
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances and challenges for experiment and theory for multi-electron multi-proton transfer at electrified solid–liquid interfaces
Ken
Sakaushi
 *a,
Tomoaki
Kumeda
a,
Sharon
Hammes-Schiffer
*b,
Marko M.
Melander
*c and
Osamu
Sugino
*d
*a,
Tomoaki
Kumeda
a,
Sharon
Hammes-Schiffer
*b,
Marko M.
Melander
*c and
Osamu
Sugino
*d
aCenter for Green Research on Energy and Environmental Materials, National Institute for Materials Science, Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan. E-mail: sakaushi.ken@nims.go.jp
bDepartment of Chemistry, Yale University, 225 Prospect Street, New Haven, CT 06520-8107, USA. E-mail: sharon.hammes-schiffer@yale.edu
cNanoscience Center, Department of Chemistry, University of Jyväskylä, P.O. Box 35 (YN), FI-40014, Jyväskylä, Finland. E-mail: marko.m.melander@jyu.fi
dThe Institute of Solid State Physics, the University of Tokyo, Kashiwanoha 5-1-5, Kashiwa, Chiba 277-8581, Japan. E-mail: sugino@issp.u-tokyo.ac.jp
First published on 1st September 2020
Abstract
Multi-electron, multi-proton transfer is important in a wide spectrum of processes spanning biological, chemical and physical systems. These reactions have attracted significant interest due to both fundamental curiosity and potential applications in energy technology. In this Perspective Review, we shed light on modern aspects of electrode processes in the 21st century, in particular on the recent advances and challenges in multistep electron/proton transfers at solid–liquid interfaces. Ongoing developments of analytical techniques and operando spectrometry at electrode/electrolyte interfaces and reliable computational approaches to simulate complicated interfacial electrochemical reactions enable us to obtain microscopic insights about these complex processes, such as the role of quantum effects in electrochemical reactions. Our motivation in this Perspective Review is to provide a comprehensive survey and discussion of state-of-the-art developments in experiments, materials, and theories for modern electrode process science, as well as to present an outlook for the future directions in this field.
 Ken Sakaushi | Ken Sakaushi is a Senior Researcher at the National Institute for Materials Science (NIMS), Tsukuba, Japan. He studied physics (BSc, 2008) and chemistry (MSc, 2010) at Keio University, and he completed his PhD at IFW Dresden/TU Dresden in 2013 with a DAAD fellowship. Then, he joined Max Planck Institute of Colloids and Interfaces on a fellowship from Max Planck Society. In 2015, Ken joined NIMS as a Tenured Member. His research focuses on various fundamental aspects of electrode process. Besides academic activities, he loves playing piano, reading books, and playing football. Ken has been recognized by awards such as the ISE Travel Award for Young Electrochemists, PCCP Prize, and PCCP Emerging Investigator. Further information: https://sites.google.com/site/sakaushiken/. |
 Tomoaki Kumeda | Tomoaki Kumeda received his PhD from Chiba University, Japan, in 2020 under the supervision of Associate Professor Masashi Nakamura. He was a recipient of a JSPS Research Fellowship for Young Scientists from 2018 to 2020. He is currently a postdoctoral researcher at National Institute for Materials Science, Japan. His research interests focus on microscopic structure of electrode/electrolyte interfaces and mechanism of electrocatalytic reactions on single crystal surfaces. |
 Sharon Hammes-Schiffer | Sharon Hammes-Schiffer received her BA in Chemistry from Princeton University in 1988 and her PhD in Chemistry from Stanford University in 1993, followed by two years at AT&T Bell Laboratories. She was the Clare Boothe Luce Assistant Professor at the University of Notre Dame from 1995–2000 and then became the Eberly Professor of Biotechnology at The Pennsylvania State University until 2012, when she became the Swanlund Professor of Chemistry at the University of Illinois Urbana-Champaign. Since 2018, she has been the John Gamble Kirkwood Professor of Chemistry at Yale University. Her research centres on the investigation of charge transfer reactions, proton-coupled electron transfer, nonadiabatic dynamics, and quantum mechanical effects in chemical, biological, and interfacial processes. Her work encompasses the development of analytical theories and computational methods, as well as applications to experimentally relevant systems. |
 Marko M. Melander | Marko M. Melander is an Academy of Finland Postdoctoral researcher at the University of Jyväskylä, Finland. Before obtaining his current position in 2017, he obtained a PhD in physical and computational chemistry from Aalto University in 2015 after which he joined the Atomic Scale Modelling and Materials group at DTU. His research focuses on development and application of theory and computational methods to study (proton-coupled) electron transfer in electrochemical systems, heterogeneous catalysis of complex systems, and multiscale modelling. |
 Osamu Sugino | Osamu Sugino was born in Fukushima Prefecture, Japan in 1961. He obtained his BSc (1984), ME (1986) and PhD (1989) degrees from the University of Tokyo. He was an employee (1989–2002) at NEC corporation, an associate professor (2002–2017) at the University of Tokyo. Since 2017, he has been a professor at the University of Tokyo. He has worked on the theory of condensed matter physics, in particular, the first-principles calculation of nonadiabatic dynamics and electrode dynamics. |
1. Scope of this perspective review
Electrode processes based on multi-electron, multi-proton transfer at solid–liquid interfaces are attracting widespread interest ranging from fundamental scientific issues to engineering aimed at establishing next-generation energy storage/conversion devices. In this Perspective Review, the cutting-edge advances and challenges of modern electrode process science are surveyed and discussed by experts in this field: Ken Sakaushi (Sections 1–3), Tomoaki Kumeda (Section 4: operando IR/Raman surface-electrochemical spectrometry), Sharon Hammes-Schiffer (Section 5: heterogeneous electrochemical proton-coupled electron transfer theory), Marko M. Melander (Section 6: grand canonical ensemble for electrochemical thermodynamics and reaction rates), and Osamu Sugino (Section 7: quantum many-body theory for understanding electrode processes).The scope of this Perspective Review focuses on the modern forms of electrode process science, including a wide range of different aspects, i.e. experimental approach, electrode material design, modeling, and first-principles calculations. We begin the discussions with the dawn of modern electrochemistry and then move to experimental approaches including kinetic analysis, material design and operando electrochemical spectrometry. After these experimental topics, we move to theoretical topics covering the recent advancements and applications of proton-coupled electron transfer theory at electrode/electrolyte interfaces, the grand canonical ensemble approach to simulate electrochemical reaction rates, and quantum many-body theories aimed toward understanding microscopic mechanisms of electrochemical reactions.
The aims in this Perspective Review are the following: (1) circulating modern knowledge of electrochemistry for a wide spectrum of scientists including graduate students, (2) summarizing the state-of-the-art developments in experimental and theoretical aspects of electrode process science, and (3) discussing current theoretical and computational approaches including classical, quantum mechanical, adiabatic, and nonadiabatic treatments in electrochemical kinetics.
2. Prologue
Nature acquired one of the most efficient energy conversion systems on the earth based on electron transfer (ET), proton transfer (PT), and proton-coupled electron transfer (PCET) (Fig. 1).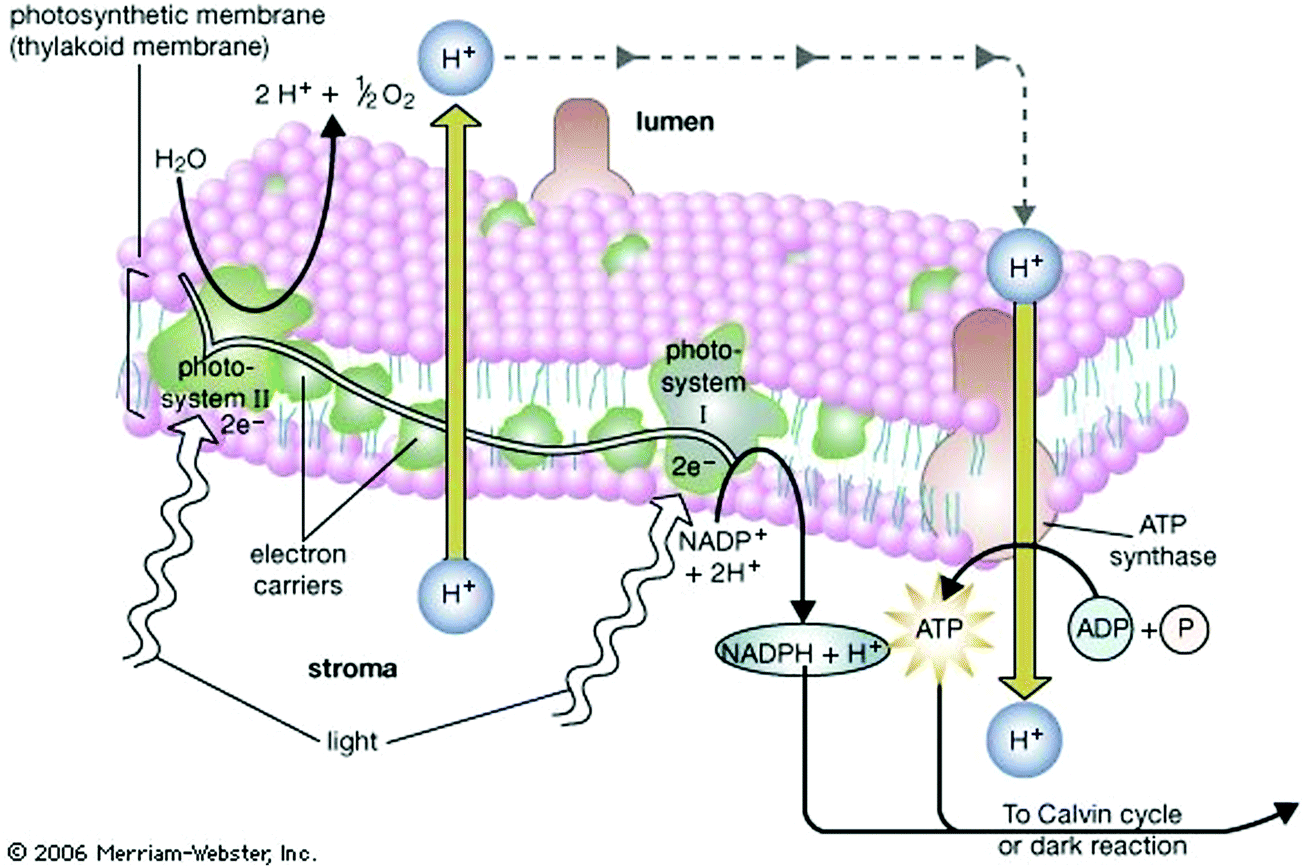 | ||
| Fig. 1 The light reaction of photosynthesis as highly efficient proton-based energy conversion system. Proton transfer is a key of this system. Photosystem II obtains replacement electrons from water molecules, resulting in their splitting into proton (H+) and oxygen atoms. The oxygen atoms combine to form molecular oxygen (O2), which is released into the atmosphere. H+ is pumped into the lumen by electron acceptor molecules. The flow of H+ back across the photosynthetic membrane provides the energy needed to drive the synthesis of the energy-rich molecule of adenosine triphosphate (ATP). High-energy electrons, which are released as photosystem I absorbs light energy, are used to drive the synthesis of nicotinamide adenine dinucleotide phosphate (NADPH). Photosystem I obtains replacement electrons from the electron transport chain. ATP provides the energy and NADPH provides the hydrogen atoms needed to drive the subsequent photosynthetic dark reaction, or Calvin cycle. The figure and caption are adopted from Encyclopædia Britannica® by courtesy of Encyclopædia Britannica, Inc., copyright 2006; used with permission: URL https://www.britannica.com/plant/plant/Photosynthesis#/media/1/463192/66097, Access Date 29th Dec. 2019. The original caption was modified by KS. | ||
As energy conversion in biological systems keeps on attracting interest since the finding of the proton transfer mechanism in the cytochrome system,1,2 it is well-known that this efficient system is a relay of multi-electron/-proton transfer reactions (Fig. 2).3–5
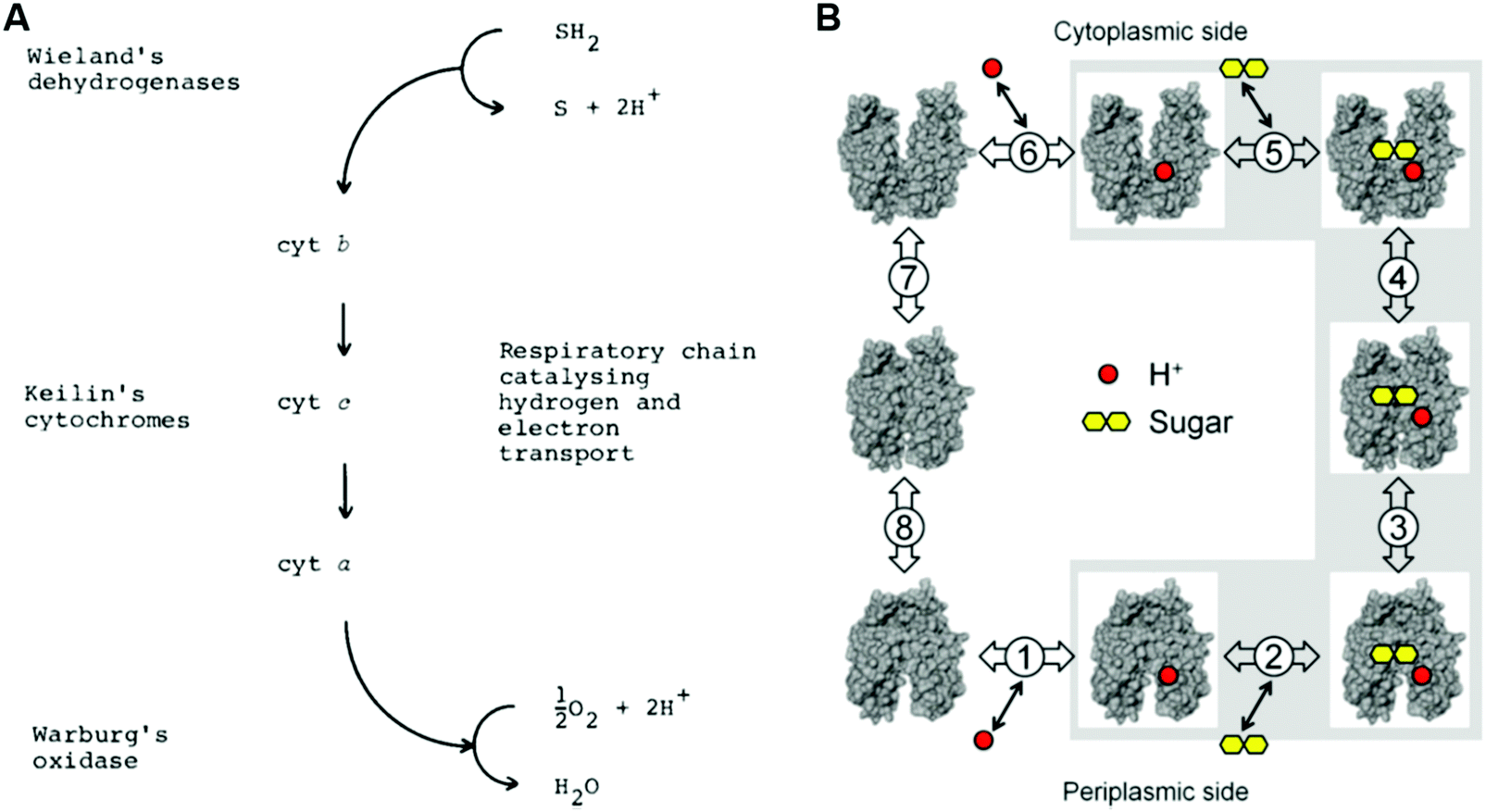 | ||
| Fig. 2 Classical and modern models for proton-related multistep reactions in proteins. (A) David Keilin's chemically simple respiratory chain concept. The panel and the caption were adopted from the ref. 4, with the permission from the Nobel Foundation. Copyright: The Nobel Foundation (2020). (B) Kinetic scheme for galactoside/H+ symport, exchange, and counterflow in LacY. The panel was reprinted from the ref. 5. Copyright (2020) National Academy of Sciences of the United States of America. | ||
Therefore, it is not surprising that there were a large number of researchers trying to understand the mechanisms of surface electrode processes involving biological synthesis of adenosine triphosphate (ATP) and adenosine diphosphate (ADP) in the early 1970s.6,7 As is obvious for ET, the modern view of PT can be described under the laws of quantum mechanics; there are active discussions on the connections between nuclear quantum effects (NQE) and microscopic mechanisms in biological systems8–13 and organic chemistry (Fig. 3).14–16
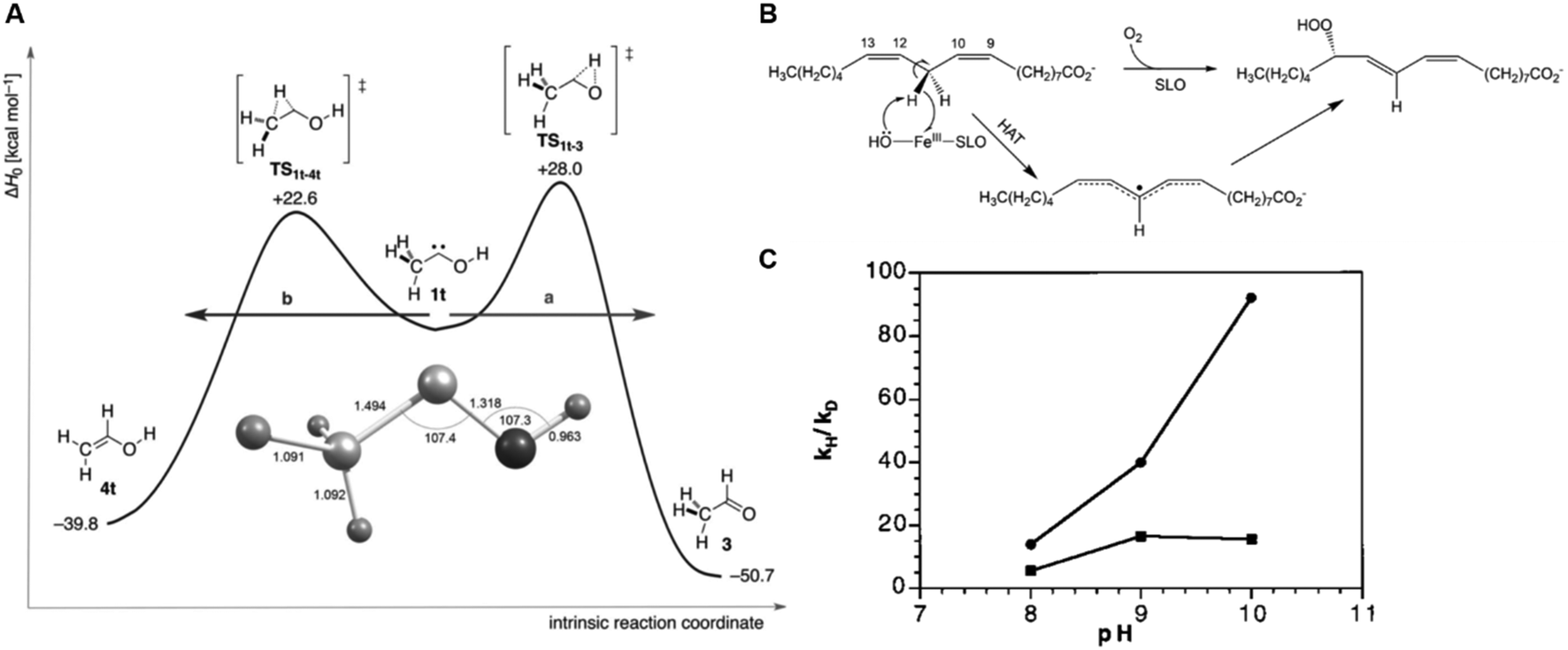 | ||
| Fig. 3 Examples of proton tunneling as key effects. (A) Tunneling control in chemical selectivity. Reprinted with the permission from the ref. 15. Reprinted with permission from the American Association for the Advancement of Science. (B and C) A strong kinetic isotopic effect in a biological reaction. Reprinted with permission from the ref. 11 and 13. Copyright (1999 and 2018) American Chemical Society. | ||
Similar to these examples from chemistry and biology, electrochemists were eager to adopt the recently developed quantum theory, and there is a rich history on utilizing quantum mechanics in electrode processes.17–31 The marriage of electrochemistry and quantum mechanics in the early 20th century was a fascinating and beautiful event in science because this opened the door to the establishment of modern electrode process science, which is the advanced framework of classical electrochemistry developed by many researchers since the 18th century.32,33 The birth of modern electrode process science was triggered by R. W. Gurney at Cavendish Laboratory/Trinity Hall, University of Cambridge in 1931: quantum mechanics was applied to interpret a microscopic mechanism of an elementary act of the hydrogen evolution reaction.17 In this report, the proton discharge at an electrode surface was described by state-of-the-art physical concepts at that time, including the Franck–Condon principle and Fermi–Dirac statistics. This arose from the communications with Condon and Morse during Gurney's stay at Palmer Physical Laboratory, Princeton University.34,35 This illustrates that the modern ideas of electrode processes emerged from strong interactions between physics and chemistry; therefore, this subject has considerable physics character by nature even in its modern form of electro“chemistry”.
After Gurney's work on applying quantum mechanics to an electrode process, a wide spectrum of quantum theories were proposed to describe microscopic electrode processes, such as bond-stretching (inner-sphere) model based theories,20,36–38 and solvent-reorganization (outer-sphere) model based theories.21–24,39–43 From this point, the application of quantum mechanics to modernize electrochemistry may have influenced the work of three Nobel Prize laureates, i.e. P. Mitchell, R. A. Marcus and J. B. Goodenough.4,44,45 Here we skip explaining the detailed history of these quantum electrochemical theories and their developments because one of the authors already summarized the aforementioned issues in recent reports.46,47 It is clear that we have plenty of basic theories to consider microscopic electrode processes; however, the development of quantum mechanical descriptions of electrode processes had become almost stagnant after the late 1970s. One of the main reasons was the extraordinarily high complexity of surface electrochemical processes at solid–liquid interfaces, requiring advanced simulation methods, theoretical concepts, and well-established surface-sensitive spectrometry to observe reaction dynamics.
Fortunately, with present technology, we have enough supercomputers and equipment to make progress, as already shown in heterogeneous surface science at solid/gas interfaces.48–52 In fact, several fascinating theoretical and computational approaches for electrode processes containing quantum effects in electron and proton transfer reactions were proposed since the late 1990s.53–66 Therefore, we believe that the time for further advancement of quantum electrode process science, a scientific topic having a long tradition, has come. It is our hope that we can show the importance, challenges, and excitement in this field to the readers in order to encourage further developments of this highly interdisciplinary topic in the 21st century. Studying and understanding how quantum mechanical effects such as nuclear and electron tunneling and non-adiabaticity control electrochemical properties will advance electrode process science and hopefully lead to materials with increased control of selectivity and activity.
3. Experimental observations of quantum effects in electrode processes
3.1. Introduction to the observation of quantum effects in multi-electron/-proton transfer electrode processes
As the lightest chemically relevant particles, electrons display quantum effects and ET is always a quantum process (Fig. 4).67 Similarly, protons should also be treated as quantum mechanical particles, and PT and concerted proton-electron transfer (CPET) are the key processes for understanding quantum effects in electrode processes (see the state-of-the-art theory on PCET in Sections 5–7). The details on CPET can be found in the previous report.68 If a PT for the slowest step in a multistep reaction displays quantum effects such as tunneling or zero-point energy effects, this reaction can be regarded as a quantum-dominated process. Therefore, quantum effects in PT and CPET are an important focus of investigations in quantum electrode processes.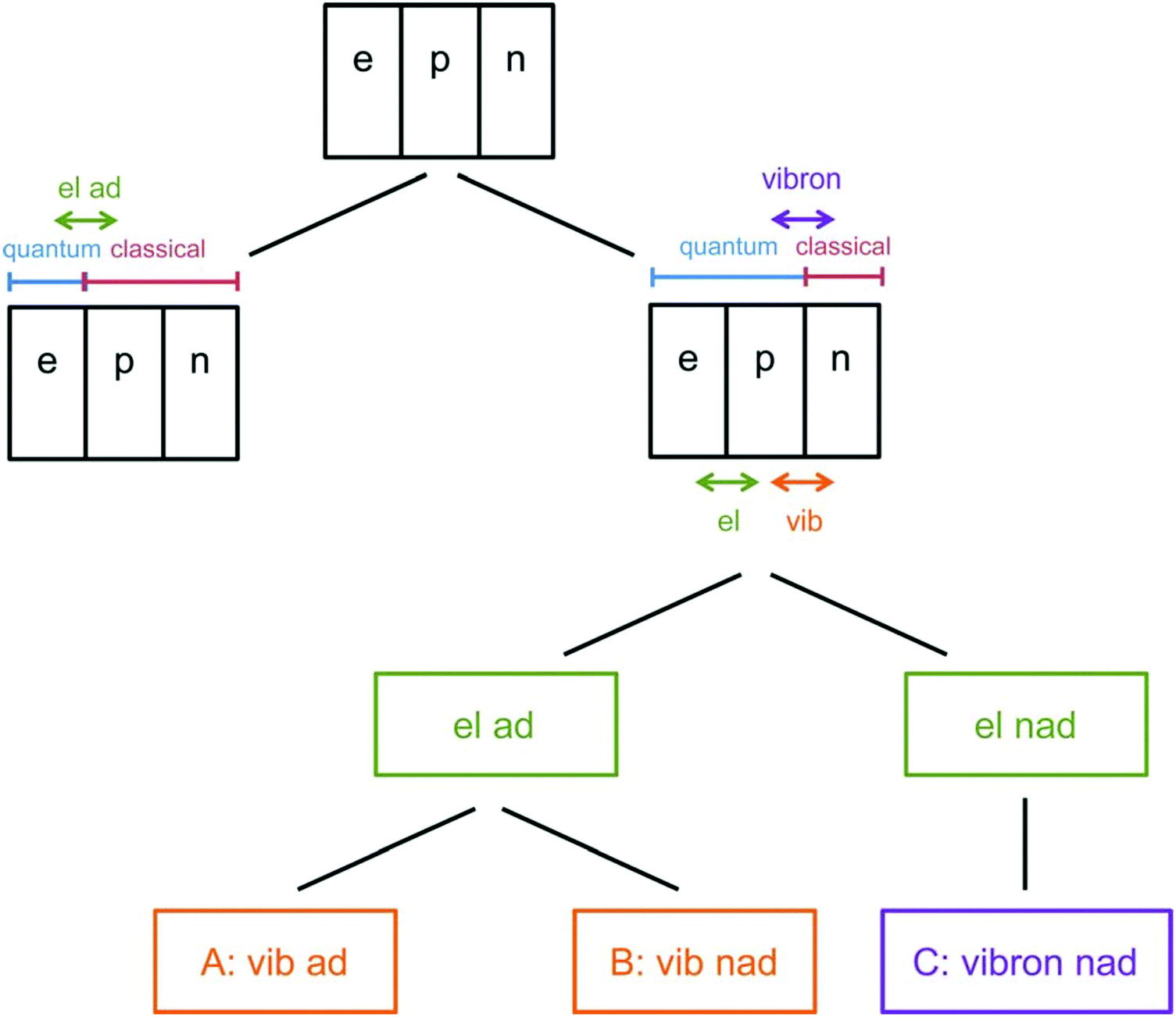 | ||
| Fig. 4 Schematic picture of the classification scheme for proton transfer and general PCET reactions. The abbreviation ‘‘el ad’’ denotes electronically adiabatic, where the electrons respond instantaneously to the motions of all nuclei. This left branch is associated with standard electronic structure calculations. The right branch depicts the situation in which the electrons and transferring proton are treated quantum mechanically, and all other nuclei are treated classically. The abbreviations are defined as follows: ‘‘el’’ denotes electronically, ‘‘vib’’ denotes vibrationally, ‘‘vibron’’ denotes vibronically, ‘‘ad’’ denotes adiabatic, and ‘‘nad’’ denotes nonadiabatic. This branch is subdivided into ‘‘el ad’’ and ‘‘el nad’’, referring to the degree of electron–proton nonadiabaticity, and the ‘‘el ad’’ case is subdivided further into ‘‘vib ad’’ and ‘‘vib nad.’’ The figure and caption were adopted with permission from the ref. 67. Copyright (2012) Royal Society of Chemistry. | ||
Although the first experimental suggestion of quantum mechanical effects in PT/CPET in an electrode process was reported by Bawn and Ogden in 1934,18 and many leading electrochemists, such as Conway,69–76 tried to confirm this phenomenon, the experimental observation of quantum electrode processes was hampered by several issues:
(1) there was no general approach to determine the rate-determining step (RDS) of multistep electrochemical reactions until the establishment of the Parsons’ relation.77
(2) analytical equations to diagnose quantum processes such as the electrochemical kinetic isotope effect (EC-KIE) were less developed.
(3) there was no easily-accessible and reliable technology to obtain ultrapure grade heavy reagents, especially water, until quite recently.78–80
Especially, the issue (3) was quite important because ultrapure heavy water is indispensable to observe a reliable EC-KIE, which is often taken as an indicator for NQEs. For these reasons, observing the signs of quantum electrode processes was enabled only quite recently.46,47,81
To investigate NQEs in electrode processes, first, we should identify the RDS in the corresponding multi-electron multi-proton transfer reaction. The first step is to classify a possible RDS as one of the following three cases: ET, PT, or CPET (Fig. 5).82–85 Practically, the classification can be possible by applying the combination of the Parsons’ relation to an experimentally obtained the transfer coefficient (α) and the EC-KIE analysis. In other words, the Parson's relation is able to check whether ET is involved in a RDS, and the EC-KIE analysis is able to indicate whether PT is involved in this RDS or not.
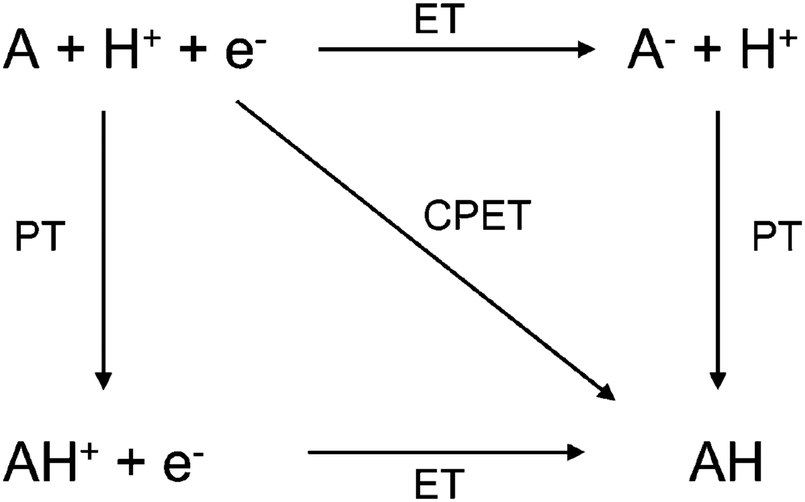 | ||
| Fig. 5 Square scheme for proton-coupled electron transfer. ET = electron transfer, PT = proton transfer, CPET = concerted proton–electron transfer. The figure and caption were adopted with permission from the ref. 85. Copyright (2013) Royal Society of Chemistry. | ||
We can confirm which reaction step is a possible RDS by checking the Tafel slope: the relation between the Tafel slope and the RDS is well studied for steps in typical reactions, such as the hydrogen evolution reaction and oxygen reduction reaction.86,87 The detailed method and equations to obtain the Tafel slope and α are summarized in the recent report.46 Furthermore, this author notes that there are issues to apply this method to several reactions so far, for instance the CO2 reduction reaction because of the difference between surface and bulk [H+].88,89 Once α is obtained, this can be substituted into the equations to calculate the EC-KIE (KH/D), which is based on the ratio of the reaction rate constant for hydrogen and deuterium.46 Typically, very large KIEs, KH/D ≫ 1 are used as indicators for NQEs. However, using the KIE as the sole indicator for discerning quantum effects is difficult as even non-adiabatic nuclear tunneling could result in KIEs close to unity if excited vibronic states play a significant role.90,91
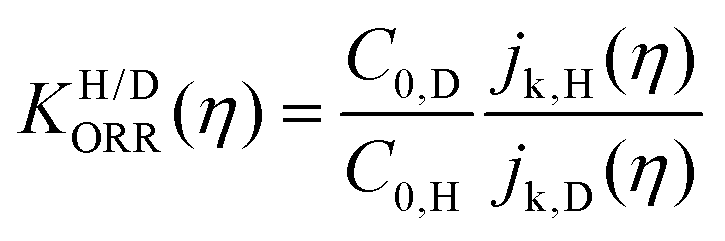 | (1) |
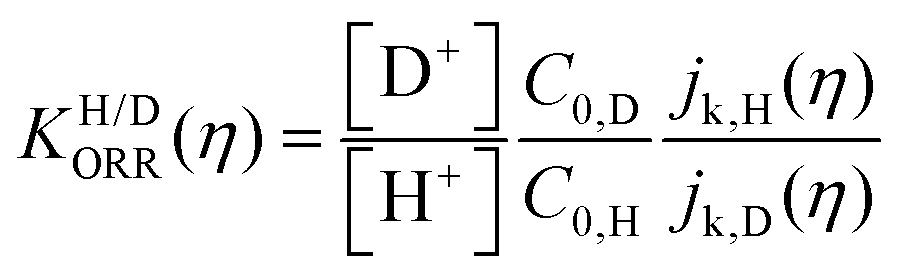 | (2) |
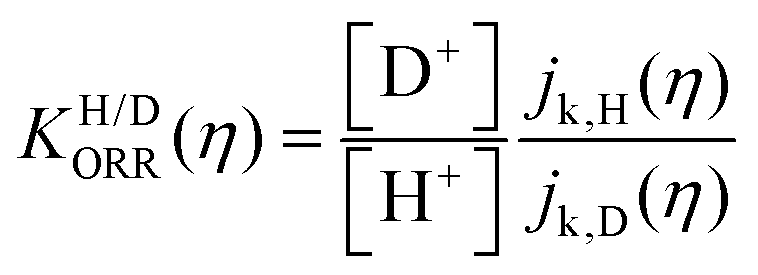 | (3) |
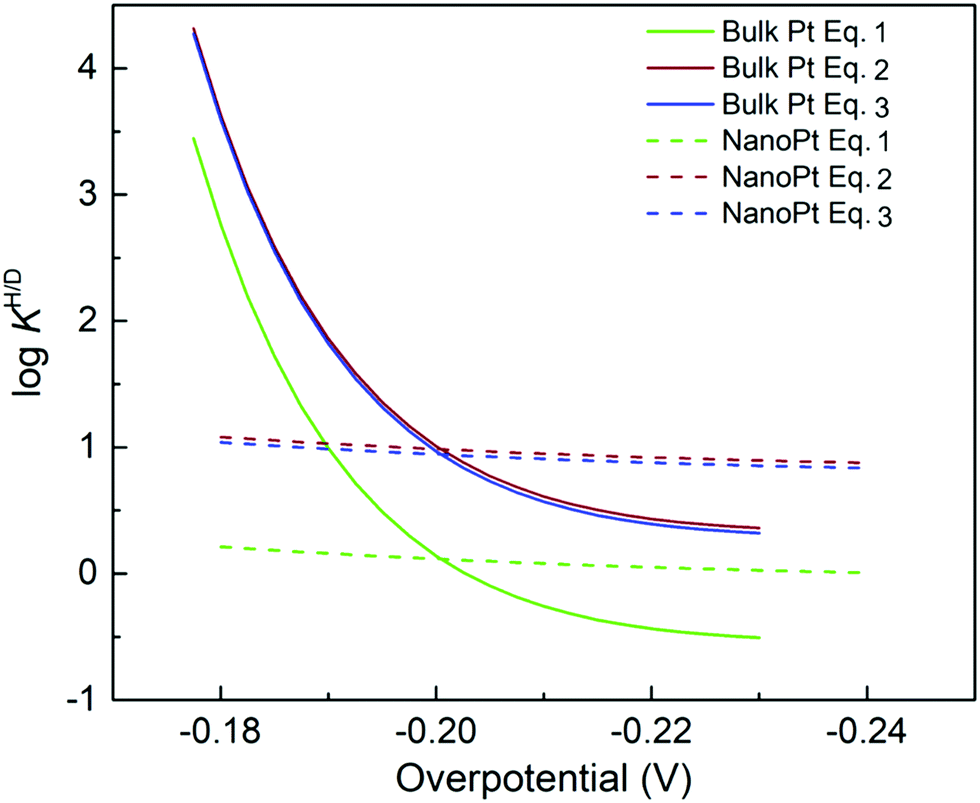 | ||
Fig. 6 log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KH/D–overpotential diagrams for ORR with different analytical equations. The solid and dotted lines respectively indicate bulk Pt and nano Pt electrodes. The different features in bulk Pt and nano Pt electrodes are obtained. This can suggest a size-dependent quantum-to-classical transition in electrode processes. The figure was adopted with permission from the ref. 92. Copyright (2020) Royal Society of Chemistry. KH/D–overpotential diagrams for ORR with different analytical equations. The solid and dotted lines respectively indicate bulk Pt and nano Pt electrodes. The different features in bulk Pt and nano Pt electrodes are obtained. This can suggest a size-dependent quantum-to-classical transition in electrode processes. The figure was adopted with permission from the ref. 92. Copyright (2020) Royal Society of Chemistry. | ||
3.2. What do we understand about quantum electrode processes so far?
Starting from the most important point, based on previous investigations, we can conclude that quantum effects, for instance zero point energy (ZPE) and tunneling, are key in electrode processes at solid–liquid interfaces.46,47,81,92 Therefore, quantum effects could be universally important for multi-electron multi-proton transfer electrode processes. For example, a strong indication of quantum proton tunneling was observed in a system composed of Pt electrode in an alkaline condition. In this system, a large EC-KIE was observed in a low overpotential region but vanished at high overpotentials. This was interpreted as a transition from a quantum proton tunneling mechanism to a classical over-the-barrier mechanism.81 This observation suggests a simple mechanistic picture, i.e. a high activation barrier allows tunneling to generate a current only at a lower overpotential condition, but this barrier height is lowered for higher overpotentials because of the Brønsted–Evans–Polanyi (BEP) principle, and therefore a current via the classical TST path will be generated. However, this phenomenon can also be explained within a theoretical framework in which the proton is fully quantized for all overpotentials.93,94Under the above simple picture, we can confirm the existence of a NQE in an electrode process. However, the important issue here is how to see a chemical or physical picture of a quantum electrode process that is solely dominated by quantum effects, especially “deep” tunneling (see Section 6 for details). As indicated by theoretical analyses (see Sections 5–7),81 the importance of quantum effects in electrode processes is valid, but confirming the microscopic mechanism and interpreting this phenomenon still presents huge challenges. Furthermore, previous reports showed the observation of a large difference in symmetry factors in H and D systems,46,95 which indicates a strong quantum effect;47 however, the reason for this observation still remains an open question. Several major challenges to understand the microscopic electrode mechanism are discussed in the next section.
