
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydration of sulfobetaine dizwitterions as a function of alkyl spacer length
Oliver S.
Hammond†
 a,
Leila
Moura
a,
Gaelle
Level
a,
Silvia
Imberti
b,
John D
Holbrey
*a and
Marijana
Blesic
*a
a,
Leila
Moura
a,
Gaelle
Level
a,
Silvia
Imberti
b,
John D
Holbrey
*a and
Marijana
Blesic
*a
aThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, Northern Ireland, UK. E-mail: j.holbrey@qub.ac.uk; m.blesic@qub.ac.uk
bISIS, Rutherford Appleton Laboratory, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 29th June 2020
Abstract
The solvation and structure of bolaform dizwitterions containing two sulfobetaine moieties in concentrated aqueous solution were determined using neutron diffraction with isotopic substitution (NDIS) combined with modelling of the measured structure factors using Empirical Potential Structure Refinement (EPSR). Strongly directional local hydration was observed in the polar regimes of the dizwitterions with 48–52 water molecules shared between dizwitterion molecules in a first shell water network around each zwitterion pair. Overall, the double zwitterions were highly hydrated, providing experimental evidence in support of the potential formation of protein-resistant hydration layers at zwitterion–water interfaces.
1 Introduction
Biofouling, the adherence of proteins to surfaces,1 manifests itself across a range of areas from marine coatings through to biomedical devices such as contact lenses, catheters, stents, grafts, and artificial organs, as well as in formulation, drug delivery, and gene therapy.2 As a consequence, controlling biofouling is of key clinical importance and direct links have been drawn between the innate hydrophilicity of materials and their ability to resist spontaneous adsorption of protein moieties.3,4It has been proposed5 that the presence of highly ordered, strongly bound near-surface water inhibits fouling due to the enthalpic penalty that would be incurred by disrupting the water, thereby repelling proteins. Numerous approaches to produce protein-resistant ‘superhydrophilic’ surfaces have been described,6,7 most commonly through incorporation of textured8 polyethylene glycol (PEG)9 polymer coatings with PEG brushes having the widest use.3,4 However PEGs are sensitive to in vivo degradation, and so there is a need to identify alternative materials with better characteristics.
Zwitterionic materials are promising in this context, offering greater biocompatibility,10 oxidation-resistance,3 increased hydrophilicity through hydration of charged sites,11 and strong resistance to fouling12 compared to non-ionic materials. For example, phosphocholine (PC) modified surfaces13 that mimic phospholipid interfaces have been shown to be effective at resisting protein adsorption.14 However, hydrolytic instability over time hampers their viability.7 Other zwitterions such as carboxybetaines (CB) and sulfobetaines (SB) that are more stable and have similar antifouling characteristics,12,15 are of interest and understanding the nature of hydration of zwitterionic groups, particularly in the context of their potential uses in antifouling materials16 is of importance.
Hydration of a range of zwitterions17,18 and their interactions at interfaces including micelles,19 lipid bilayers,20 and polymer brush surface coatings21 have been studied. Shao et al.22 explored the effects of changing the size of CB zwitterions, showing that the degree of hydration increases in response to increases in the separation between the two charge centres. These results are comparable to those found for polymer grafted CB zwitterion brushes, in which the degree of surface hydration with reaches a maximum when the charge centres are separated by 6–8 methylene groups and thereafter decreasing with a transition to hydrophobic behaviour as van der Waals interactions start to dominate.23,24
Foglia et al.25 demonstrated both hydrophilic and hydrophobic contributions to the hydration of PC, with the cationic trimethylammonium sites stabilised by the presence of additional weak H-bonding from the neighbouring methylene group which lead to a greater-than-anticipated association of the proximal hydrophobic groups of PC by water. Moreover, comparison of PC hydration states in solution and in bilayers26 has shown that solution state hydration can be used as a good proxy to inform on behaviour and properties in the interfacial region.
Dizwitterions27 occupy an intermediate position between discrete zwitterion charge pairs and polyionic materials. For example, the disulfobetaine double-zwitterions (Fig. 1) have been used in their acid forms as catalysts,28 and most recently have been shown to be promising candidates as neutral, charged materials for use in antifouling applications since they are: hydrophilic, kosmotropic, inert with both positively and negatively charged surfaces, and are protein-stabilisers.27 The degree of hydration of DSB-2 (Fig. 1) in solution was estimated from analysis of the changes in partial molal volume with concentration, giving a hydration number of 34. This corresponds to ca. 8 water molecules per charge site (neglecting van der Waals associated hydrogen-bonding in the alkyl-regions).
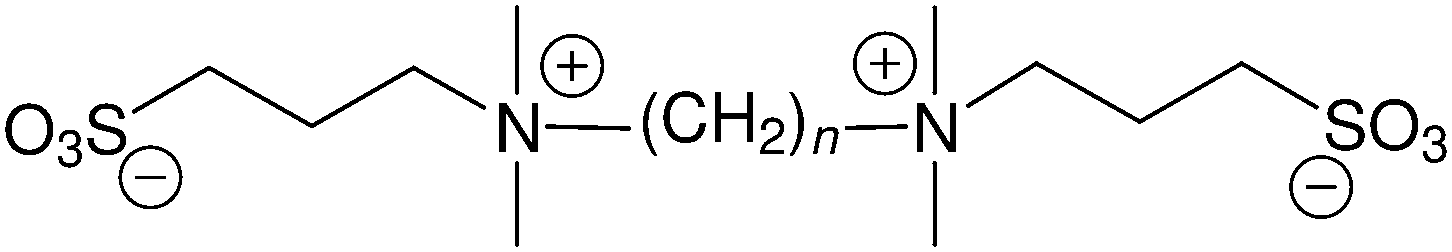 | ||
| Fig. 1 Structure of the dizwitterionic disulfobetaines under investigation: ethane-1,2-bis-N,N-dimethylammonium-N-propanesulfonate (n = 2), propane-1,3-bis-N,N-dimethylammonium-N-propanesulfonate (n = 3), and hexane-1,6-bis-N,N-dimethylammonium-N-propanesulfonate (n = 6). | ||
As part of studies exploring the characteristics of small polyzwitterions27 and zwitterionic salts29 as potential “superhydrophilic” additives, we report on the hydration of the three disulfobetaine double-zwitterions, ethane-1,2-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-2), propane-1,3-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-3), and hexane-1,6-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-6) in concentrated aqueous solution studied using neutron diffraction and isotopic substitution coupled with Empirical Potential Structure Refinement modeling (EPSR).30 These studies were prompted by the observation, described herein, that DSB-2 and DSB-6 show a comparable effect as surface tension modifiers in water, despite the changed spacers. EPSR and neutron scattering studies were therefore conducted to obtain a detailed, atomistic description of the effects on the hydration shell of changing the separation between the two tethered ‘zwitterions’, with a constant propyl (n = 3) spacer between each ammonium and sulfonate pair. The aim being, to gain insight into how potential synergistic effects can be introduced by controlling both inter- and intra-pair separations.
