IEF-PCM) and in the presence of a single explicit water molecule: ΔG‡ (C(2)–H) = 38.2 kcal mol−1 and 36.0 kcal mol−1 and ΔG‡ (C(4,6)–H) = 37.6 kcal mol−1 and 35.5 kcal mol−1, with optimization of the structures at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level, respectively (Table S2 in ESI†). In the presence of two explicit water molecules, further significant reduction in ΔG‡ values was obtained: ΔG‡ (C(2)–H) = 28.5 kcal mol−1 and 26.2 kcal mol−1 and ΔG‡ (C(4,6)–H) = 31.0 kcal mol−1 and 28.4 kcal mol−1 with optimization of the structures at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level, respectively (Table 2, Table S3 and Fig. S4 in ESI†).
The use of the empirical Grimme's dispersion correction GD3BJ51 results in further improvements of the calculated ΔG‡ values. Thus, at the B3LYP-GD3BJ/6-31+G(d) (IEF-PCM water) level, the computational values ΔG‡ (C(2)–H) = 25.6 kcal mol−1 and ΔG‡ (C(4,6)–H) = 25.7 kcal mol−1 (Table 2) are in quantitative agreement with the experimental values ΔG‡ (C(2)–H) = 23.88 kcal mol−1 and ΔG‡ (C(4,6)–H) = 23.13 kcal mol−1 (Table 1). Similar results were obtained at the PBE1PBE-GD3BJ/6-31+G(d) (IEF-PCM water) level (Table S3 in ESI†). In contrast, the computational values ΔG‡ at the M0-62X/6-31+G(d) (IEF-PCM water) and ωB97XD/6-31+G(d) (IEF-PCM water) level with van der Waals functionals were found to deviate from the experimental data (Table S4 in ESI†). The above results clearly demonstrate the advantages of the semi-empirical Grimme's dispersion correction,51 and the unique catalytic role of two water molecules in reducing the activation energies ΔG‡. In the transition state, the phenol –OH group and the two water molecules provide one proton for a hydrogen bond to C-2 carbon in a homodrome configuration.54 This imposes for the water molecules a rotation so that molecule 1 directs a proton to molecule 2 and molecule 2 to C-2 between the two –OH groups in meta position (Fig. 8).
 | ||
| Fig. 8 Aromatic C–H activation of resorcinol in the presence of two (a) and four (b) bound molecules of D2O. | ||
Mehr et al.55 investigated deuteration of aromatic rings under very mild conditions through keto-enamine tautomeric equilibrium. It was found that a chain of five water molecules has a significant role in lowering the activation barrier. This particular hydrogen bond framework, which can afford activation barriers as low as 27.3 kcal mol−1, was considered to be indispensable since several calculations using fewer than five molecules of H2O raised ΔG‡ values considerably. In order to clarify the above issue, we performed additional computations for resorcinol with four discrete molecules of H2O at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level. With both functionals, the ΔG‡ values increased relative to those with two discrete molecules of H2O. Inclusion of the semi-empirical Grimme's dispersion correction GD3BJ did not improve the results (Table 2 and Table S3 in ESI†). In order to get computational results comparable to those of the experimental data, it was necessary to use the higher energy in–in conformer (Fig. S3 in ESI†, Table 2 and Table S3 in ESI†). From the above it can be concluded that two and not four discrete molecules of H2O are catalytically important (Fig. 8).
The transition state (TS) structure of resorcinol (b) in the presence of two and four discrete water molecules is shown in Fig. 9. The geometric arrangements are the same either in the gas phase or in aqueous solution. The normal modes, corresponding to the reaction coordinate, for the TS structures with two and four discrete water molecules in aqueous solution associated with the imaginary or negative frequency were found to be ν‡ = 1001.03i cm−1 and 381.44i cm−1 respectively. By comparing the numerical values of the imaginary frequencies it can be concluded that the imaginary frequency for the transition state structure with two discrete water molecules is greater by a factor of ∼2.62 compared to the one with four water molecules. Examination of the corresponding vibrational modes for Fig. 9(a) shows that the greatest movements involve the hydrogen atoms H2, H5 and H3. This indicates that the geometric distortion of the solvation complex of resorcinol (b) + 2H2O is relatively large particularly with respect to the normal mode concerning the atom H2 which has the largest magnitude (|![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_char_0076_20d1.gif) H2| = 0.929 Å). The geometric distortions in Fig. 9(b) involve the atoms H′2, H′5 and H′3. However, inspection of all normal modes for the two transition states shows that in Fig. 9(b), the two extra water molecules (denoted O′3 and O′4) do not participate in the exchange mechanism. Hence, the first TS involves the optimal number of two water molecules, given also the fact of the greater value of the imaginary frequency. Consequently, the TS involving four water molecules becomes of secondary importance. A rigorous proof of the above transition state (TS) was obtained through an IRC (Intrinsic Reaction Coordinate) calculation,56–58 as shown in Fig. 10. This type of computation starts from the transition state and examines the reaction path on the potential energy surface (PES) on either side of the TS, leading down to reactants and products. The IRC curve was obtained using the LQA (Local Quadratic Approximation) algorithm,59,60 where the reaction path is represented as a Taylor series expansion of the arc length truncated to second order.