3.3. Key questions in experimental approach
jkversus overpotential, and Marcus-like models predict quadratic logjkversus overpotential plots. Predictions of the Tafel model are consistent with the early bond-stretching model, while Marcus-like theories build on the reorganization of the environment facilitating ET/PT/CPET via tunneling. We note that Marcus–Hush–Chidsey (MHC) formalism, which incorporates the Fermi–Dirac distribution of electrons/holes into the classical Marcus theory, can show perfect agreements between its modified rate equation and the curved Tafel plot.96,97 However, this distinction is somewhat artificial, and a more general perspective is a combination of concepts from these two models. For instance, DFT-based approaches are mainly based on models resembling the bond-stretching model and often neglect the environmental reorganization but still can exhibit a quadratic dependence of the current on the electrode potential (Fig. 7). On the other hand, models utilizing the solvent reorganization concept can result in (nearly) linear logjk-overpotential relations for large reorganization energies. Indeed, modern theoretical and computational approaches treat both the bond stretching (inner-sphere) and solvent reorganization (outer-sphere) in a consistent manner. Therefore, determining the activation mode or the “correct” theoretical framework from Tafel plots alone seems futile.
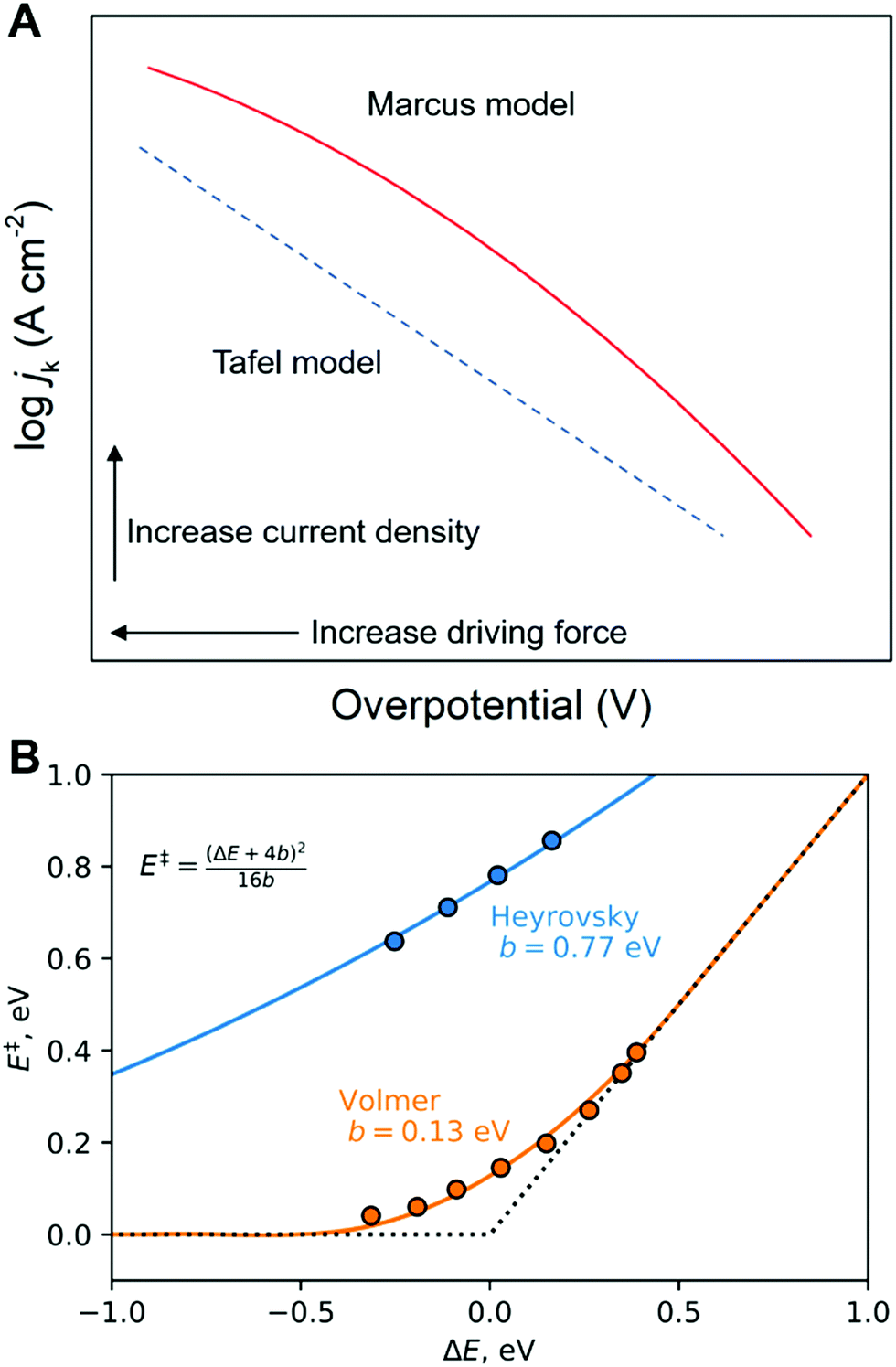 | ||
| Fig. 7 Tafel and Marcus models in logjkvs. overpotential diagrams. (A) Simulated logjk–η diagrams. The dotted blue line is calculated by the Tafel model, i.e. jk = j0exp(Aη). The solid red line is calculated by the Marcus model, i.e. jk = j0exp(B + Cη)2. The A, B, and C are arbitrary coefficients. (B) Example of Tafel-like and Marcus-like features obtained by a DFT calculation. Panel B was adopted with permission from the ref. 65. Copyright (2020) American Chemical Society. | ||
While the microscopic activation mechanism cannot be understood from Tafel plots, it is well-known that a wide spectrum of different electrocatalytic reactions show a change in Tafel slope associated with a change of potential (Fig. 8).78,81,98 This change in Tafel slope due to potential is suggested to be linked to a change of reaction mechanism, for instance, from the four-electron ORR mechanism to another mechanism (Fig. 8C and E).46,78,80,81 or potential-dependent changes in the surface coverages.99 To understand an activation mechanism and associated phenomena, we need further details from experimental and theoretical investigations at solid–liquid electrode processes at the molecular level (see Sections 5–7). Especially, the modern view of proton-coupled electron transfer theory (see Sections 5 and 6) can shed light on this fundamental yet unsolved question in electrochemistry. Moreover, a clear interpretation of activation mechanisms of electrode processes will help to discover electrocatalysts having better or comparable activity to Pt-based materials or to find a way to solve present issues in non-Pt electrocatalysts (see the “In silico electrocatalyst design” in Section 7).78,80,100–107
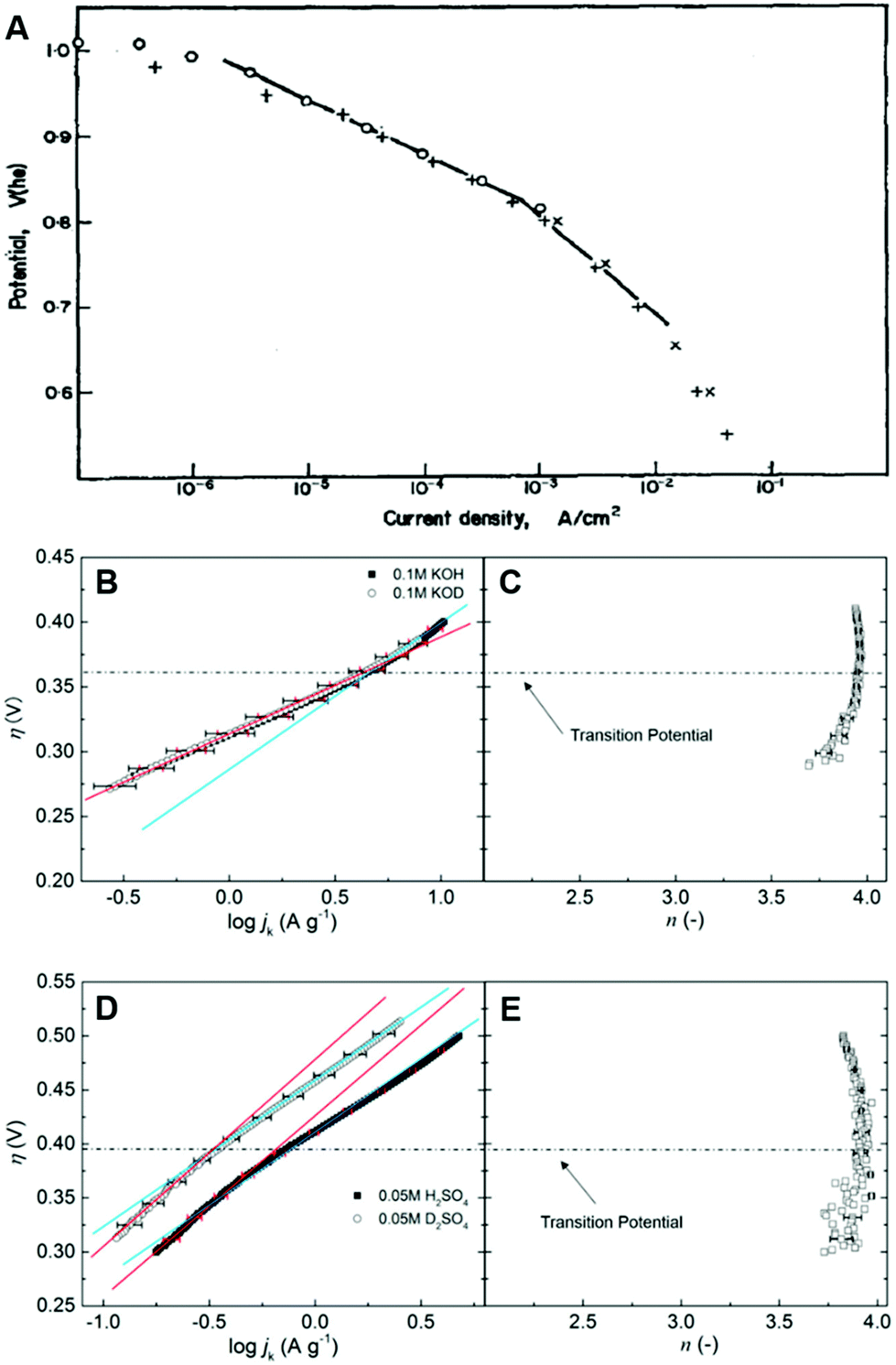 | ||
| Fig. 8 Examples for Tafel slope change as function of potential. (A) ORR on Pt electrode in acidic conditions. (B) η–logjk diagram for ORR on non-metal carbon-based catalyst in ordinary and heavy water electrolyte; (C) electron transfer number (n) according to (B) as a function of overpotential in alkaline electrolyte (0.1 M KOH in H2O and 0.1 M KOD in H2O); (D) η–logjk diagram for ORR on non-metal carbon-based catalyst in ordinary and heavy water based electrolyte; (E) n according to (D) as a function of η in acidic electrolyte (0.05 M H2SO4 in H2O and 0.05 M D2SO4 in D2O). The red and blue lines indicate fits to obtain the Tafel slope as the slope of these lines in the lower (below 0.35 and 0.4 V in alkaline and acidic solutions, respectively) and higher overpotential regions, respectively. The panel A and the corresponding caption are reprinted from the ref. 98, Copyright (1970), with permission from Elsevier. The panels B–E and the corresponding captions are reprinted and modified from the ref. 78, Copyright (2018), with permission from American Chemical Society. | ||
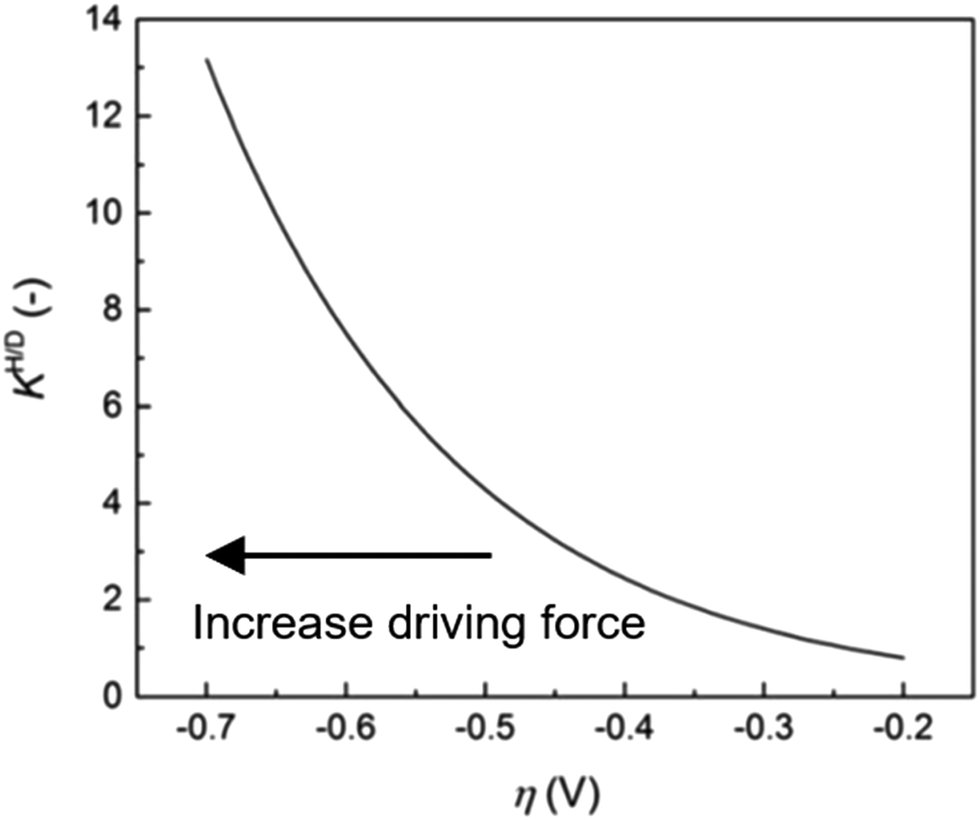 | ||
| Fig. 9 Anomalous KH/D–η diagram. KH/D increases associated with an increase of driving force. The measurements were carried out for the HER on Au electrode in 0.1 M KOH in H2O/0.1 M KOD in D2O. The figure was reprinted from the ref. 46, Copyright (2020), with permission from the Royal Society of Chemistry. | ||
The second key issue is that some theoretical suggestions are challenges to observe experimentally. Recently, a vibronically nonadiabatic process was suggested to underlie the Volmer step at a Au electrode in acidic conditions.94 The theoretical calculation of the η–logjk plot using the vibronically nonadiabatic PCET theory agreed well with the experimental observations, and therefore this theoretical work suggested that a nonadiabatic process could have an important role for electrode processes. However, the experimental η–logjk plot for a Au electrode in acidic conditions shows a linear η–logjk feature with a potential-independent KH/D of ca. 3, which traditionally has been interpreted to indicate a small contribution from nuclear quantum effects and an over-the-barrier mechanism (Fig. 10). This indicates that the EC-KIE alone cannot be used as a fully satisfactory proof of NQEs and vibronically nonadiabatic tunneling.
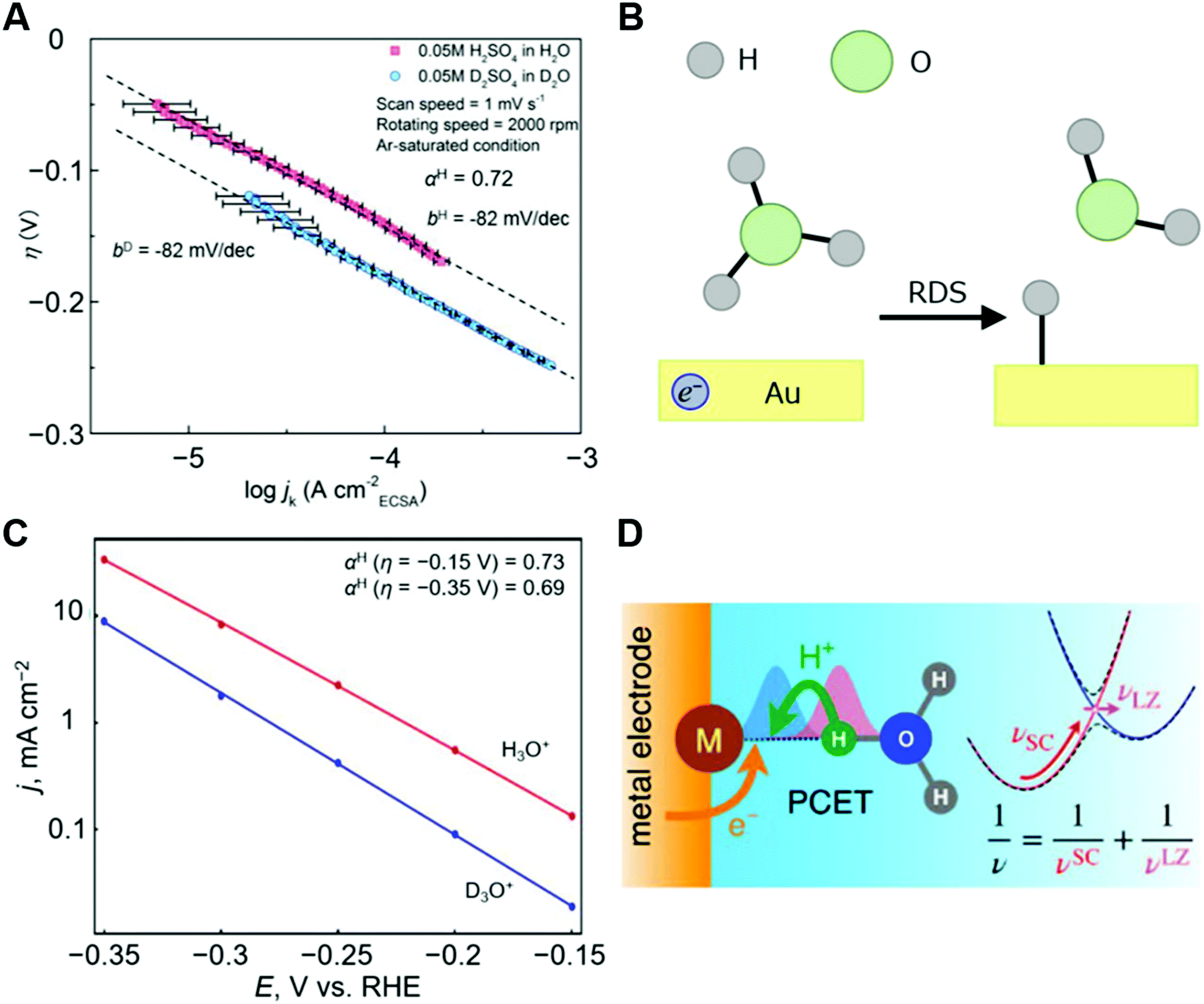 | ||
| Fig. 10 Experimental and theoretical analysis for HER. (A) η–logjk diagrams of the HER at Au electrode in 0.05 M H2SO4 in H2O/0.05 M D2SO4 in D2O. (B) Schematic illustration of the RDS for the HER at Au electrode in acidic condition (H3O+ + e− + * → H* + H2O). (C) Tafel plot (logarithm of current density versus potential) calculated by the theory and the equations in the reference for the reaction described in (B).94 The calculated data are shown as filled circles, and the lines represent linear fits to the calculated data points. The authors note here that in this case the potential (V vs. RHE) is identical to the overpotential (V). (D) Schematic illustration for the microscopic mechanism of PCET associated with vibrational nonadiabaticity and solvent dynamics. The panels A and B, and the related caption are reprinted from the ref. 47, Copyright (2020), with permission from Elsevier. The panels C and D are reprinted from the ref. 94, Copyright (2019), with permission from American Chemical Society. | ||
The third key issue is clarifying when the NQEs are important and when a classical description of electrode processes is sufficient. The importance of quantum effects is most probably system-dependent and may change depending on pH, solvent, anion additives in the electrolyte, etc. As shown in the following Sections, recent work on electrified surface systems successfully demonstrated various NQEs using computational and theoretical methods to investigate barrier structures, RDS, KIEs, and adsorption processes (see Sections 5–7 for the details).64,65,110 Therefore, these methods will play a significant role in solving the above issues in conjunction with experiments. EC-KIE experiments will likely offer much insight on mechanistic details and elementary steps of complex electrochemical reactions and provide a natural link between theory, computation, and experiments. If further advancement of computational approaches meets with operando electrochemical spectrometries (see Section 4) and state-of-the-art theories, for instance modern PCET theories (see Section 5), we will largely improve our fundamental knowledge of electrode processes.
Furthermore, recent advancements in materials science can also contribute to improving our understanding of electrode processes. For example, there are a variety of open discussions on porous electrodes. Recently, precisely prepared metallic porous electrodes were shown to be effective to study mechanisms of the CO2 reduction reaction (Fig. 11A and B).111,112 The emerging material classes of metal–organic frameworks (MOFs)113–129 and covalent-organic frameworks (COFs)100,101,130–140 have also been shown to be powerful tools to investigate fundamental features of electrochemical reactions. For example, heterojunctions of two-dimensional (2D) MOFs/COFs and metals are promising designer materials to tune the electrochemical properties of well-known electrode materials, such as Au or Pt, at solid–liquid interfaces. One of the two ideas here is based on the “classical” theories for electronic interactions at heterojunctions systems, such as the work of Bardeen,141 and of Cabrera and Mott,142 or Vol’kenshtein's theory for heterojunction catalysts,143 which has been extensively considered at solid/gas interfaces by Freund and his co-workers.48,49,52,144–147 The other idea is to use the advantage of MOFs/COFs, where electronic structures can be controlled by selections and combinations of organic monomers and metal ions.100,119,132 The proof-of-concept was first shown for HER and ORR in 2017 (Fig. 11C and D).105 In this work, a model heterojunction electrode of the 2D COF/Au was used to investigate the effect of a 2D COF as a porous-architecture to control electrocatalytic activity and showed that the additional thin COF layer is the key to improving activity or selectivity of the reactions.105 The identical concept was applied to a Au single crystal (111) surface and again confirmed that a thin COF layer can improve the HER activity.148 These advanced materials science efforts can contribute to understanding further details of microscopic mechanisms in quantum electrode processes together with other classes of well-defined modern designer materials, for instance molecular electrocatalysts (Fig. 11E and F).90,149–159
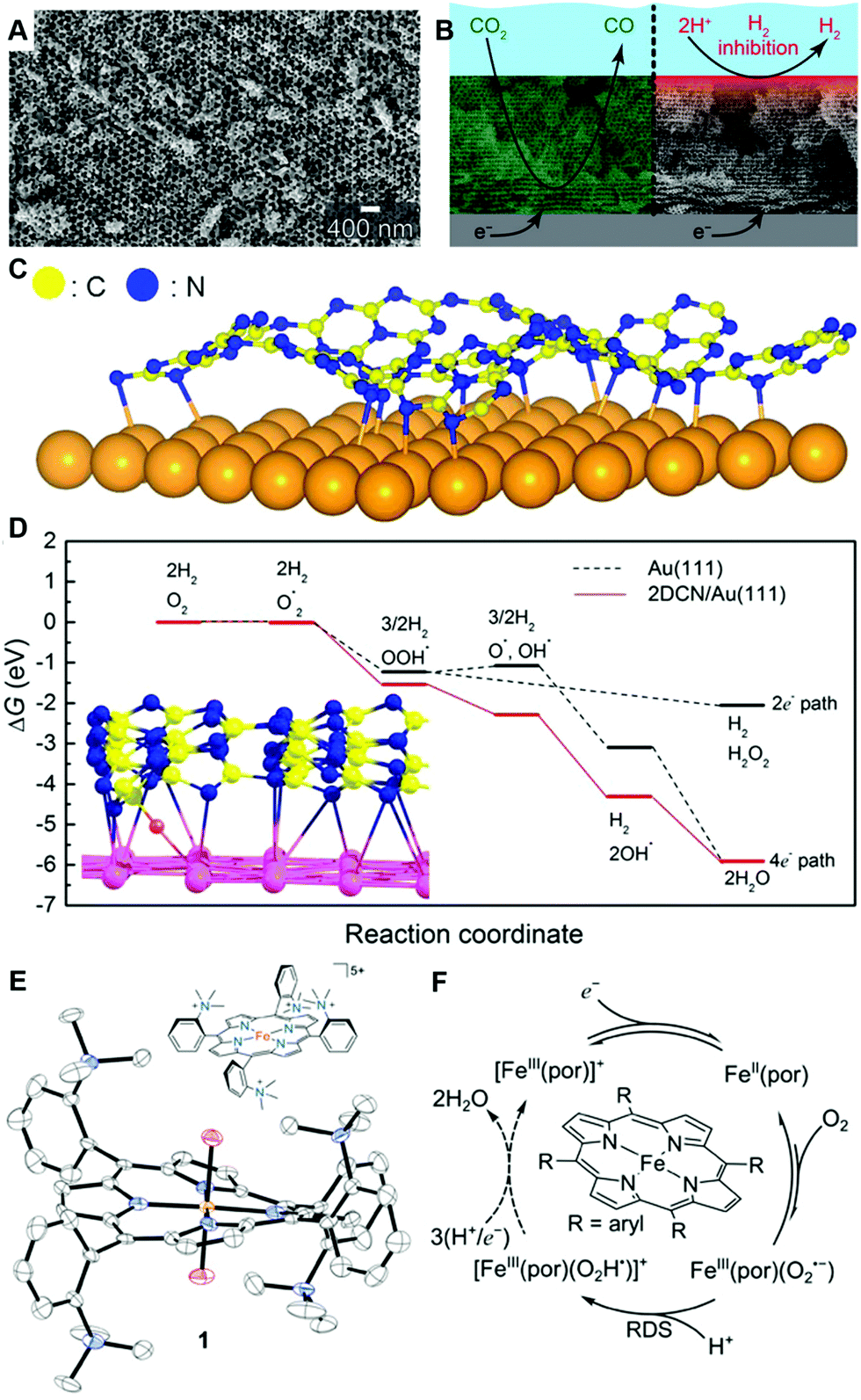 | ||
| Fig. 11 Representative modern electrocatalysts in fundamental electrode process science. (A) Scanning electron microscope image of a precise inverse-opal (IO) Au electrode and (B) application of IO Au electrode to control of the electrochemical CO2-to-CO conversion efficiency by taking the advantage of functions due to porous structure. (C) Schematic illustration of 2D COF/Au heterojunction electrode and (D) its free-energy diagram (red line) showing the alternative reaction path to obtain a four-electron transfer process which is not possible in Au electrode (black line). (E) Illustration of Fe-o-TMA of a model porphyrin-based molecular ORR electrocatalyst and the solid-state X-ray crystal structure of [Fe-o-TMA·2H2O]OTf5 (H atoms and triflates omitted, thermal ellipsoids at 50% probability). (F) ORR mechanism of Fe-porphyrin-based electrocatalysts. The panels A and B are reprinted from the ref. 111, Copyright (2015), with permission from American Chemical Society. The panels C and D are reprinted from the ref. 105, Copyright (2017), with permission from American Chemical Society. The panels E and F, and the corresponding captions are adopted from the ref. 157, Copyright (2020), with permission from American Association for the Advancement of Science. | ||
4. Operando IR and Raman electrochemical spectrometry for electrode processes at solid–liquid interfaces
4.1 Brief overview of present operando IR/Raman electrochemical spectrometry
Operando observation of electrode–electrolyte interfaces by spectroscopic methods is attracting enormous interest because of its importance in modern electrochemistry. Infrared spectroscopy and Raman spectroscopy are widely used to acquire information about various states of molecules on an electrode and electrocatalytic reaction pathways under applied potentials due to their simple set-up and high sensitivity. Application of infrared spectroscopy to electrochemistry has been developed rapidly with support by Fourier-transformed infrared spectroscopy (FT-IR) since the 1980s.160,161 The mechanism of CO adsorption and alcohol oxidation on single-crystal metal electrodes has been intensively investigated by using Infrared reflection absorption spectroscopy (IRAS) measurements because they are able to detect a wide spectrum of adsorbed chemical species.162–165 Time-resolved spectroscopic techniques combined with IRAS measurement have been also developed for the kinetic study of the electrode processes.166 The adsorption dynamics of CO on Pt electrodes were observed at millisecond-scale. However, the timelag between the real electrode potential and the applied potential caused by the external reflection configuration for IRAS complicates the kinetic analysis.167 Since the 1990s, technological improvements in equipment and software allow operando investigations with a higher sensitively and the capability for applications to a variety of different conditions. Surface-enhanced infrared absorption spectroscopy (SEIRAS) measurement by using a thin metal film electrode prepared on an attenuated-total-reflection (ATR) window has been developed by Osawa et al.168 The sensitivity of SEIRAS is higher by one digit than that of IRAS. Moreover, the internal reflection configuration for SEIRAS is compatible with time-resolved techniques. Time-resolved SEIRAS measurements have been applied to various electrode processes for elucidation of the kinetics at tens of milliseconds (rapid-scan interferometry) or submillisecond (step-scan interferometry) resolution.169–174 In recent years, the dynamics of elementary processes on electrocatalysts have been investigated by ultrafast time-resolved pump–probe IR and Raman spectroscopy at picosecond- or femtosecond-scales, which is recognized as an interesting approach.175–177 In addition to IR spectrometry, Raman spectroscopy has a long history too. As a highly sensitive method, surface-enhanced Raman spectroscopy (SERS) on a roughened substrate or nanoparticle of gold and silver was established from the 1970s to the 1980s.178 Furthermore, shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) based on silica coated gold nanoparticles was reported as an advanced approach for SERS to prevent degradation of the nanoparticles under the electrochemical environment and to utilize single crystal electrodes.179 Therefore, infrared spectroscopy and Raman spectroscopy are widely used to observe the electrode–electrolyte interface as complementary methods combined with standard electrochemical experiments to study the kinetics of electrode processes.The investigation of the KIE associated with the rate-determining step is valid for the elucidation of the PCET mechanisms (see Sections 3 and 5 for details).82,180 IR and Raman spectroscopy can identify isotopes, such as adsorbed H/D and OH/OD. For example, Pt–D stretching band (∼1500 cm−1) appears at lower frequency than that of Pt–H stretching band (∼2100 cm−1). The frequency ratio can be estimated at 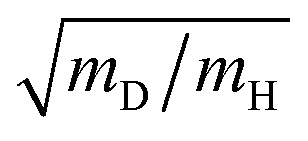 by harmonic oscillator approximation. Behm et al. revealed the potential-dependent KIE for the C–H bond breaking associated with the rate-determining step during the electro-oxidation of formic acid on a Pt-film electrode using by SEIRAS.181 In other studies, the potential dependent KIEs for ethylene glycol electro-oxidation and CO2 electro-reduction have been investigated using by SEIRAS.182,183 The combined use of KIE method and time-resolved spectroscopic techniques can strongly support the other experiments and theories for the PCET reactions.
by harmonic oscillator approximation. Behm et al. revealed the potential-dependent KIE for the C–H bond breaking associated with the rate-determining step during the electro-oxidation of formic acid on a Pt-film electrode using by SEIRAS.181 In other studies, the potential dependent KIEs for ethylene glycol electro-oxidation and CO2 electro-reduction have been investigated using by SEIRAS.182,183 The combined use of KIE method and time-resolved spectroscopic techniques can strongly support the other experiments and theories for the PCET reactions.
In recent years, spectroscopic methods have been applied to understand the mechanisms of electrocatalytic reactions, such as the hydrogen evolution reaction (HER) and oxygen reduction reaction (ORR). In particular, the development of highly active electrocatalysts for the ORR is important for fuel-cells and metal–air batteries,184 as IR and Raman spectroscopy can detect the intermediates and suggest the reaction pathway.161 However the high complexity of these reactions prevents a detailed understanding of the microscopic mechanisms (see Section 8 for details); therefore the reaction mechanism of HER, one of the simplest electrode processes, is still an open question. This Section presents the ongoing challenges for the investigation of the microscopic mechanisms of the HER and the ORR on electrified surfaces by using surface-electrochemical-spectroscopic approaches.