2 Experimental
The three dizwitterions; ethane-1,2-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-2), propane-1,3-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-3), and hexane-1,6-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-6) were synthesised from the corresponding tetramethyldiamines.Ethane-1,2-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-2, CAS 1215825-89-0)
N,N,N′,N′-Tetramethylethylenediamine (TMEDA; 26 mmol) was added to 30 mL methanol with stirring at 30 °C in an oil bath. 1,3-Propanesultone (52 mmol) was then added slowly to control the exotherm, and then the reaction mixture was stirred for 20 h. The crude product formed was recrystallised from a minimum of water adding methanol as an antisolvent, then filtered and rinsed with cold methanol. The product was then dried using a rotary evaporator and then under high vacuum at 60 °C to give 6.0 g (65%) of DSB-2. 1H NMR (400 MHz, D2O) δ 3.94 (s, 4H), 3.61–3.51 (m, 4H), 3.21 (s, 12H), 2.95 (t, 4H), 2.28–2.17 (m, 4H); 13C NMR (101 MHz, D2O) δ 63.60, 55.60, 51.30, 46.83, 18.23; elemental analysis (theoretical %/actual %) C 39.98/39.84, H 7.83/7.83, N 7.77/7.35, S 17.79/17.47; ES/MS m/z 361.1583 (M−H+, calc. 361.1467), 383.1338 (M−Na+, calc. 383.1287).Propane-1,3-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-3, CAS 1242745-61-4)
DSB-3 was prepared in an analogous manner to DSB-2 from N,N,N′,N′-tetramethylpropanediamine (TMPDA; 31 mmol) and was isolated as a colorless crystalline solid with a yield of 8.8 g (77%). 1H NMR (400 MHz, D2O) δ 3.54–3.45 (m, 4H), 3.43–3.34 (m, 4H), 3.12 (s, 12H), 2.94 (t, 4H), 2.35–2.23 (m, 2H), 2.23–2.13 (m, 4H); 13C NMR (101 MHz, D2O) δ 62.56, 59.99, 50.84, 47.04, 18.16, 16.49; elemental analysis (theoretical %/actual %) C 41.69/41.50, H 8.07/8.50, N 7.48/8.36, S 17.12/16.87; ES/MS m/z 375.1625 (M−H+, calc. 375.1624), 397.1410 (M−Na+, calc. 397.1443).Hexane-1,6-bis-N,N-dimethylammonium-N-propanesulfonate (DSB-6, CAS 53055-22-4)
DSB-6 was prepared in an analogous manner to DSB-2 from N,N,N′,N′-tetramethylhexanediamine (TMHDA; 31 mmol) and isolated after recrystallisation as a colorless crystalline solid with a yield of 9.4 g (78%). 1H NMR (400 MHz, D2O) δ 3.45–3.34 (m, 4H), 3.31–3.22 (m, 4H), 3.03 (s, 12H), 2.91 (t, 4H), 2.22–2.07 (m, 4H), 1.82–1.66 (m, 4H), 1.44–1.32 (m, 4H); 13C NMR (101 MHz, D2O) δ 63.86, 61.94, 50.70, 47.22, 25.00, 21.65, 18.13; elemental analysis (theoretical %/actual %) C 46.13/43.07, H 8.71/9.25, N 6.72/6.74, S 15.39/14.35; ES/MS m/z 417.2098 (M−H+, calc. 417.2093), 439.1916 (M−Na+, calc. 439.3913).Surface tension measurements
Aqueous solutions of the dizwitterions were prepared by diluting stock solutions previously gravimetrically prepared using an analytical balance. Doubly distilled deionised water was obtained from a Millipore Milli-Q water purification system. The surface tension of the aqueous solutions was measured with the drop shape analysis method, using a calibrated DSA25 instrument from Krüss GmbH equipped with Krüss ADVANCE software. Droplets of the relevant solution were suspended from a blunt-tipped 13 Gauge needle and the surface tension reported as an average of at least 10 measurements. For each individual measurement, the dimensions of the drop were captured and fitted using the Young–Laplace equation. Measurements were conducted at room temperature without explicit control.Neutron diffraction measurements
Neutron scattering data were collected using SANDALS, a forward-scattering time-of-flight diffractometer at the Rutherford Appleton Laboratories, Harwell, UK, measuring across a Q-range of 0.1 ≤ Q ≤ 50 Å−1 using neutrons of wavelength 0.05 ≤ λ ≤ 4.95 Å in a circularly-collimated beam of diameter 30 mm. Concentrated aqueous solutions of the isotopically substituted samples were prepared by mixing protonated and vacuum-dried DSB-2, DSB-3 or DSB-6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mole ratio (ca. 2 M) with H2O (Milli-Q), D2O (Sigma-Aldrich, 99.9 atom% D), or a 50/50 H2O:D2O mixture to produce three isotopic solute:solvent contrasts of H:H, H:D and H:H/D. Samples were sealed into Ti0.68Zr0.32 null-scattering alloy cells