H2| = 0.929 Å). The geometric distortions in Fig. 9(b) involve the atoms H′2, H′5 and H′3. However, inspection of all normal modes for the two transition states shows that in Fig. 9(b), the two extra water molecules (denoted O′3 and O′4) do not participate in the exchange mechanism. Hence, the first TS involves the optimal number of two water molecules, given also the fact of the greater value of the imaginary frequency. Consequently, the TS involving four water molecules becomes of secondary importance. A rigorous proof of the above transition state (TS) was obtained through an IRC (Intrinsic Reaction Coordinate) calculation,56–58 as shown in Fig. 10. This type of computation starts from the transition state and examines the reaction path on the potential energy surface (PES) on either side of the TS, leading down to reactants and products. The IRC curve was obtained using the LQA (Local Quadratic Approximation) algorithm,59,60 where the reaction path is represented as a Taylor series expansion of the arc length truncated to second order.
 | ||
| Fig. 9 The transition state with normal mode vectors of resorcinol (b) in the presence of two (a) and four (b) discrete water molecules. | ||
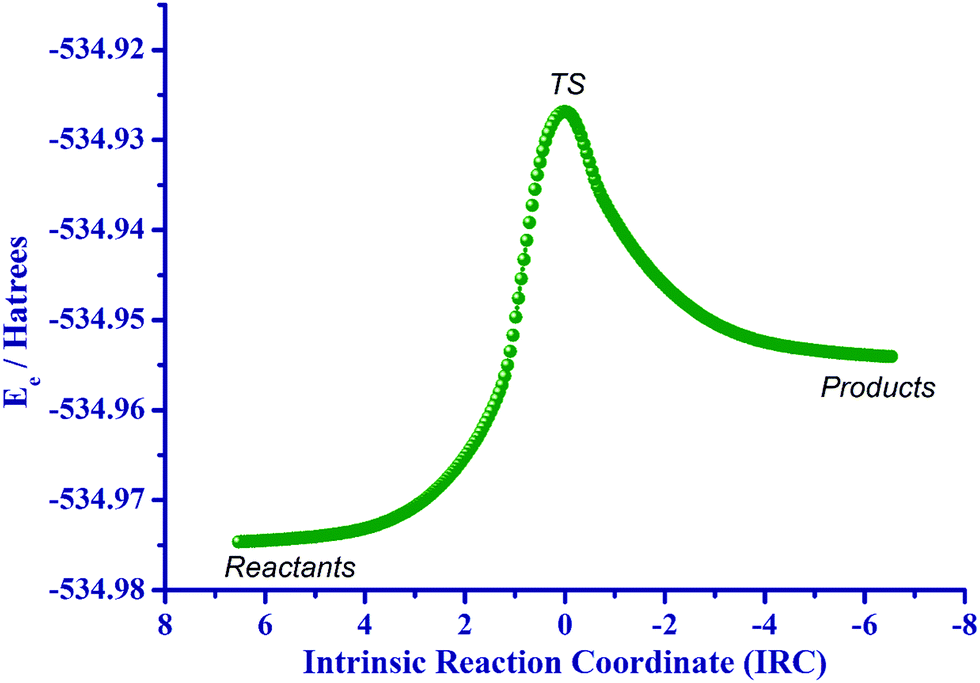 | ||
| Fig. 10 IRC path in the gaseous phase from the transition state towards reactants (resorcinol (b) plus two discrete water molecules) and products (keto product). | ||
Similar DFT calculations were carried out for phloroglucinol (c). In the lower energy ground state of phloroglucinol (c), the three –OH groups were found to be in a homodrome configuration and in the plane of the aromatic ring. As in the case of resorcinol (b), activation energies were significantly reduced in the water continuum model (IEF-PCM) and in the presence of two explicit water molecules (Fig. S5 in ESI†). Interestingly at the B3LYP-GD3BJ/6-31+G(d) and PBE1PBE-GD3BJ/6-31+G(d) level, the resulted Gibbs activation energies (Table 2 and Tables S2, S3 in ESI†) were found to be significantly lower than those of resorcinol (b), in excellent agreement with the experimental data (Table 1).
