
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO adsorption on the calcite(10.4) surface: a combined experimental and theoretical study
Tahereh Mohammadi
Hafshejani†
a,
Weijia
Wang†
a,
Jonas
Heggemann
b,
Alexei
Nefedov
 a,
Stefan
Heissler
a,
Yuemin
Wang
a,
Philipp
Rahe
b,
Peter
Thissen
a and
Christof
Wöll
*a
a,
Stefan
Heissler
a,
Yuemin
Wang
a,
Philipp
Rahe
b,
Peter
Thissen
a and
Christof
Wöll
*a
aKarlsruher Institut für Technologie (KIT), Institut für Funktionelle Grenzflächen (IFG), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: christof.woell@kit.edu
bFachbereich Physik, Universität Osnabrück, Barbarastrasse 7, 49076 Osnabrück, Germany
First published on 30th June 2020
Abstract
Detailed information on structural, chemical, and physical properties of natural cleaved (10.4) calcite surfaces was obtained by a combined atomic force microscopy (AFM) and infrared (IR) study using CO as a probe molecule under ultrahigh vacuum (UHV) conditions. The structural quality of the surfaces was determined using non-contact AFM (NC-AFM), which also allowed assigning the adsorption site of CO molecules. Vibrational frequencies of adsorbed CO species were determined by polarization-resolved infrared reflection absorption spectroscopy (IRRAS). At low exposures, adsorption of CO on the freshly cleaved (10.4) calcite surface at a temperature of 62 K led to the occurrence of a single C–O vibrational band located at 2175.8 cm−1, blue-shifted with respect to the gas phase value. For larger exposures, a slight, coverage-induced redshift was observed, leading to a frequency of 2173.4 cm−1 for a full monolayer. The width of the vibrational bands is extremely small, providing strong evidence that the cleaved calcite surface is well-defined with only one CO adsorption site. A quantitative analysis of the IRRA spectra recorded at different surface temperatures revealed a CO binding energy of −0.31 eV. NC-AFM data acquired at 5 K for sub-monolayer CO coverage reveal single molecules imaged as depressions at the position of the protruding surface features, in agreement with the IRRAS results. Since there are no previous experimental data of this type, the interpretation of the results was aided by employing density functional theory calculations to determine adsorption geometries, binding energies, and vibrational frequencies of carbon monoxide on the (10.4) calcite surface. It was found that the preferred geometry of CO on this surface is adsorption on top of calcium in a slightly tilted orientation. With increased coverage, the binding energy shows a small decrease, revealing the presence of repulsive adsorbate–adsorbate interactions.
Introduction
Calcium carbonate (CaCO3) is a widely occurring mineral in nature, constituting a substantial part of the Earth's crust.1 The most stable polymorph of CaCO3 is calcite, which is a common component of all-natural rock types.2 This ionic compound is also abounding in living organisms, is incorporated into bones, teeth, as well as in shells of mussels and plays an important role in biomineralization.2,3 Calcite has been used as construction material, for agricultural soil treatment, as sorbent, for pharmaceutical applications,4 and has been integrated in optical devices utilizing its birefringence property.5 As the largest carbon dioxide repository, this carbonate also played a significant role for the composition of the planet's atmosphere.6 Knowledge about calcite surfaces, their detailed structure, and chemical properties are thus highly relevant.Calcite is an ionic crystal consisting of calcium cations (Ca2+) and carbonate (CO32−) anions, which are held together through ionic bonding.7 The structure is trigonal (rhombohedral holohedral ![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 2/m) with a Rc space group.7c Cleavage is easiest for the thermodynamically most stable calcite(10.4) surface