with 30 × 30 mm flat-plate geometry, with path length and wall thicknesses of 1 mm, and measured for a mean of 750 μA h of integrated incident beam current. To calibrate the instrument and reduce the data, additional measurements were made of the empty cells, the empty instrument, and a 3.1 mm thick standard of pure V. Samples were held at 298 ± 0.1 K using a Julabo FP52 recirculating heater. The data were corrected using GudrunN31 for sample attenuation and multiple scattering, background contributions were removed, and normalisation to absolute units was performed by reference to the V calibration standard. Corrections were then iteratively made for inelastic scattering of 1H, producing datasets for analysis using EPSR modeling.30
:30 mole ratio (ca. 2 M) with H2O (Milli-Q), D2O (Sigma-Aldrich, 99.9 atom% D), or a 50/50 H2O:D2O mixture to produce three isotopic solute:solvent contrasts of H:H, H:D and H:H/D. Samples were sealed into Ti0.68Zr0.32 null-scattering alloy cells with 30 × 30 mm flat-plate geometry, with path length and wall thicknesses of 1 mm, and measured for a mean of 750 μA h of integrated incident beam current. To calibrate the instrument and reduce the data, additional measurements were made of the empty cells, the empty instrument, and a 3.1 mm thick standard of pure V. Samples were held at 298 ± 0.1 K using a Julabo FP52 recirculating heater. The data were corrected using GudrunN31 for sample attenuation and multiple scattering, background contributions were removed, and normalisation to absolute units was performed by reference to the V calibration standard. Corrections were then iteratively made for inelastic scattering of 1H, producing datasets for analysis using EPSR modeling.30
Empirical potential structure refinement modeling (EPSR)
EPSR is a Reverse Monte Carlo-derived atomistic modeling technique developed for ensemble structure determination and fitting of neutron diffraction data.30,32,33 In EPSR, the reference potential is constructed as a classical pair interaction for a chemical system using Lennard-Jones parameters and Coulomb interactions, with atom-centric point charges, periodic boundary conditions and Lorentz–Berthelot mixing rules, to provide, εα,β and σα,β. An empirical potential (EP) is then calculated from the residuals between the reference potential and experimental data, and refinement occurs by minimising this difference; Fourier transform artefacts are suppressed by performing this operation with serial Poisson functions.33 Thus, EPSR is distinct from the Reverse Monte Carlo (RMC) approach, which uses hard sphere potentials, and accepts or rejects refinements depending on whether the quality-of-fit has improved.34 Overall, EPSR yields good quality-of-fit due to the usage of ‘chemical sense’, that is, using numerous isotopic substitutions to increase confidence in the experimental representation of the structure, and using the physicochemical properties of a system such as the experimental density, the exact molecular structure and composition of the solvent and solutes, and in addition to this a degree of intramolecular disorder is permitted.32Models for the DSB-2, DSB-3 and DSB-6 molecules, as shown in Fig. 2, were first constructed using Jmol and parametrised. Despite many sites being chemically identical, atoms were defined and labelled according to their position along the chain, which has the side-effect of generating many partial radial distribution functions, respectively 406, 465, and 666 for DSB-2, DSB-3 and DSB-6, but allows for site-specific correlation information to be extracted from the EPSR models. For water, TIP3P parametrisation was used, whereas parameters for the sulfobetaine ions were derived from OPLS-AA and Ionic Liquid force fields,35 and are shown in Table 1.
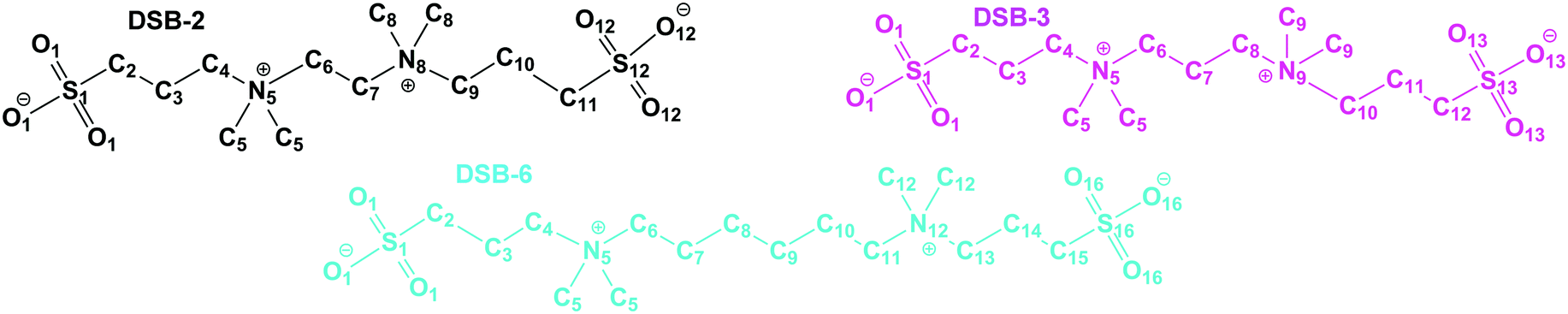 | ||
| Fig. 2 Atom labels for the double-sulfobetaine zwitterions synthesised and modeled in this study. Atoms are labelled according to their position along the major axis, though chemically identical groups such as O1, O12, O13, O16 have the same parameterisations. Water atoms (not pictured) are labelled as OW and HW. | ||