The 2R,3S configuration was used in the DFT calculations of catechin (a).61,62 The water starting arrangement was to place both water molecules either in the vicinity of the phenol C(7)–OH and C(6)–H groups or in the vicinity of C(9)–OH and C(8)–H groups. After optimization, the water molecules turn out to be hydrogen bonded to each other. The DFT structures of the ground states of catechin (a) with respect to its transition state with two discrete molecules of H2O are illustrated in Fig. 11. Again, a significant reduction in ΔG‡ values was obtained with respect to ΔG‡ values in the absence of discrete molecules of H2O. The overall excellent agreement of computational and experimental ΔG‡ values, therefore, demonstrates a unique catalytic effect of H2O63 of over 23–30 kcal mol−1. Interestingly, the synergistic effect of two OH groups in meta position, the presence of an additional electron donating oxygen group, and the deprotonation of OH groups result only in a moderate reduction of ΔG‡ values by a factor of 0.4 to 4.5 kcal mol−1.
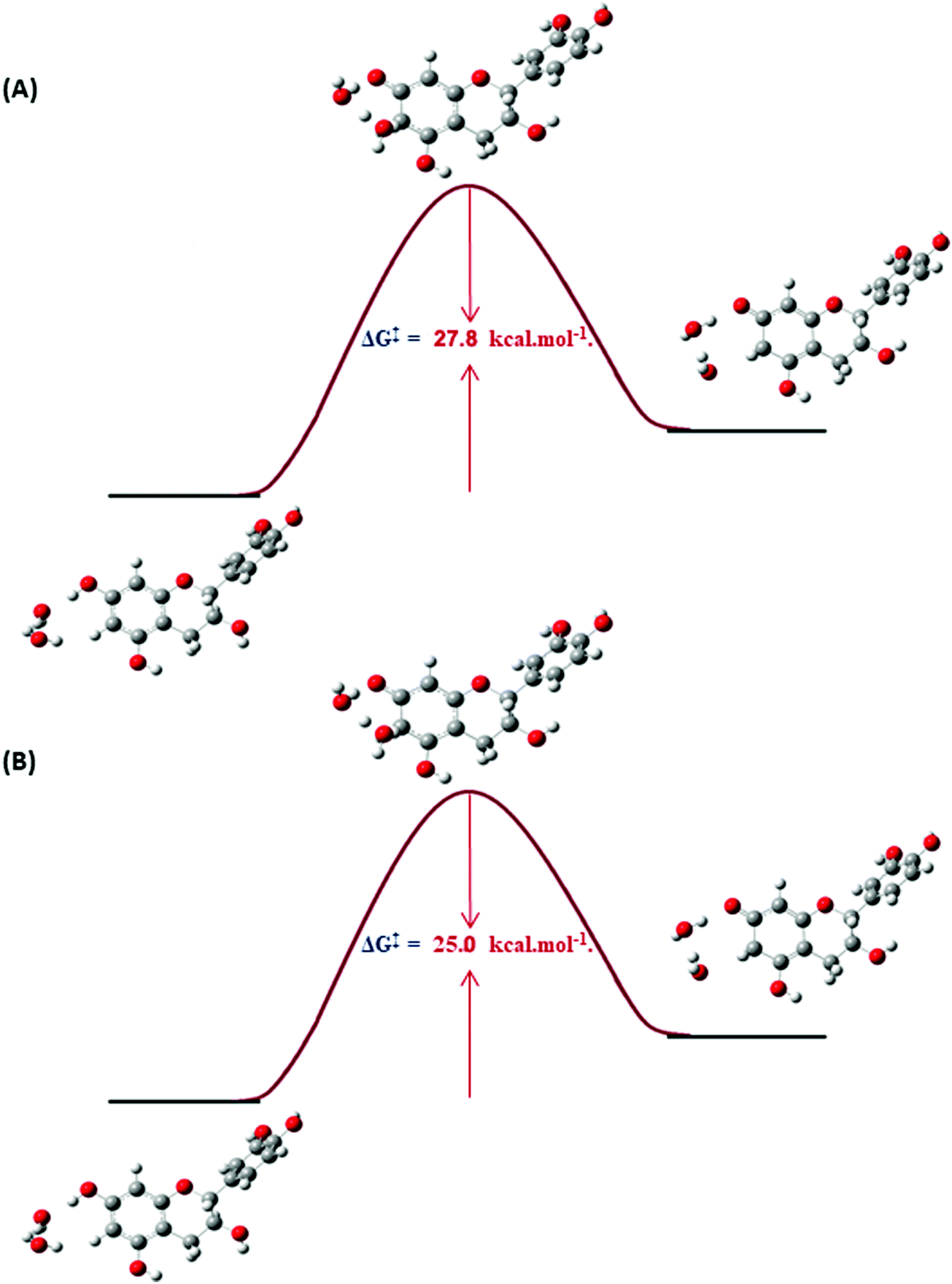 | ||
| Fig. 11 DFT structures of the ground states of catechin (a) with respect to its transition state with two discrete molecules of water at the C(6)–H proton with minimization of the structures at the DFT/B3LYP/6-31+G(d) (IEF-PCM Water) (A) and at the DFT/PBE1PBE/6-31+G(d) (IEF-PCM Water) level of theory (B). | ||
The computed values of the different contributions to the total activation entropy, ΔS‡, are shown in Table S5 (ESI†). The vibration entropy, ΔSVib, makes a major contribution to the activation energy with translational entropy, ΔSTrans, and rotational entropy, ΔSRot, playing a negligible role at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level of theory. The significant effect of vibrational entropy is in agreement with the previous analysis of Fig. 9. A natural bond orbital (NBO) analysis of catechin (a), resorcinol (b), and phloroglucinol (c) was carried out to explain the effect of charge transfer and delocalization on the H/D exchange process (Table S6, ESI†). The NBO charges, at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level, indicate a significant variation of the charges of the carbon atoms, involved in the H/D exchange process. Thus, the charge density of C(2), C(4), and C(6) of phloroglucinol (c) (−0.413) shows a significant increase with respect to C(2) (−0.380), and C(4), C(6) (−0.368) of resorcinol (b). The plot of the NBO charge densities of the aromatic carbons involved in H/D exchange vs. the respective computational or experimental Gibbs activation energies for catechin (a), resorcinol (b), and phloroglucinol (c), however, does not show any functional dependence.
Materials
All chemicals were of reagent grade and were purchased commercially.NMR spectroscopy
1H NMR spectra were obtained on Bruker AV-500 and AV-400 NMR instruments using 2.5 mM catechin (a), resorcinol (b), and phloroglucinol (c) dissolved in 25 mM phosphate buffer in D2O solutions at specified pD values.Computational details