with a surface energy calculated around 0.59 J m−2.8 The calcite(10.4) surface is furthermore nonpolar, namely a type I surface according to Tasker's classification, and thus bears zero charge (q = 0) and zero dipole moment (μ = 0).9 Accordingly, most previous surface studies reported for this material focused on this surface. The calcite(10.4) surface (a structural model is reproduced in Fig. 1) contains a total of two calcium atoms and two carbonate groups within the (1 × 1) unit cell (with a unit cell size of 5.0 × 8.1 Å2), both are present in the top layer. Note that the two carbonate groups are rotated with respect to each other and, as a consequence, the upmost oxygen atom points either to the left or to the right along the [42
2/m) with a Rc space group.7c Cleavage is easiest for the thermodynamically most stable calcite(10.4) surface with a surface energy calculated around 0.59 J m−2.8 The calcite(10.4) surface is furthermore nonpolar, namely a type I surface according to Tasker's classification, and thus bears zero charge (q = 0) and zero dipole moment (μ = 0).9 Accordingly, most previous surface studies reported for this material focused on this surface. The calcite(10.4) surface (a structural model is reproduced in Fig. 1) contains a total of two calcium atoms and two carbonate groups within the (1 × 1) unit cell (with a unit cell size of 5.0 × 8.1 Å2), both are present in the top layer. Note that the two carbonate groups are rotated with respect to each other and, as a consequence, the upmost oxygen atom points either to the left or to the right along the [42![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] direction.7b,10
] direction.7b,10
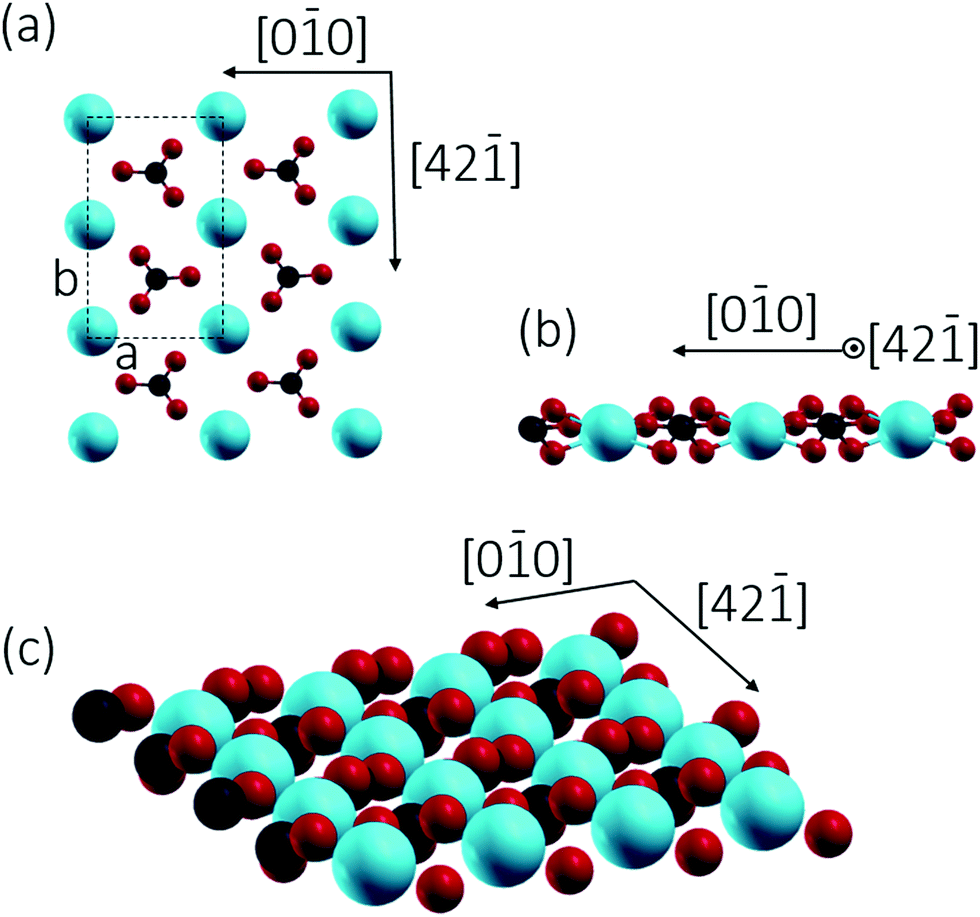 | ||
| Fig. 1 Top (a) and side (b) views of the (1 × 1) calcite(10.4) surface model (a = 5.0 Å and b = 8.1 Å). Bottom image (c) shows the atoms exposed at the surface, revealing that the oxygens are located at two different but equivalent positions. (red: oxygen, black: carbon, blue: calcium). | ||
The microscopic nature of the calcite(10.4) surface has intensively been studied by various experimental10,11 and theoretical methods.12 Two deviations from the bulk-truncated (1 × 1) structure have been reported with their origins and geometry still being under discussion. First, a loss of the pg surface symmetry (also referred to as “row-pairing”) has been observed within atomic force microscopy (AFM) data,10,11,13 yet, this surface relaxation can also be explained by an asymmetric tip probing an unrelaxed surface. Second, the existence of a (2 × 1) reconstruction has been proposed on the basis of low energy electron diffraction (LEED)11d and AFM data.11b,14 This reconstruction is characterised by a rectangular supercell with dimensions of 10.0 Å and 8.1 Å oriented along the [00] and [42] directions, respectively.
Here, the surface structure of the UHV-cleaved calcite(10.4) surface is investigated by polarization-resolved infrared reflection absorption spectroscopy (IRRAS) using the surface-ligand IR (SLIR) spectroscopy approach,15 with CO as the probe molecule, and complemented by non-contact atomic force microscopy (NC-AFM) experiments and density functional theory (DFT) calculations. We would like to point out that there are no previous IRRAS studies of adsorbates on well-defined surfaces of macroscopic calcite single crystals available. This lack of experimental data is due to technical problems in recording IR-data using reflection geometries for dielectric substrates, which could only recently be overcome.15,16 The IRRAS results of CO on calcite(10.4) surfaces obtained at low temperature show only one sharp absorption band, providing strong evidence that this surface exhibits only one type of Ca2+ binding site. To our knowledge, these are the first IRRAS data for any adsorbates bound to a calcite single crystal substrate.