| Atom types | σ/Å | ε/kJ mol−1 | Charge/e |
|---|---|---|---|
| DSB-2 | |||
| S1, S12 | 3.55 | 1.04600 | 1.180 |
| O1, O12 | 3.15 | 0.83700 | −0.680 |
| C2, C11 | 3.50 | 0.27614 | −0.140 |
| C3, C10 | 3.50 | 0.27614 | −0.120 |
| C4, C6, C7, C9 | 3.50 | 0.27614 | 0.205 |
| C5, C8 | 3.50 | 0.27614 | 0.145 |
| N5, N8 | 3.25 | 0.71128 | −0.300 |
| H2, H11 | 2.50 | 0.12600 | 0.000 |
| H3–H10 | 2.50 | 0.12600 | 0.060 |
| DSB-3 | |||
| S13 | 3.55 | 1.04600 | 1.180 |
| O13 | 3.15 | 0.83700 | −0.680 |
| C12 | 3.50 | 0.27614 | −0.140 |
| C7, C11 | 3.50 | 0.27614 | −0.120 |
| C8, C10 | 3.50 | 0.27614 | 0.205 |
| C9 | 3.50 | 0.27614 | 0.145 |
| N9 | 3.25 | 0.71128 | −0.300 |
| H12 | 2.50 | 0.12600 | 0.000 |
| H7–H11 | 2.50 | 0.12600 | 0.060 |
| DSB-6 | |||
| S16 | 3.55 | 1.04600 | 1.180 |
| O16 | 3.15 | 0.83700 | −0.680 |
| C15 | 3.50 | 0.27614 | −0.140 |
| C7–C10, C14 | 3.50 | 0.27614 | −0.120 |
| C11, C13 | 3.50 | 0.27614 | 0.205 |
| C12 | 3.50 | 0.27614 | 0.145 |
| N12 | 3.25 | 0.71128 | −0.300 |
| H15 | 2.50 | 0.12600 | 0.000 |
| H7–H14 | 2.50 | 0.12600 | 0.060 |
| Water | |||
| Ow | 3.15 | 0.63400 | −0.834 |
| Hw | 0.00 | 0.00000 | 0.417 |
Densities of the fully-protonated solutions of DSB-2, DSB-3 and DSB-6 were averaged from 3 measurements using an Anton Paar DMA4100M vibrating tube densitometer, giving experimental densities of 1.1411, 1.1316 and 1.1065 g cm−3 respectively, corresponding to atomic densities of 0.10678, 0.106496, and 0.10582 atoms Å−3. Simulation boxes were then constructed using 1500 water molecules and 50 DSB, at 1/20 of the experimental density values. A number of MC refinement cycles were then run, where for each cycle EPSR attempts to move, rotate and translate every eligible atom in the simulation box, until the model had equilibrated; the boxes were compressed to the experimental densities en route by 10% per cycle which reduced the energy to a net attractive potential. This resulted in simulation boxes of side length 40.32, 40.64 and 41.57 Å respectively for DSB-2, DSB-3 and DSB-6, sufficient for the determination of ‘long-range’ structure of up to ca. 2 nm. After equilibration, the EPSR model was refined by introducing the EP to sample against the neutron scattering data, where each refinement cycle comprises 5 individual MC cycles and a recalculation of the EP. The models are finally accumulated to gather statistics and calculate partial radial distribution functions, intermolecular coordination numbers and spatial density functions.
3 Results and discussion
Interfacial surface tension of aqueous DSB-2 and DSB-6 solutions
The subsequent investigation of hydration of the three disulfobetaine double zwitterions was prompted by initial screening of the surface activity of the zwitterions in water. The influence of DSB-2 and DSB-6 on the interfacial surface tension of aqueous solutions as a function of concentration is shown in Fig. 3. Despite the change in separation of the two zwitterionic moieties from ca. 4 Å to 9 Å on replacement of the ethyl DSB-2 with a hexyl DSB-6 spacer, a remarkable and unexpected similarity in the effect of addition of the two double zwitterions to surface tension reduction is observed. Neither DSB-2 or DSB-6 show evidence of aggregation in solution, with a constant, and common degree of reduction in surface tension with increasing concentration of either double zwitterion. An increase in amphiphilicity was anticipated with DSB-6 due to the longer, and more hydrophobic, hexyl-spacer, however the similar results from the surface tension measurements suggest that any changes in DSB-6 are insufficient to contribute to additional disruption of the hydrogen bonds in water.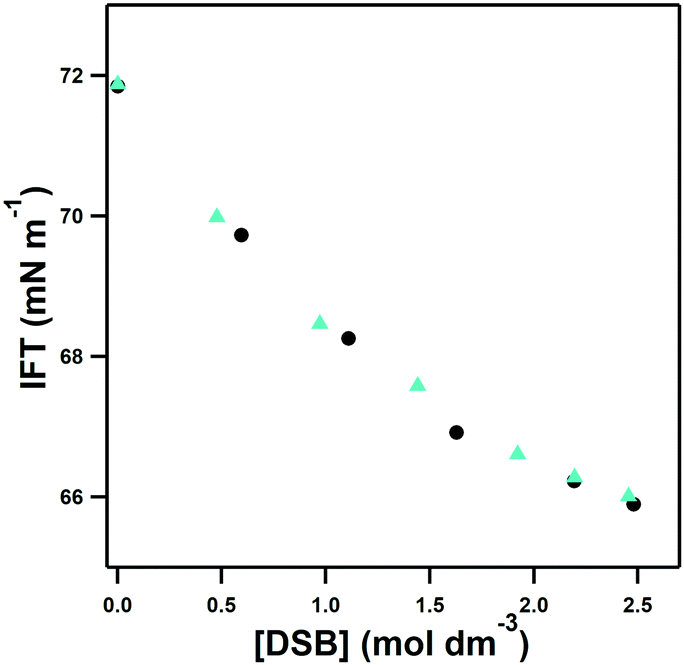 | ||
| Fig. 3 Interfacial surface tension data from aqueous solutions of DSB-2 (black circles) and DSB-6 (cyan triangles) showing equal effectiveness in the reduction of surface tension and lack of aggregation. | ||
This appears to contrast with the effect of changing the separation of the positive and negative charge centres of zwitterionic groups, as previously reported,23,24 and prompted the atomistic investigations by neutron scattering described here.