4.2 Operando surface-electrochemical spectrometry for hydrogen evolution reaction
The HER is a cathodic process for water electrolysis and also the reverse process of the hydrogen oxidation reaction (HOR) in the working reaction of fuel-cell. Three elementary steps are suggested for the HER.185–187 The first step is the adsorption of a hydrogen atom from the hydronium ion in the solution onto the electrode (Volmer step). The second step is the association and desorption reaction between the adsorbed hydrogen atom and the hydronium ion (Heyrovsky step). The other possible second step is the association and desorption reaction between two adsorbed hydrogen atoms (Tafel step). The kinetics of the HER has been widely studied by electrochemical analysis. For example, on the Pt electrode, the HER proceeds by the Volmer–Tafel process, and the rate-determining step is the Tafel step.185 However, recent studies proposed the surface-orientation dependence of the HER process on the Pt electrode, i.e. the Volmer–Tafel process proceeds on Pt(110), while the Volmer–Heyrovsky process proceeds on Pt(100).185To investigate the orientation-dependent-mechanism, operando infrared spectroscopy is used to acquire information on the HER mechanism on the Pt electrode by monitoring the adsorbed intermediate associated with a change of potential. The results of SEIRAS measurements indicated a band around 2100 cm−1 assigned to the Pt–H stretching vibration adsorbed at atop sites (atop H) on the Pt polycrystalline electrode in the HER potential range (<0.1 V vs. RHE).188,189 The band intensity corresponding to the atop H increases in parallel with the HER current density at more negative potentials. These results indicated that the rate-determining step is the Tafel step. A dependence of the intermediate on the surface-orientation of the Pt has been investigated by using single-crystal electrodes.190 The band of the atop H was observed on Pt(110) and high index planes with (110) step structure. In contrast, the band around 1630 cm−1 assigned to the Pt–H stretching vibration adsorbed at the asymmetric bridge site (bridge H) was observed on Pt(100) and high index planes with (100) terrace or a step structure. On Pt(111), a high-resolution electron energy loss spectrometry (HR-EELS) and first-principles calculations suggest that a hydrogen atom adsorbs at the fcc hollow site (fcc H).191 Recent years, Sugino et al. suggested the coexistence of both atop H and fcc H on Pt(111) using by the random phase approximation for the correlation functional of DFT (see Section 7 for details).192 This insight is consistent with operando sum-frequency generation (SFG) spectroscopy.193 Unfortunately, IRAS bands originated in fcc H on Pt(111) have never been reported under electrochemical conditions because of the optical restriction for the lower frequency. Raman spectroscopy measurement may provide a breakthrough in the structure and kinetics of hydrogen atom adsorbed on Pt(111). The HER/HOR exchange current density on Pt(110) is higher than that of Pt(100) and Pt(111).185 These results suggest the HER kinetics may be enhanced through the atop H.
4.3 Operando surface-electrochemical spectrometry for oxygen reduction reaction
The oxygen reduction reaction (ORR) occurs as two- or four-electrons processes depending on pH and/or the specific electrocatalyst. For a long time, it was generally accepted based on experimental results that the four- and the two-electron processes, respectively, occur on Pt and Au.194 In addition to experimental results, DFT calculations were used to compute the so-called volcano relationship, a diagram that correlates the ORR activity and the oxygen binding energy (ΔEO).194 As this diagram is based on the previous approaches of Parsons based on the Sabatier principle, a better electrocatalyst has an optimal adsorption energy of a key intermediate in the corresponding reaction, i.e. a high enough adsorption energy to follow a favorable reaction path but not too high to release the product efficiently. In this computationally-calculated diagram, the Pt(111) surface was suggested to be located at the position to have the highest ORR activity among other typical precious metals for the four-electron process. On the other hand, the Au(111) surface has a higher ΔEO than Pt. The adsorbed oxygen atom on the Au(111) surface is unstable, and this instability was suggested to be the reason for the incapability of the Au(111) surface to transfer four protons and four electrons to the oxygen at this electrocatalyst under acidic conditions.Aiming to obtain further insights of the ORR mechanism, infrared spectroscopy and Raman spectroscopy have been applied to the ORR mechanism on the Au and Pt electrode through the adsorbed intermediate. By using SEIRAS the intermediate during the ORR on Au polycrystalline electrode was observed in both alkaline and acidic solutions.195 The asymmetric bending mode of the adsorbed OOH (OOHad), which is suggested as an intermediate during the four-electron process, appeared between 0.1 and −0.6 V vs. Ag/AgCl in an O2-saturated alkaline solution. The previous studies of the ORR on Au single-crystal electrodes suggested that the four-electron process occurs only on the Au(100) surface in alkaline solutions.196,197 In contrast, the band intensity of OOHad in acidic solutions is weaker than that in alkaline solutions. These studies suggested that the key intermediate for the four-electron process of OOHad is unlikely to be the main product, and therefore the two-electron process occurs on the Au surface in acidic solutions. In another study, the two bands assigned to the O–O stretching mode of adsorbed O2− (O2−ad) and the O–O stretching mode of HOOH (HOOHad) on the Au roughened surface were observed in alkaline solutions below 0.85 V vs. RHE by a SERS measurement.198 This observation supports the four-electron process on Au electrodes in alkaline media. On the other hand, the band intensity of O2−ad was weak and HOOHad was not observed in acidic solution. This fact suggests that the one-electron process mainly occurs as the RDS, and some oxygen atoms are reduced afterwards through the two-electron process via O2−ad in acidic solutions. Furthermore, another study based on a SERS measurement indicated that the four-electron process occurs on the Au surface modified with Bi even in acidic solution.199
On Pt electrodes, the band corresponding to the O–O stretching mode of O2−ad was observed by a SEIRA measurement in alkaline solutions, and this report claimed that the HOOHad is related to the RDS of the four-electron process.200 In acidic solutions, attenuated total reflection infrared (ATR-IR) spectrometry was used to observe the ORR intermediates on Pt/C nanoparticles.201Fig. 12A shows the ATR-IR spectra on Pt/C nanoparticles during ORR and under Ar in a HClO4 solution. In this study, three intermediates were observed during the ORR, and a possible mechanism was proposed based on this observation (Fig. 12B). The observation of the O–O stretching mode of OOHad and the OOH bending mode of HOOHad indicates that the O–O bond breaking occurs after the protonation of the OOHad, and therefore the ORR mainly proceeds through the associative pathway. However, the band of OOHad appears at 0.9 V (RHE), whereas HOOHad appears only below 0.7 V. This suggests that the ORR mainly proceeds through the dissociative pathway at higher potentials. The dependence of the ORR mechanism on the surface-orientation has been investigated using SHINERS measurement. The ORR occurs via OOHad on Pt(111), whereas it occurs via OHad on Pt(100) and Pt(110).202 SHINERS was used to observe the O–O stretching mode of OOHad and the Pt–OH bending mode of OHad on high-index planes of Pt.203
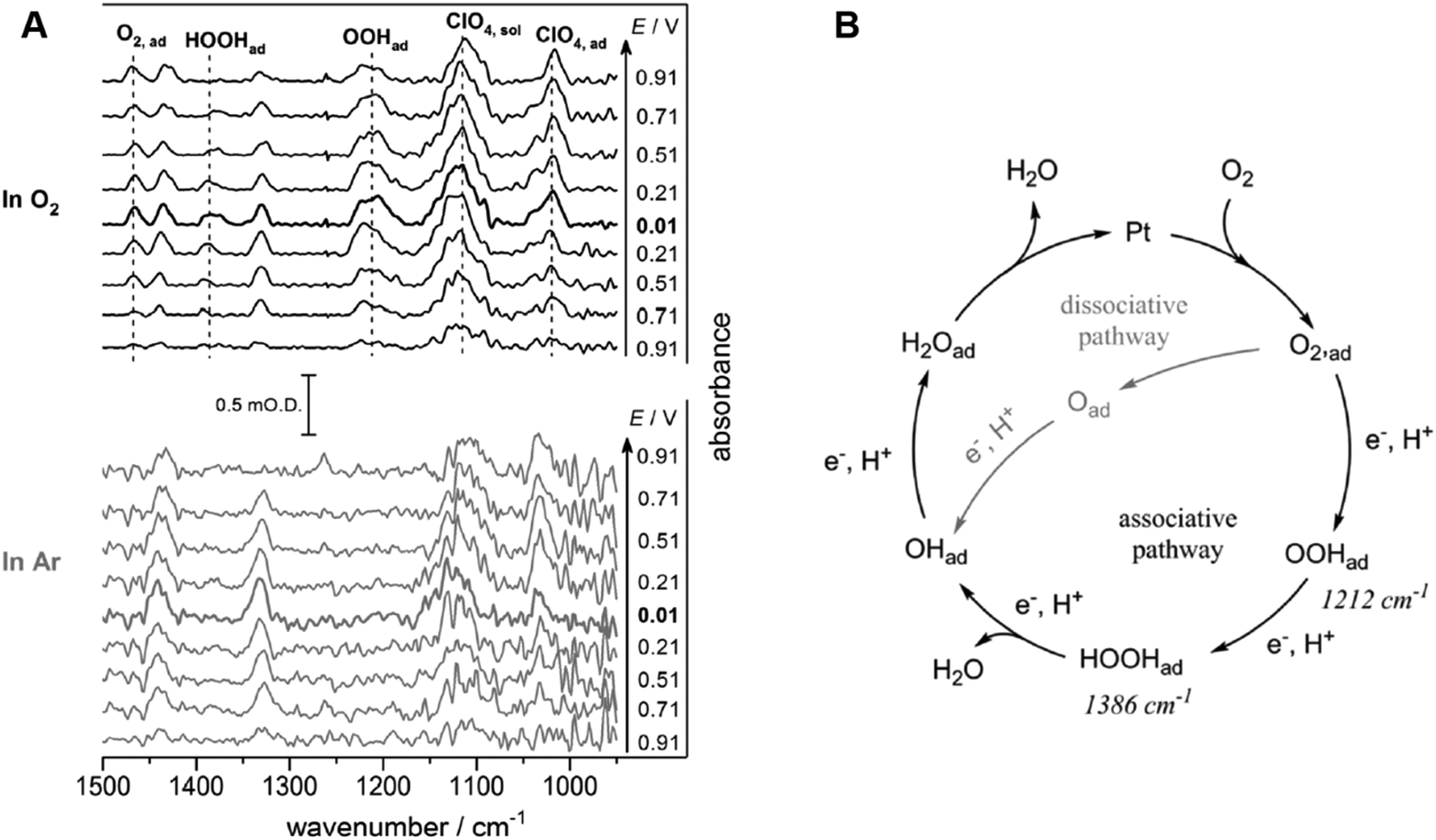 | ||
| Fig. 12 (A) ATR-IR spectra on Pt/C nanoparticle during ORR and under Ar in 0.1 M HClO4. The reference spectra are obtained at 1.01 V vs. SHE. (B) Proposed mechanism of ORR at a Pt/C nanoparticle catalyst in 0.1 M HClO4. The figure and caption were adopted from the reference with permission from the ref. 201. Copyright (2018) Wiley-VCH. | ||
In addition to the mechanism, the activity of the ORR on Pt is also affected by surface adsorbed species such as specifically-adsorbed anions and adsorbed hydroxide (OHad). Sulfate and bisulfate anions specifically adsorb on the Pt(111) surface, and therefore the ORR activity in H2SO4 solutions is much lower than that in HClO4 solutions.204–206 An IRAS measurement was used to investigate the SO3 symmetric stretching mode of adsorbed (bi)sulfate anion at the three-fold site of the Pt(111) surface via three oxygen atoms.207,208 Perfluorosulfonic acids (PFSA) such as Nafion used for polymer electrolytes also specifically adsorb on the Pt(111) surface and decrease the ORR activity.209–211 The IRAS and SEIRAS measurements suggested that the sulfonate group in PFSA adsorbs on Pt via one or two oxygen atoms, and the ether group also interacts with the surface.211,212 The adsorbed species formed during the surface oxidation such as OHad is a key factor in the ORR activity as well as the specifically-adsorbed electrolyte species. The equation of OHad formation is as follows:
| Pt + H2O → Pt–OH + H+ + e− |
The ORR activity of Pt low-index planes at 0.9 V (RHE) increases by following the order of Pt(110) > Pt(111) > Pt(100) in HClO4.213 The band assigned to the Pt–OH bending vibration band was observed by an IRAS measurement, and this band intensity at 0.9 V increased by following the order of Pt(100) > Pt(111) > Pt(110).214 Similarly, there was an inverse relationship between the band intensity and the ORR activity on high-index planes of Pt.215 Based on the these investigations, the stability of OHad was suggested to be dependent on the surface orientation of the electrocatalysts and therefore may determine the ORR activity as a similar feature is well-known in the field of solid–gas catalysis in ultrahigh vacuum (UHV) conditions.216 Not only surface orientation but also the OHad formation and the ORR activity of Pt can depend on electrolyte cations. At the electrode–electrolyte interface, electrolyte cations are hydrated and behave as non-specifically adsorbed species but weakly interact with Pt surfaces through hydrogen bonds and electrostatic interactions, i.e., so-called non-covalent interactions. In alkaline solutions, the ORR activity of Pt(111) is related to electrolyte cations as follows: Li+ < Na+ < K+ < Cs+.217 The Pt–OH band in LiOH containing solution on Pt(111) was observed by an IRAS measurement, whereas no bands were observed in CsOH218 These results suggested that OHad is a blocking species toward the ORR, and the formation of OHad is dependent on the electrolyte cation. The hydration water around hydrophilic cations such as Li+ is coordinated with the dipole moment pointed outward, and the hydrogen atoms of the hydration water can link with the outer oxygen species.219 Hydrated Li+ has a stronger non-covalent interaction with the Pt(111) surface to stabilize OHad than hydrated Cs+, and therefore the ORR activity in LiOH solutions is lower than that in CsOH solutions.
In addition to the above cations based on the alkaline metals, recent studies showed that organic cations, i.e., quaternary ammonium cations with long alkyl chains, enhanced the ORR activity of Pt.220,221 The ORR activity of Pt(111) in HClO4 containing tetrahexylammonium cation (THA+) at 0.9 V is eight times higher than that without this organic cation. Fig. 13A shows the IRA spectra of Pt(111) and THA+ modified Pt(111) surfaces in acidic solution. In this observation, the Pt–OH bending mode around 1050 cm−1 was observed above 0.6 V, which suggests that the addition of THA+ inhibits the OH adsorption on the Pt(111) surface. Fig. 13B shows the potential dependence of the charge density of Pt surface oxidation estimated from cyclic voltammetry studies and the band intensity of Pt–OH.221 The onset potential of OH adsorption shifts positively and the band intensity of Pt–OH decreases due to the presence of THA+. These results are consistent with the charge density of Pt surface oxidation and suggests that THA+ has some effects of destabilizing and decreasing OHad on the Pt(111) surface. The positive- and negative-going bands around 1650 and 1610 cm−1 are assigned to the HOH bending mode of non-adsorbed and adsorbed hydrogen-bonded water, respectively. Similar multiple bands appeared around 1500 cm−1 on THA+-modified Pt(111). These bands can be assigned to the HOH bending mode of non-adsorbed and adsorbed water monomers. The adsorbed water monomer has been observed under UHV conditions.222 However, at the electrode–electrolyte interface, the water molecules are cross-linked via hydrogen bonding, and therefore the adsorbed water monomer is unstable. Unlike the hydration water shell around hydrophilic cations, the shell around hydrophobic cations such as THA+ exhibits restricted coordination with the outer oxygen species because of the complete hydrogen-bonding network within the shell.219 This specific hydration structure may stabilize the adsorbed water monomer. The enhancement effect for the ORR activity due to THA+ did not appear on Pt(100) and Pt(110). The hydrogen-bonded co-adsorption layer of OHad and water forms on the Pt(111) surface because the symmetry and the hydrogen bonding distance fit with the Pt(111) lattice. Hydrophobic species destabilize the hydrogen bonding network and form an efficient interface for the ORR. On Pt(100) and Pt(110), the hydrogen bonding network is weak due to the lattice mismatch. THA+ specifically destabilizes the hydrogen bonding network and decreases the coverage of the OHad species on the Pt(111) surface. As a result, an efficient electrode–electrolyte interface for the ORR is formed. The ORR kinetics cannot be explained sufficiently by the thermodynamics of adsorbates. The above cations and water molecule may influence the quantum processes, such as proton and electron transfer. Kinetic studies at the characteristic electrode/electrolyte interface using by spectroscopic measurements combined the KIE method elucidate the microscopic mechanism for the efficient ORR.
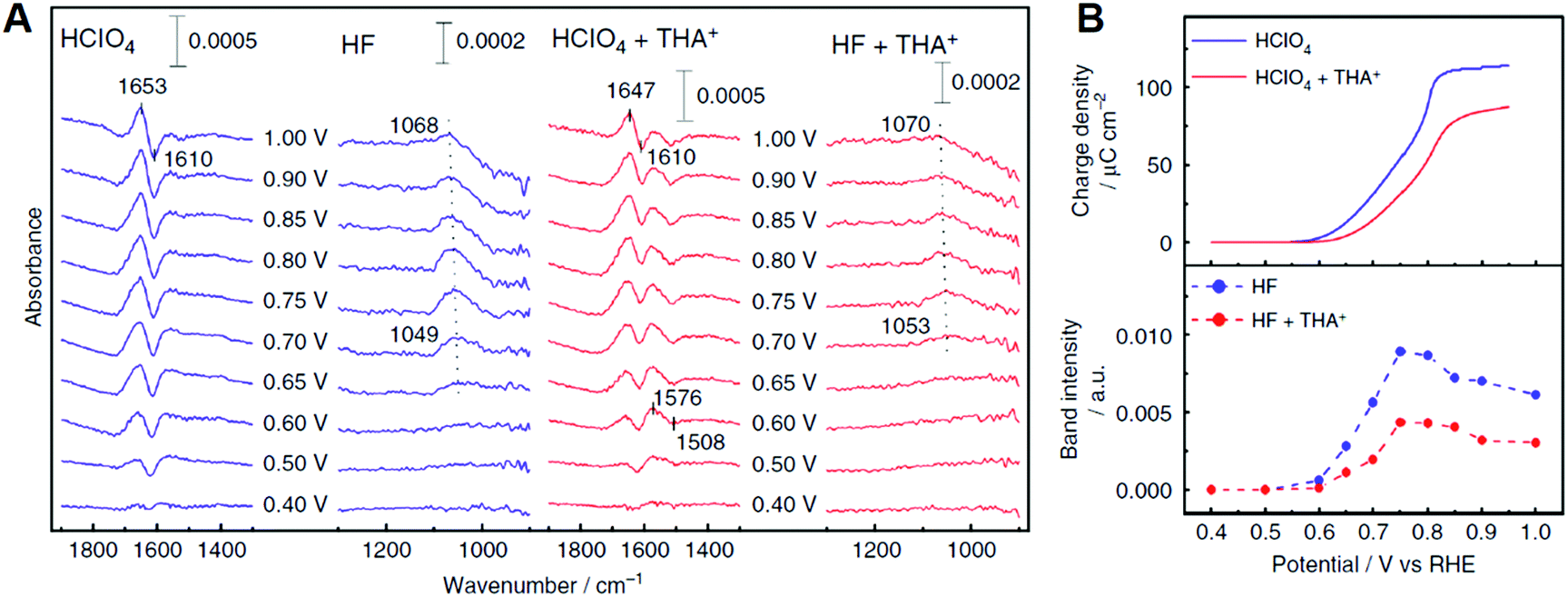 | ||
| Fig. 13 (A) Infrared reflection absorption spectra on Pt(111) and Pt(111) modified with tetrahexylammonium cation (THA+) in 0.1 M acidic solutions saturated with Ar. The reference spectra are obtained at 0.30 V vs. RHE. The potentials are stepped in the positive direction. (B) Potential dependence of the charge density of Pt oxidation and the band intensity of Pt–OH. The figure and caption were adopted from the ref. 221. | ||
Recent years, non-Pt based catalysts such as iron and cobalt porphyrin complexes for the ORR have been widely investigated.223 Living things convert the energy from the ORR efficiently using by these complex catalysts. Cytochrome is an heme protein containing iron porphyrin complex, which acts as the ORR catalyst and the proton pump.224 The PCET mechanisms in the ORR by various types of cytochrome have been investigated using by IR and Raman spectroscopy.225,226 Naumann et al. found the potential- and time-dependent band intensity assigned to C–C stretching mode of the porphyrine ring using by time-resolved SERS measurement.227 The band intensity is correlated to the transition from the reduced to the oxidized state of cytochrome via heterogeneous electron transfer. The rate constants of the electron transfer was estimated at kox = 46 ± 7 s−1 and kred = 84 ± 20 s−1. The same group also estimated the rate constant from the band intensity assigned to the amide region using by time-resolved SEIRAS measurement.228,229 Weidinger et al. revealed the KIE for the electron transfer of iron porphyrin complexes from the band intensity assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of the protonated carboxylic acid using by the SERS and SEIRAS measurements.230 The heterogeneous electron transfer of the porphyrin complex is strongly modulated by the protonation state of the acidic group via hydrogen bond interactions.
O stretching mode of the protonated carboxylic acid using by the SERS and SEIRAS measurements.230 The heterogeneous electron transfer of the porphyrin complex is strongly modulated by the protonation state of the acidic group via hydrogen bond interactions.
4.4 Outlooks for the future of operando surface-electrochemical spectrometry
Operando observations using infrared spectroscopy and Raman spectroscopy are useful to elucidate electrocatalytic processes and molecular structures at electrode–electrolyte interfaces. The spectroscopic study combined with time-resolved techniques and the KIE method will strongly support the microscopic kinetics of the HER and the ORR. In addition to the HER and the ORR, the photocatalytic hydrogen production and the CO2 electroreduction are also interesting subjects for the spectroscopic study.231–234 Non-linear spectroscopy methods such as sum-frequency generation (SFG) spectroscopy and stimulated Raman scattering (SRS) can be applied to analyze the dynamics of electrode processes on the pico or femtosecond timescale.235–239 Single-molecule spectroscopy and imaging techniques such as tip-enhanced Raman spectroscopy (TERS) will enable monitoring of electrochemical reactions with nanoscale resolution.240 These approaches are expected to provide further detailed information on complicated phenomena at electrified solid–liquid interfaces, such as elucidating the role of quantum effects.5. Heterogeneous PCET theory applied to the Volmer reaction
5.1 Basics of general PCET theory
As discussed above, many electrochemical processes involve PCET. A general theoretical formulation for PCET that treats the active electrons and transferring protons quantum mechanically and includes the reorganization of the solvent environment, as well as the proton donor–acceptor motion, has been developed.241–244 In the context of Fig. 4,67 this theoretical formulation covers the entire right branch, in which the electrons and protons are treated quantum mechanically. If the process is fully electronically and vibrationally adiabatic, where the electrons and protons respond instantaneously to the other nuclei, the transition state theory formulation is often applicable (case A in Fig. 4). If the process is vibrationally or vibronically nonadiabatic, in the absence of this instantaneous response, the Fermi golden rule nonadiabatic formulation is often applicable (cases B and C in Fig. 4),242,243 although the vibronic coupling has a different form in the electronically adiabatic and nonadiabatic regimes.243,245,246 In the more complex case of slow solvent dynamics, the rate constant expressions must also account for the solvent-controlled regime.247–250 This section will discuss the rate constants for all of these limits as well as the intermediate regimes, focusing primarily on electrochemical PCET.5.2 Concepts of heterogeneous electrochemical PCET theory
Electrochemical PCET can be divided into homogeneous and heterogeneous processes (Fig. 14). In homogeneous electrochemical PCET, the electrode serves as a reservoir for donating and accepting electrons but is not involved in the chemical bond breaking and forming. In this case, the electron transfers between the solvated redox molecule and the electrode, and the proton transfer reaction occurs within the molecular complex. Homogeneous electrochemical PCET is described in detail elsewhere251–254 and will not be discussed further herein. In heterogeneous electrochemical PCET, the electrode participates in the chemical bond breaking and forming process. In the prototypical Volmer reaction, the proton is transferred from the molecular system, which could be specifically adsorbed on the surface, to the electrode, forming a metal–hydrogen covalent bond. The remainder of this section will focus on heterogeneous electrochemical PCET in the context of the Volmer reaction.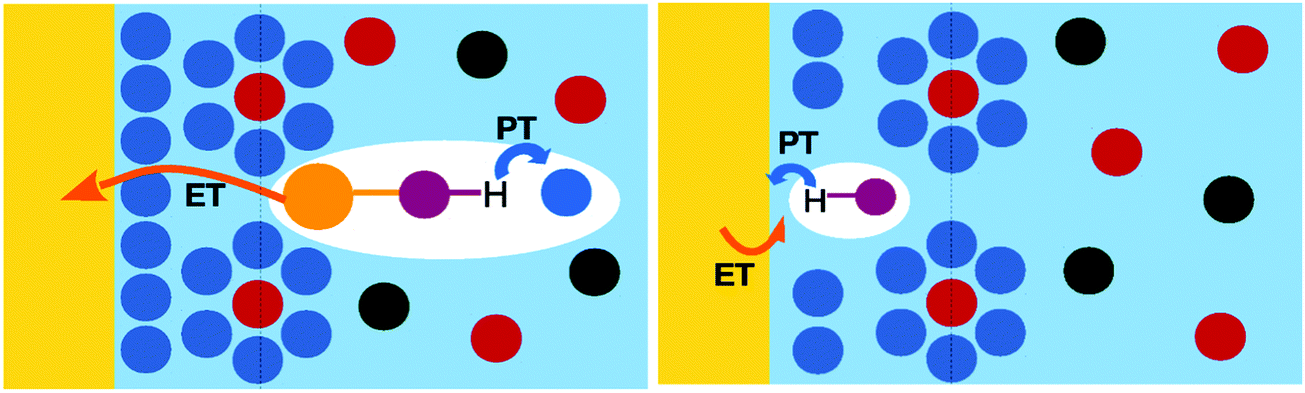 | ||
| Fig. 14 Schematic depiction of homogeneous electrochemical PCET (left) and heterogeneous electrochemical PCET (right). In homogeneous PCET, the electrode serves as a reservoir for electrons but is not directly involved in the chemistry; the electron transfers between the molecule and the electrode, and the proton transfers within the molecular complex. In heterogeneous PCET, the electrode participates in the chemistry; in the Volmer reaction, the proton transfers from the molecule to the electrode, forming a metal–hydrogen bond. | ||
In the acidic Volmer reaction, a proton is transferred from an acid, AH+, to a metal surface M, forming the conjugate base A and a metal–hydrogen (MH) bond via an electron from the electrode:
| M + AH+ + e− ⇌ MH + A. | (4) |
In both regimes, the resulting rate constant k(R,E) depends on the distance R from the electrode and the applied electrode potential E. The current density is obtained by averaging this rate constant over the distance R, weighting by the concentration cHA+(R) of the acid:
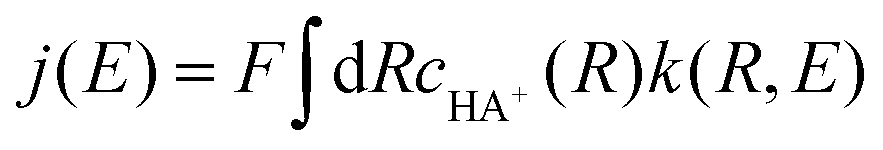 | (5) |
The form of the cathodic rate constant in the vibronically nonadiabatic regime for heterogeneous electrochemical PCET can be derived using Fermi's golden rule, leading to the following expression:93,242,243,251
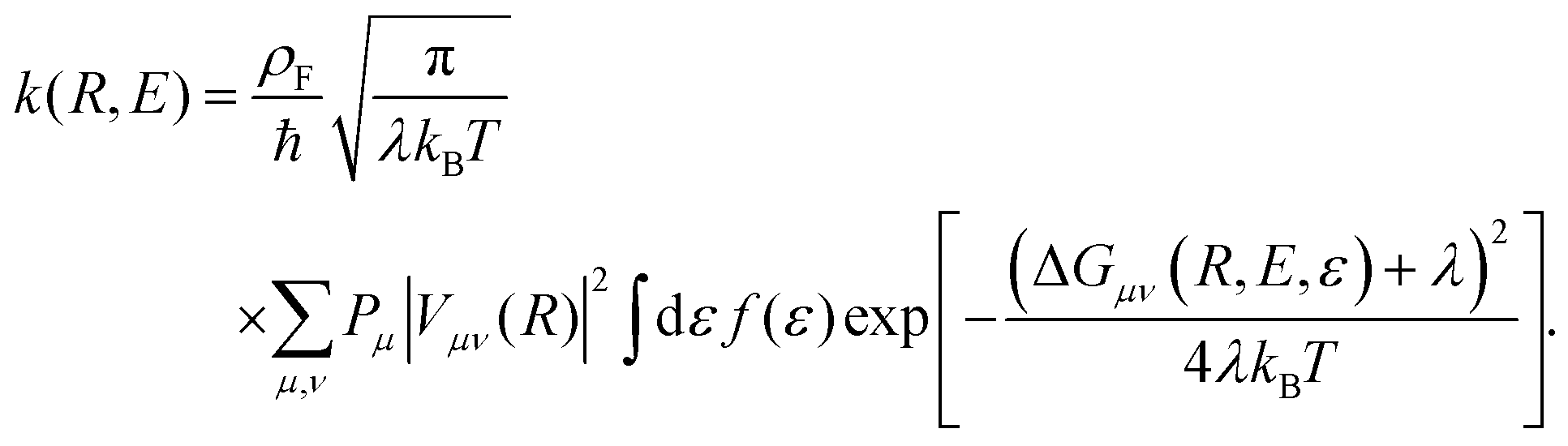 | (6) |
| ΔGμν(R,E,ε) = ΔG° + Δεμν − ε + e(E + ERef) − eψ(R,E) − ΔW(R) | (7) |
The electrostatic potential ψ(R,E) can be determined using a suitable model for the electrical double layer (EDL). A practical approach that includes the key physical properties of the EDL is a recently developed extended Guoy–Chapman–Stern model.93,256–259 This extended model describes the electrode–solvent interface with three regions that each have different dielectric constants.93 The inner Helmholtz layer is assigned a relatively small dielectric constant to describe the constrained mobility of the first layer of solvent molecules, the diffuse layer is assigned the solvent dielectric constant, and the intervening outer Helmholtz layer is assigned a variable dielectric constant that depends on the applied electrode potential.259 These types of dielectric continuum models provide qualitatively reasonable descriptions of the EDL but do not describe the detailed behavior of the electrostatic potential at the interface. The electrostatic potential at a gold electrode surface has been shown to be inhomogeneous and to depend strongly on the applied electrode potential.260 Explicit solvent molecules and ions must be included to capture the detailed interfacial behavior. Nevertheless, the simple dielectric continuum models that incorporate the essential physical properties of the EDL are useful for computing heterogeneous PCET rate constants.
The form of the vibronic coupling Vμν(R) depends on the degree of electron–proton nonadiabaticity.245,246 Within the vibronically nonadiabatic regime, the electronically adiabatic and nonadiabatic limits correspond to cases B and C, respectively, in Fig. 4. In the electronically nonadiabatic limit, Vμν(R) is the product of the electronic coupling and the overlap integral between the reactant and product proton vibrational wavefunctions associated with vibronic states μ and ν. In the electronically adiabatic limit, Vμν is half the tunneling splitting Δμν between the delocalized proton vibrational states computed for the electronically adiabatic proton potential curve associated with alignment of the energies of vibronic states μ and ν. The general expression for the vibronic coupling spanning both regimes is245,246
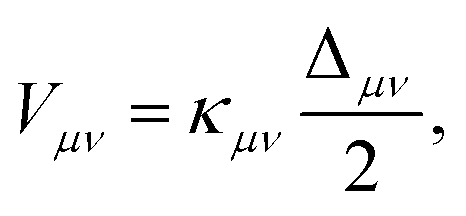 | (8) |
5.3 Application of heterogeneous electrochemical PCET theory to uncover microscopic mechanism of Volmer reaction
This vibronically nonadiabatic approach was used to investigate the Volmer reaction corresponding to proton discharge from triethylammonium (TEAH+) to a gold surface in acetonitrile (Fig. 15).93 Experimental studies of this process revealed significantly different Tafel slopes for TEAH+ and its deuterated counterpart, TEAD+, leading to a potential-dependent kinetic isotope effect (KIE) for the hydrogen evolution reaction.95 A theoretical analysis of this process suggested that it is vibronically nonadiabatic, enabling the use of the rate constant in eqn (6), and is predominantly but not fully electronically adiabatic, necessitating the use of the general expression for the vibronic coupling given in eqn (8) to describe the intermediate regime.93 Application of this theoretical formulation in conjunction with a physically reasonable model reproduced the experimental observations (Fig. 15).93,95 The physical explanation for the potential-dependent KIE emerging from this study was that different pairs of reactant and product vibronic states dominate for hydrogen and deuterium with relative contributions that depend on the applied potential. In particular, the smaller energy splittings between the vibrational states for deuterium result in greater contributions from the excited reactant vibronic states for TEAD+ compared to TEAH+.93 This application highlights the importance of nuclear quantum effects, nonadiabaticity, and excited vibronic states in heterogeneous PCET. It also provides an illustration of how PCET reactions proceeding by the same fundamental mechanism can exhibit different Tafel slopes.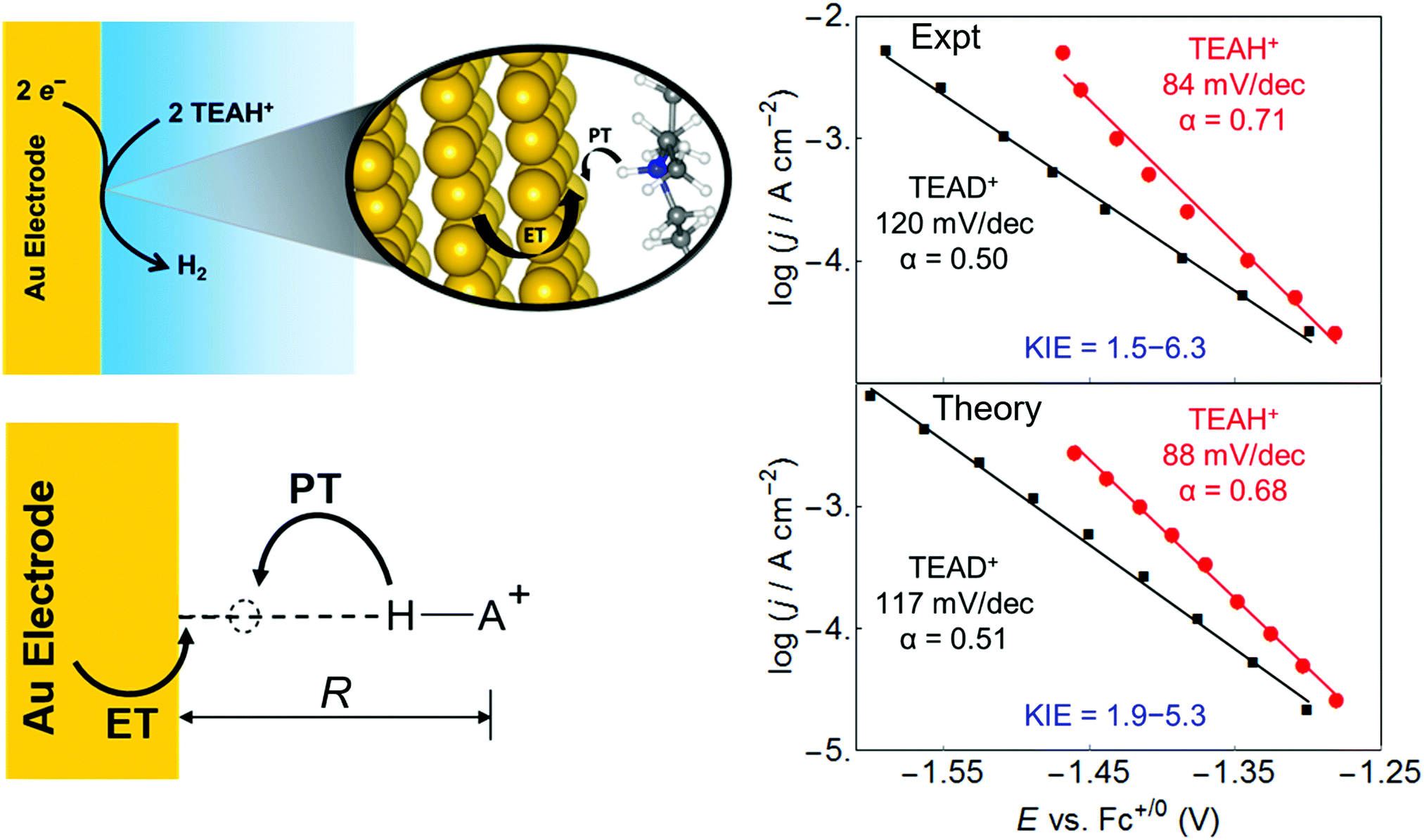 | ||
| Fig. 15 Left: Schematic depiction of the PCET reaction associated with proton discharge from TEAH+ to a gold electrode, corresponding to the Volmer reaction that is a key step in the hydrogen evolution reaction. Right: Tafel plots, which reflect the current density versus the applied potential, obtained from experimental data (upper panel) and theoretical calculations (lower panel) for the PCET reaction associated with proton discharge from TEAH+ (red circles) or TEAD+ (black, squares) to a gold electrode in acetonitrile. The reported Tafel slopes and electrochemical transfer coefficients, α, are obtained from linear fits to the data points shown. The smallest and largest KIEs reported are the ratios between the TEAH+ and TEAD+ linear fits at −1.30 V and −1.46 V, respectively. Figures adapted with permission from ref. 93. Copyright 2018 American Chemical Society. | ||
For the Volmer reaction in aqueous solution, the proton is typically presumed to be donated directly from H3O+, which is hydrogen bonded to other water molecules in some type of protonated water cluster. Because the proton-donating acid is located near or adsorbed to the surface, leading to relatively strong electronic coupling and short proton donor–acceptor distances, the aqueous Volmer reaction is often assumed to be fully adiabatic (i.e., in the vibronically adiabatic limit). In this case, the reaction occurs on the ground state free energy surface, which can be described as a function of r, X, and R (Fig. 16).255 Quantization of the transferring proton coordinate results in a vibronic free energy surface that depends on only X and R, as well as the applied electrode potential E. For each value of E, the free energy barrier corresponding to the saddle point relative to the reactant minimum on this two-dimensional vibronic free energy surface can be computed.255 Within a transition state theory framework, the rate constant depends exponentially on this free energy barrier, and the transfer coefficients can be computed.