Methods
Infrared reflection absorption spectroscopy
All IRRAS experiments were performed in a multi-technique UHV apparatus (Prevac, Poland). A more comprehensive description of the experimental setup can be found elsewhere.16 Large blocks of calcite(10.4) single crystal (8 × 8 × 12 mm3) with optical quality were purchased from Korth Kristalle GmbH (Altenholz, Germany). The blocks were cut into small pieces in atmosphere before attaching them to the sample holder. Cleavage of the calcite single crystal in the UHV chamber was carried out using a scalpel blade attached to a wobble stick. A photograph of the sample holder with a piece of a calcite single crystal is shown in Fig. 2. The central part of the base plate can be rotated in clockwise and anticlockwise directions using the wobble stick inside the UHV chamber, so that the azimuthal orientation of the specimen can be changed. After the single crystal was introduced into the UHV system, it was first briefly annealed before cleavage. After cleavage the samples were quickly transferred to the IRRAS chamber. The IRRAS data were acquired using polarized infrared light at a grazing incidence angle (80°) with a spectral resolution of 1 cm−1. The base pressure of the UHV chamber amounted to 4.0 × 10−11 mbar. Dosing of CO was performed using a capillary tube ending in front of the sample with the temperature of the sample held at 62 K. The temperature of the sample was monitored by a K-type thermocouple that was attached to the edge of the sample holder. An exposure of one Langmuir (L) corresponds to 1.33 × 10−6 mbar s. | ||
| Fig. 2 Photograph of a calcite (CaCO3) single crystal mounted in the sample holder (designed for UHV cleavage) with the (10.4) surface pointing up. Size of the base plate of the sample holder is 8 × 8 mm2. | ||
Non-contact atomic force microscopy (NC-AFM)
Sample preparation and NC-AFM experiments were performed under ultra-high vacuum conditions in a system separate from the IRRAS setup. Calcite crystals (Korth Kristalle, Altenholz, Germany) were cleaved in-situ to produce a (10.4) surface by using a scalpel blade attached to a wobble stick.17 The crystal was mounted on a double sample holder supporting an additional Ag(111) surface for tip preparation and was heated for 1.5 h to about 440 K to remove residual charges.18 NC-AFM experiments were performed with an LT qPlus gen.III microscope (ScientaOmicron, Taunusstein, Germany) operated by a MATRIX controller and using W-tips attached to qPlus sensors19 as supplied by the manufacturer. Tips were prepared by common scanning tunnelling microscopy strategies on the Ag(111) surface mounted nearby the calcite sample. CO was dosed directly into the scan head (Tsample < 10 K while opening the shields) at background pressures around p ∼ 5 × 10−8 mbar. Metal tips were partly terminated with CO molecules for improving the imaging contrast.20 The surface orientation has been determined ex-situ after the NC-AFM experiments by optically identifying the orientation of the ordinary and extraordinary ray of the birefringence.21Computational details
DFT calculations were performed using the generalized gradient approximation (GGA) and the PBE functional, as implemented in the Vienna ab initio simulation package (VASP).22 The electron–ion interaction was described within the projector-augmented wave (PAW) scheme.23 The electronic wave functions were expanded into plane waves up to a kinetic energy of 400 eV.The calculations reported here were carried out for a slab (thickness of 4 layers) exposing a (10.4) surface with a (1 × 1) unit cell and containing a total number of 40 atoms (Ca: 8, C: 8, O: 24), see also Fig. 1. This model is similar to that used in earlier studies.8b,24 A vacuum region of 20 Å was found to be large enough to avoid interactions between the surface and adsorbed molecules and their replica images. We define coverage as the ratio of the number of CO molecules to the number of surface Ca ions. All degrees of freedom, except the bottom layer, were allowed to relax until the forces on the atoms were below 1 meV Å−1. The optimization of the atomic coordinates (and unit cell size for the bulk materials) was performed through a conjugate gradient technique, which utilizes the total energy and the Hellmann–Feynman forces on the atoms (and stresses on the unit cell). The structures were considered to be fully relaxed when the forces on the atoms were smaller than 1 meV Å−1. The Brillouin zone integration was performed using a 4 × 4 × 1 mesh within the Monkhorst–Pack scheme.25 Vibrational eigenmodes and frequencies were calculated by the force-constant approach. The start geometry for the optimization was a CO molecule (bond length 1.14 Å) positioned on top of Ca2+ in an upright orientation.