Calculations were performed with the DFT method, with the B3LYP and PBE0 (PBE1PBE) hybrid functional as implemented in the Gaussian 09W Package.49 The molecular structures of catechin (a), resorcinol (b), and phloroglucinol (c) without and with one and two discrete water molecules were optimized at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) basis set level in the gas phase, as well as using the IEF-PCM model (Integral Equation Formalism-Polarizable Continuum Model). For resorcinol, further computations were performed at the M0-62X/6-31+G(d) and ωB97XD/6-31+G(d) level with two discrete H2O molecules and at the B3LYP-GD3BJ/6-31+G(d) and PBE1PBE-GD3BJ/6-31+G(d) level with two and four discrete H2O molecules. For phloroglucinol, further computations were performed at the B3LYP-GD3BJ/6-31+G(d) and PBE1PBE-GD3BJ/6-31+G(d) level. In order to investigate the transition state, as well as the structure of the products, calculations were carried out at the same level of theory using gas phase and IEF-PCM models without and with one, two, and four discrete water molecules. The computed geometries were verified as minima by frequency calculations at the same level of theory (no imaginary frequencies).Conclusions
We have reported that aromatic H/D substitution reactions can be performed in water at neutral pH and at near ambient temperatures by the use of water catalysis under unprecedented mild conditions. The experimental ΔG‡ values of catechin and phloroglucinol were found to be in the range of 21–19.5 kcal mol−1 at pD values of 6.9–8.8. The enthalpic ΔH‡ term was found to be the major contributor to the ΔG‡ values with a minor contribution (up to 3.7 kcal mol−1) of the entropic TΔS‡ term. In contrast, the exchange rates for H/D exchange were significantly slower in resorcinol with ΔG‡ values in the range of 22.5 to 23.9 kcal mol−1, at pD values 8.9–9.9. This demonstrates that the acceleration of the H/D exchange rate and, thus, reduction in ΔG‡ values of catechin and phloroglucinol are not only due to the synergistic effect of the two –OH groups in meta position, but also due to the presence of an additional electron donating –OH group. The experimental data were compared with DFT calculations at the B3LYP/6-31+G(d) and PBE1PBE/6-31+G(d) level of theory using the IEF-PCM model. Without explicit molecules of water, ΔG‡ values were found to be in the range of 62 to 50 kcal mol−1. In contrast, inclusion of two discrete solvation molecules of H2O results in a significant reduction of ΔG‡ values which were found to be in the range of 30 to 23 kcal mol−1. The use of the empirical Grimme's dispersion correction GD3BJ51 results in further improvement of the calculated ΔG‡ values for resorcinol and phloroglucinol which are in quantitative agreement with the experimental values. This study demonstrates that the activation enthalpy (ΔH‡), activation entropy (ΔS‡) and Gibbs activation free energy (ΔG‡) can be of predictive value for the aromatic C–H activation in polyphenolic compounds, making their physicochemical interpretation an invaluable tool for the understanding of mechanisms and reactivity. This approach is meaningful for several reasons:(i) the present data necessitate the incorporation of water catalysis (Fig. 8) in undergraduate textbooks in addition to the acid/base catalyzed H/D exchange mechanism (Fig. 2);