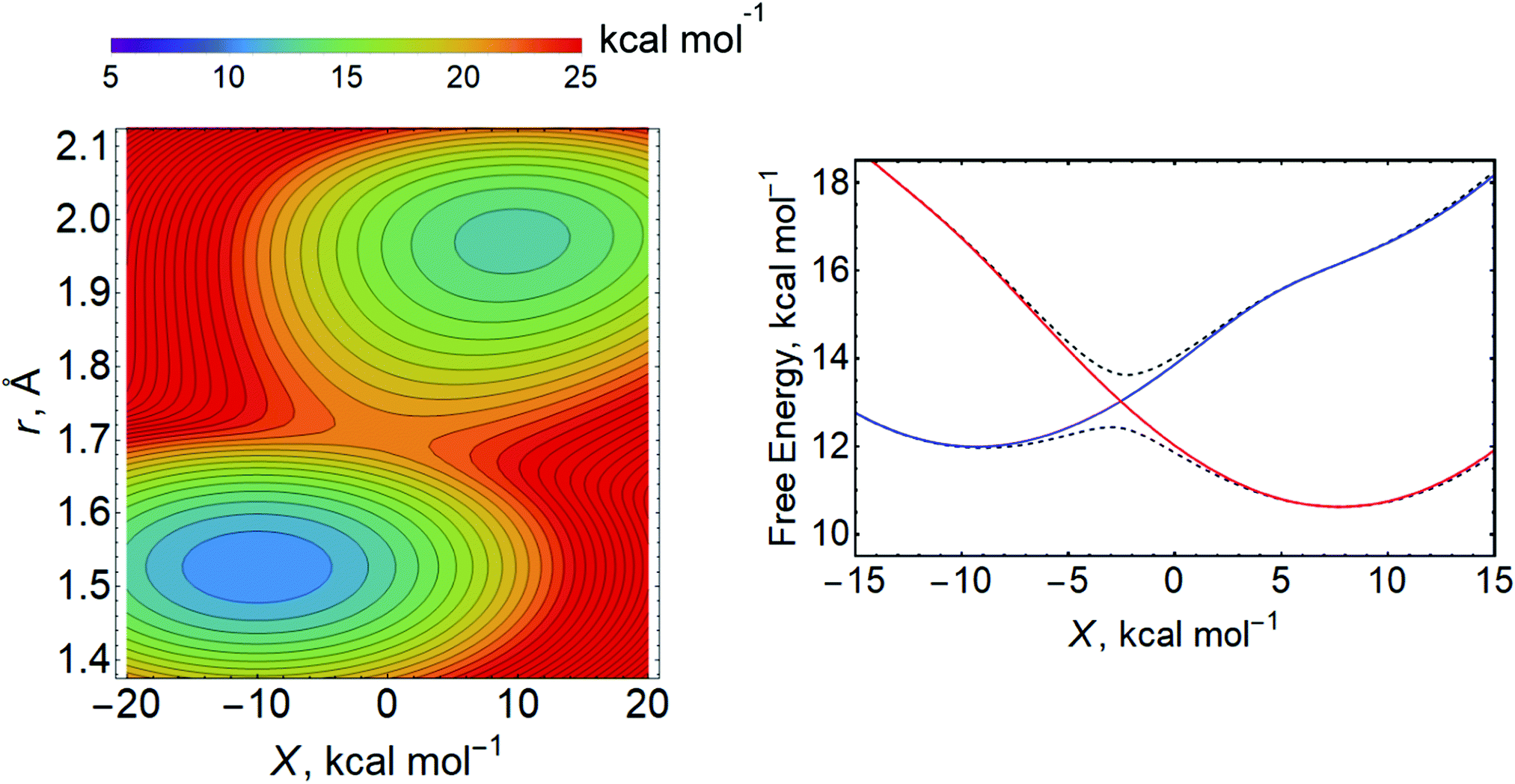 | ||
| Fig. 16 Left: Example of two-dimensional free energy surface as function of the proton coordinate r and the collective solvent coordinate X computed at a fixed distance R between the acid and the electrode surface. Right: Example of free energy surfaces for the lowest two adiabatic vibronic states (dashed black lines) and associated diabatic vibronic states (solid lines, reactant state in red, product state in blue) as a function of the collective solvent coordinate X at a fixed R obtained by quantization of the proton coordinate. Left figure adapted with permission from ref. 255. Copyright 2019 American Chemical Society. Right figure adapted with permission from ref. 94. Copyright 2019 American Chemical Society. | ||
This vibronically adiabatic approach was applied to the Volmer reaction in acidic aqueous solution.255 In this application, an empirical valence bond model was developed to generate the ground electronic state free energy surfaces at each applied potential E, followed by quantization of the proton and calculation of the free energy barrier on the vibronic ground state free energy surface. Most of the parameters in this model can be computed using first-principles simulation methods, although in some cases experimental data may be used as a guide. Typically the qualitative trends are not sensitive to the specific values of the parameters within physically reasonable ranges. Within this model, the calculated transfer coefficients were consistent with experimental data, but the H/D KIEs were lower than the experimental values. Analysis of the vibronic free energy surfaces indicated that the splitting between the ground and first excited proton vibrational states on the electronic ground state was small enough at large R to indicate a significant degree of vibrational nonadiabaticity.255 Neglect of the vibrational nonadiabaticity was proposed to be a plausible explanation for the underestimation of the H/D KIEs.
Thus, the vibronically adiabatic framework was modified to include the effects of the first excited adiabatic proton vibrational state.94 A unified formulation was developed to describe the Volmer reaction in terms of a curve crossing between the two diabatic vibronic states associated with the two lowest adiabatic proton vibrational states.94 In this formulation, the rate constant is expressed in the form
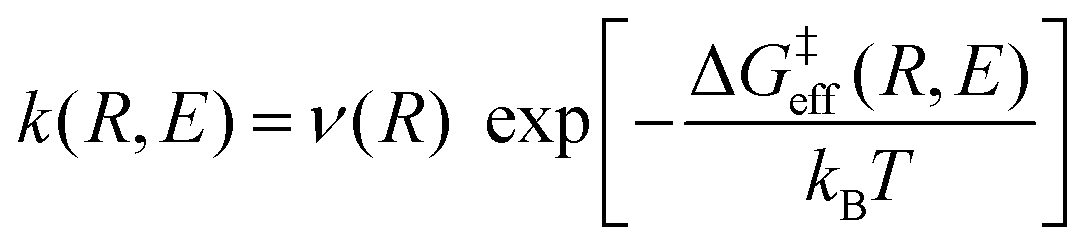 | (9) |
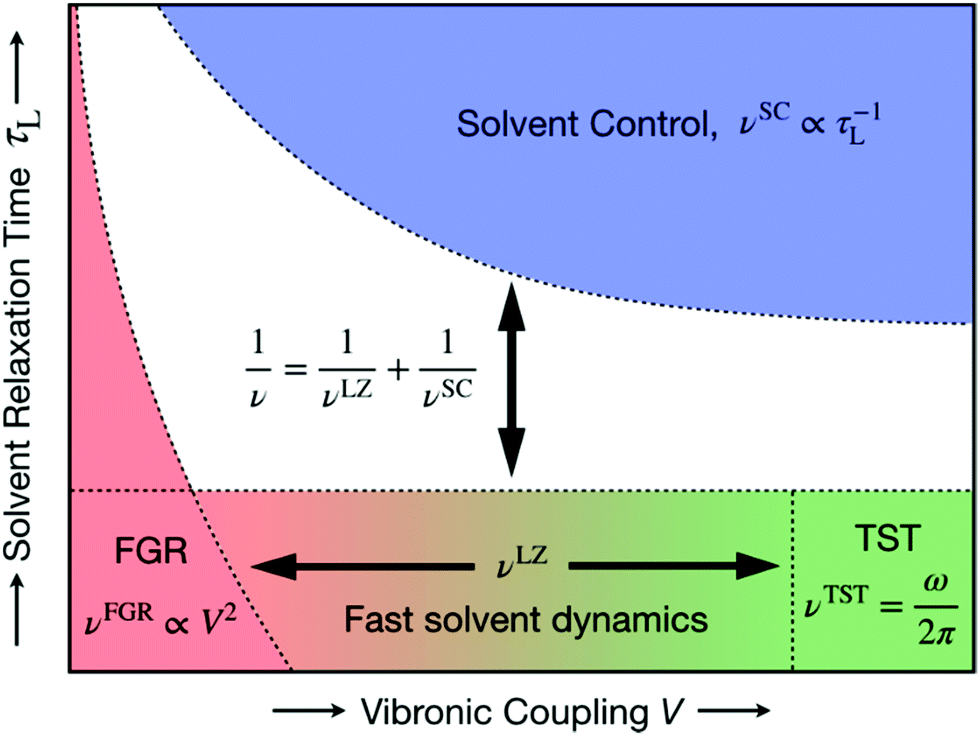 | ||
| Fig. 17 Schematic representation of the unified formulation that accounts for both nonadiabaticity and solvent dynamics. In the limit of fast solvent dynamics (bottom of figure), the pre-exponent is given by νLZ, which in the normal region interpolates between the nonadiabatic Fermi golden rule (FGR) limit νFGR (bottom left) and the adiabatic transition state theory (TST) limit νTST (bottom right). In the limit of slow solvent dynamics (top), the pre-exponent is given by νSC. The model is valid for thermally activated processes in the two-state curve-crossing formulation. Reproduced with permission from ref. 94. Copyright 2019 American Chemical Society. | ||
The interpolated curve-crossing approach was applied to the Volmer reaction in acidic aqueous solution using the empirical valence bond model described above.94 The lowest two adiabatic vibronic free energy surfaces along the collective solvent coordinate X were diabatized for each distance R between the acid and the electrode surface (Fig. 16), allowing the calculation of the interpolated prefactor ν(R), as illustrated in Fig. 10D. Subsequently, the rate constant k(R,E) was calculated from eqn (9), and then the current density was calculated as a function of applied potential E using eqn (5), as depicted in Fig. 10C. Analysis of the results indicated that vibrational nonadiabaticity and solvent dynamics play significant roles in this reaction.94 The calculated transfer coefficients of 0.69–0.73 and kinetic isotope effects of 3.8–7.1 are in reasonable agreement with experimental data46,261,262 for the Volmer reaction in acidic aqueous solution. Applications to the Volmer reaction in basic aqueous solution may require the development of additional methodology because of contributions from excited electronic states and thermodynamic effects from the formation of the hydroxide ion at the surface.
6. Grand canonical ensemble DFT for electrochemical rate computation
6.1 Recent issues in DFT calculation for electrochemical reactions
The electrode potential is at the heart of all electrochemistry as it offers a direct way to control electrochemical thermodynamics and kinetics. In principle, explicit control of the electrode potential is the only factor separating electrochemical and electrocatalytic reactions from all other “normal” chemical reactions. At the microscopic level, the electrode potential gives direct access to changing the chemical potential of electrons (μ) which in turn is directly proportional to the Fermi-level: μ ∝ −EF.62 Given the importance of the electrode potential, it has been widely acknowledged that computational studies of electrochemical systems should be conducted at fixed electron chemical potential conditions. As discussed shortly below in Section 6.2, first principles calculations on electrochemical thermodynamics can be rigorously defined in the grand canonical ensemble (GCE), and a variety of approaches have been developed to realize or approximate the GCE.While GCE has emerged as the gold standard in first principles computational studies on electrochemical thermodynamics, only a handful of works have focused on kinetics. The scarcity of studies on electrochemical kinetics can probably be attributed to both methodological and theoretical complexities.263 In particular, a generally valid rate theory for electrochemical systems has only recently been developed66 showing how electrochemical rate constants can be computed within the GCE. The GCE rate theory (GCE-RT) formalism allows inclusion of non-adiabatic and nuclear tunneling contributions as a function of the electrode potential using general first principles Hamiltonians. It has also been shown that GCE-RT enables the extension of all theoretical methods developed for studying tunneling in “normal” chemical reactions to be utilized for electrochemical reactions.
The emphasis of this chapter is on showing how first-principles methods, especially GCE-DFT (see Section 6.2), can be utilized to address reaction rates and nuclear quantum effects (NQE) as a function of the electrode potential using GCE-RT. The discussion presented here serves both as a perspective as well as a review and tutorial of promising methods to address nuclear quantum effects at electrochemical interfaces using first principles Hamiltonians. The perspective part of this section is that electrochemical rates are obtained by extending the canonical rate theories to the GCE. This provides a theoretically rigorous way to compute rate constants from first principles as a function of the electrode potential. Starting from this general GCE rate theory it is shown how well-defined approximations to treating nuclear tunnelling as a function of the electrode potential can be developed. Instead of giving a historical account, this section aims to review and connect different theoretical approaches in a pedagogical manner. The treated methods have been utilized to study condensed phase chemistry but many of them have not yet been applied to electrochemical reactions and it is shown here how this extension is achieved within GCE-RT. More traditional model Hamiltonian-based approaches are discussed in other parts of the work (Sections 5 and 7). This section will serve a three-fold purpose: (1) to introduce general rate theory to researchers working on experimental or computational electrochemistry, (2) to show how nuclear quantum effects can be addressed with computational tools used in heterogeneous (electro)catalysis, and (3) to inspire joint experimental–computational studies of quantum phenomena at electrochemical interfaces.
6.2 The grand canonical ensemble for electrochemistry
Properties of electrochemical interfaces are controlled by the electrode potential and the solvent or equivalently the (electro)chemical potential of electrons and the electrolyte, respectively.62 In macroscopic systems both the electrode potential and the solvent chemical potentials are constant, which means that electrochemical interfaces work under fixed electrochemical potentials. However, most of computational chemistry, such as density functional theory (DFT) calculations, are performed in the canonical or the NVT ensemble characterized by constant temperature (T), particle number (N), and volume (V) which lead to the Helmholtz free energy.264 In this NVT ensemble the chemical potentials fluctuate, which makes it unsuitable for constant potential calculations. To achieve constant potential calculations, a Legendre transformation from the canonical ensemble to the GCE is performed to change the thermodynamic variables from NVT to μVT.It has been shown62 that in the μVT ensemble, the thermodynamic state at fixed chemical potential is obtained rigorously from the GCE partition function Ξ at chemical potential μ and nuclear-electronic Hamiltonian Ĥtot as Ξ = Tr[exp[−Ĥtot + μ![[N with combining circumflex]](https://www.rsc.org/images/entities/i_char_004e_0302.gif) ]] (Tr denotes a quantum mechanical trace i.e. quantum mechanical analogue of phase-space averaging). As Ĥtot is the full Hamiltonian, all quantum and non-adiabatic effects of electrons and nuclei are included in this multicomponent definition. In the GCE all expectation values are given in terms of the GCE density operator
]] (Tr denotes a quantum mechanical trace i.e. quantum mechanical analogue of phase-space averaging). As Ĥtot is the full Hamiltonian, all quantum and non-adiabatic effects of electrons and nuclei are included in this multicomponent definition. In the GCE all expectation values are given in terms of the GCE density operator ![[small rho, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b7.gif) as 〈O〉 = Tr[Ô]. In particular, the constant potential free energy Ω is a functional of the GCE density operator62 and the equilibrium free energy Ω[μ,T,V] = Ω[] of the nuclear-electronic quantum system is uniquely obtained by minimizing Ω[] over all densities at constant chemical potentials leading to GCE-DFT. In this μTV ensemble the number of species is allowed to fluctuate and the corresponding relevant thermodynamic free energy is the grand or Landau free energy Ω. Methods for approximating the exact electrochemical free energy from atomistic DFT simulations as a function of the electrode potential have been devised and include self-consistent field method,265 iterative approaches,64,266 and the potentiostat scheme.267 Many of these techniques have been reviewed in the recent reports,268 and in the present work formally exact approaches are treated in Section 7 while model Hamiltonian-based approaches are utilized in Section 5.
as 〈O〉 = Tr[Ô]. In particular, the constant potential free energy Ω is a functional of the GCE density operator62 and the equilibrium free energy Ω[μ,T,V] = Ω[] of the nuclear-electronic quantum system is uniquely obtained by minimizing Ω[] over all densities at constant chemical potentials leading to GCE-DFT. In this μTV ensemble the number of species is allowed to fluctuate and the corresponding relevant thermodynamic free energy is the grand or Landau free energy Ω. Methods for approximating the exact electrochemical free energy from atomistic DFT simulations as a function of the electrode potential have been devised and include self-consistent field method,265 iterative approaches,64,266 and the potentiostat scheme.267 Many of these techniques have been reviewed in the recent reports,268 and in the present work formally exact approaches are treated in Section 7 while model Hamiltonian-based approaches are utilized in Section 5.
6.3 Direct computation of rate constants
As discussed above, GCE provides a rigorous basis for first principles computation of electrochemical thermodynamics at fixed potentials. To extend the GCE treatment to kinetics as well, it has been recently shown that equilibrium rate constants at fixed potential kinetics can be exactly derived using a GCE extension to the canonical rate theory.66 It is well known that the exact thermal rate constant in the canonical ensemble is given by ref. 66, 264, 269 and 270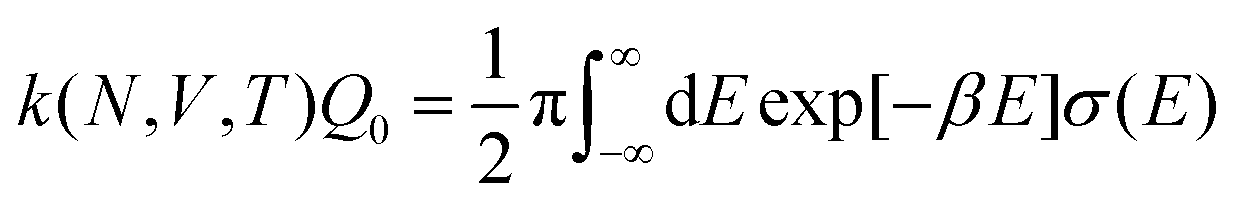 | (10) |
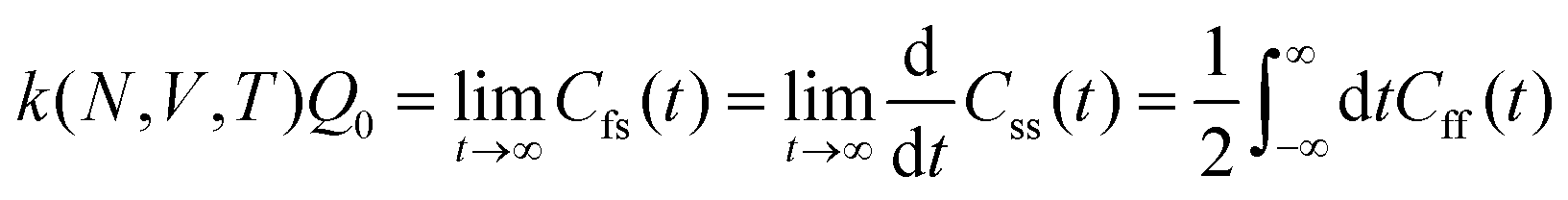 | (11) |
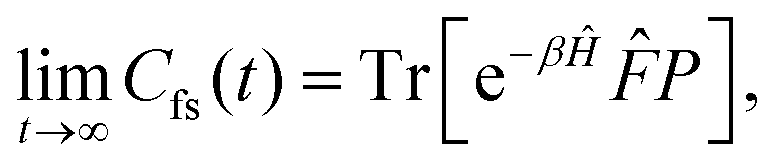 | (12) |
![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) is the flux operator, and P the projector operator. In analogy with the classical rate theory discussed below, eqn (12) can be understood as shown in Fig. 18 and as follows. One starts a quantum mechanical “trajectory” from a multidimensional dividing surface ĥ[q‡], which separates the reactants and products, according to a quantum mechanical thermal distribution (e−βĤ). The flux through ĥ[q‡] towards the product is measured by . The projector operator
is the flux operator, and P the projector operator. In analogy with the classical rate theory discussed below, eqn (12) can be understood as shown in Fig. 18 and as follows. One starts a quantum mechanical “trajectory” from a multidimensional dividing surface ĥ[q‡], which separates the reactants and products, according to a quantum mechanical thermal distribution (e−βĤ). The flux through ĥ[q‡] towards the product is measured by . The projector operator 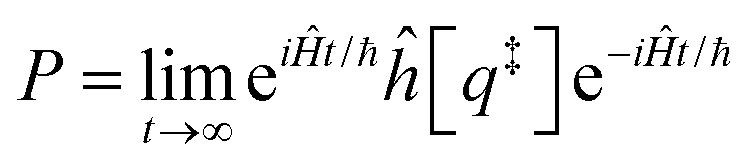 indicates the Heisenberg time-evolution of the dividing surface measuring how many “trajectories” initialized at ĥ[q‡] end up in the product region as time is advanced.
indicates the Heisenberg time-evolution of the dividing surface measuring how many “trajectories” initialized at ĥ[q‡] end up in the product region as time is advanced.
 | ||
| Fig. 18 A 2D free energy surface depicted with contours: the darker colors correspond to lower energies, purple is the product region, green is the reactant region and the thick black line is the dividing surface h[q‡] between the reactants and products. A trajectory is initiated at the dividing surface at t = 0 with a flux towards the products with momentum denoted by the arrow. The particle follows the trajectory as denoted with the grey dash-dotted line. At t1 the particle is on the product side and the side function h = 1. As time evolves the particle recrosses the dividing surface reactant side (h = 0 at t2), and finally returns to the product side (h = 1) at t3 making the trajectory a reactive one. | ||
Eqn (10) and (11) form the formal starting point for developing well-defined approximations to the exact rate constant in both the canonical ensemble264,269,270 and the electrochemically relevant GCE.66 The explicit forms of the cumulative reaction probability and the correlation functions depend on the Hamiltonian (classical or quantum) and the intended application (adiabatic, non-adiabatic, deep or shallow tunneling, etc.). Below, the general equations are connected to various approximations and schemes for estimating nuclear quantum effects.
To transfer the machinery developed for canonical rate constants to the electrochemical GCE setting, the following generalization is made66
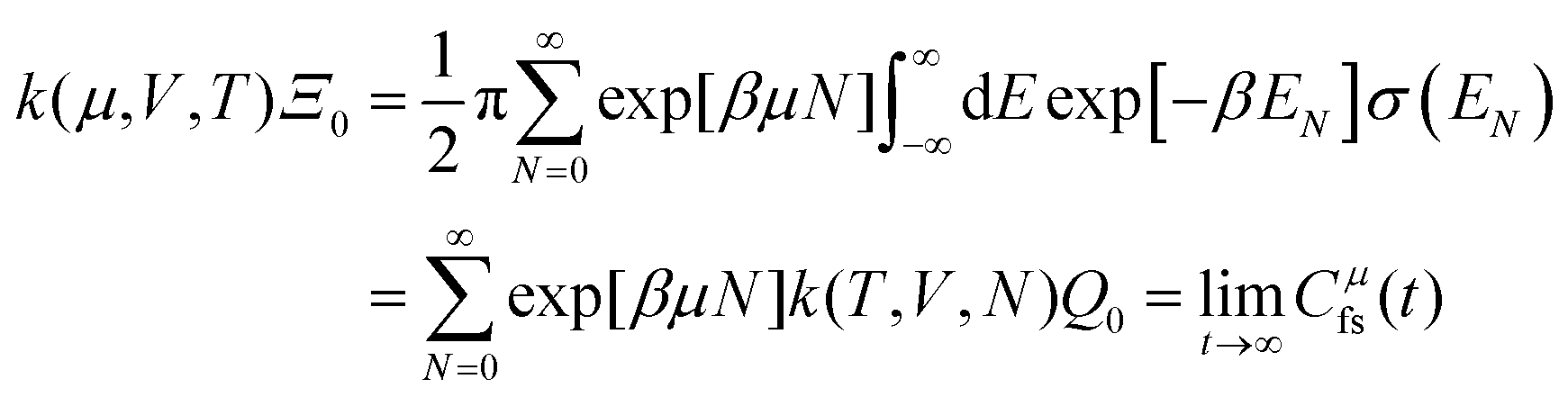 | (13) |
6.4 Adiabatic reactions
In this subsection the focus is on reactions taking place on a single ground state potential energy surface (PES) and all excited states are neglected. To enable this, the Born–Oppenheimer approximation (see Section 7) is assumed and applied to separate nuclear and electron movement – the electrons are assumed to respond instantaneously to the nuclear displacement. In the treatment of proton-coupled electron transfer reactions where the protons are quantized, sometimes a double-adiabatic approximation is invoked (see Section 7) – the electrons and protons are considered to respond instantaneously to the movement of heavier nuclei. As discussed in Section 5, often the Born–Oppenheimer approximation is not valid for proton-coupled electron transfer reactions, and a vibronically nonadiabatic treatment is required. However, the Born–Oppenheimer approximation is the underlying assumption for all the rate theories covered in this section.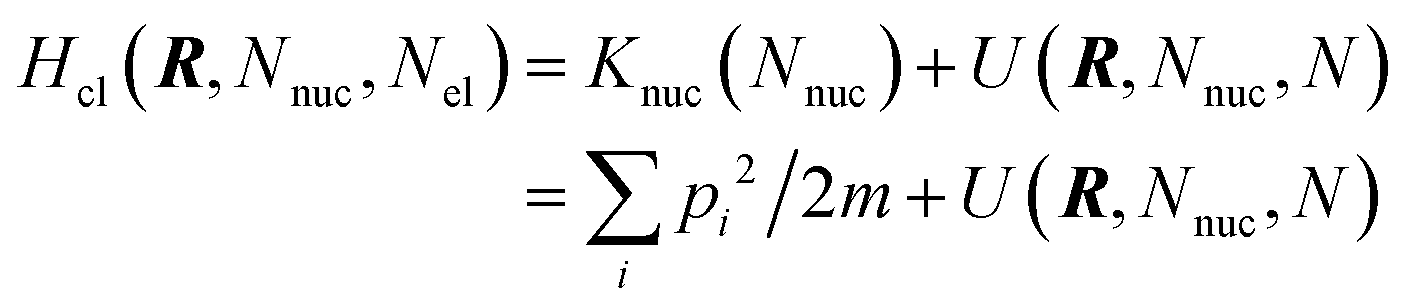 and the dependence on the nuclear positions R and of the number of nuclei (Nnuc) and electrons (Nel) has been explicitly noted. In density functional theory or other first-principles calculations U(R,Nnuc,Nel) is solved directly as a function of nuclear positions and number of electrons, see Section 7. After introducing the classical Hamiltonian, flux and projections in eqn (12) gives the classical rate constant as264
and the dependence on the nuclear positions R and of the number of nuclei (Nnuc) and electrons (Nel) has been explicitly noted. In density functional theory or other first-principles calculations U(R,Nnuc,Nel) is solved directly as a function of nuclear positions and number of electrons, see Section 7. After introducing the classical Hamiltonian, flux and projections in eqn (12) gives the classical rate constant as264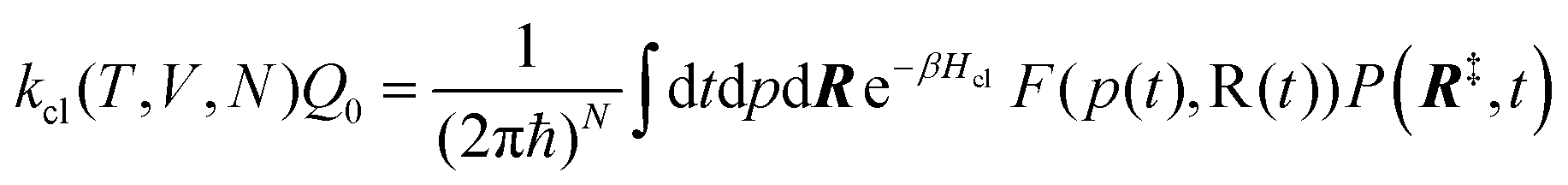 | (14) |
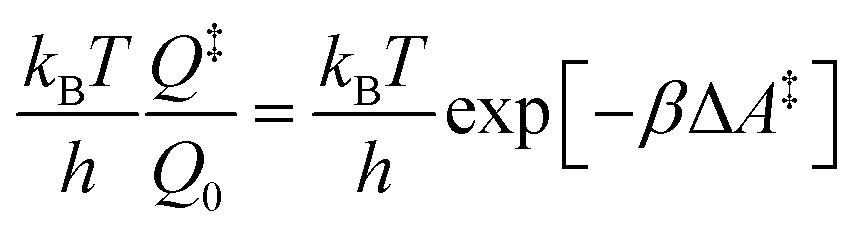 | (15) |
| kcl,hTST(T,V,N) = vnexp[−β(ΔE‡ + TΔSvib)] | (16) |
Extension of the canonical classical rate theory to electrochemical and electrocatalytic situation is rather straight-forward.66 The formalism remains exactly the same apart from a modified PES and classical Hamiltonian; the potential energy becomes dependent on the fluctuation number of electrons at a fixed chemical potential, U(R,Nnuc,μ). While this extension seems simple, in fact an additional Born–Oppenheimer-like approximation is needed as the change in number of electrons is assumed to be instantaneous and the electron transfer rates within the electrode or from the bulk to surface the are assumed to be infinitely fast. As such, the extension to GCE may break down for e.g. poorly conduction semiconductor electrodes. If the electron transfer within the electrode is fast, the fixed potential TST is simply in terms of GCE partition functions or the change in the grand canonical energy ΔΩ66
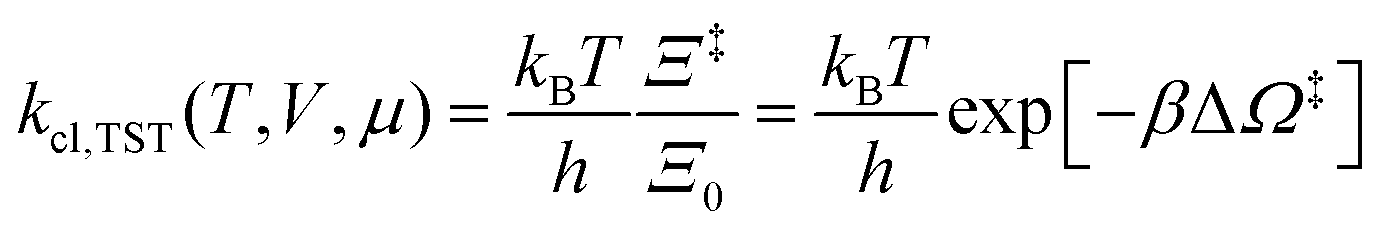 | (17) |
A coarse division in treating tunneling is based on the notion of deep and shallow tunneling.285,286 The former can be considered as genuine through the barrier tunneling from a single quantum level and the reaction pathway is different than from classical nuclei – referred to as corner-cutting. Shallow tunneling, on the other hand, can be viewed as a quantum mechanical correction due to tunneling near the classical pathway due to nuclear delocalization at the ensemble level.285 The division between deep and shallow tunneling can be based on the instanton cross-over temperature Tc = ħωn/2πkb which depends on the imaginary saddle point angular frequency ωn;287,288 at temperatures below Tc deep tunneling is operational while at higher temperatures the shallow tunneling description is appropriate. To get a feel for the range of vibrational frequencies to enter the deep tunneling regime at room temperature, the imaginary wave number at the barrier top, ṽn = ωn/2πc, should be larger than 1400 cm−1. However, this division into tunneling regimes should be viewed with caution, and general approaches will cover all regimes – the division is used as a modelistic tool to aid making approximations to the true quantum solution.
Below, some of the currently most promising approaches for treating both shallow and deep tunneling in condensed phases and matter are discussed. The included methods have been demonstrated to perform well in characterizing NQEs in canonical condensed phase systems. Their expected performance, range of validity, and attainable insight is then placed in an electrochemical perspective. However, before diving into theoretical and computational approaches, some mathematical and physical background on necessary concepts is introduced.