The surface energy is defined as the difference in total energy between the bulk and the surface per unit area and, for a stoichiometric surface, can be obtained by the following equation:26
| Esurf = (Eslab − NEbulk)/2A |
The adsorption energy (Eads) can be calculated as a function of the coverage of the CO molecules:28
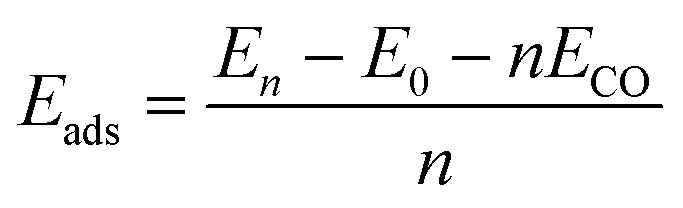
Results and discussion
The polarization-resolved IRRAS data shown in Fig. 3a were recorded for p-polarized IR light incident along the surface after varying exposures of the freshly cleaved calcite(10.4) surface to CO at a substrate temperature of 62 K. For an exposure of 0.05 L, an asymmetric single band located at 2175.8 cm−1 is observed, which is assigned to the stretching vibration of CO molecules bound with the C atom to Ca2+-cations at the calcite surface. When increasing the exposure to 0.1 L, the band increased in intensity and, at the same time, shifted slightly to lower frequency, 2174.4 cm−1. With increasing exposures, the lowering of frequency continued until a value of 2173.4 cm−1 was reached. For exposures higher than 2.0 L no further increases of the band intensity or changes of the band shape were observed, indicating that the calcite(10.4) surface was saturated with CO and no multilayer formation occurred at this substrate temperature.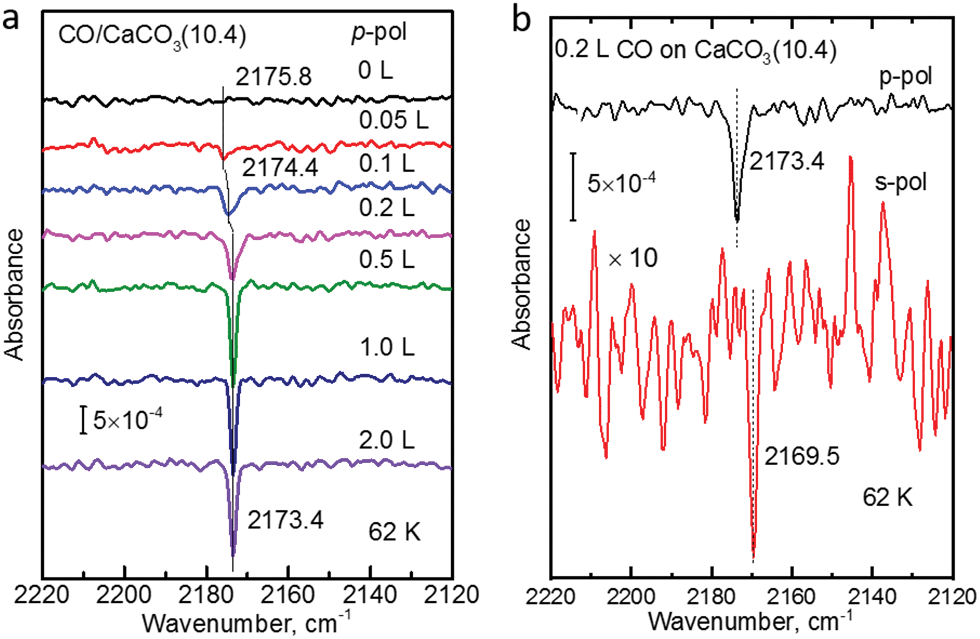 | ||
| Fig. 3 (a) IRRAS spectra with p-polarized light for CO adsorbed onto the calcite(10.4) surface at 62 K with increasing exposures. (b) IRRAS spectra with p-polarized light (top) and s-polarized light (bottom) for 0.2 L CO adsorbed onto the calcite(10.4) surface at 62 K. | ||
The CO vibration was also observed in IRRAS data recorded with s-polarized light (Fig. 3b, bottom). However, the intensity is much smaller, by an order of magnitude, compared to that excited by p-polarized light (Fig. 3b, top). The occurrence of a vibrational feature also for s-polarization reveals that there must be a static or dynamic tilt angle, because only vibrational modes with a component of their transition dipole moment (TDM) orientated parallel to the surface can be excited by s-polarized light.16
In order to determine the binding energy of the CO adlayers we recorded IRRAS-data after heating to different temperatures, see Fig. 4a. In Fig. 4b we present the variation of the integrated band intensities as a function of surface temperature. The position (Fig. 4a) and the intensity (Fig. 4b) of the CO–Ca2+ band stays constant for temperatures below 92 K. Upon further annealing to 100 K, CO starts to desorb from the surface, yielding a decrease in band intensity accompanied by a slight blue shift of the vibrational frequency. A simple quantitative analysis of the IR band intensity evolution, using the Redhead equation,29 yields a binding energy of −0.31 eV for CO on the calcite(10.4) surface.