(ii) aqueous chemistry predominates in biological processes and, thus, the catalytic role of water may have implications for biocatalytic reactions and artificial biomimetic systems;
(iii) water catalysis by definition is a green chemistry and environmentally friendly method, since it avoids the use of dipolar aprotic solvents;
(iv) water catalysis may require lower reaction temperatures given the high concentration of D2O as a cheap and environmentally benign deuterium source which can potentially translate into large scale processes.
Author contributions
AF conducted the NMR experiments, AF, MGS and PV conducted the computations, and MGS, MIC, A-t-W and IPG conceived and designed the project, analyzed the data and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.References
- M. Siskin and A. R. Katritzky, Chem. Rev., 2001, 101, 825–835 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083–4091 CrossRef CAS.
- X. Ribas, R. Xifra, T. Parella, A. Poater, M. Solà and A. Llobet, Angew. Chem., Int. Ed., 2006, 45, 2941–2944 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2011, 110, 624–655 CrossRef PubMed.
- D. M. Marcus, K. A. McLachlan, M. A. Wildman, J. O. Ehresmann, P. W. Kletnieks and J. F. Haw, Angew. Chem., Int. Ed., 2006, 45, 3133–3136 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed.
- I. V. Tetko, P. Bruneau, H. W. Mewes, D. C. Rohrer and G. I. Poda, Drug Discovery Today, 2006, 11, 700–707 CrossRef CAS PubMed.
- R. P. Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef PubMed.
- P. M. Yavorsky and E. Gorin, J. Am. Chem. Soc., 1962, 84, 1071–1072 CrossRef CAS.
- P. McGeady and R. A. Croteau, J. Chem. Soc., Chem. Commun., 1993, 9, 774–776 RSC.
- P. S. Kiuru and K. Wähälä, Steroids, 2006, 71, 54–60 CrossRef CAS PubMed.
- P. Ryberg and O. Matsson, J. Org. Chem., 2002, 67, 811–814 CrossRef CAS PubMed.
- C. Berthelette and J. Scheigetz, J. Labelled Compd. Radiopharm., 2004, 47, 891–894 CrossRef CAS.
- T. Furuta, A. Suzuki, M. Matsuzawa, H. Shibasaki and Y. Kasuya, Steroids, 2003, 68, 693–703 CrossRef CAS PubMed.
- A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS PubMed.
- R. Y. Tang, G. Li and J. Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. A. White, Nat. Chem., 2015, 7, 987–994 CrossRef CAS PubMed.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- J. L. Garnett and R. J. Hodges, J. Am. Chem. Soc., 1967, 89, 4546–4547 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
- S. S. Stahl, J. A. Labinger and J. E. Bercaw, Angew. Chem., Int. Ed., 1998, 37, 2180–2192 CrossRef PubMed.
- B. A. Arndtsen and R. G. Bergman, Science, 1995, 270, 1970–1973 CrossRef CAS.