6.4.2.1 Concepts of path-integrals and semi-classical approximations. Given the impossibility of solving the Schrödinger equation for complex condensed phase systems, several alternatives, both exact and approximate, have been developed. Among the most suitable exact methods are Feynman path integrals289–292 (PI) which are also amenable to both numerical and semi-classical approximations. In particular, PIs enable an alternative expression for the time-evolution of quantum systems as discussed in detail in Section 7. Using a PI propagator for a particle moving from xi to xf during time t, the quantum mechanical solution (propagation) and PIs are related as
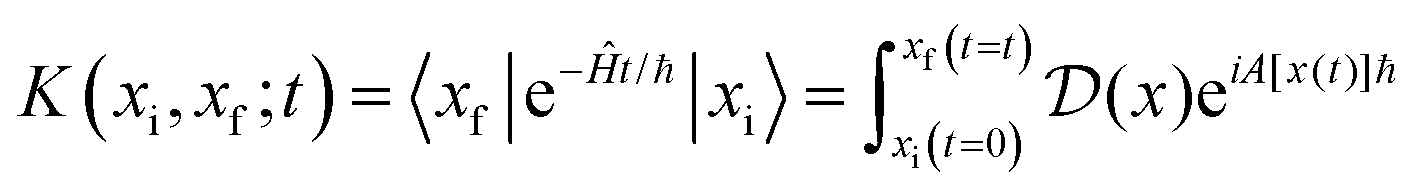 | (18) |
 denotes all possible classical pathways s between xi and xf. K is interpreted as the amplitude of the transition and is related to the probability as P(xi → xf) = |K|2. The weight or importance of each pathway depends on the classical action along pathway s on A[x(s)] of eqn (18) and is highly useful for obtaining physical insight into tunnelling processes. First, in classical mechanics, extremum of the action defines the classical pathway. This is equivalent to Newtonian, Lagrangian, or Hamiltonian mechanics. Second, quantum particles can take any pathway between the end points. Third, it can be shown that the most important tunneling paths are those whose action changes very little between two distinct pathways. Fourth, the most important (tunneling) pathways are obtained from extremization of the action. These observations lead to import conclusions: (1) pathways close to classical ones can be considered as quantum corrections to classical rates and (2) non-classical pathways with extremized actions correspond to tunneling pathways through barriers.289
denotes all possible classical pathways s between xi and xf. K is interpreted as the amplitude of the transition and is related to the probability as P(xi → xf) = |K|2. The weight or importance of each pathway depends on the classical action along pathway s on A[x(s)] of eqn (18) and is highly useful for obtaining physical insight into tunnelling processes. First, in classical mechanics, extremum of the action defines the classical pathway. This is equivalent to Newtonian, Lagrangian, or Hamiltonian mechanics. Second, quantum particles can take any pathway between the end points. Third, it can be shown that the most important tunneling paths are those whose action changes very little between two distinct pathways. Fourth, the most important (tunneling) pathways are obtained from extremization of the action. These observations lead to import conclusions: (1) pathways close to classical ones can be considered as quantum corrections to classical rates and (2) non-classical pathways with extremized actions correspond to tunneling pathways through barriers.289
Besides dynamical properties, PIs can be utilized for computing quantum mechanical (grand) canonical partition functions. This is related to the mathematical similarity between the propagator exp[−iHtħ] and the Boltzmann operator exp[−βH] which can be interpreted in terms of imaginary time t = −iβħ. This property leads to the definition of Euclidian action289
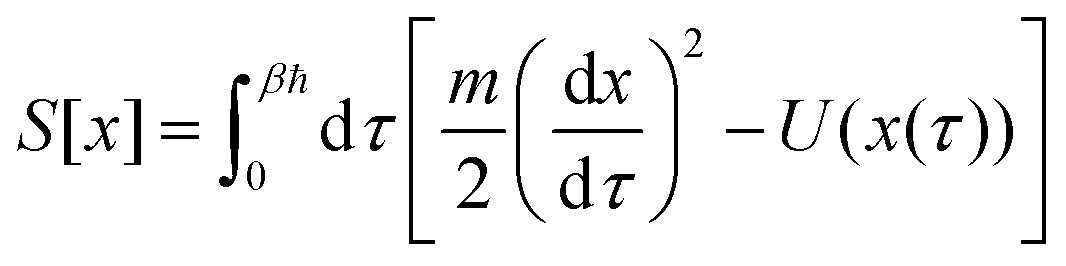 | (19) |
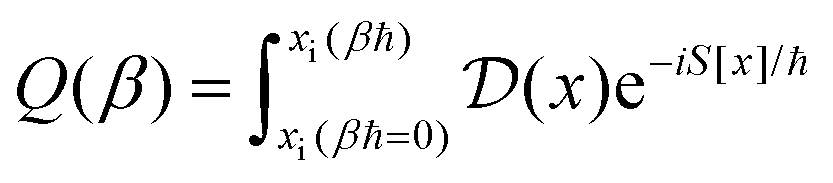 . The sampled paths are cyclic such that the initial and final points are identical. Euclidian path integrals lend well to numerical simulations and are among the most useful concepts for evaluating quantum effects in condensed phases,289,293–295 and for instance, a recent PI molecular dynamics (MD) study has shown that water dissociation on Pt is greatly enhanced by quantum effects.296
. The sampled paths are cyclic such that the initial and final points are identical. Euclidian path integrals lend well to numerical simulations and are among the most useful concepts for evaluating quantum effects in condensed phases,289,293–295 and for instance, a recent PI molecular dynamics (MD) study has shown that water dissociation on Pt is greatly enhanced by quantum effects.296
Euclidian action is also particularly suitable for analytic approximations at the semiclassical limit for capturing the most important tunnelling phenomena as discussed below. As a particular example, expanding the action of a one-dimensional system to second order and using stationary phase integration to the PI leads to the well-known WKB transmission probability292,297,298
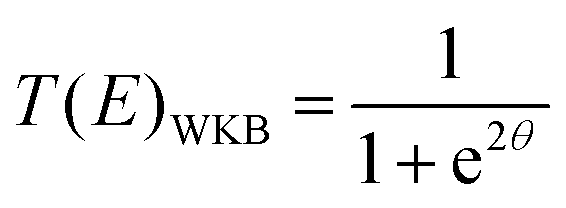 | (20) |
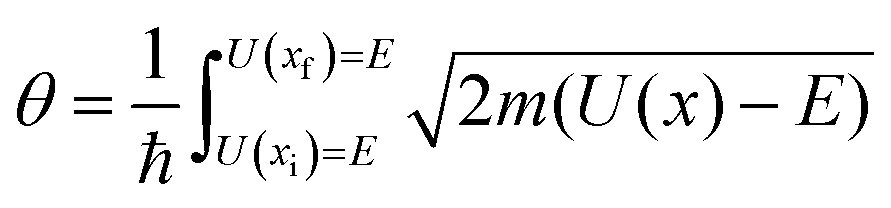 at constant energy.
at constant energy.
6.4.2.2 Shallow tunneling. The shallow tunneling describes nuclear quantum effects around the classical pathway. Here we take the well-defined semi-classical transition state theory264,269,270,297,299,300 (SC-TST) as the starting point to arrive at various approximations for treating quantum mechanical effects near the classical pathway. While SC-TST can in some cases be extended to treat deep tunneling,301,302 only shallow tunneling contributions are achievable from fully numerical first-principles approaches using perturbative approaches.
The formulation of SC-TST starts from eqn (10) and approximates the cumulative reaction probability with the semi-classical WKB-like transmission probability of eqn (20). Unlike the one-dimensional WKB-approximation, the general SC-TST works for multidimensional systems in which different coordinates are non-separable and the barrier penetration integral in SC-TST should be viewed as generalization of the WKB integral. For electrochemical reactions the equivalent of the SC-TST rate constant is obtained from GCE-RT and takes the form
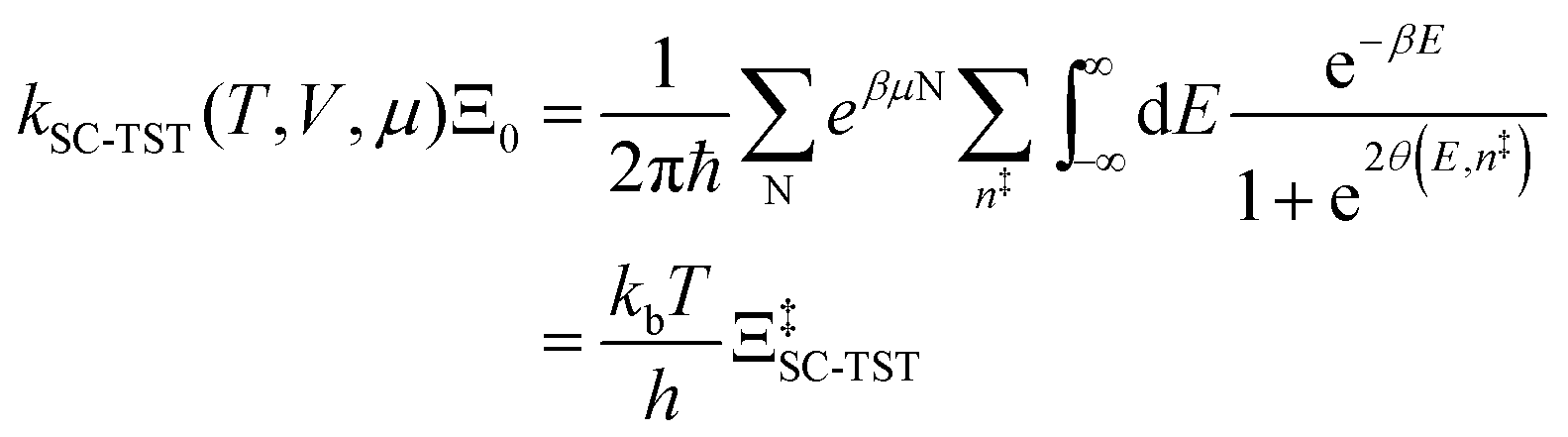 | (21) |
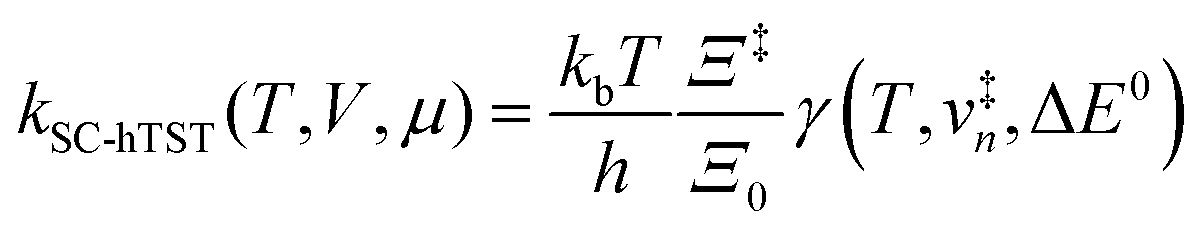 | (22) |
An even simpler analytic form that can be derived from SC-TST is the Wigner correction, which in quantum chemistry is taken as the first approximation of shallow tunnelling effects. The Wigner correction is derived for a one-dimensional parabolic barrier at temperatures well above Tc and given by 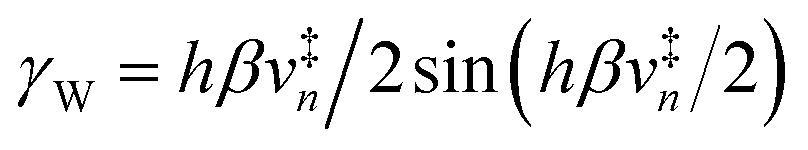 . The Wigner correction diverges as Tc is approached from above, and close to Tc SC-(h)TST provides a more robust alternative.
. The Wigner correction diverges as Tc is approached from above, and close to Tc SC-(h)TST provides a more robust alternative.
6.4.2.3 Deep tunneling. In the deep tunneling regime, the reaction pathway differs from the classical path and takes place through the barrier. This section focuses on two widely used PI-based methods which can describe electrochemical deep tunneling when combined with GCE rate theory: a semi-classical approximation to the propagator K resulting in the instanton rate theory and ring-polymer molecular dynamics (RP-MD) which enables numerical evaluation of correlation functions from PI-based molecular dynamics. It is noteworthy that both approaches lead to similar rate expressions in the deep-tunnelling region and are equivalent to genuine quantum mechanical TST.306–308
The instanton rate theory297 can be derived using a semi-classical approximation to the PI propagator.298,309,310 Starting from eqn (11) the time-evolution is replaced by the imaginary time as discussed in Section 6.2.1. The initial and final positions are equal at the dividing surface, but the pathway connecting them is chosen to go through the classical barrier which is described as through barrier tunneling. The imaginary time trajectories are approximated by a semi-classical stationary phase approximation to the PI propagator which is exact for parabolic functions. This results in the instanton rate constant
| k(T,V,N)instantonQ0 = Ae−S[x]/ℏ, | (23) |
In recent years, instanton calculations combined with first principles electronic structure methods have started to become more frequent. This adaptation has been enabled by more robust numerical implementations describing the action and optimizing MAPs. The numerical presentation requires a discretization of the action, and currently the most robust discretization is based on a ring polymer representation of the cyclic instanton path308,312 (see below). Based on the RP discretization effective algorithms for locating MAPs have been developed.313–316
The instanton rate constant is appropriate for deep tunneling only well below Tc. Another limitation is the harmonic MAP approximation, which precludes the use of the instanton theory on highly anharmonic systems or liquids. Note that a similar restriction applies also to hTST which is the de facto standard for heterogeneous (electro)catalytic systems. Therefore, the instanton theory is expected to work well for describing deep tunneling in surface catalyzed reactions. Indeed, one of the first applications of the instanton rate theory with DFT calculations was H2 dissociation on Cu at low temperatures.317 Other solid-state examples of the method include kinetics of e.g. hydrogen diffusion in metals318 and double-hydrogen transfer on metal supported porphycene.319 Extending the canonical instanton theory to electrochemical systems is straightforward – the only needed modification is to use the effective constant electrode potential PES given by U(R,Nnuc,μ) as in the classical TST case.
To address deep tunneling in liquid environments and to account for anharmonicities, the instanton approach can be replaced with another PI method, namely the ring-polymer (RP) approach. RP methods rely on the isomorphism between ring-like polymers and PIs and enable one to use an extended phase where each point-like classical nucleus is allowed to delocalize to represent the uncertainty principle. This delocalization is achieved by splitting a nucleus into several pieces (beads) connected to each other with harmonic springs so that the beads form a closed loop. With this presentation, the classical Hamiltonian is transformed to the RP Hamiltonian of the form
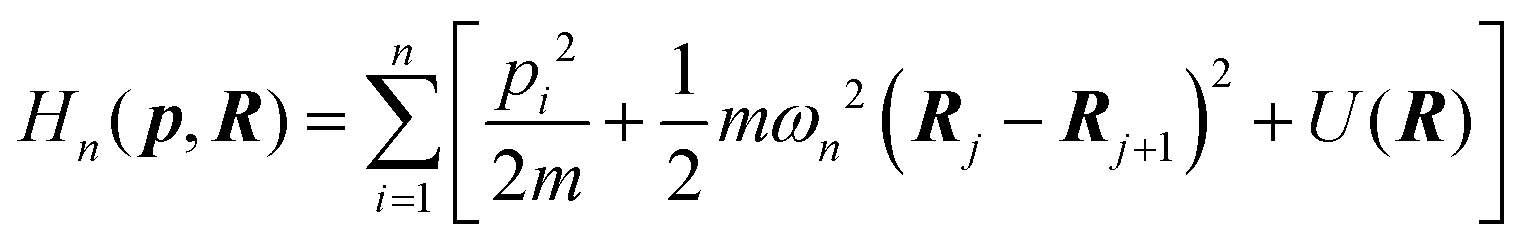 | (24) |
The RPMD method has proven to be a powerful method for studying quantum effects of liquids.294,295 Also, several proton and proton-coupled electron transfer reaction have been addressed in the liquid phase by using RPMD.324–326 In the solid state, hydrogen diffusion in318 and on327 metals have been studied. Given the applicability of RPMD for modelling metals, surfaces, and liquids it is presumable that RPMD will also emerge as a powerful technique for studying electrochemical and electrocatalytic reactions. Again, the generalization to electrochemical reactions is accomplished by using the effective GCE PES given by U(R,Nnuc,μ) or from computing the flux–side of eqn (13) with the extended RP Hamiltonian. While RPMD, WKB, and instanton rate theories are yet to be applied to electrochemical reactions, other PI-based methods have recently provided insight into the importance of nuclear quantum effects at electrochemical interfaces.296
6.5 HER from first principles
After reviewing the GCE-DFT and GCE-RT approaches to adiabatic classical and quantum mechanical rate constants, utility of this machinery is demonstrated for the hydrogen evolution reaction (HER) – arguably the simplest and most important electrocatalytic PCET reaction. Yet, its atomic-level mechanism and kinetics still remain elusive despite numerous experimental, computational, and theoretical studies. As discussed in Section 6.2, constant electrode potential first-principles simulations enable the study of electrochemical reactions at complicated electrified solid–liquid interfaces without empirical parameters. Thus far, simulations have focused largely on electrocatalytic thermodynamics, but lately also studies on kinetics have started to appear.A highlight on what can be achieved using the GCE-RT and GCE-DFT methods in electrocatalysis has been recently provided by the Peterson group.64,65 In their work, the GCE-DFT method implemented using the surface-jellium model and the NEB276 method were utilized to understand the acidic HER on smooth gold and platinum surfaces with classical GCE-hTST rates. Besides demonstrating that it is technically possible to address complex and complete electrocatalytic pathways at electrochemical interfaces as a function of the electrode potential, these studies have shown that even the prototypical Pt-catalyzed HER cannot be fully understood from a thermodynamic perspective. Instead, the excellent performance of Pt as compared to e.g. Au can result from potential-dependent kinetics. Inclusion of kinetics has also enabled new mechanistic insight into the role of top and hollow bound hydrogens on both Au and Pt. Using a novel GC-formulation of the Marcus-like empirical valence bond (EVB) model, Melander has shown66 that potential-dependent reaction barriers obey a Marcus-like dependency on the reaction grand canonical energy. Besides the barrier, the GCE-EVB model was also successfully parametrized using constrained DFT328,329 to compute the solvent reorganization energy at the water–gold interface and estimate the TST geometry as a function of the electrode potential using just a single NEB calculation as a reference.
As a PCET reaction HER is a potential candidate for observing NQEs. We address treating hydrogen tunneling in Pt-catalyzed HER in acidic solutions using the methodology adopted from ref. 64 and 65. For the first i.e. Volmer step, the proton vibrational frequencies at the transition state geometry are very low, around 50–450 cm−1 depending on the potential.64,65,330 Based on the vibrational frequencies, the crossover temperatures are around 40 K and therefore deep tunneling is not expected at room temperature. At room temperature shallow tunneling contributions based on either the SC-hTST or Wigner model are also negligibly small. Also, the KIE computed from the Wigner correction and harmonic TST is small, below 1.1 for all electrode potentials. The second HER step is the Heyrovsky reaction, which is predicted65,331 as the rate limiting step for Pt-catalyzed HER. The Heyrovsky reaction exhibits somewhat larger imaginary vibrational frequencies which are in the range of 400–800 cm−1 depending on the potential. The reaction is still not in the deep tunnelling regime as Tc is below 200 K. Using the Wigner expression, the KIE for the Pt-Heyrovsky reaction is around 1.4–1.5 in nice agreement with the experiments65,331 with Heyrovsky as the rate limiting step. The nuclear quantum effects in acidic Pt-catalyzed HER seem negligible and can likely be understood from the adiabatic shallow tunneling perspective.
While the above discussion provides evidence that HER on Pt can be understood from classical TST and that NQEs seem negligible for reaction kinetics, results from PIMD simulations show that NQEs might be important for water structures and interfacial pH.296 In particular, water dissociation is greatly facilitated on Pt but not on Au surfaces. As a result, H3O+ and OH− ions concentrate on the Pt surface. Interestingly enough, deuterated water does not exhibit markedly-increased ionization upon inclusion of NQE. Furthermore, kinetic isotope effects for water dissociation were obtained from PIMD evaluated partition functions to parametrized PI-based quantum TST332 (also known as centroid TST333). This study demonstrated the applicability of PI techniques on realistic electrochemical solid–liquid interfaces as well as the complex and unexpected nuclear quantum phenomena on surface- and isotope-dependent solvent effects at room temperature.
Application of electrocatalytic rate theory has enabled a first-principles computational approach to study reaction trends, mechanisms, and kinetics as a function of the electrode potential. In the Pt-catalyzed HER, electronic quantum effects are crucial while the nuclear degrees of freedom can be treated classically, and small NQEs can be understood using the adiabatic shallow tunneling perspective. Yet, the dynamic water structure and dissociation exhibit NQEs, and their importance depends on the surface. These observations highlight the complexity of solid–liquid interfaces and surprising sources of quantum effects.
6.6. Summary and remarks for this section
In this section we have shown how well-defined electrochemical rate constants at a fixed electrode potential are achieved by extending canonical rate theories to the GCE. This facilitates following well-defined route to classical TST as well as to treating nuclear quantum effects from a set of fundamental equations and concepts. The GCE rate theory and approximations to it can be combined with general first principles Hamiltonians and DFT methods as the formalism is not tied to any model Hamiltonians. Different approximations to the GCE-RT to treat classical nuclei and nuclear tunneling within shallow and deep tunneling conditions were discussed and the validity of different approximations are recapped in Fig. 19. The discussion has focused on electronically adiabatic reactions, but non-adiabatic reactions can also be treated within the general GCE rate theory.66 Novel rate theories for including non-adiabatic effects in instanton theory334,335 and RPMD284,293,336 exist but thus far they have been applied only to model systems. Therefore, the non-adiabatic reactions are presented within the more established curve-crossing picture in Sections 5 and 7.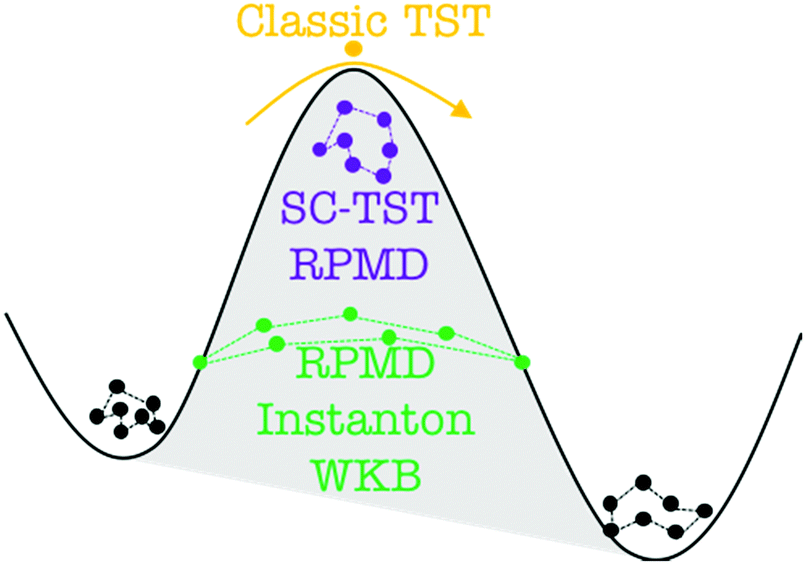 | ||
| Fig. 19 Overview of the adiabatic tunnelling methods. The ring polymer size is proportional to the anticipated quantum tunnelling effects and the grey area is the classically forbidden area under the barrier. | ||
7. First-principle calculations of quantum effects in proton transfer electrode processes
7.1. Quantum many-body theories in electrode process science
The fuel cell reaction, a process to generate current from hydrogen and dioxygen (2H2 + O2 → 2H2O), is comprised of charge transfer processes that occur at the electrode. A typical example is the electroadsorption of a proton described by H3O+ + e− → H* + H2O called the Volmer step of the hydrogen evolution reaction (HER), which is a PCET reaction (see Fig. 4 in Section 3 for a schematic image and Section 5 for the basic concepts and classifications for PCET).337–339 HER is obviously the simplest and the most studied reaction, but the understanding remains incomplete. This reflects the fact that the reaction cannot be simply captured by the formation and breaking of the chemical bond but needs to be captured with the strongly coupled environment. When the adsorbed hydrogen atoms, H*'s, subsequently associate on the surface as 2H* → H2, it is called the Tafel step of HER (Fig. 20). When another electroadsorption occurs on the adsorbed H* as H* + H3O+ + e− → H2 + H2O, it is called the Heyrovsky step of HER. These steps compete delicately on different surfaces. When the reaction occurs reversely on the hydrogen fuel cells, it is called hydrogen oxidation reaction that occurs at the anode side. At the cathode side, oxygen molecules are successively reduced by charge transfer processes via the reaction intermediates, O2*, OH*, O2H*, and O*. Their balance needs to be understood carefully. Despite these difficulties, it has long been the dream of theoreticians to establish the theory based on the sophisticated quantum many-body theory. Toward this goal, theory has advanced step by step, and at this stage, it will be important to review how the theory has advanced and how it should be developed in the future. | ||
| Fig. 20 Hydrogen evolution reaction on Pt(111). Hydrated proton is exchanged with electron in the electrode in the Volmer step (left). In the Tafel step, the adsorbed hydrogen atoms associate (middle), and in the Heyrovsky step, the Volmer step occurs on the adsorbed hydrogen atom (right). Filled red circles and yellow circles respectively indicate the adsorbed proton at the different position, i.e. ontop and hollow. The importance of adsorption sites are discussed later with Fig. 21. | ||
7.2 Theoretical picture of electrochemical reaction dynamics at solid/liquid interfaces
to the reactant state Ψr weakly coupled to the product state Ψp. This is a general quantum formula for the reactant (r) → product (p) transition probability (R). Here V is the energy of the perturbation to cause the transition, and the δ function with an energy difference Ep − Er expresses the energy conservation law in the transition. Note that the wave functions, Ψr and Ψp, contain full electronic and nuclear degrees of freedom and are extremely complex to handle. Practically, the wave function was simply decomposed into a product of the electronic Φ and nuclear χ parts, and the latter was subsequently approximated as a direct product of the wave function of the reacting proton and that of other nuclei. The resulting “double adiabatic” approximation neglects the coupling among electrons, reacting proton, and the environmental atoms and thus is insufficient for the electrochemical reactions. The coupling is overwhelmingly complex, as can be found in the study of PCET shown in Section 5.
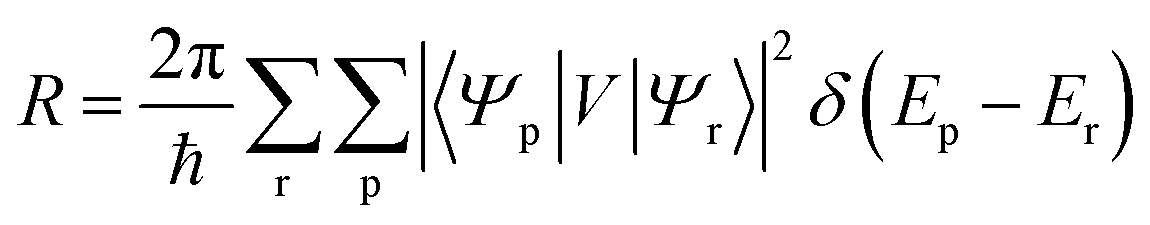
Formal theory of the reaction dynamics can be provided from first principles within the path integral (PI) scheme.340 The reaction rate can be given as 〈Qp|e−Hτ|Qr〉, where Qr and Qp are the initial and final configurations, respectively, and H is the full Hamiltonian H(r,R) for all the electronic r and nuclear R degrees of freedom. The rate is evaluated in PI by the set of nuclear paths R(t) for a time interval τ, which is discretized into pieces Δt as R(kΔt) ≡ Rk with 0 ≤ k ≤ p. The formulation is done by decomposing H(r,R) into the nuclear part
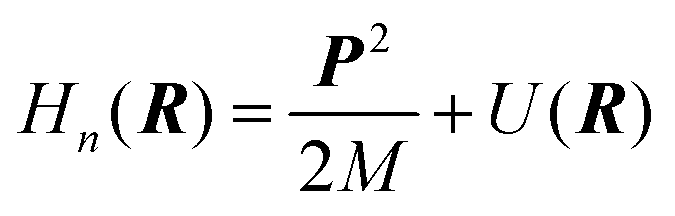
and by extracting the operator to be applied to the reactant Φr and product Φp states of the electrons as

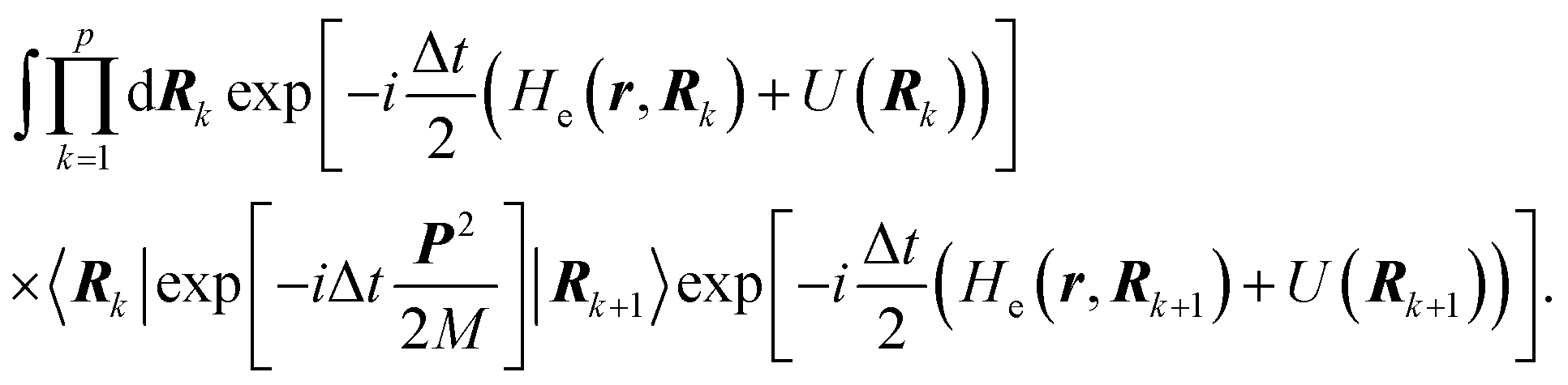
This can be recast into the typical PI form, or the integration over all nuclear paths

and the potential part
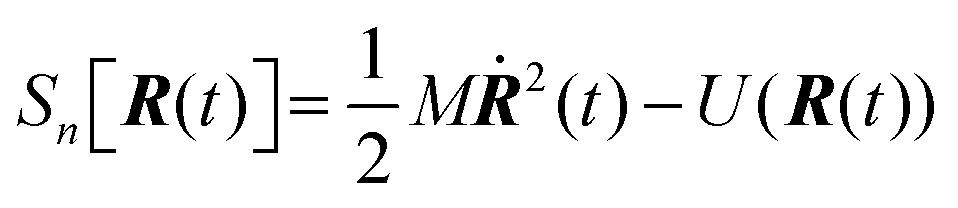
To evaluate G[R], a complete set is inserted between the exponentials. To allow nuclei to mix different Born–Oppenheimer (BO) states beyond the BO-PI scheme,341 it is convenient to use the Slater determinant consisting of nonorthogonal orbitals ϕ(r,Rk), that satisfy the coherent state condition342
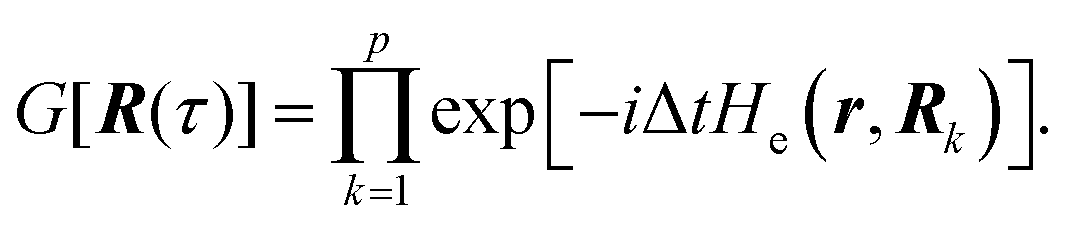
| 〈r|ϕ(Rk)〉 = exp[ϕ(r,Rk)a+]|0〉. |
With the complete set relation described as an integration over the orbitals
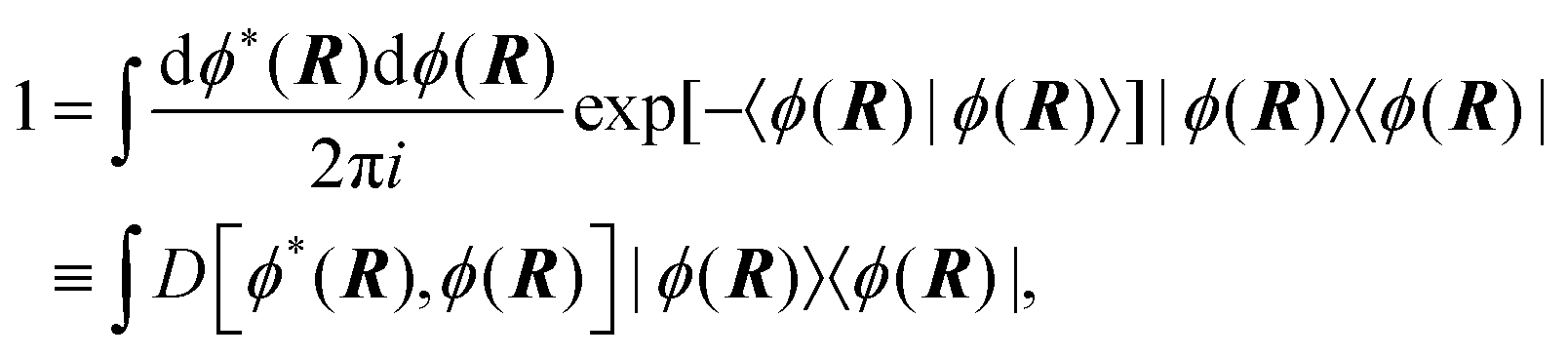
using the electronic action
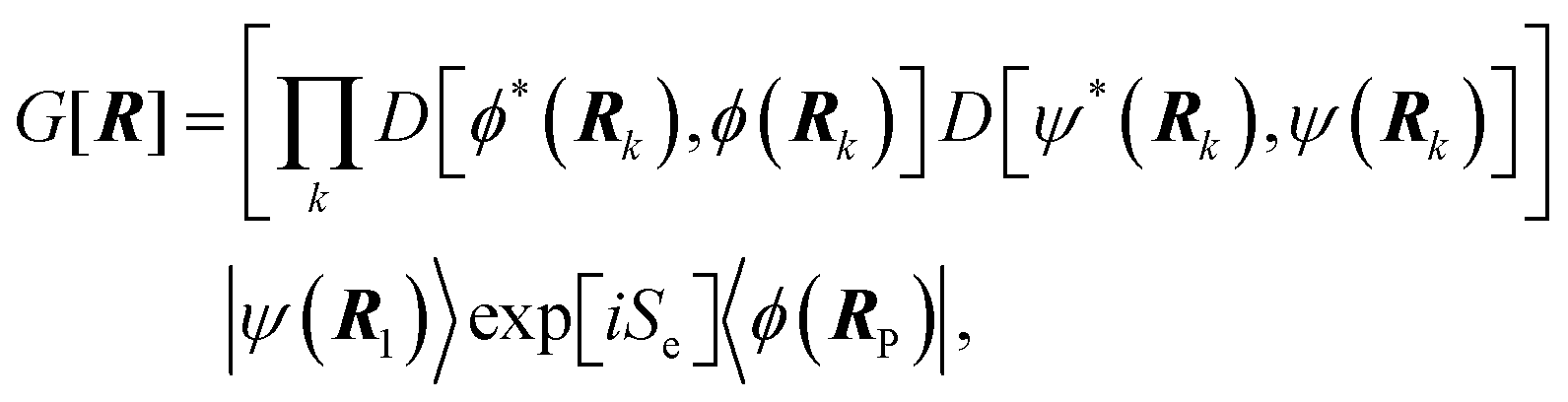
with
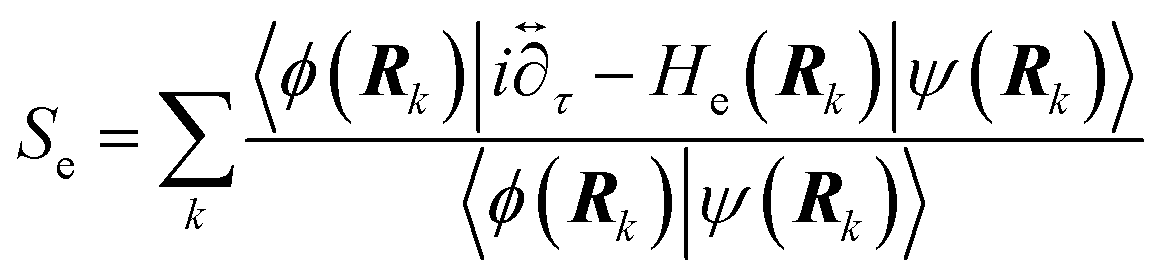
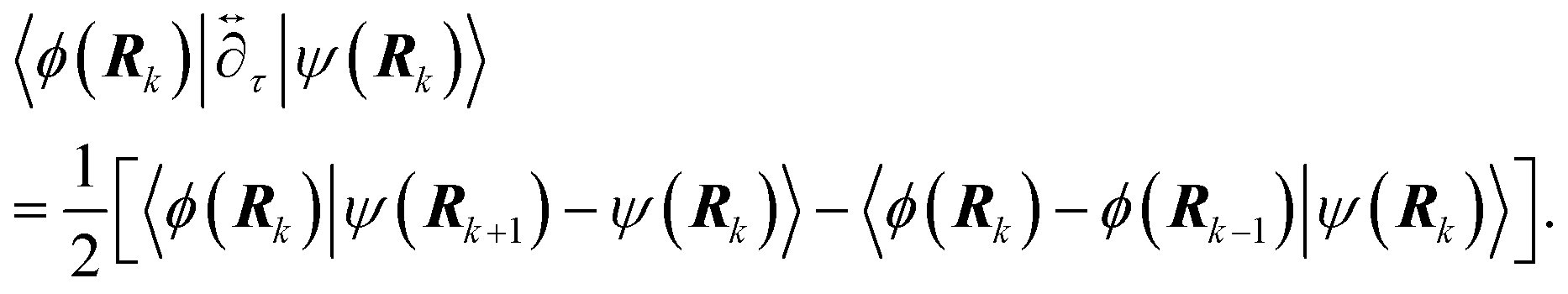
Therefore, in the limit of an infinitesimal time interval, one can describe the action more symmetrically for nuclei and electrons
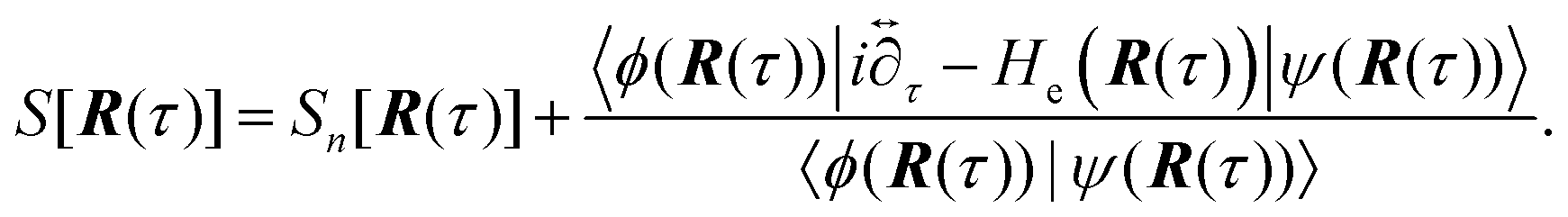
If the coherent states are approximately replaced by the Born–Oppenheimer (BO) state Φ, the action becomes
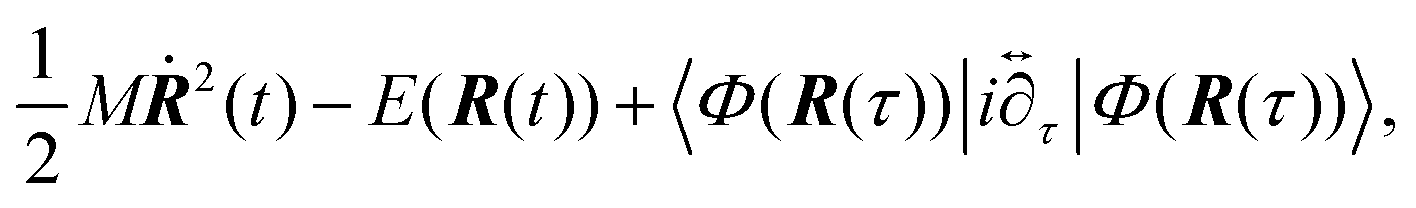
M![[R with combining umlaut]](https://www.rsc.org/images/entities/b_i_char_0052_0308.gif) = −∇RE(R) + (nonadiabatic terms). = −∇RE(R) + (nonadiabatic terms). |
Before mentioning the adiabatic dynamics, the model-based approach for the rate of electron transfer reactions is worth discussion. In the Newns–Anderson–Schmickler model,345 the electrochemical interface is characterized by the density of states of the electrode and a level of the redox center. The model is found to reasonably well describe the electrochemical reactions including the Volmer step346 despite its simplicity. On this basis, Tanaka et al.347 derived a field theoretical formulation to account for the reaction rate within the linear coupling regime of the redox level and the displacement of environmental coordinates. The derived formula, in the weak coupling limit, clearly demonstrates the importance of quantum mechanical effects. The model-based approach will play a role complementary to the fully ab initio methods in the future in view of the recent development of mapping density functional calculations to the Hubbard model systematically.348
Electrochemical interfaces have been studied with increasingly elaborate description of the electric double layer, as reviewed comprehensively in recent literature.358–363 To avoid duplication, here we just summarize recent application of our method. The ab initio molecular dynamics (AIMD) simulation was combined with ESM model to the problem of oxygen reduction reaction or water splitting on Pt,277,364–366 hydrogen evolution on Ag,367 and an ionic liquid on Li.368 The constant chemical potential scheme was applied to the problem of redox level,369 Pt electrode,110 and oxygen evolution reaction pathways.370 The ESM-RISM was applied to an oxidized Pt,371 and a graphitic surface.372 We point out that simple solvation models are preferred even today,373–375 while there is increasing number of researches that adopt the conventional periodic-interface-model where the associated boundary problem is reduced by incorporating more water molecules and solvated ions into the computational cell.64,376–378
7.3 Quantum effect on hydrogen adsorption
In addition to an accurate description of the EDL, the accuracy of sorption phenomena is very important. The potential energy surface on the metal surface is generally flat on the order of 10–100 meV for different adsorption sites on Pt(111) (Fig. 21), which is comparable to the thermal energy at room temperature, 26 meV. The electronic structure calculation should be accurate to this energy scale, but it is not so easy to achieve this. It was shown that the adsorption of CO is inaccurate by 0.1 eV on Pt(111) when using the generalized gradient approximation (GGA) or a semilocal approximation to the exchange–correlation energy. The problem apparent in the popular DFT simulations was overcome by using an advanced density functional called random phase approximation (RPA).379 When this calculation was applied to H/Pt(111), hydrogen atoms more favorably occupy the fcc site at the full monolayer coverage condition, but the adsorption energy is comparable to the atop site within the energy scale of room temperature. This coexisting behavior of the adsorbed hydrogen atoms contrasts with the view provided by a GGA calculation that indicates the fcc site is considerably more stable. The qualitative difference in the adsorption feature should affect the kinetics of HER and HOR. This is because the hydrogen adsorption is considered to occur preferentially on the atop site so that the whole reaction is influenced by the subsequent hopping. If the fcc hydrogen predominantly exist as a stable species, it will exist as a spectator of the reaction. If the atop is predominant on the contrary, it will block the reaction as a poison. The coexistence means a possible flow of the atop to the fcc depending on the barrier height existing in between.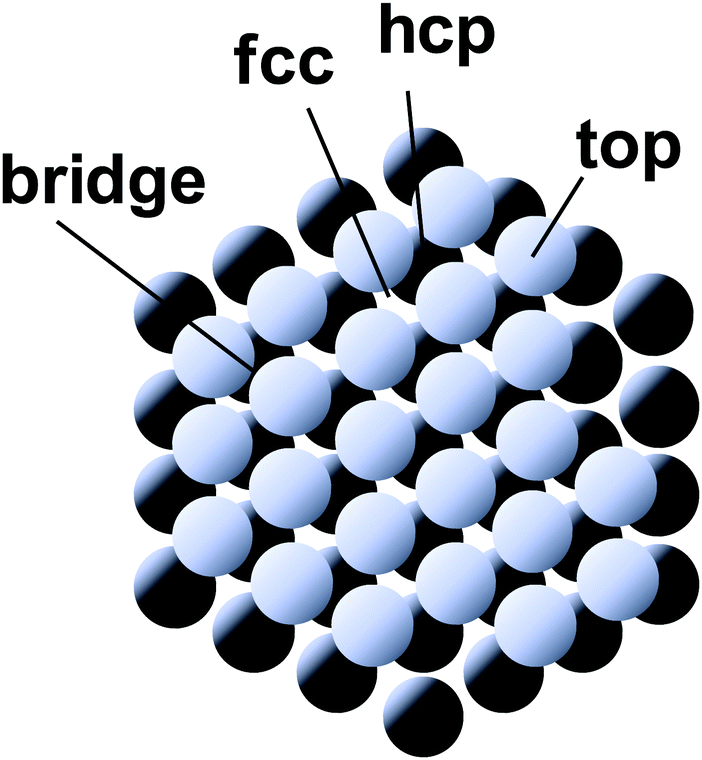 | ||
| Fig. 21 Symmetric adsorption sites on Pt(111). | ||
Quantum confinement is another important factor. This has been understood from the role of the zero-point energy (ZPE) in determining the most stable adsorption site.380,381 On the Pt(111) site under the monolayer coverage condition, hydrogen forms a monocoordinated Pt–H bond on the atop site that is stronger than that formed on the three-fold coordinated fcc sites; however, the stability on the atop is less than that on the fcc because of the larger ZPE effect on the atop that overcompensates for the effect of classical binding energy.