 | ||
| Fig. 4 (a) IRRAS spectra for 2.0 L of CO adsorbed onto calcite(10.4) surfaces at T = 62 K and after annealing to indicated temperatures. (b) Integrated intensity evolution of the spectra as a function of temperature. | ||
Next, we recorded AFM micrographs to investigate the adsorption position and CO arrangement after dosing CO into the cold scan head on calcite(10.4) for three different total times of 25 s, 50 s, and 150 s, resulting in a coverage of about 0.006, 0.013 and 0.067 monolayers (ML), respectively. 1 ML is defined as a coverage of two CO molecules per (1 × 1) unit cell. Note that the pressure was slightly higher in the third experiment. The corresponding results are shown in Fig. 5. The calcite surface structure is in all images apparent in the form of four bright protrusions per (2 × 1) unit cell, having unit cell dimensions of 10 × 8.1 Å2 as indicated by white rectangles. The contrast in Fig. 5a, b and d represents a staggered (zig-zag) structure along the [42] direction, which can be related to the different orientation of the two carbonate groups within the surface unit cell. Additionally, the (2 × 1) reconstruction is visible as a modulation along the [010] direction of the dark rows in all micrographs. As no row-pairing reconstruction is apparent, the pg surface symmetry is maintained.
 | ||
| Fig. 5 Imaging single CO molecules on calcite(10.4) with NC-AFM at 5 K using total deposition times of (a) 25 s, (b and d) 50 s, and (c) 150 s. Data in (b and d) were acquired after dosing further 25 s onto the sample presented in (a). Data in (c) are acquired after dosing for 25 s and additional 125 s. Single CO molecules are marked by an arrow in all figures, molecules slightly off-center with white circles in (b). The (2 × 1) unit cell is marked by white rectangles in all micrographs. | ||
Individual dark depressions, which are not observed on pristine calcite(10.4) surfaces, are visible and identified as single CO molecules (examples are marked by white arrows in Fig. 5). These depressions are well separated from each other, but are often manipulated while scanning (see Fig. 5c). We did not observe the formation of clusters or adsorbate islands. As apparent from the detailed micrograph in Fig. 5d, the molecules are located at the position of calcite surface features imaged bright in NC-AFM. Despite the sharp single band measured with IRRAS, we also observe molecules positioned slightly off-center the bright calcite surface feature (examples are marked by white circles in Fig. 5b). However, the difference between these orientations might be thermally averaged at the IRRAS measurement temperature of 62 K, while the barrier is too high at the NC-AFM imaging temperature of 5 K. Based on the IRRAS results we can furthermore conclude that the bright protrusions in these NC-AFM micrographs are representing the locations of the surface Ca2+ cations as the CO molecules are located at these positions. Two possibilities including a “positive” or “negative potential nanotip” imaging model were suggested for NC-AFM measurements of calcite(10.4) surfaces before.30 The imaging was explained to be dominated by electrostatic interactions, where either the oxygen or calcium sublattices are imaged as “bright” due to attraction with a positive or negative ion located at the tip apex, respectively.30 Thus, we conclude that the data herein were acquired with a probing tip with a negative termination.
Finally, we studied two different CO/calcite configurations by DFT, namely (i) a single CO molecule adsorbed within the (1 × 1) cell, corresponding to a coverage of 0.5 ML, and (ii) two CO molecules adsorbed within the (1 × 1) cell, corresponding to a complete monolayer coverage (1.0 ML). In all cases, CO was adsorbed on-top of a surface Ca site and initially positioned perpendicular to the surface with the carbon pointing downwards. This geometry corresponds to the common coordination of CO on metals and metal oxides.31 Optimization with DFT yielded a tilt of 14.42° for 0.5 ML and tilts of 14.57° and 15.14° for the two CO molecules within the (1 × 1) unit cell at 1 ML. The optimized geometries obtained for the different configurations are depicted in Fig. 6 and 7, according parameters are shown in Table 1. The slight increase in the tilt angle with coverage reveals the influence of lateral interactions between the CO molecules.32
 | ||
| Fig. 6 Side views of CO/calcite(10.4) after energy minimization for two different CO coverages of 0.5 ML (a) and 1.0 ML (b and c). CO tilt angles are included. | ||
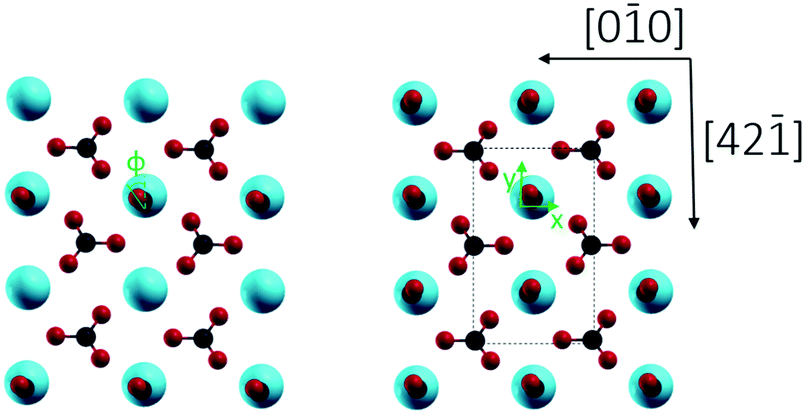 | ||
| Fig. 7 Top view of CaCO3 surface unit cell with one carbon monoxide molecule (0.5 ML coverage, left) and a complete monolayer of CO (right) on the energetically favored adsorption position. Φ represents the azimuthal angle of adsorbed CO after their optimized configurations. | ||
| 0.5 ML | 1.0 ML | ||
|---|---|---|---|
| CO | CO | (CO)2 | |
| C–O/Å | 1.14 | 1.14 | 1.14 |
| x(C–Ca(surface))/Å | 0.04 | 0.06 | 0.05 |
| y(C–Ca(surface))/Å | 0.02 | 0.02 | 0.02 |
| z(C–Ca(surface))/Å | 2.78 | 2.78 | 2.79 |
| Ca(surface)–O(subsurface) | 2.33 | 2.33 | 2.33 |
| Θ/(deg) | 14.42 | 14.57 | 15.14 |
| Φ/(deg) | 36.49 | 36.18 | 38.34 |
The calculated adsorption energies and stretching frequencies of CO molecules at different coverages are reported in Table 2. For the isolated CO gas phase molecule, a stretch frequency of 2123 cm−1 was obtained, showing the frequency shift (Δν) of 20 cm−1 compared to the experimental value of 2143 cm−1.32 In the following, we will use the relative shifts to the free molecule, Δν, when comparing experimental and theoretical results.