- M. Beller and C. Bolm, Transition metals for organic synthesis, Wiley-VCH, Verlag GmbH, 1998 Search PubMed.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159–164 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751–2772 CrossRef PubMed.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS PubMed.
- T. G. Kim, Y. Kim and D.-J. Jang, J. Phys. Chem. A, 2001, 105, 4328–4332 CrossRef CAS.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS.
- S. Otto and J. B. Engberts, Org. Biomol. Chem., 2003, 1, 2809–2820 RSC.
- N. A. Isley, R. T. Linstadt, S. M. Kelly, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 4734–4737 CrossRef CAS PubMed.
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed.
- M. Pohjoispää, E. Leppälä and K. Wähälä, J. Labelled Compd. Radiopharm., 2007, 50, 521–522 CrossRef.
- S. Rasku and K. Wähälä, Tetrahedron, 2000, 56, 913–916 CrossRef CAS.
- S. Rasku, K. Wähälä, J. Koskimies and T. Hase, Tetrahedron, 1999, 55, 3445–3454 CrossRef CAS.
- H. Jiang, H. Fang, L. Jiang, L. Zheng, N. Wang, A. Zheng, F. Deng and M. Liu, Chin. J. Chem., 2010, 28, 2281–2286 CrossRef CAS.
- M. Jordheim, T. Fossen, J. Songstad and O. M. Andersen, J. Agric. Food Chem., 2007, 55, 8261–8268 CrossRef CAS PubMed.
- M. Lohrie and W. Knoche, J. Am. Chem. Soc., 1993, 115, 919–924 CrossRef CAS.
- E. S. Hand and R. M. Horowitz, J. Am. Chem. Soc., 1964, 86, 2084–2085 CrossRef CAS.
- A. J. Kresge and Y. Chiang, J. Am. Chem. Soc., 1961, 83, 2877–2885 CrossRef CAS.
- J. M. Herrero-Martínez, M. Sanmartin, M. Rosés, E. Bosch and C. Ràfols, Electrophoresis, 2005, 26, 1886–1895 CrossRef PubMed.
- S. E. Blanco, M. C. Almandoz and F. H. Ferretti, Spectrochim. Acta, Part A, 2005, 61, 93–102 CrossRef CAS PubMed.
- D. Wang, K. Hildenbrand, J. Leitich, H. P. Schuchmann and C. von Sonntag, Z. Naturforsch., B: J. Chem. Sci., 1992, 48, 478–482 Search PubMed.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
- N. Mandal, A. K. Pal, P. Gain and A. Datta, J. Am. Chem. Soc., 2020, 142, 5331–5337 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, B. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- S. Grimme, S. Ehrlish and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem., 2006, 110, 5121–5129 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. Michaelides and K. Morgenstern, Nat. Mater., 2007, 6, 597–601 CrossRef CAS PubMed.
- S. H. M. Mehr, K. Fukuyama, S. Bishop, F. Lelj and M. J. MacLachlan, J. Org. Chem., 2015, 80, 5144–5150 CrossRef CAS PubMed.
- F. Jensen, Introduction to Computational Chemistry, Wiley, Chichester, 2nd edn, 2007 Search PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- K. Fukui, Int. J. Quantum Chem., 1981, 15, 633–642 CAS.
- M. Page and J. W. McIver Jr, J. Chem. Phys., 1988, 88, 922–935 CrossRef CAS.
- M. Page, C. Doubleday and J. W. McIver Jr, J. Chem. Phys., 1990, 93, 5634–5642 CrossRef CAS.
- J. K. Harper, J. A. Doebbler, E. Jacques, D. M. Grant and R. B. Von Dreele, J. Am. Chem. Soc., 2010, 132, 2928–2937 CrossRef CAS PubMed.
- F. R. Fronczek, R. W. Hemingway, G. W. Mcgraw, J. P. Steynberg, C. A. Helfer and W. L. Mattice, Biopolymers, 1993, 33, 275–282 CrossRef CAS.
- G. Alagona, C. Ghio and P. I. Nagy, Phys. Chem. Chem. Phys., 2010, 12, 10173–10188 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: pKa values, NBO charges, H/D exchange of catechin and resorcinol, isomers of resorcinol, DFT structures of the ground and transition states of resorcinol and phloroglucinol, computed ΔH‡, TΔS‡ and ΔG‡ values. See DOI: 10.1039/d0cp02826f |
| This journal is © the Owner Societies 2020 |