The ZPE effect is included in the abovementioned calculations, but the confinement potential is quite anharmonic.382 The anharmonicity is considerably larger under sub-monolayer conditions than under the full monolayer condition because of the existence of a ‘vacancy’. The vacancy also facilitates hopping of a hydrogen atom to a site nearby. According to the ab initio path integral molecular dynamics (PIMD) simulation done at room temperature (Fig. 22),382 the hopping occurs frequently under a 2/3 monolayer, θ = 2/3, causing a delocalized feature in the density distribution. The density is maximum on the fcc and hcp hollow sites and is slightly smaller in the region in between, in sharp contrast to the density at the monolayer where the maximum in the density appears both at the atop and the fcc. This was interpreted using a lattice gas model augmented with the hopping between the nearest neighbor fcc and hcp sites. The model is characterized by the strong H–H repulsion and the nearly degenerate nature of the adsorption sites and thus has the property typical of strongly correlated many-body systems. The delocalization with reduced coverage and the resulting kinetic energy lowering are the typical property of the system; indeed, a similar property of hydrogen was found also on other metal surfaces (also see the references in ref. 324).383 In the PIMD simulation, the hydrogen put on the atop immediately hops to the hollow sites and is delocalized,384 so that the hollow sites should play a participating role in the Volmer step.
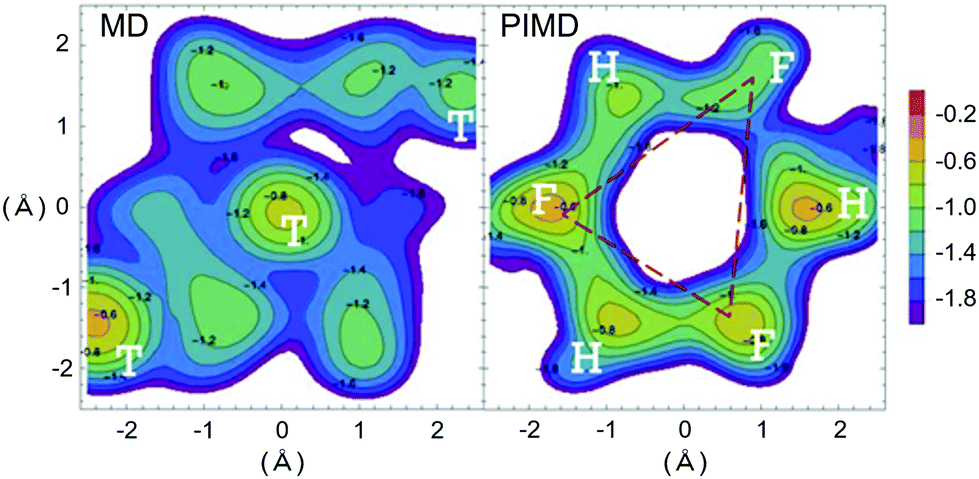 | ||
| Fig. 22 Density distribution of H on Pt(111). The density has a maximum on the atop (T) site when simulated classically (a), while it has a maximum at the hollow sites (F and H) then simulated quantum mechanically (b). The figure was adopted with permission from the ref. 382. Copyright (2020) American Physical Society. | ||
Although the research has been mostly limited to static properties like the adsorption site, the PI approach should be able to reveal dynamic properties as well. The role of quantum tunneling was studied mostly for surface diffusion,385 but such simulations can clarify the role of tunneling on the reaction.
7.4 In silico electrocatalyst design
To design the most efficient electrocatalysts, it is not practical to apply the sophisticated theory. For the purpose of screening, the most familiar method is the method based on the computational electrode model.386 Therein, the pristine surface is prepared to calculate the adsorption energy of the reaction intermediates: H* for the anode and O*, OH*, O2H* for the cathode. The energy of the hydrated proton is taken to be equal to the free energy of H2 in the gas phase at 1.23 V vs. SHE and is increased by x volt as the bias potential is reduced by x volt. This allows one to approximately relate the adsorption energy with the reversible electrode potential. The theory explains simply the peak of the electrocatalytic activity versus the adsorption energy, known as the volcano relation, which is considered as a manifestation of the Sabatier principle; for higher activity the adsorption should not be too strong and not be too weak. The theory was used to explain why platinum is more active than other noble metals.386 The reason why it is difficult to overcome platinum was ascribed to the linear relation of the adsorption energy between O* and OH*. The relation was shown to be applicable to many surfaces where the reaction intermediates take an adsorption structure common to the intermediates. There are, however, exceptions in some cases, including the doped TiO2 surface,387 which was expected to be a candidate for a superior electrocatalyst. The simple screening method thus has been successfully applied to many materials, but the success does not automatically justify the assumptions made in the theory; indeed, there are critical arguments.388 In this context a trial was made to replace the adsorption on a pristine surface with the average taken over possible configurations of the environment.389 It is now possible to do the thermodynamic sampling without mapping to a model owing to the development of ab initio Monte Carlo simulation.390 It is worth pointing out that such simulation should enable one to describe the space charge layer, or the EDL, formed on a semiconductor-based-electrode, although such electrode materials have only infrequently been studied theoretically. An important target of the theory will be therefore to make feasible the elaborate environmental description and then finally to enable simulation of the reaction dynamics.8. General conclusion and future outlook
We have discussed a wide spectrum of recent advances and challenges in modern electrode process science at solid–liquid interfaces. As shown in the overall sections, the challenges in these systems are extraordinarily difficult and complicated, and on the other hand, the advancements still seem too little to understand the complicated microscopic mechanism of multi-electron/-proton transfer electrode processes at solid–liquid interfaces. However, we have an advantage: the key research concepts and philosophy of fundamental electrode process science at solid–liquid interfaces are greatly affected by the surface science at solid–gas interfaces with model systems.50,391–396 It is well-known that these solid–gas systems brought greater understanding of a variety of basic catalytic mechanisms at single crystal and well-ordered thin film surfaces, as highlighted by the Nobel Prize for Gerhard Ertl in 2007.391 Therefore, we already have cumulative knowledge since the Ostwald era to import to solid–liquid systems.397 The well-established guidelines developed to tackle complicated solid–gas systems combined with experimental and theoretical approaches shown in the previous review49 allow us to investigate the microscopic principles of charge transfer reactions at solid–liquid interfaces using model systems combined with theoretical and spectroscopic approaches (Fig. 23). However, it is important to note that solid–liquid interfaces present additional challenges, particularly in terms of the EDL, as discussed in this review.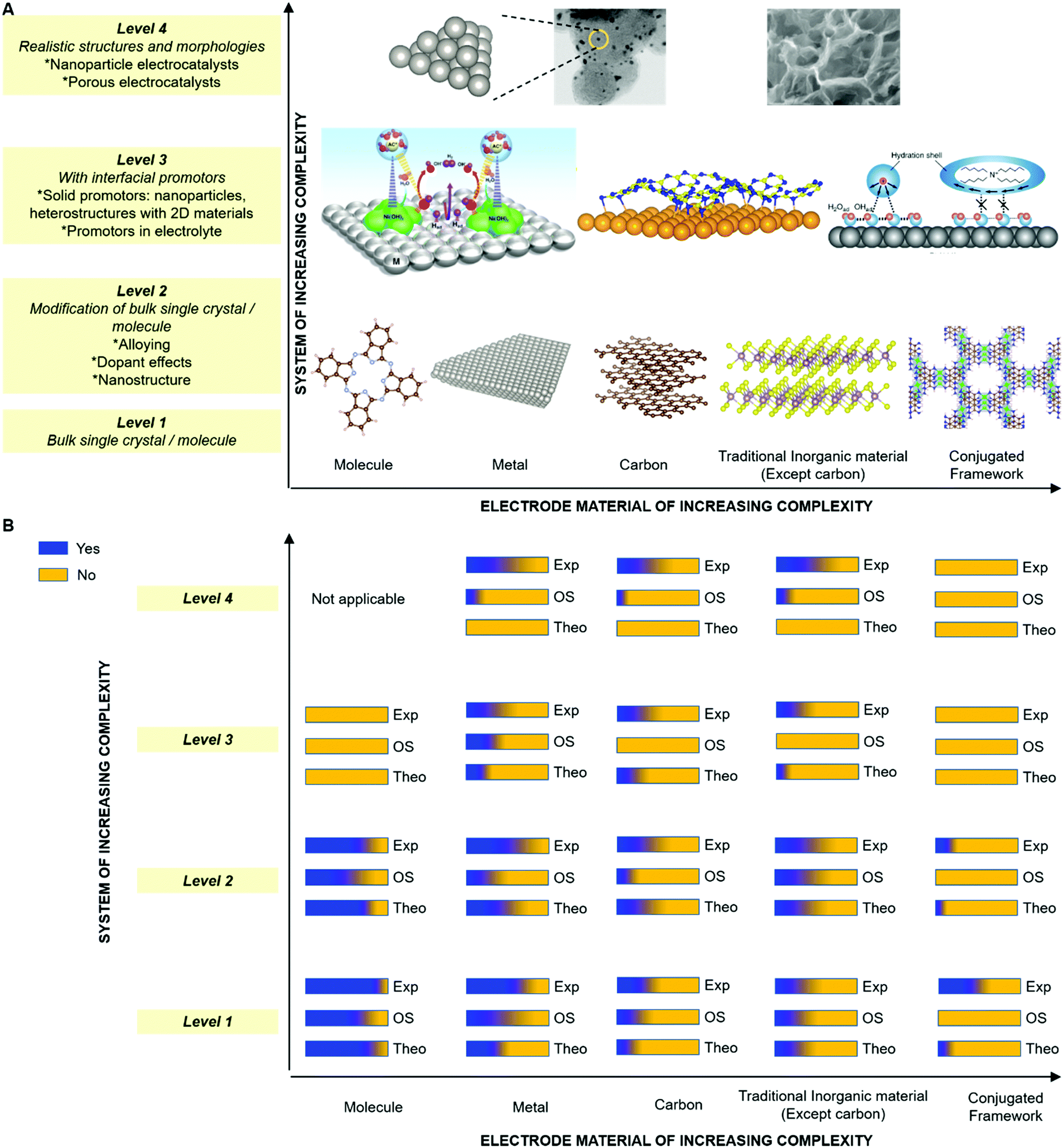 | ||
| Fig. 23 Schematic representation for increasing complexity of model solid–liquid systems as functions of increasing complexity of electrode materials to capture the essential features of a working catalyst. (A) System complexity versus electrode materials complexity diagram. The complexity of model electrode materials increases from metal (generally the simplest) to conjugated frameworks, such as MOFs/COFs, (generally the most complicated). The complexity of model solid/liquid systems increase from Level 2 (alloying, doping, nanostructuring, or other modifications),398–439 Level 3 (addition of interfacial promotors on electrode surfaces and/or in electrolyte),221,440–444 to Level 4.445–453 VESTA was used to prepare several structure images in the panel A.454 (B) Evaluations of present level of achievement for each combination of an electrode-system complexity. Each level is evaluated with respect to standard electrochemical experiment (Exp),25,87,337,455–466operando surface-electrochemical spectrometry, microscopy or other analytical methods (OS),467–495 and theory (Theo),64,65,107,351,352,386,496–508 and with respect to progress that has been made to achieve a proper representation/description of each level for basic multi-electro/-proton transfer electrode processes, for instance, HER, HOR, ORR and CO oxidation reaction. The evaluation was made based on the discussions and references in this Perspective Review. Blue: yes, achieved; orange: no, not achieved. This figure was prepared by referencing the schematic and pictorial representation of model solid/gas systems as a function of increasing complexity to capture the essential features of a working catalyst.49 The panel A is partially adopted from the ref. 443, Copyright (2011), with permission from American Association for the Advancement of Science, the ref. 105, Copyright (2017), with permission from American Chemical Society, and the ref. 221. | ||
Although we still need basic understanding about electrocatalytic reactions at single crystals, operando electrochemical techniques have already been applied to investigate these reactions not only for Pt nanoparticles and nanostructured MoS2,509–512 but also for metal-free carbon catalysts.513 Interestingly, most material systems with realistic structures and/or morphologies (Level 4 in Fig. 23) have already been studied to evaluate their electrochemical properties, possibly because of recent interest in electrochemical energy devices. However, it was clarified that fundamental investigations before going to Level 4 are lacking. This indicated that fundamental research of Levels 1–3 will be necessary to bring the reliable impact at Level 4 (see Fig. 23A the descriptions about these levels). This is especially necessary for carbon-based catalysts, where materials are very sensitive to impurities and/or exhibit complicated doping effects.514–517 Therefore, fundamental studies are very important to uncover the complicated characteristics of this catalyst system.518–521 The electrode process science at solid–liquid interfaces has another advantage in being able to incorporate ongoing advanced technology, such as operando spectrometries using X-rays at ambient conditions, i.e. X-ray free electron laser and resonance inelastic X-ray scattering,522–527 or other surface sensitive operando spectrometries such as two-dimensional heterodyne-detected vibrational sum-frequency generation239,528 and advanced scanning probe microscopies.529–533
Computational studies have also benefitted greatly from approaches developed to study solid–gas interfaces. However, most computational electrocatalysis studies have treated the electrochemical interface using approaches adopted from computational surface science and heterogeneous catalysis without self-consistently accounting for the liquid electrolyte nor the electrode potential, the defining factors of an electrochemical system. With recent advances in theory and computational implementations, both the liquid environment and the electrode potential can now be treated self-consistently from first principles as discussed in Sections 6 and 7. In particular, the electrode potential can be included using GCE-DFT. The liquid environment is, in fact, more challenging, and several approaches exist. The most primitive models based on dielectric continuum theory are being replaced by more refined models from statistical theory of liquids using RISM or classical DFT.534,535
With advances in modelling the electrochemical interface, the computational community is also moving from thermodynamics to addressing electrocatalytic and electrochemical kinetics. Thus far, the kinetics have been addressed for inner-sphere reactions using classical, adiabatic harmonic TST using tools adopted from computational surface science and heterogeneous catalysis. While classical inner-sphere reactions are important for some catalytic reactions, there are many reactions that display outer-sphere character, non-adiabaticity, and/or nuclear tunneling. In addition to accounting for these reaction mechanisms, approaches beyond the harmonic approximation are needed to describe kinetics at strongly anharmonic solid–liquid interfaces. Another open question relates to the valid reaction coordinate; approaches developed for solid–gas interfaces use bond distances as reaction coordinates, while electrochemical rate theories (i.e., Marcus, Newns–Anderson–Schmickler, or Soudackov–Hammes–Schiffer approaches) are based on the environment reorganization as the reaction coordinate. The reorganization coordinate used in the general GCE-RT66 by combining GCE-DFT with constrained DFT328,329 has been demonstrated to give reliable estimates of electron/hole transfer in solid battery materials.536 Combining first principles methods with GCE-RT outlined in Section 6, as well as the (non-adiabatic) quantum or semiclassical dynamics, present promising approaches to address electrochemical kinetics beyond classical harmonic TST and the inner-sphere reaction coordinate. These approaches are also derived from exact rate theory and quantum dynamics, making them suitable for well-defined approximations to address quantum effects, solvent reorganization and stabilization, and different reaction mechanisms in electrochemical processes.
Combinations of these techniques, as well as the analytical methods/theories described in Section 5, will bring a clear picture of real-time reaction dynamics at electrochemical solid–liquid interfaces. As described in Fig. 23, the general understanding of electrochemical solid–liquid systems is still in a preliminary phase compared to the level for solid–gas systems. An example to describe the present stage of electrochemical solid–liquid interfaces is that we still do not understand the microscopic mechanism of HER at Pt(111), one of the simplest multi-electron/-proton transfer reactions, the complexity of which can be comparable to the CO oxidation reaction at Pt(110) in UHV solid–gas systems.537–542 Therefore, we still need to use the above mentioned advanced theoretical and experimental techniques to study very basic model systems, i.e. single crystal surfaces. Although nowadays there is a wide spectrum of “advanced” materials for electrocatalysis, the “classical” single crystal electrodes are now even more important than before to advance the field of electrode process science.467,543–568 In these above works, the close collaborations between theory and experiment are key, as already well discussed.362,398,569–574 We hope that these fundamental investigations will bring knowledge to enable the study of more complicated model systems, such as MOFs/COFs and model electrocatalysts having porous structures or heterojunctions (Fig. 12) or much more realistic structures/morphologies.445,446,575–578 Of course, there are much more complicated systems, such as microbes.579–588 These systems are so complicated that it is challenging to unveil their microscopic working principles. However, after acquiring the fundamental knowledge from the above systems at several levels of complexity, there will be a possibility to uncover the microscopic reaction mechanisms in these more complex systems and to develop highly efficient electrochemical energy technology by taking advantages of quantum phenomena.
In summary, we note here the key issues required to bring the present level of understanding of quantum electrochemistry at solid–liquid systems into the next level of complexity:
(1) An advanced experimental approach to indentify a variety of quantum effects in electrode processes, such as vibronically non-adiabatic tunneling, is essential. This issue is inseparable from issue (2) given below.
(2) Theoretical and computational methods should be further developed and applied to understand fundamental electrode processes and to provide experimentally verifiable predictions. In particular, comparisons of KIEs and spectroscopies between theory and experiment will provide significant insights into the microscopic mechanisms and the important fundamental interactions at solid–liquid interfaces.
(3) Understanding the dynamic interfacial evolution effects for quantum electrode processes by combinations of experimental and theoretical studies. For example, geometries of electrified surfaces589,590 and water orientations are dynamically changed by the applied potential. Including these dynamical effects in our interpretation of experimental data and theoretical calculations will enable us to understand complex quantum phenomena in electrode processes.
Conflicts of interest
The authors have no conflict to declare.Acknowledgements
KS is thankful for MMM and SHS for their fruitful discussions, comments and modifications for Sections 1–3 and 8. KS and TK are indebted to the supports from National Institute for Materials Science for fundamental science. This work was partially supported by JSPS KAKENHI grant numbers 19K15527 and 19H05460, and TIA collaborative research program [Kakehashi] Program No. TK19-002 and TK20-11. MMM acknowledges financial support by the Academy of Finland (project 307853) and computational resources provided by CSC – IT Services for Science. MMM also thanks Georg Kastlunger and Per Lindgren for providing the vibrational frequencies for Pt-catalyzed HER. SHS acknowledges financial support from the Center for Alkaline-Based Energy Solutions, an Energy Frontier Research Center, funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences as well as helpful discussions with Alexander Soudackov and Yan-Choi Lam.References
- D. Keilin, Proc. R. Soc. London, Ser. B, 1925, 98, 312–339 Search PubMed.
- D. Keilin, Proc. R. Soc. London, Ser. B, 1929, 104, 206–252 CAS.
- P. Mitchell, Nature, 1961, 191, 144–148 CrossRef CAS PubMed.
- P. Mitchell, Presented in part at the Nobel Lecture, Stockholm Concert Hall, 1978.
- X. Jiang, N. Ermolova, J. Lim, S. W. Choi and H. R. Kaback, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 977–981 CrossRef CAS PubMed.
- J. W. Woodbury, S. H. White, M. C. Mackey, W. L. Hardy and D. B. Chang, in Physical Chemistry An Advanced Treatise, ed. H. Eyring, D. Henderson and W. Jost, Academic Press, New York, USA, 1970, ch. 11, vol. IXB, pp. 904–983 Search PubMed.
- D. M. Dražić, in MTP International Review of Science Physical Chemistry Series One, ed. A. D. Buckingham, Electrochmistry, Butterworths, UK, 1973, vol. 6, pp. 287–332 Search PubMed.
- Y. Cha, C. Murray and J. Klinman, Science, 1989, 243, 1325–1330 CrossRef CAS PubMed.
- B. J. Bahnson, T. D. Colby, J. K. Chin, B. M. Goldstein and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12797–12802 CrossRef CAS.
- A. Kohen, R. Cannio, S. Bartolucci, J. P. Klinman and J. P. Klinman, Nature, 1999, 399, 496–499 CrossRef CAS.
- K. W. Rickert and J. P. Klinman, Biochemistry, 1999, 38, 12218–12228 CrossRef CAS PubMed.
- M. J. Knapp, K. Rickert and J. P. Klinman, J. Am. Chem. Soc., 2002, 124, 3865–3874 CrossRef CAS PubMed.
- J. P. Klinman and A. R. Offenbacher, Acc. Chem. Res., 2018, 51, 1966–1974 CrossRef CAS PubMed.
- P. R. Schreiner, H. P. Reisenauer, F. C. Pickard Iv, A. C. Simmonett, W. D. Allen, E. Mátyus and A. G. Császár, Nature, 2008, 453, 906–909 CrossRef CAS PubMed.
- P. R. Schreiner, H. P. Reisenauer, D. Ley, D. Gerbig, C.-H. Wu and W. D. Allen, Science, 2011, 332, 1300–1303 CrossRef CAS.
- P. R. Schreiner, J. Am. Chem. Soc., 2017, 139, 15276–15283 CrossRef CAS.
- R. W. Gurney, Proc. R. Soc. London, Ser. A, 1931, 134, 137–154 Search PubMed.
- C. E. H. Bawn and G. Ogden, Trans. Faraday Soc., 1934, 30, 432–443 RSC.
- J. Horiuti and M. Polanyi, Acta Physicochim. URSS, 1935, 2, 505–532 Search PubMed.
- J. A. V. Butler, Proc. R. Soc. London, Ser. A, 1936, 157, 423–433 CAS.
- W. F. Libby, J. Phys. Chem., 1952, 56, 863–868 CrossRef CAS.
- J. Weiss, Proc. R. Soc. London, Ser. A, 1954, 222, 128–141 CAS.
- V. Levich and R. Dogonadze, Dokl. Akad. Nauk SSSR, 1959, 124, 123–126 CAS.
- R. Dogonadze, A. Kuznetsov and V. Levich, Elektrokhimiya, 1967, 3, 739–742 CAS.
- A. N. Frumkin, V. S. Bagotskii, Z. A. Iofa and B. N. Kobanov, Kinetics of Electrode Processes, Moscow State University, Moscow, USSR, 1967 Search PubMed.
- J. Horiuti, in Physical Chemistry An Advanced Treatise, ed. H. Eyring, D. Henderson and W. Jost, Academic Press, New York, USA, 1970, ch. 6, vol. IXB, pp. 543–610 Search PubMed.
- L. G. Levich, in Physical Chemistry An Advanced Treatise, ed. H. Eyring, D. Henderson and W. Jost, Academic Press, New York, USA, 1970, ch. 12, vol. IXB, pp. 985–1074 Search PubMed.
- R. R. Dogonadze, in Reactions of Molecules at Electrodes, ed. N. S. Hush, John Wiley & Sons Ltd., Bath, UK, 1971, ch. 3, pp. 135–227 Search PubMed.
- A. J. Appleby, J. O. M. Bockris, R. K. Sen and B. E. Conway, in MTP International Review of Science Physical Chemistry Series One, ed. A. D. Buckingham, Electrochemistry, Butterworths, UK, 1973, vol. 6, pp. 1–40 Search PubMed.
- J. O. M. Bockris and S. U. Khan, Quantum Electrochemistry, Plenum Press, USA, 1979 Search PubMed.
- A. M. Kuznetsov, Electrochim. Acta, 1987, 32, 1271–1278 CrossRef CAS.
- A. Volta, Philos. Trans. R. Soc. London, 1800, 90, 403–431 CrossRef.
- M. Faraday, Philos. Trans. R. Soc. London, 1832, 122, 125–162 CrossRef.
- R. W. Gurney and E. U. Condon, Phys. Rev., 1929, 33, 127–140 CrossRef CAS.
- R. W. Gurney and P. M. Morse, Phys. Rev., 1929, 33, 789–799 CrossRef CAS.
- H. Gerischer, Z. Phys. Chem., 1960, 26, 223–247 CrossRef CAS.
- H. Gerischer, Z. Phys. Chem., 1960, 26, 325–338 CrossRef CAS.
- H. Gerischer, Z. Phys. Chem., 1961, 27, 48–79 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- R. Dogonadze, Y. A. Chizmadzhev and A. Kuznetsov, Zh. Fiz. Khim., 1964, 38, 1194–1202 Search PubMed.
- R. R. Dogonadze, A. M. Kuznetsov and V. G. Levich, Electrochim. Acta, 1968, 13, 1025–1044 CrossRef CAS.
- W. Schmickler, Ber. Bunsen-Ges., 1973, 77, 991–994 CAS.
- W. Schmickler and W. Vielstich, Electrochim. Acta, 1973, 18, 883–888 CrossRef CAS.
- R. A. Marcus, Presented in part at the Nobel Lecture, Stockholm Concert Hall, 1992.
- J. B. Goodenough, Presented in part at the Nobel Lecture, Stockholm Concert Hall, 2019.
- K. Sakaushi, Faraday Discuss., 2020, 221, 428–448 RSC.
- K. Sakaushi, A. Lyalin and T. Taketsugu, Curr. Opin. Electrochem., 2020, 19, 96–105 CrossRef CAS.
- X. Lin, N. Nilius, M. Sterrer, P. Koskinen, H. Häkkinen and H.-J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 153406 CrossRef.
- J. Sauer and H.-J. Freund, Catal. Lett., 2015, 145, 109–125 CrossRef CAS.
- S. Schauermann and H.-J. Freund, Acc. Chem. Res., 2015, 48, 2775–2782 CrossRef CAS PubMed.
- H.-J. Freund, J. Am. Chem. Soc., 2016, 138, 8985–8996 CrossRef CAS PubMed.
- H.-J. Freund, Rendiconti Lincei, 2017, 28, 5–18 CrossRef.
- A. Soudackov and S. Hammes-Schiffer, J. Chem. Phys., 1999, 111, 4672–4687 CrossRef CAS.
- A. Soudackov and S. Hammes-Schiffer, J. Chem. Phys., 2000, 113, 2385–2396 CrossRef CAS.
- S. Hammes-Schiffer, Acc. Chem. Res., 2001, 34, 273–281 CrossRef CAS PubMed.
- J. Grimminger, S. Bartenschlager and W. Schmickler, Chem. Phys. Lett., 2005, 416, 316–320 CrossRef CAS.
- M. Otani and O. Sugino, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 115407 CrossRef.
- J. Grimminger and W. Schmickler, Chem. Phys., 2007, 334, 8–17 CrossRef CAS.
- M. Otani, I. Hamada, O. Sugino, Y. Morikawa, Y. Okamoto and T. Ikeshoji, J. Phys. Soc. Jpn., 2008, 77, 024802 CrossRef.
- M. Otani, I. Hamada, O. Sugino, Y. Morikawa, Y. Okamoto and T. Ikeshoji, Phys. Chem. Chem. Phys., 2008, 10, 3609–3612 RSC.
- J. Haruyama, T. Ikeshoji and M. Otani, Phys. Rev. Mater., 2018, 2, 095801 CrossRef.
- M. M. Melander, M. J. Kuisma, T. E. K. Christensen and K. Honkala, J. Chem. Phys., 2019, 150, 041706 CrossRef PubMed.
- O. Sugino, J. Phys. Soc. Jpn., 2020, 89, 051013 CrossRef.