Neutron diffraction fits and data
All three systems contain a 1:30 molar ratio of the dizwitterion (DSB-2, DSB-3 or DSB-6) to water, corresponding to concentrations of approximately 2 M. Lower concentrations than the 1:30 (2 M) solutions studied were avoided because, even though it was considered possible that the DSBs would not be completely hydrated at 1:30 DSB:water given their large size (22–26 non-hydrogen atoms), higher dilutions would lead to even greater contribution from bulk water to the F(Q) data, potentially masking information from water–DSB contributions. Even at these high substrate concentrations, the scattering patterns are dominated by contributions from the aqueous component with only subtle observable differences in the data in the atomic-scale region (1–3 Å).
Fig. 4 shows the experimental neutron scattering datasets collected for the solutions of the three zwitterions DSB-2, DSB-3 and DSB-6 in the three isotopic contrasts of water (H2O, D2O, and HOD) in both Q-space (Fig. 4a) and real space (Fig. 4b). With the exception of the region ≤1 Å−1 where data show slight over-subtraction of the inelastic hydrogen background, particularly in the H:H contrast (Fig. 4a, top set) where this is most challenging,36 the fits align very closely with the data for all of the measured systems. Despite only having isotopic contrast in the aqueous component of the samples, the EPSR fit residuals are very small particularly when considering the real-space comparison (Fig. 4b) which provides confidence in the models. It is noteworthy that the experimental data showed systematically slightly lower (ca. 10%) scattering cross-sections than calculated, which was hypothesised to be due to the absorption of atmospheric CO2 facilitated by interactions with sulfonate groups.37 Therefore, there is overall a good degree of confidence that the model is representative of the experimentally-determined structure.
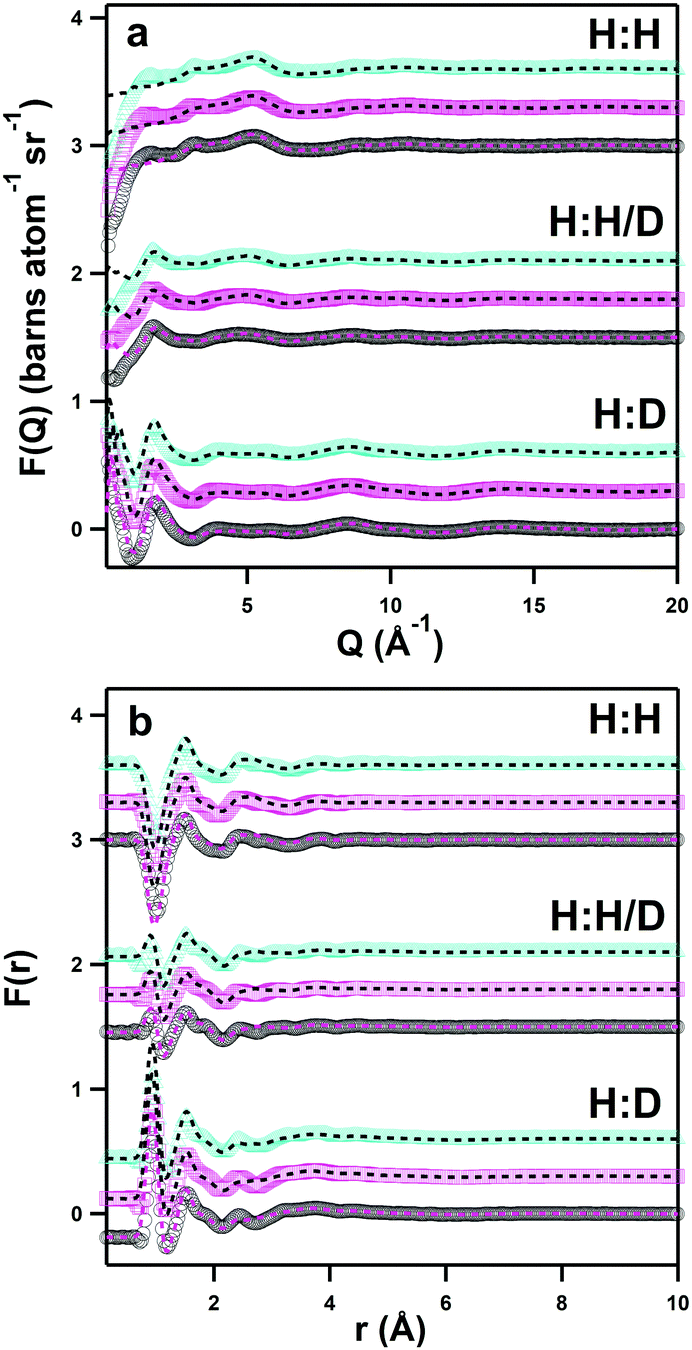 | ||
| Fig. 4 Experimental neutron scattering data for 1:30 DSB:water solutions of DSB-2 (black circles, bottom of each set), DSB-3 (magenta squares, middle of each set) and DSB-6 (cyan triangles, top of each set) and fits to the data using EPSR (black/magenta, dashed lines), for the H:H, H:H/D, and H:D contrasts, shown as a function of Q-space (a, top) and r-space (b, bottom). | ||
Hydration of the zwitterions
Fig. 5 shows a collated set of representative partial radial distribution functions (pRDFs) derived from the EPSR models that describe the overall hydration of each DSB molecule by water hydrogen bond donor (HW) and hydrogen bond acceptor (OW) sites to the atomic sites on the DSBs defined in Fig. 2. The corresponding integrated intermolecular coordination numbers (Ncoord) for a selection of these peaks are shown in Table 2. Due to the molecular symmetry, only one of each example of nitrogen and sulfonate is shown, as the values and pRDFs calculate to be equivalent and indistinguishable within the experimental error. As suggested by the data, there are only very subtle differences between the pRDFs and in some case no discernible difference at all, indicating that the structure in this regime is dominated by the pair of zwitterionic moieties. For the zwitterion-water pRDFs, there is generally no long-range order, with only minor oscillations seen about unity at ≥1 nm, confirming that the hydration is solely local.38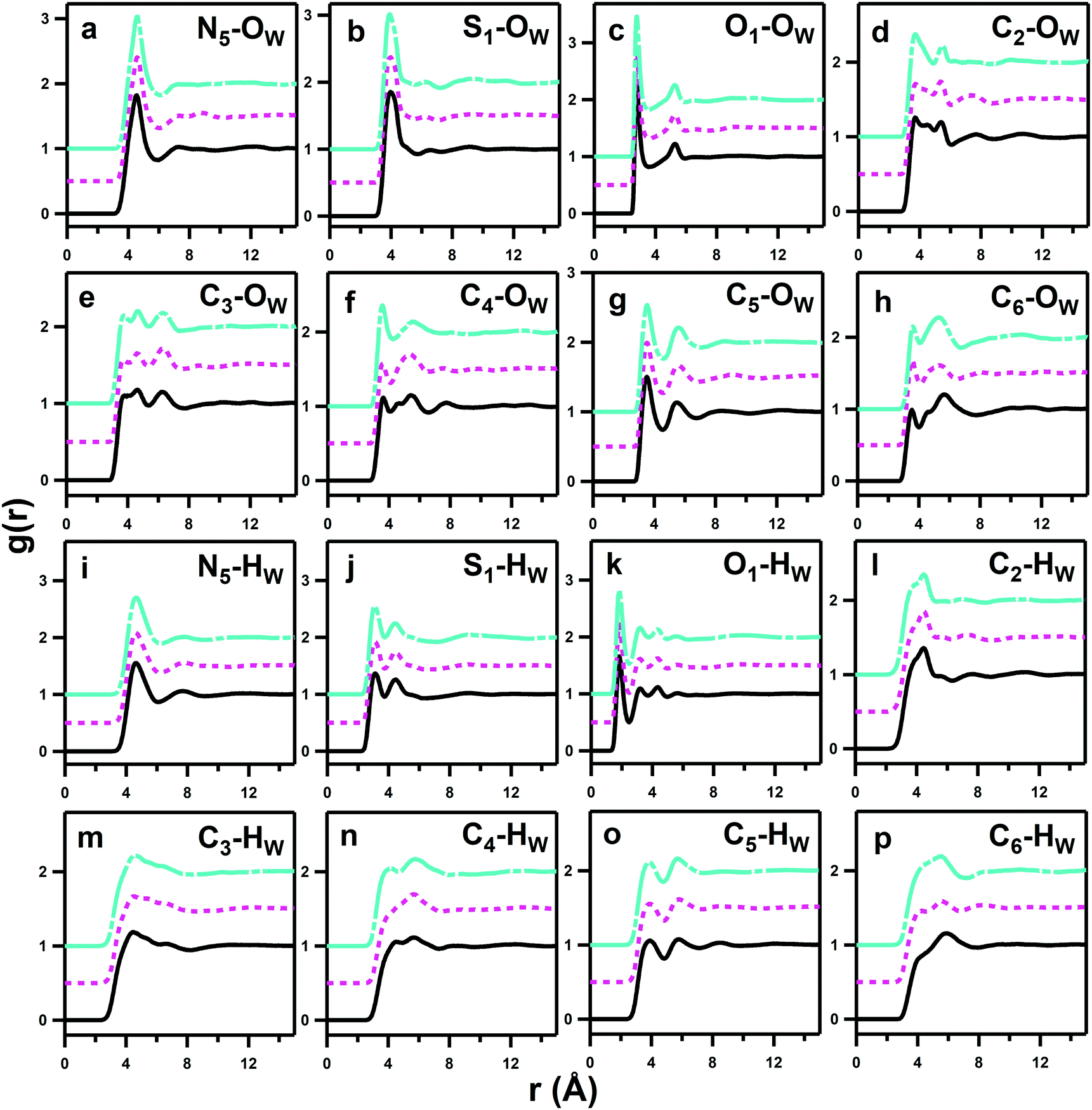 | ||