For a single CO molecule at 0.5 ML coverage, the adsorption energy is calculated as −0.216 eV, and the CO stretching frequency is blue shifted by 27 cm−1 relative to the gas phase value. For the full monolayer, the binding energy is lowered to −0.210 eV and the blue shift is reduced to 21 cm−1. The lowering of binding energy is attributed to repulsive adsorbate–adsorbate interactions, as observed for CO adsorption on other oxide surfaces (e.g. TiO233). The absence of attractive adsorbate–adsorbate interactions also explains why no island formation is observed when increasing exposures. A simple rationalization of the changes in frequency between low and high coverage are difficult, since the overall blue shift (relative to the free molecule) is a result of a combination of several mechanisms: (1) the interaction between the surface electric field and the CO dipole moment (Stark effect), and (2) the repulsive potential (Pauli repulsion) due to the fact that the CO molecule vibrates against the rigid surface (also called wall effect).31,33 Yet, intermolecular repulsive interactions are in agreement with the NC-AFM data where no clustering of the CO molecules is observed.
Conclusions
Polarization-resolved IRRAS and NC-AFM were used to study the adsorption of CO molecules on freshly cleaved calcite(10.4) surfaces. The NC-AFM data demonstrated a high structural quality of the surface. Adsorption of CO resulted in the occurrence of features appearing dark, located predominantly on-top of bright surface features. The latter were identified as the surface Ca2+ cations. The IRRAS data recorded with p-polarized incident light reveal that on the cleaved calcite(10.4) surface only one adsorbate species is present, in accord with the NC-AFM findings.DFT calculations for CO adsorbed on the (10.4) surface of calcite were carried out for two different surface coverages (0.5 ML and 1.0 ML). According to the calculations, CO adopts a slightly tilted geometry for the two different coverages studied here. In accord with the IRRAS experiments, a blue shift of the vibrational frequencies relative to the free molecule is obtained. The difference between experimental and theoretical shifts is less than 2 cm−1 for 0.5 ML, revealing a very good agreement. For 1.0 ML the difference between experimental and theoretical shifts increases to 12 cm−1. A missing vdW-potential in the DFT functional can partially explain the discrepancy. The theoretical value for the adsorption energy of −0.210 eV for 1.0 ML is good agreement with the experimental value of −0.31 eV. The binding energy decreased for higher coverages (1.0 ML relative to 0.5 ML), demonstrating the presence of repulsive interactions between adjacent CO adsorbates. This observation is in accord with the NC-AFM data indicating the absence of CO island formation.
Conflicts of interest
The authors declare no competing financial interest.Note added after first publication
This article replaces the version published on 09 July 2020, which contained errors in Fig. 5 and an incorrect image for Fig. 3.Acknowledgements
PR gratefully acknowledges funding by the German Research Foundation via grant RA2832/1-1. W. W. is grateful for a postdoctoral fellowship donated by the Helmholtz Association and China Postdoctoral Council (OCPC). The results presented in this paper have been gained within the DFG-funded project TH1566/3-2. We thank Ralf Bechstein and Angelika Kühnle for providing us with a suitable sample holder.References
- J. W. Morse and F. T. Mackenzie, Geochemistry of sedimentary carbonates. Elsevier, 1990, vol. 48 Search PubMed.
- M. E. Tucker and V. P. Wright, Carbonate sedimentology. John Wiley & Sons, 2009 Search PubMed.
- (a) M. S. Reddy, Biomineralization of calcium carbonates and their engineered applications: a review, Front. Microbiol., 2013, 4, 314 Search PubMed; (b) L. Addadi and S. Weiner, Control and design principles in biological mineralization, Angew. Chem., Int. Ed. Engl., 1992, 31(2), 153–169 CrossRef.