- G. Kastlunger, P. Lindgren and A. A. Peterson, J. Phys. Chem. C, 2018, 122, 12771–12781 CrossRef CAS.
- P. Lindgren, G. Kastlunger and A. A. Peterson, ACS Catal., 2020, 10, 121–128 CrossRef CAS.
- M. M. Melander, J. Electrochem. Soc., 2020, 167, 116518 CrossRef.
- S. Hammes-Schiffer, Energy Environ. Sci., 2012, 5, 7696–7703 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Rev., 2010, 110, PR1–PR40 CrossRef PubMed.
- B. E. Conway, Can. J. Chem., 1959, 37, 178–189 CrossRef CAS.
- E. Conway Brian, Proc. R. Soc. London, Ser. A, 1960, 256, 128–144 Search PubMed.
- B. E. Conway and M. Salomon, Ber. Bunsen-Ges., 1964, 68, 331–340 CrossRef CAS.
- B. E. Conway and M. Salomon, Electrochim. Acta, 1964, 9, 1599–1615 CrossRef CAS.
- B. E. Conway and M. Salomon, J. Chem. Phys., 1964, 41, 3169–3177 CrossRef CAS.
- M. Salomon and B. E. Conway, Ber. Bunsen-Ges., 1965, 69, 669–674 CrossRef CAS.
- M. Salomon and B. E. Conway, Discuss. Faraday Soc., 1965, 39, 223–238 RSC.
- J. O. M. Bockris and D. B. Matthews, J. Chem. Phys., 1966, 44, 298–309 CrossRef CAS.
- R. Parsons, Trans. Faraday Soc., 1951, 47, 1332–1344 RSC.
- K. Sakaushi, M. Eckardt, A. Lyalin, T. Taketsugu, R. J. Behm and K. Uosaki, ACS Catal., 2018, 8, 8162–8176 CrossRef CAS.
- K. Sakaushi, J. Electroanal. Chem., 2019, 849, 113372 CrossRef CAS.
- M. Eckardt, K. Sakaushi, A. Lyalin, M. Wassner, N. Hüsing, T. Taketsugu and R. J. Behm, Electrochim. Acta, 2019, 299, 736–748 CrossRef CAS.
- K. Sakaushi, A. Lyalin, T. Taketsugu and K. Uosaki, Phys. Rev. Lett., 2018, 121, 236001 CrossRef CAS PubMed.
- C. Costentin, Chem. Rev., 2008, 108, 2145–2179 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2010, 43, 1019–1029 CrossRef CAS PubMed.
- M. T. M. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- M. T. M. Koper, Chem. Sci., 2013, 4, 2710–2723 RSC.
- D. S. Gnanamuthu and J. V. Petrocelli, J. Electrochem. Soc., 1967, 114, 1036 CrossRef CAS.
- J. O. M. Bockris and A. K. N. Reddy, Modern Electrochemistry: An Introduction to an interdisciplinary area, Plenum Press, 1970 Search PubMed.
- M. Dunwell, X. Yang, B. P. Setzler, J. Anibal, Y. Yan and B. Xu, ACS Catal., 2018, 8, 3999–4008 CrossRef CAS.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- G. A. Parada, Z. K. Goldsmith, S. Kolmar, B. Pettersson Rimgard, B. Q. Mercado, L. Hammarström, S. Hammes-Schiffer and J. M. Mayer, Science, 2019, 364, 471–475 CrossRef CAS PubMed.
- M. T. Huynh, S. J. Mora, M. Villalba, M. E. Tejeda-Ferrari, P. A. Liddell, B. R. Cherry, A.-L. Teillout, C. W. Machan, C. P. Kubiak, D. Gust, T. A. Moore, S. Hammes-Schiffer and A. L. Moore, ACS Cent. Sci., 2017, 3, 372–380 CrossRef CAS PubMed.
- K. Sakaushi, Phys. Chem. Chem. Phys., 2020, 22, 11219–11243 RSC.
- Z. K. Goldsmith, Y. C. Lam, A. V. Soudackov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2018, 141, 1084–1090 CrossRef PubMed.
- Y.-C. Lam, A. V. Soudackov and S. Hammes-Schiffer, J. Phys. Chem. Lett., 2019, 10, 5312–5317 CrossRef CAS PubMed.
- M. N. Jackson and Y. Surendranath, J. Am. Chem. Soc., 2016, 138, 3228–3234 CrossRef CAS.
- N. S. Hush, J. Chem. Phys., 1958, 28, 962–972 CrossRef CAS.
- C. E. D. Chidsey, Science, 1991, 251, 919–922 CrossRef CAS PubMed.
- A. Damjanovic and M. A. Genshaw, Electrochim. Acta, 1970, 15, 1281–1283 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS PubMed.
- K. Sakaushi and M. Antonietti, Bull. Chem. Soc. Jpn., 2015, 88, 386–398 CrossRef CAS.
- K. Sakaushi, T.-P. Fellinger and M. Antonietti, ChemSusChem, 2015, 8, 1156–1160 CrossRef CAS PubMed.
- K. Sakaushi, S. J. Yang, T. P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2015, 3, 11720–11724 RSC.
- K. Sakaushi and K. Uosaki, ChemNanoMat, 2016, 2, 99–103 CrossRef CAS.
- K. Sakaushi, A. Lyalin, S. Tominaka, T. Taketsugu and K. Uosaki, ACS Nano, 2017, 11, 1770–1779 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- N. Brüniche-Olsen and J. Ulstrup, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 205–226 RSC.
- S. J. Edwards, A. V. Soudackov and S. Hammes-Schiffer, J. Phys. Chem. A, 2009, 113, 2117–2126 CrossRef CAS PubMed.
- A. Bouzid and A. Pasquarello, J. Phys. Chem. Lett., 2018, 9, 1880–1884 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed.
- T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J.-H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- T. Yamada, K. Otsubo, R. Makiura and H. Kitagawa, Chem. Soc. Rev., 2013, 42, 6655–6669 RSC.
- E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Nat. Commun., 2016, 7, 10942 CrossRef CAS PubMed.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- R. Dong, T. Zhang and X. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS.
- K. Sakaushi, in Functional Organic and Hybrid Nanostructured Materials, ed. Q. Li, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, ch. 11, pp. 419–444 DOI:10.1002/9783527807369.ch11.
- K. Sakaushi, in Materials Nanoarchitectonics, ed. K. Ariga and M. Ebara, John Wiley and Sons, 2018, ch. 8, pp. 155–164 DOI:10.1002/9783527808311.ch8.
- K. Wada, K. Sakaushi, S. Sasaki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 8886–8890 CrossRef CAS.
- M. L. Aubrey, B. M. Wiers, S. C. Andrews, T. Sakurai, S. E. Reyes-Lillo, S. M. Hamed, C.-J. Yu, L. E. Darago, J. A. Mason, J.-O. Baeg, F. Grandjean, G. J. Long, S. Seki, J. B. Neaton, P. Yang and J. R. Long, Nat. Mater., 2018, 17, 625–632 CrossRef CAS PubMed.
- Z. Liang, C. Qu, W. Guo, R. Zou and Q. Xu, Adv. Mater., 2018, 30, 1702891 CrossRef PubMed.
- N. Hosono and S. Kitagawa, Acc. Chem. Res., 2018, 51, 2437–2446 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00766.
- M. Amores, K. Wada, K. Sakaushi and H. Nishihara, J. Phys. Chem. C, 2020, 124, 9215–9224 CrossRef CAS.
- K. Wada, H. Maeda, T. Tsuji, K. Sakaushi, S. Sasaki and H. Nishihara, Inorg. Chem., 2020, 59, 10604–10610 CrossRef CAS PubMed.
- C.-W. Kung, S. Goswami, I. Hod, T. C. Wang, J. Duan, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2020, 53, 1187–1195 CrossRef CAS PubMed.
- D.-W. Lim and H. Kitagawa, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00842.
- K. Sakaushi, G. Nickerl, F. M. Wisser, D. Nishio-Hamane, E. Hosono, H. Zhou, S. Kaskel and J. Eckert, Angew. Chem., Int. Ed., 2012, 51, 7850–7854 CrossRef CAS.
- K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef.
- K. Sakaushi, G. Nickerl, H. C. Kandpal, L. Cano-Cortés, T. Gemming, J. Eckert, S. Kaskel and J. van den Brink, J. Phys. Chem. Lett., 2013, 4, 2977–2981 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- K. Sakaushi, E. Hosono, G. Nickerl, H. Zhou, S. Kaskel and J. Eckert, J. Power Sources, 2014, 245, 553–556 CrossRef CAS.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed.
- X. Li, P. Yadav and K. P. Loh, Chem. Soc. Rev., 2020, 49, 4835–4866 RSC.
- A. Savateev, N. V. Tarakina, V. Strauss, T. Hussain, K. ten Brummelhuis, J. M. Sánchez Vadillo, Y. Markushyna, S. Mazzanti, A. P. Tyutyunnik, R. Walczak, M. Oschatz, D. M. Guldi, A. Karton and M. Antonietti, Angew. Chem., Int. Ed. DOI:10.1002/anie.202004747.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00550.
- J. Bardeen, Phys. Rev., 1947, 71, 717–727 CrossRef.
- N. Cabrera and N. F. Mott, Rep. Prog. Phys., 1949, 12, 163–184 CrossRef CAS.
- F. F. Vol'kenshtein, Russ. Chem. Rev., 1966, 35, 537–546 CrossRef.
- X. Lin, B. Yang, H.-M. Benia, P. Myrach, M. Yulikov, A. Aumer, M. A. Brown, M. Sterrer, O. Bondarchuk, E. Kieseritzky, J. Rocker, T. Risse, H.-J. Gao, N. Nilius and H.-J. Freund, J. Am. Chem. Soc., 2010, 132, 7745–7749 CrossRef CAS PubMed.
- C. Stiehler, F. Calaza, W.-D. Schneider, N. Nilius and H.-J. Freund, Phys. Rev. Lett., 2015, 115, 036804 CrossRef PubMed.
- C. P. O'Brien, K. H. Dostert, M. Hollerer, C. Stiehler, F. Calaza, S. Schauermann, S. Shaikhutdinov, M. Sterrer and H. J. Freund, Faraday Discuss., 2016, 188, 309–321 RSC.
- F. Calaza, C. Stiehler, Y. Fujimori, M. Sterrer, S. Beeg, M. Ruiz-Oses, N. Nilius, M. Heyde, T. Parviainen, K. Honkala, H. Häkkinen and H.-J. Freund, Angew. Chem., Int. Ed., 2015, 54, 12484–12487 CrossRef CAS PubMed.
- P. Alexa, J. M. Lombardi, P. Abufager, H. F. Busnengo, D. Grumelli, V. S. Vyas, F. Haase, B. V. Lotsch, R. Gutzler and K. Kern, Angew. Chem., Int. Ed., 2020, 59, 8411–8415 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef CAS PubMed.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- M. L. Pegis, C. F. Wise, B. Koronkiewicz and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 11000–11003 CrossRef CAS PubMed.
- Y.-H. Wang, M. L. Pegis, J. M. Mayer and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 16458–16461 CrossRef CAS.
- M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 8315–8326 CAS.
- D. J. Martin, B. Q. Mercado and J. M. Mayer, Sci. Adv., 2020, 6, eaaz3318 CrossRef PubMed.
- D. J. Martin, C. F. Wise, M. L. Pegis and J. M. Mayer, Acc. Chem. Res., 2020, 53, 1056–1065 CrossRef CAS PubMed.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2020, 5, 72–78 CrossRef CAS.
- T. Iwasita and F. C. Nart, Prog. Surf. Sci., 1997, 55, 271–340 CrossRef CAS.
- W. Chen, A. Yu, Z.-J. Sun, B.-Q. Zhu, J. Cai and Y.-X. Chen, Curr. Opin. Electrochem., 2019, 14, 113–123 CrossRef CAS.
- F. Kitamura, M. Taiseda, M. Takahashi and M. Ito, Chem. Phys. Lett., 1987, 142, 318–322 CrossRef CAS.
- S.-C. Chang, L.-W. H. Leung and M. J. Weaver, J. Phys. Chem., 1989, 93, 5341–5345 CrossRef CAS.
- Y. Kinomoto, S. Watanabe, M. Takahashi and M. Ito, Surf. Sci., 1991, 242, 538–543 CrossRef CAS.
- E. Morallon, A. Rodes, J. L. Vazquez and J. M. Perez, J. Electroanal. Chem., 1995, 391, 149–157 CrossRef.
- M. Nakamura, H. Ogasawara, J. Inukai and M. Ito, Surf. Sci., 1993, 283, 248–254 CrossRef CAS.
- B. O. Budevska and P. R. Griffiths, Anal. Chem., 1993, 65, 2963–2971 CrossRef CAS.
- M. Osawa, K.-I. Ataka, K. Yoshii and T. Yotsuyanagi, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 371–379 CrossRef CAS.
- G. Samjeske and M. Osawa, Angew. Chem., Int. Ed., 2005, 44, 5694–5698 CrossRef CAS PubMed.
- G. Samjeske, A. Miki and M. Osawa, J. Phys. Chem. C, 2007, 111, 15074–15083 CrossRef CAS.
- G. Samjeske, A. Miki, S. Ye and M. Osawa, J. Phys. Chem. B, 2006, 110, 16559–16566 CrossRef CAS PubMed.
- G. Samjeske, A. Miki, S. Ye, A. Yamakata, Y. Mukouyama, H. Okamoto and M. Osawa, J. Phys. Chem. B, 2006, 109, 23509–23516 CrossRef PubMed.
- M. Osawa, K. Yoshii, K.-I. Ataka and T. Yotsuyanagi, Langmuir, 1994, 10, 640–642 CrossRef CAS.
- H. Noda, K.-I. Ataka, L.-J. Wan and M. Osawa, Surf. Sci., 1999, 427–428, 190–194 CrossRef CAS.
- A. Yamakata and M. Osawa, J. Electroanal. Chem., 2017, 800, 19–24 CrossRef CAS.
- D. Palecek, G. Tek, J. Lan, M. Iannuzzi and P. Hamm, J. Phys. Chem. Lett., 2018, 9, 1254–1259 CrossRef CAS PubMed.
- H. Abramczyk, B. Brozek-Pluska, K. Kurczewski, M. Kurczewski, I. Szymczyk, P. Krzyczmonik, T. Blaszczyk, H. Scholl and W. Czajkowski, J. Phys. Chem. A, 2006, 110, 8627–8636 CrossRef CAS PubMed.
- R. L. Garrell, Anal. Chem., 1989, 61, 401–411 CrossRef.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- Y.-X. Chen, M. Heinen, Z. Jusys and R. J. Behm, ChemPhysChem, 2007, 8, 380–385 CrossRef CAS PubMed.
- J. Schnaidt, M. Heinen and R. J. Behm, Catal. Today, 2013, 202, 154–162 CrossRef CAS.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS PubMed.
- H. Lv, D. Li, D. Strmcnik, A. P. Paulikas, N. M. Markovic and V. R. Stamenkovic, Nano Energy, 2016, 29, 149–165 CrossRef CAS.
- N. M. Markovic, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1997, 101, 5405–5413 CrossRef CAS.
- M. R. Generro de Chialvo and A. C. Chialvo, Electrochem. Commun., 1999, 1, 379–382 CrossRef.
- A. Lasia, Int. J. Hydrogen Energy, 2019, 44, 19484–19518 CrossRef CAS.
- K. Kunimatsu, T. Senzaki, M. Tsushima and M. Osawa, Chem. Phys. Lett., 2005, 401, 451–454 CrossRef CAS.
- K. Kunimatsu, T. Senzaki, G. Samjeské, M. Tsushima and M. Osawa, Electrochim. Acta, 2007, 52, 5715–5724 CrossRef CAS.
- M. Nakamura, T. Kobayashi and N. Hoshi, Surf. Sci., 2011, 605, 1462–1465 CrossRef CAS.
- Ş. C. Bădescu, K. Jacobi, Y. Wang, K. Bedürftig, G. Ertl, P. Salo, T. Ala-Nissila and S. C. Ying, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 205401 CrossRef.
- L. Yan, Y. Sun, Y. Yamamoto, S. Kasamatsu, I. Hamada and O. Sugino, J. Chem. Phys., 2018, 149, 164702 CrossRef PubMed.
- A. Peremans and A. Tadjeddine, J. Chem. Phys., 1995, 103, 7197 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- M. H. Shao and R. R. Adzic, J. Phys. Chem. B, 2005, 109, 16563–16566 CrossRef CAS.
- R. R. Adzic, N. M. Markovic and V. B. Vesovic, J. Electroanal. Chem., 1984, 165, 105–120 CrossRef CAS.
- S. Strbac, N. A. Anastasijevic and R. R. Adzic, J. Electroanal. Chem., 1992, 323, 179–195 CrossRef CAS.
- H. Y. Yu, X. F. Li, T. H. Zhang, J. Liu, J. H. Tian and R. Yang, ChemSusChem, 2020, 13, 2702–2708 CrossRef CAS PubMed.
- X. Li and A. A. Gewirth, J. Am. Chem. Soc., 2005, 127, 5252–5260 CrossRef CAS PubMed.
- M.-h. Shao, P. Liu and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 7408–7409 CrossRef CAS PubMed.
- S. Nayak, I. J. McPherson and K. A. Vincent, Angew. Chem., Int. Ed., 2018, 57, 12855–12858 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2018, 4, 60–67 CrossRef.
- J. C. Dong, M. Su, V. Briega-Martos, L. Li, J. B. Le, P. Radjenovic, X. S. Zhou, J. M. Feliu, Z. Q. Tian and J. F. Li, J. Am. Chem. Soc., 2020, 142, 715–719 CrossRef CAS PubMed.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1995, 99, 3411–3415 CrossRef CAS.
- J. X. Wang, N. M. Markovic and R. R. Adzic, J. Phys. Chem. B, 2004, 108, 4127–4133 CrossRef CAS.
- J. Tymoczko, V. Colic, A. Ganassin, W. Schuhmann and A. S. Bandarenka, Catal. Today, 2015, 244, 96–102 CrossRef CAS.
- H. Ogasawara, Y. Sawatari, J. Inukai and M. Ito, J. Electroanal. Chem., 1993, 358, 337–342 CrossRef CAS.
- Y. Sawatari, J. Inukai and M. Ito, J. Electron Spectrosc. Relat. Phenom., 1993, 64, 515–522 CrossRef.
- K. Kodama, A. Shinohara, N. Hasegawa, K. Shinozaki, R. Jinnouchi, T. Suzuki, T. Hatanaka and Y. Morimoto, J. Electrochem. Soc., 2014, 161, F649–F652 CrossRef CAS.
- J. Tymoczko, F. Calle-Vallejo, V. Colic, M. T. M. Koper, W. Schuhmann and A. S. Bandarenka, ACS Catal., 2014, 4, 3772–3778 CrossRef CAS.
- K. Kodama, K. Motobayashi, A. Shinohara, N. Hasegawa, K. Kudo, R. Jinnouchi, M. Osawa and Y. Morimoto, ACS Catal., 2018, 8, 694–700 CrossRef CAS.
- A. M. Gómez-Marín, A. Berná and J. M. Feliu, J. Phys. Chem. C, 2010, 114, 20130–20140 CrossRef.
- N. M. Markovic, R. R. Adzic, B. D. Cahan and E. B. Yeager, J. Electroanal. Chem., 1994, 377, 249–259 CrossRef CAS.
- H. Tanaka, S. Sugawara, K. Shinohara, T. Ueno, S. Suzuki, N. Hoshi and M. Nakamura, Electrocatalysis, 2015, 6, 295–299 CrossRef CAS.
- T. Ueno, H. Tanaka, S. Sugawara, K. Shinohara, A. Ohma, N. Hoshi and M. Nakamura, J. Electroanal. Chem., 2017, 800, 162–166 CrossRef CAS.
- F. Zaera, Prog. Surf. Sci., 2001, 69, 1–98 CrossRef CAS.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2009, 1, 466–472 CrossRef CAS PubMed.
- M. Nakamura, Y. Nakajima, N. Hoshi, H. Tajiri and O. Sakata, ChemPhysChem, 2013, 14, 2426–2431 CrossRef CAS PubMed.
- A. Yamakata and M. Osawa, J. Phys. Chem. Lett., 2010, 1, 1487–1491 CrossRef CAS.
- T. Kumeda, R. Kubo, N. Hoshi and M. Nakamura, ACS Appl. Energy Mater., 2019, 2, 3904–3909 CrossRef CAS.
- T. Kumeda, H. Tajiri, O. Sakata, N. Hoshi and M. Nakamura, Nat. Commun., 2018, 9, 4378 CrossRef PubMed.
- S. Yamamoto, A. Beniya, K. Mukai, Y. Yamashita and J. Yoshinobu, J. Phys. Chem. B, 2005, 109, 5816–5823 CrossRef CAS PubMed.
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CrossRef CAS.
- S. Chatterjee, K. Sengupta, S. Samanta, P. K. Das and A. Dey, Inorg. Chem., 2015, 54, 2383–2392 CrossRef CAS PubMed.
- C. Zou, M. Larisika, G. Nagy, J. Srajer, C. Oostenbrink, X. Chen, W. Knoll, B. Liedberg and C. Nowak, J. Phys. Chem. B, 2013, 117, 9606–9614 CrossRef CAS PubMed.
- S. Chatterjee, K. Sengupta, S. Samanta, P. K. Das and A. Dey, Inorg. Chem., 2013, 52, 9897–9907 CrossRef CAS PubMed.
- M. Grosserueschkamp, M. G. Friedrich, M. Plum, W. Knoll and R. L. C. Naumann, J. Phys. Chem. B, 2009, 113, 2492–2497 CrossRef CAS PubMed.
- C. Nowak, C. Luening, D. Schach, D. Baurecht, W. Knoll and R. L. C. Naumann, J. Phys. Chem. C, 2009, 113, 2256–2262 CrossRef CAS.
- A. Schwaighofer, C. Steininger, D. M. Hildenbrandt, J. Srajer, C. Nowak, W. Knoll and R. L. C. Naumann, Biophys. J., 2013, 105, 2706–2713 CrossRef CAS PubMed.
- H. K. Ly, P. Wrzolek, N. Heidary, R. Gotz, M. Horch, J. Kozuch, M. Schwalbe and I. M. Weidinger, Chem. Sci., 2015, 6, 6999–7007 RSC.
- T. Chen, Z. Feng, G. Wu, J. Shi, G. Ma, P. Ying and C. Li, J. Phys. Chem. C, 2007, 111, 8005–8014 CrossRef CAS.
- S. Zhu, T. Li, W.-B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- J. Heyes, M. Dunwell and B. Xu, J. Phys. Chem. C, 2016, 120, 17334–17341 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- Q. Cheng, L. Wei, Z. Liu, N. Ni, Z. Sang, B. Zhu, W. Xu, M. Chen, Y. Miao, L.-Q. Chen, W. Min and Y. Yang, Nat. Commun., 2018, 9, 2942 CrossRef PubMed.
- P. Guyot-Sionnest and A. Tadjeddine, Chem. Phys. Lett., 1990, 172, 341–345 CrossRef CAS.
- A. Peremans and A. Tadjeddine, Phys. Rev. Lett., 1994, 73, 3010–3013 CrossRef CAS PubMed.
- W. Zhou, S. Inoue, T. Iwahashi, K. Kanai, K. Seki, T. Miyamae, D. Kim, Y. Katayama and Y. Ouchi, Electrochem. Commun., 2010, 12, 672–675 CrossRef CAS.
- A. Sayama, S. Nihonyanagi, Y. Ohshima and T. Tahara, Phys. Chem. Chem. Phys., 2020, 22, 2580–2589 RSC.
- X. Wang, S.-C. Huang, T.-X. Huang, H.-S. Su, J.-H. Zhong, Z.-C. Zeng, M.-H. Lia and B. Ren, Chem. Soc. Rev., 2017, 46, 4020–4041 RSC.
- A. Soudackov and S. Hammes-Schiffer, J. Chem. Phys., 1999, 111, 4672–4687 CrossRef CAS.
- A. Soudackov and S. Hammes-Schiffer, J. Chem. Phys., 2000, 113, 2385–2396 CrossRef CAS.
- S. Hammes-Schiffer and A. V. Soudackov, J. Phys. Chem. B, 2008, 112, 14108–14123 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed.
- Y. Georgievskii and A. A. Stuchebrukhov, J. Chem. Phys., 2000, 113, 10438–10450 CrossRef CAS.
- J. H. Skone, A. V. Soudackov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2006, 128, 16655–16663 CrossRef CAS PubMed.
- D. F. Calef and P. G. Wolynes, J. Phys. Chem., 1983, 87, 3387–3400 CrossRef CAS.
- J. T. Hynes, J. Phys. Chem., 1986, 90, 3701–3706 CrossRef CAS.
- J. E. Straub and B. J. Berne, J. Chem. Phys., 1987, 87, 6111–6116 CrossRef CAS.
- D. E. Makarov and M. Topaler, Chem. Phys. Lett., 1995, 245, 343–350 CrossRef CAS.
- C. Venkataraman, A. V. Soudackov and S. Hammes-Schiffer, J. Phys. Chem. C, 2008, 112, 12386–12397 CrossRef CAS.
- I. Navrotskaya, A. V. Soudackov and S. Hammes-Schiffer, J. Chem. Phys., 2008, 128, 244712 CrossRef PubMed.
- B. Auer, L. E. Fernandez and S. Hammes-Schiffer, J. Am. Chem. Soc., 2011, 133, 8282–8292 CrossRef CAS PubMed.
- M. T. Huynh, S. J. Mora, M. Villalba, M. E. Tejeda-Ferrari, P. A. Liddell, B. R. Cherry, A.-L. Teillout, C. W. Machan, C. P. Kubiak and D. Gust, ACS Cent. Sci., 2017, 3, 372–380 CrossRef CAS PubMed.
- Y.-C. Lam, A. V. Soudackov, Z. K. Goldsmith and S. Hammes-Schiffer, J. Phys. Chem. C, 2019, 123, 12335–12345 CrossRef CAS.
- G. Guoy, J. Phys., 1910, 9, 457–467 Search PubMed.
- D. L. Chapman, London Edinburgh Philos. Mag. J. Sci., 1913, 25, 475–481 CrossRef.
- O. Stern, Z. Elektrochem., 1924, 30, 1014–1020 Search PubMed.
- A. Velikonja, E. Gongadze, V. Kralj-Iglic and A. Iglic, Int. J. Electrochem. Sci., 2014, 9 Search PubMed.
- Z. K. Goldsmith, M. Secor and S. Hammes-Schiffer, ACS Cent. Sci., 2020, 6, 304–311 CrossRef CAS PubMed.
- A. Hamelin and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1987, 223, 171–184 CrossRef CAS.
- B. E. Conway, Proc. R. Soc. London, Ser. A, 1960, 256, 128–144 CAS.
- Z.-D. He, Y.-X. Chen, E. Santos and W. Schmickler, Angew. Chem., Int. Ed., 2018, 57, 7948–7956 CrossRef CAS PubMed.
- W. H. Miller, J. Chem. Phys., 1974, 61, 1823–1834 CrossRef CAS.
- R. Sundararaman, W. A. Goddard III and T. A. Arias, J. Chem. Phys., 2017, 146, 114104 CrossRef.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- N. Bonnet, T. Morishita, O. Sugino and M. Otani, Phys. Rev. Lett., 2012, 109, 266101 CrossRef PubMed.
- R. Jinnouchi, K. Kodama and Y. Morimoto, Curr. Opin. Electrochem., 2018, 8, 103–109 CrossRef CAS.
- W. H. Miller, S. D. Schwartz and J. W. Tromp, J. Chem. Phys., 1983, 79, 4889–4898 CrossRef CAS.
- W. H. Miller, J. Phys. Chem. A, 1998, 102, 793–806 CrossRef CAS.
- F. Studt, J. K. Nørskov, F. Abild-Pedersen and T. Bligaard, Fundamental concepts in heterogeneous catalysis, John Wiley & Sons, Inc., 2014 Search PubMed.
- Y. Huang, R. J. Nielsen and W. A. Goddard, J. Am. Chem. Soc., 2018, 140, 16773–16782 CrossRef CAS PubMed.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2016, 7, 1686–1690 CrossRef CAS PubMed.
- M. J. Janik and M. Neurock, Electrochim. Acta, 2007, 52, 5517–5528 CrossRef CAS.
- M. Van den Bossche, E. Skúlason, C. Rose-Petruck and H. Jónsson, J. Phys. Chem. C, 2019, 123, 4116–4124 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- T. Ikeshoji and M. Otani, Phys. Chem. Chem. Phys., 2017, 19, 4447–4453 RSC.
- R. Kronberg, H. Lappalainen and K. Laasonen, Phys. Chem. Chem. Phys., 2020, 22, 10536–10549 RSC.
- M. Sprik and G. Ciccotti, J. Chem. Phys., 1998, 109, 7737–7744 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS.
- H. S. Pillai and H. Xin, Ind. Eng. Chem. Res., 2019, 58, 10819–10828 CrossRef CAS.
- L.-Y. Chen, T.-C. Kuo, Z.-S. Hong, M.-J. Cheng and W. A. Goddard, Phys. Chem. Chem. Phys., 2019, 21, 17605–17612 RSC.
- J. E. Lawrence and D. E. Manolopoulos, Faraday Discuss., 2020, 221, 9–29 RSC.
- Y. Zhang, J. B. Rommel, M. T. CvitaÅ and S. C. Althorpe, Phys. Chem. Chem. Phys., 2014, 16, 24292–24300 RSC.
- J. O. Richardson, PhD thesis, University of Cambridge, 2012.
- I. Affleck, Phys. Rev. Lett., 1981, 46, 388–391 CrossRef.
- M. J. Gillan, J. Phys. C: Solid State Phys., 1987, 20, 3621–3641 CrossRef.
- M. Tuckerman, Statistical Mechanics: Theory and Molecular Simulations, Oxford University Press, 2010 Search PubMed.
- R. P. Feynman, Rev. Mod. Phys., 1948, 20, 367–387 CrossRef.
- R. P. Feynman and A. R. Hibbs, Quantum Mechanics and Path Integrals, McGraw-Hill, 1965 Search PubMed.
- S. Coleman, The Uses of Instantons, in The Whys of Subnuclear Physics, ed. A. Zichichi, The Subnuclear Series, Springer, Boston, MA, 1979, vol. 15 Search PubMed.
- P. G. Wolynes, J. Chem. Phys., 1987, 87, 6559–6561 CrossRef.
- M. Ceriotti, W. Fang, P. G. Kusalik, R. H. McKenzie, A. Michaelides, M. A. Morales and T. E. Markland, Chem. Rev., 2016, 116, 7529–7550 CrossRef CAS PubMed.
- W. Fang, J. Chen, Y. Feng, X.-Z. Li and A. Michaelides, Int. Rev. Phys. Chem., 2019, 38, 35–61 Search PubMed.
- J. Lan, V. V. Rybkin and M. Iannuzzi, J. Phys. Chem. Lett., 2020, 11, 3724–3730 Search PubMed.
- W. H. Miller, J. Chem. Phys., 1975, 62, 1899–1906 CrossRef CAS.
- E. J. Heller, The Semiclassical Way to Dynamics and Spectroscopy, Princeton University Press, 2018 Search PubMed.
- R. Hernandez and W. H. Miller, Chem. Phys. Lett., 1993, 214, 129–136 CrossRef CAS.
- W. H. Miller, Faraday Discuss. Chem. Soc., 1977, 62, 40–46 RSC.
- A. F. Wagner, J. Phys. Chem. A, 2013, 117, 13089–13100 CrossRef CAS PubMed.
- X. Shan, T. A. H. Burd and D. C. Clary, J. Phys. Chem. A, 2019, 123, 4639–4657 CrossRef CAS PubMed.
- J. T. Fermann and S. Auerbach, J. Chem. Phys., 2000, 112, 6787–6794 CrossRef CAS.
- K. Heinola and T. Ahlgren, J. Appl. Phys., 2010, 107, 113531 CrossRef.
- K. Mudiyanselage, M. Trenary and R. J. Meyer, J. Phys. Chem. C, 2007, 111, 7127–7136 CrossRef CAS.
- S. C. Althorpe and T. J. H. Hele, J. Chem. Phys., 2013, 139, 084115 CrossRef PubMed.
- T. J. H. Hele and S. C. Althorpe, J. Chem. Phys., 2013, 138, 084108 CrossRef PubMed.
- J. O. Richardson and S. C. Althorpe, J. Chem. Phys., 2009, 131, 214106 CrossRef.
- J. O. Richardson, J. Chem. Phys., 2016, 144, 114106 CrossRef PubMed.
- J. O. Richardson, Int. Rev. Phys. Chem., 2018, 37, 171–216 Search PubMed.
- W. H. Miller, Faraday Discuss. Chem. Soc., 1977, 62, 40–46 RSC.
- S. Andersson, G. Nyman, A. Arnaldsson, U. Manthe and H. Jónsson, J. Phys. Chem. A, 2009, 113, 4468–4478 CrossRef CAS PubMed.
- V. Àsgeirsson, A. Arnaldsson and H. Jònsson, J. Chem. Phys., 2018, 148, 102334 CrossRef PubMed.
- J. B. Rommel, T. P. M. Goumans and J. Kästner, J. Chem. Theory Comput., 2011, 7, 690–698 CrossRef CAS PubMed.
- G. Laude, D. Calderini, D. P. Tew and J. O. Richardson, Faraday Discuss., 2018, 212, 237–258 RSC.
- M. Kryvohuz, J. Chem. Phys., 2012, 137, 234304 CrossRef PubMed.
- G. Mills, G. K. Schenter, D. E. Makarov and H. Jónsson, Chem. Phys. Lett., 1997, 278, 91–96 CrossRef CAS.
- W. Fang, J. O. Richardson, J. Chen, X.-Z. Li and A. Michaelides, Phys. Rev. Lett., 2017, 119, 126001 CrossRef PubMed.
- Y. Litman, J. O. Richardson, T. Kumagai and M. Rossi, J. Am. Chem. Soc., 2019, 141, 2526–2534 CrossRef CAS PubMed.
- D. Chandler and P. G. Wolynes, J. Chem. Phys., 1981, 74, 4078–4095 CrossRef CAS.
- I. R. Craig and D. E. Manolopoulos, J. Chem. Phys., 2004, 121, 3368–3373 CrossRef CAS PubMed.
- I. R. Craig and D. E. Manolopoulos, J. Chem. Phys., 2005, 122, 084106 CrossRef PubMed.
- I. R. Craig and D. E. Manolopoulos, J. Chem. Phys., 2005, 123, 034102 CrossRef PubMed.
- R. Collepardo-Guevara, I. R. Craig and D. E. Manolopoulos, J. Chem. Phys., 2008, 128, 144502 CrossRef PubMed.
- J. S. Kretchmer and T. F. Miller, J. Chem. Phys., 2013, 138, 134109 CrossRef PubMed.
- J. S. Kretchmer and T. F. Miller, Inorg. Chem., 2016, 55, 1022–1031 CrossRef CAS PubMed.
- Y. V. Suleimanov, J. Phys. Chem. C, 2012, 116, 11141–11153 CrossRef CAS.
- B. Kaduk, T. Kowalczyk and T. Van Voorhis, Chem. Rev., 2012, 112, 321–370 CrossRef CAS PubMed.
- M. Melander, E. Ö. Jónsson, J. J. Mortensen, T. Vegge and J. M. García-Lastra, J. Chem. Theory Comput., 2016, 12, 5367–5378 CrossRef CAS PubMed.
- G. Kastlunger, personal communication.
- K. Sakaushi, Faraday Discuss., 2020, 221, 428–448 RSC.
- G. A. Voth, D. Chandler and W. H. Miller, J. Chem. Phys., 1989, 91, 7749–7760 CrossRef CAS.