| Fig. 5 Partial Radial Distribution Functions (pRDFs) showing site–site interactions for the water oxygen OW (a–h) and water hydrogen sites HW (i–p) around DSB-2 (black solid lines, bottom), DSB-3 (magenta dotted lines, middle) and DSB-6 (cyan dashed lines, top). Interactions are shown for hydration of the DSB nitrogen (a and i) and sulfonate group (b, c, j and k), and common molecules in the carbon backbone (d–h and l–p). Data are offset to facilitate viewing. | ||
| A | B | R min/Å | R max/Å | DSB-2 | DSB-3 | DSB-6 |
|---|---|---|---|---|---|---|
| N5 | OW | 1.00 | 6.00 | 18.55 ± 2.62 | 18.68 ± 2.97 | 18.17 ± 2.62 |
| N5 | HW | 1.00 | 6.20 | 39.76 ± 4.83 | 40.00 ± 5.75 | 39.59 ± 5.29 |
| S1 | OW | 1.00 | 5.60 | 16.15 ± 2.93 | 15.83 ± 2.87 | 15.15 ± 2.68 |
| S1 | OW | 5.60 | 7.20 | 18.24 ± 3.72 | 17.72 ± 3.90 | 16.77 ± 3.64 |
| S1 | HW | 1.00 | 3.78 | 7.76 ± 1.80 | 7.79 ± 1.73 | 7.79 ± 1.67 |
| S1 | HW | 3.78 | 6.45 | 42.02 ± 7.33 | 41.40 ± 7.49 | 38.82 ± 6.65 |
| O1 | OW | 1.00 | 3.51 | 3.16 ± 1.08 | 3.17 ± 1.05 | 3.05 ± 1.04 |
| O1 | OW | 3.51 | 5.94 | 16.08 ± 3.31 | 15.77 ± 3.32 | 15.02 ± 2.92 |
| O1 | HW | 1.00 | 2.49 | 2.16 ± 0.82 | 2.18 ± 0.81 | 2.15 ± 0.78 |
| O1 | HW | 2.49 | 3.75 | 6.65 ± 2.18 | 6.62 ± 2.11 | 6.39 ± 2.11 |
| O1 | HW | 3.75 | 4.98 | 13.87 ± 3.63 | 13.72 ± 3.53 | 12.96 ± 3.20 |
| O1 | HW | 4.98 | 6.36 | 25.25 ± 5.37 | 24.68 ± 5.43 | 23.03 ± 4.87 |
| N5 | S1 | 1.00 | 6.87 | 1.71 ± 0.66 | 1.76 ± 0.90 | 1.55 ± 0.63 |
As anticipated, the ammonium group displays strong hydration with a prominent and sharp pRDF for the N5–OW interaction (Fig. 5a). The first solvation shell extends to 6 Å from the N5 site with water coordination numbers of 18.55 ± 2.62, 18.68 ± 2.97 and 18.17 ± 2.62 for DSB-2, DSB-3 and DSB-6, respectively. The preferred dipole orientation of water can be confirmed by comparison of the pRDFs from the water OW and HW sites to both the quaternary nitrogen and the adjacent methyl groups. The first minima in the N5–HW pRDFs for each system occur at marginally longer distances of 6.2 Å, rather than 6 Å for N5–OW, and the N5–HW peaks are less intense and broader, which is a sign of more disorder. Similarly, the C5–OW pRDFs show less diffuse and closer-range interactions than the C5–HW set, which further suggests that the water dipoles preferentially orient with the hydrogen atoms pointing away from the central zwitterion. This means that the electrostatic contribution of the ammonium group is likely dominant, and this orientation is further stabilised by weak H-bonding between methyl protons and the water OW sites.