- H. Cölfen, Precipitation of carbonates: recent progress in controlled production of complex shapes, Curr. Opin. Colloid Interface Sci., 2003, 8(1), 23–31 CrossRef.
- P. Hlubina and W. Urbańczyk, Dispersion of the group birefringence of a calcite crystal measured by white-light spectral interferometry, Meas. Sci. Technol., 2005, 16(6), 1267 CrossRef CAS.
- J. M. Matter and P. B. Kelemen, Permanent storage of carbon dioxide in geological reservoirs by mineral carbonation, Nat. Geosci., 2009, 2(12), 837–841 CrossRef CAS.
-
(a) R. Innocenti Malini, A. R. Finney, S. A. Hall, C. L. Freeman and J. H. Harding, The Water–Amorphous Calcium Carbonate Interface and Its Interactions with Amino Acids, Cryst. Growth Des., 2017, 17(11), 5811–5822 CrossRef CAS;
(b) N. H. de Leeuw and S. C. Parker, Surface Structure and Morphology of Calcium Carbonate Polymorphs Calcite, Aragonite, and Vaterite:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) An Atomistic Approach, J. Phys. Chem. B, 1998, 102(16), 2914–2922 CrossRef CAS;
(c)
R. Howie; J. Zussman and W. Deer, An introduction to rock-forming minerals. Longman, 1992 Search PubMed.
An Atomistic Approach, J. Phys. Chem. B, 1998, 102(16), 2914–2922 CrossRef CAS;
(c)
R. Howie; J. Zussman and W. Deer, An introduction to rock-forming minerals. Longman, 1992 Search PubMed. - (a) N. H. de Leeuw and S. C. Parker, Surface structure and morphology of calcium carbonate polymorphs calcite, aragonite, and vaterite: an atomistic approach, J. Phys. Chem. B, 1998, 102(16), 2914–2922 CrossRef CAS; (b) S. Kerisit, S. C. Parker and J. H. Harding, Atomistic simulation of the dissociative adsorption of water on calcite surfaces, J. Phys. Chem. B, 2003, 107(31), 7676–7682 CrossRef CAS.
- P. Tasker, The stability of ionic crystal surfaces, J. Phys. C: Solid State Phys., 1979, 12(22), 4977 CrossRef CAS.
- P. Rahe, J. Schütte and A. Kühnle, NC-AFM contrast formation on the calcite (10.4) surface, J. Phys.: Condens. Matter, 2012, 24(8), 084006 CrossRef PubMed.
-
(a) S. Rode, N. Oyabu, K. Kobayashi, H. Yamada and A. Kühnle, True atomic-resolution imaging of (1014) calcite in aqueous solution by frequency modulation atomic force microscopy, Langmuir, 2009, 25(5), 2850–2853 CrossRef CAS PubMed;
(b) J. Schütte, P. Rahe, L. Tröger, S. Rode, R. Bechstein, M. Reichling and A. Kühnle, Clear signature of the (2 × 1) reconstruction of calcite (104), Langmuir, 2010, 26(11), 8295–8300 CrossRef PubMed;
(c) P. Fenter, P. Geissbühler, E. DiMasi, G. Srajer, L. Sorensen and N. Sturchio, Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity, Geochim. Cosmochim. Acta, 2000, 64(7), 1221–1228 CrossRef CAS;
(d) S. L. Stipp and M. F. Hochella Jr, Structure and bonding environments at the calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy electron diffraction (LEED), Geochim. Cosmochim. Acta, 1991, 55(6), 1723–1736 CrossRef CAS.
- (a) S. Kerisit, A. Marmier and S. C. Parker, Ab initio surface phase diagram of the {1014} calcite surface, J. Phys. Chem. B, 2005, 109(39), 18211–18213 CrossRef CAS PubMed; (b) A. Silvestri, A. Budi, E. Ataman, M. Olsson, M. Andersson, S. Stipp, J. Gale and P. Raiteri, A Quantum Mechanically Derived Force Field To Predict CO2 Adsorption on Calcite {10.4} in an Aqueous Environment, J. Phys. Chem. C, 2017, 121(43), 24025–24035 CrossRef CAS; (c) M. P. Andersson, K. Dideriksen, H. Sakuma and S. L. S. Stipp, Modelling how incorporation of divalent cations affects calcite wettability-implications for biomineralisation and oil recovery, Sci. Rep., 2016, 6, 28854 CrossRef CAS PubMed.
-
(a) G. Raina, R. Gauldie, S. K. Sharma and C. E. Helsley, A study of the calcite cleavage plane using the atomic force microscope, Ferroelectr., Lett. Sect., 1994, 17(3-4), 65–72 CrossRef CAS;
(b) J. Schütte, P. Rahe, L. Tröger, S. Rode, R. Bechstein, M. Reichling and A. Kühnle, Clear Signature of the (2 × 1) Reconstruction of Calcite (104), Langmuir, 2010, 26(11), 8295–8300 CrossRef PubMed.