- S. Habershon, D. E. Manolopoulos, T. E. Markland and T. F. Miller, Annu. Rev. Phys. Chem., 2013, 64, 387–413 CrossRef CAS PubMed.
- E. R. Heller and J. O. Richardson, J. Chem. Phys., 2020, 152, 034106 CrossRef CAS PubMed.
- M. J. Thapa, W. Fang and J. O. Richardson, J. Chem. Phys., 2019, 150, 104107 CrossRef PubMed.
- J. E. Lawrence and D. E. Manolopoulos, J. Chem. Phys., 2019, 151, 244109 CrossRef PubMed.
- J. O. M. Bockris and S. U. Khan, Surface electrochemistry: a molecular level approach, Plenum Press, New York, USA, 1993 Search PubMed.
- N. M. Marković and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef.
- G. Jerkiewicz, Electrocatalysis, 2010, 1, 179–199 CrossRef CAS.
- V. Krishna, J. Chem. Phys., 2007, 126, 134107 CrossRef PubMed.
- D. Marx and M. Parrinello, Z. Phys. B: Condens. Matter, 1994, 95, 143–144 CrossRef CAS.
- J. P. Blaizot and H. Orland, Phys. Rev. C: Nucl. Phys., 1981, 24, 1740–1761 CrossRef CAS.
- J. C. Tully and R. K. Preston, J. Chem. Phys., 1971, 55, 562–572 CrossRef CAS.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS.
- W. Schmickler, Interfacial electrochemistry, Oxford University Press, The United States of America, 1996 Search PubMed.
- E. Santos, P. Quaino and W. Schmickler, Phys. Chem. Chem. Phys., 2012, 14, 11224–11233 RSC.
- S. Tanaka and C.-P. Hsu, J. Chem. Phys., 1999, 111, 11117–11137 CrossRef CAS.
- K. Nakamura, Y. Nohara, Y. Yosimoto and Y. Nomura, Phys. Rev. B, 2016, 93, 085124 CrossRef.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- P. S. Rice, Y. Mao, C. Guo and P. Hu, Phys. Chem. Chem. Phys., 2019, 21, 5932–5940 RSC.
- R. Jinnouchi and A. B. Anderson, J. Phys. Chem. C, 2008, 112, 8747–8750 CrossRef CAS.
- R. Jinnouchi and A. B. Anderson, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 245417 CrossRef.
- G. Fisicaro, L. Genovese, O. Andreussi, N. Marzari and S. Goedecker, J. Chem. Phys., 2016, 144, 014103 CrossRef CAS PubMed.
- D. Chandler and H. C. Andersen, J. Chem. Phys., 1972, 57, 1930–1937 CrossRef CAS.
- A. Kovalenko and F. Hirata, J. Chem. Phys., 1999, 110, 10095–10112 CrossRef CAS.
- S. Nishihara and M. Otani, Phys. Rev. B, 2017, 96, 115429 CrossRef.
- Y. Noguchi, M. Hiyama, M. Shiga, H. Akiyama and O. Sugino, J. Chem. Theory Comput., 2019, 15, 5474–5482 CrossRef CAS PubMed.
- M. J. Eslamibidgoli, J. Huang, T. Kadyk, A. Malek and M. Eikerling, Nano Energy, 2016, 29, 334–361 CrossRef CAS.
- S. Maheshwari, Y. Li, N. Agrawal and M. J. Janik, in Advances in Catalysis, ed. C. Song, Academic Press, 2018, vol. 63, pp. 117–167 Search PubMed.
- M. J. Eslamibidgoli and M. H. Eikerling, Curr. Opin. Electrochem., 2018, 9, 189–197 CrossRef CAS.
- A. Groß and S. Sakong, Curr. Opin. Electrochem., 2019, 14, 1–6 CrossRef.
- O. M. Magnussen and A. Groß, J. Am. Chem. Soc., 2019, 141, 4777–4790 CrossRef CAS PubMed.
- K. Schwarz and R. Sundararaman, Surf. Sci. Rep., 2020, 75, 100492 CrossRef CAS.
- T. Ikeshoji, M. Otani, I. Hamada and Y. Okamoto, Phys. Chem. Chem. Phys., 2011, 13, 20223–20227 RSC.
- T. Ikeshoji, M. Otani, I. Hamada, O. Sugino, Y. Morikawa, Y. Okamoto, Y. Qian and I. Yagi, AIP Adv., 2012, 2, 032182 CrossRef.
- Y. Qian, I. Hamada, M. Otani and T. Ikeshoji, Catal. Today, 2013, 202, 163–167 CrossRef CAS.
- T. Ikeshoji, T. Uchida, M. Otani and M. Osawa, J. Electroanal. Chem., 2017, 800, 13–18 CrossRef CAS.
- Y. Ando, Y. Kawamura, T. Ikeshoji and M. Otani, Chem. Phys. Lett., 2014, 612, 240–244 CrossRef CAS.
- A. Bouzid and A. Pasquarello, J. Chem. Theory Comput., 2017, 13, 1769–1777 CrossRef CAS PubMed.
- A. Bouzid, P. Gono and A. Pasquarello, J. Catal., 2019, 375, 135–139 CrossRef CAS.
- V. M. Fernandez-Alvarez and M. H. Eikerling, ACS Appl. Mater. Interfaces, 2019, 11, 43774–43780 CrossRef CAS PubMed.
- C. Zhan, M. R. Cerón, S. A. Hawks, M. Otani, B. C. Wood, T. A. Pham, M. Stadermann and P. G. Campbell, Nat. Commun., 2019, 10, 4858 CrossRef PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- J. A. Gauthier, C. F. Dickens, H. H. Heenen, S. Vijay, S. Ringe and K. Chan, J. Chem. Theory Comput., 2019, 15, 6895–6906 CrossRef CAS PubMed.
- H. H. Heenen, J. A. Gauthier, H. H. Kristoffersen, T. Ludwig and K. Chan, J. Chem. Phys., 2020, 152, 144703 CrossRef CAS PubMed.
- S. Sakong and A. Groß, J. Chem. Phys., 2018, 149, 084705 CrossRef PubMed.
- T. Cheng, L. Wang, B. V. Merinov and W. A. Goddard, J. Am. Chem. Soc., 2018, 140, 7787–7790 CrossRef CAS PubMed.
- H. H. Kristoffersen, K. Chan, T. Vegge and H. A. Hansen, Chem. Commun., 2020, 56, 427–430 RSC.
- L. Schimka, J. Harl, A. Stroppa, A. Grüneis, M. Marsman, F. Mittendorfer and G. Kresse, Nat. Mater., 2010, 9, 741–744 CrossRef CAS.
- T. L. Tan, L.-L. Wang, D. D. Johnson and K. Bai, J. Phys. Chem. C, 2013, 117, 22696–22704 CrossRef CAS.
- T. T. T. Hanh, Y. Takimoto and O. Sugino, Surf. Sci., 2014, 625, 104–111 CrossRef CAS.
- L. Yan, Y. Yamamoto, M. Shiga and O. Sugino, Phys. Rev. B, 2020, 101, 165414 CrossRef CAS.
- M. Nishijima, H. Okuyama, N. Takagi, T. Aruga and W. Brenig, Surf. Sci. Rep., 2005, 57, 113–156 CrossRef CAS.
- L. Yan, Y. Yamamoto and O. Sugino, Phys. Rev. B, 2020, 101, 165414 CrossRef CAS.
- W. Fang, J. O. Richardson, J. Chen, X.-Z. Li and A. Michaelides, Phys. Rev. Lett., 2017, 119, 126001 CrossRef PubMed.
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC.
- Y. Yamamoto, S. Kasamatsu and O. Sugino, J. Phys. Chem. C, 2019, 123, 19486–19492 CrossRef CAS.
- A. R. Zeradjanin, J.-P. Grote, G. Polymeros and K. J. J. Mayrhofer, Electroanalysis, 2016, 28, 2256–2269 CrossRef CAS.
- S. Ringe, H. Oberhofer and K. Reuter, J. Chem. Phys., 2017, 146, 134103 CrossRef PubMed.
- S. Kasamatsu and O. Sugino, J. Phys.: Condens. Matter, 2019, 31, 085901 CrossRef PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- G. A. Somorjai and Y. Li, Introduction to surface chemistry and catalysis, John Wiley & Sons, 2010 Search PubMed.
- H.-J. Freund, H. Kuhlenbeck and V. Staemmler, Rep. Prog. Phys., 1996, 59, 283–347 CrossRef CAS.
- H.-J. Freund and G. Pacchioni, Chem. Soc. Rev., 2008, 37, 2224–2242 RSC.
- S. Schauermann, N. Nilius, S. Shaikhutdinov and H.-J. Freund, Acc. Chem. Res., 2013, 46, 1673–1681 CrossRef CAS PubMed.
- G. Pacchioni and H. Freund, Chem. Rev., 2013, 113, 4035–4072 CrossRef CAS PubMed.
- W. Ostwald, Über Katalyse, Akademische Verlagsgesellschaft, Leipzig, 1923 Search PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC.
- R. R. Adžić and A. R. Despić, J. Chem. Phys., 1974, 61, 3482–3483 CrossRef.
- R. R. Adžić and A. R. Despić, Z. Phys. Chem., 1975, 98, 95 CrossRef.
- R. R. Adžić, D. N. Simić, A. R. Despić and D. M. Dražić, J. Electroanal. Chem. Interfacial Electrochem., 1975, 65, 587–601 CrossRef.
- R. R. Adžić, A. V. Tripković and W. E. O'Grady, Nature, 1982, 296, 137–138 CrossRef.
- R. R. Adžić, A. V. Tripković and N. M. Marković, J. Electroanal. Chem. Interfacial Electrochem., 1983, 150, 79–88 CrossRef.
- N. A. Anastasijević, V. Vesović and R. R. Adžić, J. Electroanal. Chem. Interfacial Electrochem., 1987, 229, 305–316 CrossRef.
- M. Baldauf and D. M. Kolb, J. Phys. Chem., 1996, 100, 11375–11381 CrossRef CAS.
- L. A. Kibler, A. M. El-Aziz, R. Hoyer and D. M. Kolb, Angew. Chem., Int. Ed., 2005, 44, 2080–2084 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- R. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082–5104 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, F. J. Perez-Alonso, F. Calle-Vallejo, L. Bech, T. P. Johansson, A. K. Jepsen, R. Frydendal, B. P. Knudsen, J. Rossmeisl and I. Chorkendorff, J. Am. Chem. Soc., 2011, 133, 5485–5491 CrossRef CAS PubMed.
- W. Yang, T.-P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Grønbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 CrossRef CAS PubMed.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- P. Chen, T.-Y. Xiao, Y.-H. Qian, S.-S. Li and S.-H. Yu, Adv. Mater., 2013, 25, 3192–3196 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- T. Fukushima, W. Drisdell, J. Yano and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 10926–10929 CrossRef CAS PubMed.
- S. J. Yang, M. Antonietti and N. Fechler, J. Am. Chem. Soc., 2015, 137, 8269–8273 CrossRef CAS PubMed.
- M.-R. Gao, J.-X. Liang, Y.-R. Zheng, Y.-F. Xu, J. Jiang, Q. Gao, J. Li and S.-H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- N. D. Ricke, A. T. Murray, J. J. Shepherd, M. G. Welborn, T. Fukushima, T. Van Voorhis and Y. Surendranath, ACS Catal., 2017, 7, 7680–7687 CrossRef CAS.
- M. N. Jackson, S. Oh, C. J. Kaminsky, S. B. Chu, G. Zhang, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2018, 140, 1004–1010 CrossRef CAS PubMed.
- R. Wu, B. Xiao, Q. Gao, Y.-R. Zheng, X.-S. Zheng, J.-F. Zhu, M.-R. Gao and S.-H. Yu, Angew. Chem., Int. Ed., 2018, 57, 15445–15449 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 8921–8926 CrossRef CAS PubMed.
- Y. Xiong, Y. Yang, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2018, 140, 7248–7255 CrossRef CAS PubMed.
- Y. Duan, Z.-Y. Yu, S.-J. Hu, X.-S. Zheng, C.-T. Zhang, H.-H. Ding, B.-C. Hu, Q.-Q. Fu, Z.-L. Yu, X. Zheng, J.-F. Zhu, M.-R. Gao and S.-H. Yu, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed.
- S. Li, C. Cheng, A. Sagaltchik, P. Pachfule, C. Zhao and A. Thomas, Adv. Funct. Mater., 2019, 29, 1807419 CrossRef.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS.
- H.-H. Li and S.-H. Yu, Adv. Mater., 2019, 31, 1803503 CrossRef.
- Y. Lin, H. Wan, D. Wu, G. Chen, N. Zhang, X. Liu, J. Li, Y. Cao, G. Qiu and R. Ma, J. Am. Chem. Soc., 2020, 142, 7317–7321 CrossRef CAS.
- P. Xiong, B. Sun, N. Sakai, R. Ma, T. Sasaki, S. Wang, J. Zhang and G. Wang, Adv. Mater., 2020, 32, 1902654 CrossRef CAS PubMed.
- M.-Y. Ye, S. Li, X. Zhao, N. V. Tarakina, C. Teutloff, W. Y. Chow, R. Bittl and A. Thomas, Adv. Mater., 2020, 32, 1903942 CrossRef CAS.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- N. M. Marković, H. A. Gasteiger, B. N. Grgur and P. N. Ross, J. Electroanal. Chem., 1999, 467, 157–163 CrossRef.
- D. Strmcnik, D. F. van der Vliet, K. C. Chang, V. Komanicky, K. Kodama, H. You, V. R. Stamenkovic and N. M. Marković, J. Phys. Chem. Lett., 2011, 2, 2733–2736 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- P. P. Lopes, D. Strmcnik, J. S. Jirkovsky, J. G. Connell, V. Stamenkovic and N. Markovic, Catal. Today, 2016, 262, 41–47 CrossRef CAS.
- J. Knossalla, P. Paciok, D. Göhl, D. Jalalpoor, E. Pizzutilo, A. M. Mingers, M. Heggen, R. E. Dunin-Borkowski, K. J. J. Mayrhofer, F. Schüth and M. Ledendecker, J. Am. Chem. Soc., 2018, 140, 15684–15689 CrossRef CAS PubMed.
- D. Göhl, A. Garg, P. Paciok, K. J. J. Mayrhofer, M. Heggen, Y. Shao-Horn, R. E. Dunin-Borkowski, Y. Román-Leshkov and M. Ledendecker, Nat. Mater., 2020, 19, 287–291 CrossRef PubMed.
- M. Uchida, Y. Aoyama, N. Eda and A. Ohta, J. Electrochem. Soc., 1995, 142, 4143 CrossRef CAS.
- M. Uchida, Y. Fukuoka, Y. Sugawara, H. Ohara and A. Ohta, J. Electrochem. Soc., 1998, 145, 3708 CrossRef CAS.
- M. Lee, M. Uchida, D. A. Tryk, H. Uchida and M. Watanabe, Electrochim. Acta, 2011, 56, 4783–4790 CrossRef CAS.
- K. Shinozaki, H. Yamada and Y. Morimoto, J. Electrochem. Soc., 2011, 158, B467 CrossRef CAS.
- M. Uchida, Y.-C. Park, K. Kakinuma, H. Yano, D. A. Tryk, T. Kamino, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2013, 15, 11236–11247 RSC.
- Y.-C. Park, K. Kakinuma, M. Uchida, H. Uchida and M. Watanabe, Electrochim. Acta, 2014, 123, 84–92 CrossRef CAS.
- K. Kodama, R. Jinnouchi, A. Shinohara and Y. Morimoto, R&D Rev. Toyota CRDL, 2018, 49, 1–11 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. Adzic, in Electrocatalysis, ed. J. Lipkowski and P. N. Ross, Wiley-VCH, USA, 1998, vol. 197, pp. 197–242 Search PubMed.
- B. E. Conway, in Interfacial Electrochemistry: Theory, Experiment, and Applications, ed. A. Wieckowski, Marcel Dekker, USA, 1999, ch. 9, pp. 131–150 Search PubMed.
- N. M. Marković, R. R. Adžić, B. D. Cahan and E. B. Yeager, J. Electroanal. Chem., 1994, 377, 249–259 CrossRef.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1995, 99, 3411–3415 CrossRef CAS.
- N. M. Marković, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715–6721 CrossRef.
- N. M. Markovića, S. T. Sarraf, H. A. Gasteiger and P. N. Ross, J. Chem. Soc., Faraday Trans., 1996, 92, 3719–3725 RSC.
- N. M. Marković, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1997, 101, 5405–5413 CrossRef.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- K. Ojha, N. Arulmozhi, D. Aranzales and M. T. M. Koper, Angew. Chem., Int. Ed., 2020, 59, 711–715 CrossRef CAS PubMed.
- Y. Fang, S. Y. Ding, M. Zhang, S. N. Steinmann, R. Hu, B. W. Mao, J. M. Feliu and Z. Q. Tian, J. Am. Chem. Soc., 2020, 142, 9439–9446 CrossRef CAS PubMed.
- B. M. Ocko, J. Wang, A. Davenport and H. Isaacs, Phys. Rev. Lett., 1990, 65, 1466–1469 CrossRef CAS.
- J. Wang, B. M. Ocko, A. J. Davenport and H. S. Isaacs, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 10321–10338 CrossRef CAS PubMed.
- J. Lipkowski, L. Stolberg, D. F. Yang, B. Pettinger, S. Mirwald, F. Henglein and D. M. Kolb, Electrochim. Acta, 1994, 39, 1045–1056 CrossRef CAS.
- K.-i. Ataka, T. Yotsuyanagi and M. Osawa, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- K.-i. Ataka and M. Osawa, Langmuir, 1998, 14, 951–959 CrossRef CAS.
- O. M. Magnussen, B. M. Ocko, M. J. Regan, K. Penanen, P. S. Pershan and M. Deutsch, Phys. Rev. Lett., 1995, 74, 4444–4447 CrossRef CAS PubMed.
- M. J. Regan, E. H. Kawamoto, S. Lee, P. S. Pershan, N. Maskil, M. Deutsch, O. M. Magnussen, B. M. Ocko and L. E. Berman, Phys. Rev. Lett., 1995, 75, 2498–2501 CrossRef CAS.
- M. R. Vogt, A. Lachenwitzer, O. M. Magnussen and R. J. Behm, Surf. Sci., 1998, 399, 49–69 CrossRef CAS.
- I. Burgess, C. A. Jeffrey, X. Cai, G. Szymanski, Z. Galus and J. Lipkowski, Langmuir, 1999, 15, 2607–2616 CrossRef CAS.
- A. Chen and J. Lipkowski, J. Phys. Chem. B, 1999, 103, 682–691 CrossRef CAS.
- O. M. Magnussen, J. Scherer, B. M. Ocko and R. J. Behm, J. Phys. Chem. B, 2000, 104, 1222–1226 CrossRef CAS.
- O. M. Magnussen, L. Zitzler, B. Gleich, M. R. Vogt and R. J. Behm, Electrochim. Acta, 2001, 46, 3725–3733 CrossRef CAS.
- O. M. Magnussen, Chem. Rev., 2002, 102, 679–726 CrossRef CAS.
- M. Nakamura, O. Endo, T. Ohta, M. Ito and Y. Yoda, Surf. Sci., 2002, 514, 227–233 CrossRef CAS.
- Z.-Q. Tian and B. Ren, Annu. Rev. Phys. Chem., 2004, 55, 197–229 CrossRef CAS.
- T. Tansel and O. M. Magnussen, Phys. Rev. Lett., 2006, 96, 026101 CrossRef CAS PubMed.
- D.-Y. Wu, J.-F. Li, B. Ren and Z.-Q. Tian, Chem. Soc. Rev., 2008, 37, 1025–1041 RSC.
- B. J. Trześniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. M. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef PubMed.
- M. C. Figueiredo, I. Ledezma-Yanez and M. T. M. Koper, ACS Catal., 2016, 6, 2382–2392 CrossRef CAS.
- J. H. K. Pfisterer, Y. Liang, O. Schneider and A. S. Bandarenka, Nature, 2017, 549, 74–77 CrossRef CAS PubMed.
- K. Wonner, M. V. Evers and K. Tschulik, J. Am. Chem. Soc., 2018, 140, 12658–12661 CrossRef CAS PubMed.
- Z. Jusys and R. J. Behm, ChemPhysChem, 2019, 20, 3276–3288 CrossRef CAS PubMed.
- Y. Yang, Y. Wang, Y. Xiong, X. Huang, L. Shen, R. Huang, H. Wang, J. P. Pastore, S.-H. Yu, L. Xiao, J. D. Brock, L. Zhuang and H. D. Abruña, J. Am. Chem. Soc., 2019, 141, 1463–1466 CrossRef CAS PubMed.
- M. V. Evers, M. Bernal, B. Roldan Cuenya and K. Tschulik, Angew. Chem., Int. Ed., 2019, 58, 8221–8225 CrossRef CAS PubMed.
- J.-C. Dong, M. Su, V. Briega-Martos, L. Li, J.-B. Le, P. Radjenovic, X.-S. Zhou, J. M. Feliu, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2020, 142, 715–719 CrossRef CAS PubMed.
- Z. Su, M. Karaskiewicz, J. Rogalski, R. Bilewicz and J. Lipkowski, J. Electroanal. Chem., 2020, 113820, DOI:10.1016/j.jelechem.2020.113820.
- J. Klein, V. Chesnyak, M. Löw, M. Schilling, A. K. Engstfeld and R. J. Behm, J. Am. Chem. Soc., 2020, 142, 1278–1286 CrossRef CAS PubMed.
- F. Buchner, M. Eckardt, T. Bohler, J. Kim, J. Gerlach, J. Schnaidt and R. J. Behm, ChemSusChem, 2020, 13, 3199–3211 CrossRef CAS PubMed.
- A. B. Anderson and T. V. Albu, Electrochem. Commun., 1999, 1, 203–206 CrossRef CAS.
- A. B. Anderson and T. V. Albu, J. Am. Chem. Soc., 1999, 121, 11855–11863 CrossRef CAS.
- A. B. Anderson, N. M. Neshev, R. A. Sidik and P. Shiller, Electrochim. Acta, 2002, 47, 2999–3008 CrossRef CAS.
- A. B. Anderson, Curr. Opin. Electrochem., 2017, 1, 27–33 CrossRef CAS.
- M. Andersen, S. V. Levchenko, M. Scheffler and K. Reuter, ACS Catal., 2019, 9, 2752–2759 CrossRef CAS.
- F. Tian, R. Jinnouchi and A. B. Anderson, J. Phys. Chem. C, 2009, 113, 17484–17492 CrossRef CAS.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS.
- A. B. Anderson, J. Uddin and R. Jinnouchi, J. Phys. Chem. C, 2010, 114, 14946–14952 CrossRef CAS.
- A. B. Anderson, R. Jinnouchi and J. Uddin, J. Phys. Chem. C, 2013, 117, 41–48 CrossRef CAS.
- R. Jinnouchi, K. Kodama, T. Hatanaka and Y. Morimoto, Phys. Chem. Chem. Phys., 2011, 13, 21070–21083 RSC.
- A. B. Anderson, Phys. Chem. Chem. Phys., 2012, 14, 1330–1338 RSC.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS.
- Y. Takahashi, A. Kumatani, H. Shiku and T. Matsue, Anal. Chem., 2017, 89, 342–357 CrossRef CAS PubMed.
- Y. Takagi, T. Uruga, M. Tada, Y. Iwasawa and T. Yokoyama, Acc. Chem. Res., 2018, 51, 719–727 CrossRef CAS PubMed.
- L. Yu, Y. Takagi, T. Nakamura, T. Sakata, T. Uruga, M. Tada, Y. Iwasawa, S. Masaoka and T. Yokoyama, J. Phys. Chem. C, 2019, 123, 603–611 CrossRef CAS.
- Y. Takahashi, Y. Kobayashi, Z. Wang, Y. Ito, M. Ota, H. Ida, A. Kumatani, K. Miyazawa, T. Fujita, H. Shiku, Y. E. Korchev, Y. Miyata, T. Fukuma, M. Chen and T. Matsue, Angew. Chem., Int. Ed., 2020, 59, 3601–3608 CrossRef CAS PubMed.
- H. Kiuchi, H. Niwa, M. Kobayashi, Y. Harada, M. Oshima, M. Chokai, Y. Nabae, S. Kuroki, M.-A. Kakimoto, T. Ikeda, K. Terakura and S. Miyata, Electrochim. Acta, 2012, 82, 291–295 CrossRef CAS.
- M. Pumera and H. Iwai, J. Phys. Chem. C, 2009, 113, 4401–4405 CrossRef CAS.
- A. Ambrosi, S. Y. Chee, B. Khezri, R. D. Webster, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2012, 51, 500–503 CrossRef CAS PubMed.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
- L. Wang, Z. Sofer and M. Pumera, ACS Nano, 2020, 14, 21–25 CrossRef CAS PubMed.
- E. Yeager, P. Krouse and K. V. Rao, Electrochim. Acta, 1964, 9, 1057–1070 CrossRef CAS.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527–1537 CrossRef CAS.
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- Y. Harada, J. Miyawaki, H. Niwa, K. Yamazoe, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 5487–5491 CrossRef CAS PubMed.
- K. H. Kim, H. Pathak, A. Späh, F. Perakis, D. Mariedahl, J. A. Sellberg, T. Katayama, Y. Harada, H. Ogasawara, L. G. M. Pettersson and A. Nilsson, Phys. Rev. Lett., 2017, 119, 075502 CrossRef PubMed.
- A. Nilsson, J. LaRue, H. Öberg, H. Ogasawara, M. Dell'Angela, M. Beye, H. Öström, J. Gladh, J. K. Nørskov, W. Wurth, F. Abild-Pedersen and L. G. M. Pettersson, Chem. Phys. Lett., 2017, 675, 145–173 CrossRef CAS.
- C. W. Nicholson, A. Lücke, W. G. Schmidt, M. Puppin, L. Rettig, R. Ernstorfer and M. Wolf, Science, 2018, 362, 821–825 CrossRef CAS PubMed.
- H. Ali-Löytty, M. Hannula, M. Valden, A. Eilert, H. Ogasawara and A. Nilsson, J. Phys. Chem. C, 2019, 123, 25128–25134 CrossRef.
- D. Friebel, H. Ogasawara and A. Nilsson, in Surface and Interface Science, ed. K. Wandelt, John Wiley and Sons, Weinheim, 2020, ch. 58, pp. 733–772 DOI:10.1002/9783527680603.ch58.
- M. Ahmed, K. I. Inoue, S. Nihonyanagi and T. Tahara, Angew. Chem., Int. Ed., 2020, 59, 9498–9505 CrossRef CAS PubMed.
- S. Kawai, T. Glatzel, S. Koch, B. Such, A. Baratoff and E. Meyer, Phys. Rev. Lett., 2009, 103, 220801 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed.
- H. Imada, K. Miwa, M. Imai-Imada, S. Kawahara, K. Kimura and Y. Kim, Nature, 2016, 538, 364–367 CrossRef CAS PubMed.
- E. Kazuma, J. Jung, H. Ueba, M. Trenary and Y. Kim, Science, 2018, 360, 521–526 CrossRef CAS PubMed.
- R. Evans, Adv. Phys., 1979, 28, 143–200 CrossRef CAS.
- R. Evans, in Fundamentals of Inhomogeneous Fluids, ed. D. Henderson, Dekker, New York, 1992, ch. 3, pp. 85–175 Search PubMed.
- H. Park, N. Kumar, M. Melander, T. Vegge, J. M. Garcia Lastra and D. J. Siegel, Chem. Mater., 2018, 30, 915–928 CrossRef CAS.
- T. Engel and G. Ertl, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weez, Academic Press, 1979, vol. 28, pp. 1–78 Search PubMed.
- M. Eiswirth and G. Ertl, Surf. Sci., 1986, 177, 90–100 CrossRef CAS.
- G. Ertl, Adv. Catal., 1990, 37, 213–277 CAS.
- K. Krischer, M. Eiswirth and G. Ertl, J. Chem. Phys., 1992, 96, 9161–9172 CrossRef CAS.
- R. Imbihl and G. Ertl, Chem. Rev., 1995, 95, 697–733 CrossRef CAS.
- M. Kim, M. Bertram, M. Pollmann, A. V. Oertzen, A. S. Mikhailov, H. H. Rotermund and G. Ertl, Science, 2001, 292, 1357–1360 CrossRef CAS PubMed.
- J. Clavilier, R. Faure, G. Guinet and R. Durand, J. Electroanal. Chem. Interfacial Electrochem., 1980, 107, 205–209 CrossRef CAS.
- J. Clavilier, J. Electroanal. Chem. Interfacial Electrochem., 1980, 107, 211–216 CrossRef CAS.
- J. Clavilier, D. Armand, S. G. Sun and M. Petit, J. Electroanal. Chem. Interfacial Electrochem., 1986, 205, 267–277 CrossRef CAS.
- J. Clavilier, R. Albalat, R. Gomez, J. M. Orts, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1992, 330, 489–497 CrossRef CAS.
- J. Hotlos, O. M. Magnussen and R. J. Behm, Surf. Sci., 1995, 335, 129–144 CrossRef CAS.
- F. A. Möller, O. M. Magnussen and R. J. Behm, Phys. Rev. Lett., 1996, 77, 5249–5252 CrossRef PubMed.
- F. A. Möller, O. M. Magnussen and R. J. Behm, Phys. Rev. Lett., 1996, 77, 3165–3168 CrossRef PubMed.
- F. A. Möller, J. Kintrup, A. Lachenwitzer, O. M. Magnussen and R. J. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 12506–12518 CrossRef.
- M. T. M. Koper and R. A. van Santen, J. Electroanal. Chem., 1999, 476, 64–70 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, E. Herrero, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2000, 487, 37–44 CrossRef CAS.
- E. Herrero, L. J. Buller and H. D. Abruña, Chem. Rev., 2001, 101, 1897–1930 CrossRef CAS PubMed.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 12938–12947 CrossRef CAS.
- M. D. Maciá, J. M. Campiña, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2004, 564, 141–150 CrossRef.
- T. E. Shubina, C. Hartnig and M. T. M. Koper, Phys. Chem. Chem. Phys., 2004, 6, 4215–4221 RSC.
- A. Kuzume, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2007, 599, 333–343 CrossRef CAS.
- J. Solla-Gullón, P. Rodríguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359–1373 RSC.
- S. C. S. Lai and M. T. M. Koper, Phys. Chem. Chem. Phys., 2009, 11, 10446–10456 RSC.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- E. Herrero and J. M. Feliu, Curr. Opin. Electrochem., 2018, 9, 145–150 CrossRef CAS.
- L. Jacobse, Y.-F. Huang, M. T. M. Koper and M. J. Rost, Nat. Mater., 2018, 17, 277–282 CrossRef CAS PubMed.
- V. Briega-Martos, E. Herrero and J. M. Feliu, Curr. Opin. Electrochem., 2019, 17, 97–105 CrossRef CAS.
- R. Martínez-Hincapié, V. Climent and J. M. Feliu, Curr. Opin. Electrochem., 2019, 14, 16–22 CrossRef.
- V. Climent and J. Feliu, Annu. Rev. Anal. Chem., 2020, 13, 201–222 CrossRef CAS PubMed.
- F. Calle-Vallejo, M. T. M. Koper and A. S. Bandarenka, Chem. Soc. Rev., 2013, 42, 5210–5230 RSC.
- A. S. Bandarenka, A. S. Varela, M. Karamad, F. Calle-Vallejo, L. Bech, F. J. Perez-Alonso, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 11845–11848 CrossRef CAS PubMed.
- V. Climent and J. M. Feliu, J. Solid State Electrochem., 2011, 15, 1297 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. Hammes-Schiffer, Faraday Discuss., 2020, 221, 582–588 RSC.
- S. C. Althorpe, W. Barford, J. Blumberger, C. Bungey, I. Burghardt, A. Datta, S. Ghosh, S. Giannini, T. Grünbaum, S. Habershon, S. Hammes-Schiffer, S. Hay, S. Iyengar, G. Jones, A. Kelly, K. Komarova, J. Lawrence, Y. Litman, J. Mannouch, D. Manolopoulos, C. Martens, R. J. Maurer, M. Melander, M. Rossi, K. Sakaushi, M. Saller, A. Schile, S. Sturniolo, G. Trenins and G. Worth, Faraday Discuss., 2020, 221, 564–581 RSC.
- A. M. Alvertis, W. Barford, S. Bourne Worster, I. Burghardt, A. Chin, A. Datta, A. Dijkstra, T. Fay, H. Fielding, T. Grünbaum, S. Habershon, S. Hammes-Schiffer, S. Iyengar, A. R. Jones, K. Komarova, J. Léonard, Y. Litman, D. Picconi, D. Plant, A. Schile, G. D. Scholes, J. Segarra-Martí, F. Segatta, A. Troisi and G. Worth, Faraday Discuss., 2020, 221, 322–349 RSC.
- A. M. Alvertis, W. Barford, S. Bourne Worster, I. Burghardt, A. Datta, A. Dijkstra, T. Fay, S. Ghosh, T. Grünbaum, S. Habershon, P. J. Hore, D. Hutchinson, S. Iyengar, A. R. Jones, G. Jones, K. Komarova, J. Lawrence, J. Léonard, Y. Litman, J. Mannouch, D. Manolopoulos, C. Martens, M. Mondelo-Martell, D. Picconi, D. Plant, K. Sakaushi, M. A. C. Saller, A. Schile, G. D. Scholes, J. Segarra-Martí, F. Segatta, A. Troisi and G. Worth, Faraday Discuss., 2020, 221, 168–201 RSC.
- S. C. Althorpe, A. M. Alvertis, W. Barford, R. L. Benson, I. Burghardt, S. Giannini, S. Habershon, S. Hammes-Schiffer, S. Hay, S. Iyengar, A. Kelly, K. Komarova, J. Lawrence, Y. Litman, C. Martens, R. J. Maurer, D. Plant, M. Rossi, K. Sakaushi, A. Schile, S. Sturniolo, D. P. Tew, G. Trenins and G. Worth, Faraday Discuss., 2020, 221, 478–500 RSC.
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- Z. He, S. D. Minteer and L. T. Angenent, Environ. Sci. Technol., 2005, 39, 5262–5267 CrossRef CAS PubMed.
- D. R. Lovley and K. P. Nevin, Curr. Opin. Biotechnol., 2011, 22, 441–448 CrossRef CAS PubMed.
- K. Dolch, J. Danzer, T. Kabbeck, B. Bierer, J. Erben, A. H. Förster, J. Maisch, P. Nick, S. Kerzenmacher and J. Gescher, Bioresour. Technol., 2014, 157, 284–292 CrossRef CAS PubMed.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS.
- K. K. Sakimoto, N. Kornienko and P. Yang, Acc. Chem. Res., 2017, 50, 476–481 CrossRef CAS PubMed.
- Z. Ji, H. Zhang, H. Liu, O. M. Yaghi and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10582–10587 CrossRef CAS PubMed.
- K. K. Sakimoto, N. Kornienko, S. Cestellos-Blanco, J. Lim, C. Liu and P. Yang, J. Am. Chem. Soc., 2018, 140, 1978–1985 CrossRef CAS PubMed.
- N. Heidary, N. Kornienko, S. Kalathil, X. Fang, K. H. Ly, H. F. Greer and E. Reisner, J. Am. Chem. Soc., 2020, 142, 5194–5203 CrossRef CAS PubMed.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem., 2020, 4, 6–21 CrossRef CAS.
- E. M. Gaffney, M. Grattieri, Z. Rhodes and S. D. Minteer, J. Electrochem. Soc., 2020, 167, 065502 CrossRef.
- D. M. Kolb and J. Schneider, Electrochim. Acta, 1986, 31, 929–936 CrossRef CAS.
- D. M. Kolb, Prog. Surf. Sci., 1996, 51, 109–173 CrossRef CAS.
| This journal is © the Owner Societies 2020 |