Overall, there are only very minor differences in the hydration of the cationic ammonium sites in the three DSBs, especially in terms of coordination number. DSB-6 is slightly less hydrated, and a correspondingly higher hydration and disorder is observed for DSB-3, which is possibly due to different flexibility between trans and gauche conformers between the two nitrogen centres in the presence of the propyl spacer. On the other hand, as the spacer increases from ethyl (DSB-2) to hexyl (DSB-6), the onset of the ammonium hydration peaks N5–OW and N5–HW is reduced by ca. 0.1 Å which indicates an increase in the strength of hydration. These small differences in the hydration behaviour patterns as the alkyl chain is lengthened in the three disulfobetaines are more easily visualised in the spatial density function (SDF) plots in Fig. 6. For DSB-2, a hydration band is observed around the ammonium group in a Reuleaux triangle, with distortions from a pure radial geometry to accommodate the hydrophobic methyl groups. Hydration of these groups is less favourable, with gaps in the hydration shell for regions most proximal to the methyl locations. Similarly, water molecules are less likely to occupy sites close to the alkyl chain between N and S, or N and N. The same general structural hierarchy can also be observed for DSB-3 and DSB-6, but with the preferred correlation sites less diffuse as the chain length increases, with more significant voids in occupancy in areas where the alkyl chain may rotate. Therefore, while the hydration number slightly drops, the spatial distribution becomes slightly more well-defined, which is similar to the difference between SB and CB zwitterions.22
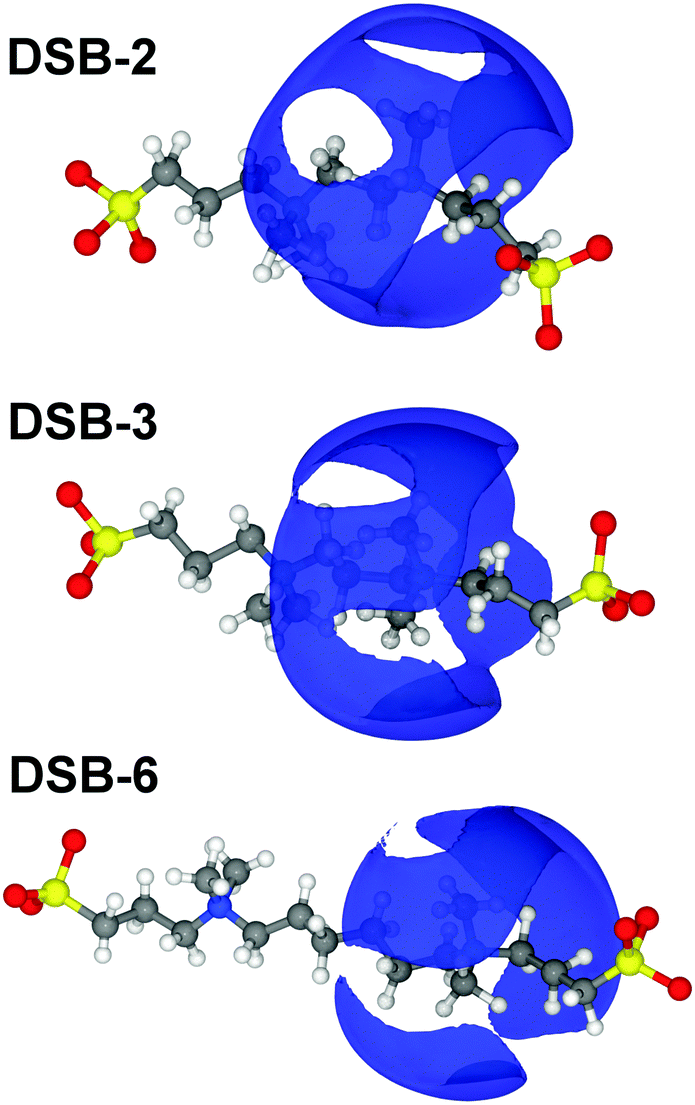 | ||
| Fig. 6 SDF plots around the three disulfobetaines, where the blue isosurface represents the top 15% most likely configurations of the water OW atom around the N5 atom of the ammonium group between 1–6 Å, i.e. the primary hydration sphere. | ||
The calculated hydration numbers of 18–19 around each nitrogen atom are noticeably lower than those observed in studies of dilute (0.4 M) aqueous solutions of tetramethylammonium bromide (TMABr) and tetrapropylammonium bromide (TPABr) where hydration numbers of, respectively, 22.0 ± 3.8 and 31.0 ± 6.0 were found.39 However, the values are consistent with previous calculations from Shao et al.22 who observed a coordination number of 18.64 ± 0.01 for the ammonium group of a monosulfobetaine containing a C2 N–S spacer, and is additionally relatable to the degree of hydration reported from previous neutron scattering studies on aqueous acetylcholine and phosphocholine solutions.25,40