- A. L. Rachlin, G. S. Henderson and M. C. Goh, An atomic force microscope (AFM) study of the calcite cleavage plane: image averaging in Fourier space, Am. Mineral., 1992, 77(9-10), 904–910 CAS.
- C. Wöll, Structure and Chemical Properties of Oxide Nanoparticles Determined by Surface-Ligand IR Spectroscopy, ACS Catal., 2019, 10(1), 168–176 CrossRef.
- Y. Wang and C. Wöll, IR spectroscopic investigations of chemical and photochemical reactions on metal oxides: bridging the materials gap, Chem. Soc. Rev., 2017, 46(7), 1875–1932 RSC.
- L. Tröger, J. Schütte, F. Ostendorf, A. Kühnle and M. Reichling, Concept for support and cleavage of brittle crystals, Rev. Sci. Instrum., 2009, 80(6), 063703 CrossRef PubMed.
- C. Barth and C. R. Henry, Kelvin probe force microscopy on surfaces of UHV cleaved ionic crystals, Nanotechnology, 2006, 17(7), S155 CrossRef CAS PubMed.
- F. J. Giessibl, High-speed force sensor for force microscopy and profilometry utilizing a quartz tuning fork, Appl. Phys. Lett., 1998, 73(26), 3956–3958 CrossRef CAS.
- L. Bartels, G. Meyer and K.-H. Rieder, Controlled vertical manipulation of single CO molecules with the scanning tunneling microscope: a route to chemical contrast, Appl. Phys. Lett., 1997, 71(2), 213–215 CrossRef CAS.
- S. Kuhn, M. Kittelmann, Y. Sugimoto, M. Abe, A. Kühnle and P. Rahe, Identifying the absolute orientation of a low-symmetry surface in real space, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90(19), 195405 CrossRef CAS.
- (a) G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed; (b) M. Izadifar, F. Königer, A. Gerdes, C. Wöll and P. Thissen, Correlation between Composition and Mechanical Properties of Calcium Silicate Hydrates Identified by Infrared Spectroscopy and Density Functional Theory, J. Phys. Chem. C, 2019, 123(17), 10868–10873 CrossRef CAS.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- (a) S. Hwang, M. Blanco and W. A. Goddard, Atomistic simulations of corrosion inhibitors adsorbed on calcite surfaces I. Force field parameters for calcite, J. Phys. Chem. B, 2001, 105(44), 10746–10752 CrossRef CAS; (b) J. S. Lardge, D. M. Duffy and M. J. Gillan, Investigation of the Interaction of Water with the Calcite (10.4) Surface Using Ab Initio Simulation, J. Phys. Chem. C, 2009, 113(17), 7207–7212 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192 CrossRef.
- N. Giraudo, P. G. Weidler, F. Laye, M. Schwotzer, J. Lahann, C. Wöll and P. Thissen, Corrosion of Concrete by Water-Induced Metal-Proton Exchange, J. Phys. Chem. C, 2016, 120(39), 22455–22459 CrossRef CAS.
- M. Bruno, E. Bittarello, F. Massaro and D. Aquilano, The effect of impurities on the structure and energy of a crystal surface: Mg impurities in calcite as a case study, CrystEngComm, 2018, 20(32), 4556–4564 RSC.
- S. Sanna, W. G. Schmidt and P. Thissen, Formation of Hydroxyl Groups at Calcium–Silicate–Hydrate (C–S–H): Coexistence of Ca–OH and Si–OH on Wollastonite (001), J. Phys. Chem. C, 2014, 118(15), 8007–8013 CrossRef CAS.
- P. A. Redhead, Thermal desorption of gases, Vacuum, 1962, 12(4), 203–211 CrossRef CAS.
- A. S. Foster, A. L. Shluger and R. M. Nieminen, Quantitative modelling in scanning force microscopy on insulators, Appl. Surf. Sci., 2002, 188(3-4), 306–318 CrossRef CAS.
- G. Pacchioni, A. M. Ferrari and P. S. Bagus, Cluster and band structure ab initio calculations on the adsorption of CO on acid sites of the TiO2(110) surface, Surf. Sci., 1996, 350(1–3), 159–175 CrossRef CAS.
- J. P. P. Ramalho, F. Illas and J. R. B. Gomes, Adsorption of CO on the rutile TiO2(110) surface: a dispersion-corrected density functional theory study, Phys. Chem. Chem. Phys., 2017, 19(3), 2487–2494 RSC.
- M. Setvin, M. Buchholz, W. Y. Hou, C. Zhang, B. Stoger, J. Hulva, T. Simschitz, X. Shi, J. Pavelec, G. S. Parkinson, M. C. Xu, Y. M. Wang, M. Schmid, C. Wöll, A. Selloni and U. Diebold, A Multitechnique Study of CO Adsorption on the TiO2 Anatase (101) Surface, J. Phys. Chem. C, 2015, 119(36), 21044–21052 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Owner Societies 2021 |