Hydration of the anionic sulfonate groups in the DSBs is also significant. The S1–OW pRDFs for each system (Table 2) show that the first solvation shell spans up to 5.6 Å. This is closer to the central sulphur site than the approach of water to the ammonium sites (N5–OW) that are sterically blocked by the presence of methyl groups on the ammonium center. The hydration numbers of the DSB sulfonate groups decrease progressively as the alkyl chain length of the spacer is increased, from 16.15 ± 2.93 in DSB-2 to 15.83 ± 2.87 in DSB-3 and 15.15 ± 2.68 for DSB-6, and a more extreme reduction in coordination in the second hydration shell of S1–OW can also be observed. However, given the proximity of the anionic sulfonate and cationic ammonium sites, it is likely that the S1–OW pRDF also partially integrates the primary hydration shell of the ammonium group, which is suggested by the shorter length scale and lower coordination numbers for the S1–HW interactions, and can be observed from the proximity of the isosurfaces in the spatial distribution functions (SDFs) around the ammonium center shown in Fig. 6 (N5–OW) and the sulfonate group in Fig. 7 (S1–OW). It is therefore more useful to consider the O1–OW and O1–HW pRDFs to determine the headgroup hydration and indeed, both sets of pRDFs show narrow and intense peaks, corresponding with low disorder.
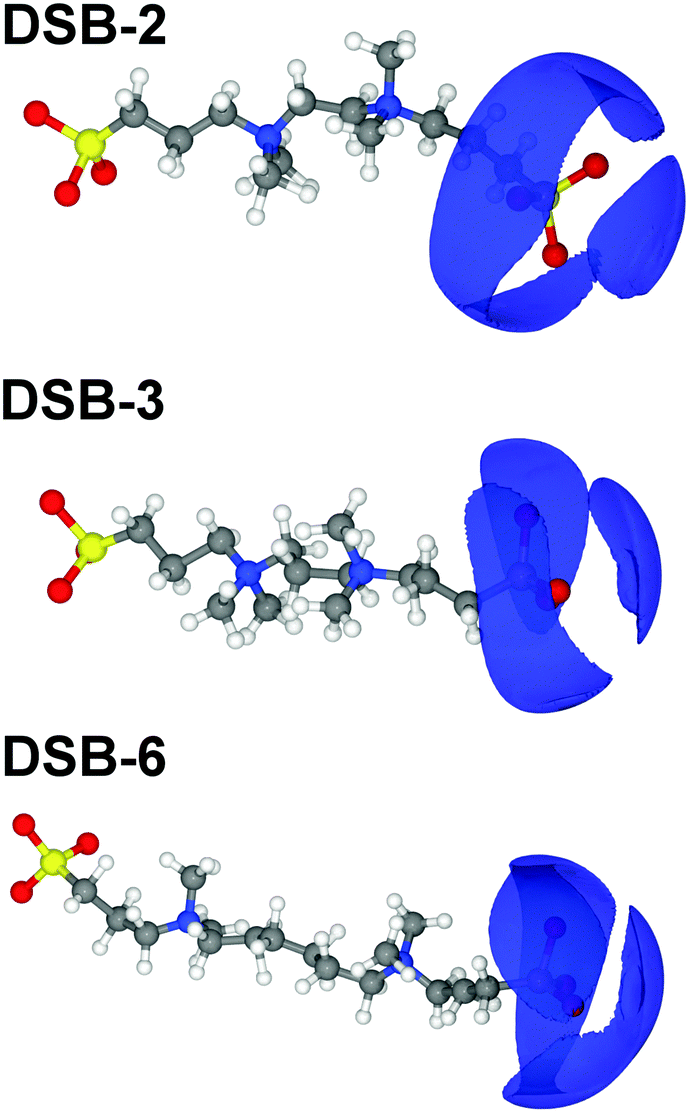 | ||
| Fig. 7 SDF plots around the three disulfobetaines, where the blue isosurface represents the top 15% most likely configurations of the water OW atom around the S1 atom of the sulfonate group between 1–5.6 Å, i.e. the primary hydration sphere. | ||
Changing the alkyl chain length of the spacer between the two zwitterionic components in the DSBs has relatively little effect on the S1–HW interactions, with the only discernible difference being a slightly closer onset and sharper peak as the spacer was increased to C6, signifying marginally less disorder. The SDF plots in Fig. 7 show the slight differences in orientational ordering of water around DSB-2, DSB-3 and DSB-6 sulfonate groups. Being more distal from the core alkyl spacer group, it follows that the hydration of the sulfonate group is less affected by the alkyl chain length.
The first minima of the O1–HW interactions occur at 2.49 Å compared to 3.51 Å for O1–OW, signifying opposite water dipole orientations to those at the ammonium group, as may be expected for the respective charges on the two ionic moieties. The net hydration numbers for each sulfonate oxygen remains essentially invariant as the alkyl chain length increases, with Ncoord for O1–HW of 2.16 ± 0.82, 2.18 ± 0.81 and 2.15 ± 0.78 respectively for DSB-2, DSB-3 and DSB-6, and for O1–OW of 3.16 ± 1.08, 3.17 ± 1.05, and 3.05 ± 1.04. Considering the closer-range O1–HW interactions only to avoid double-counting, this suggests that each sulfonate oxygen is the recipient of two H-bonds from water HW 5/6 of the time and 3 H-bonds 1/6 of the time, making the average hydration number for the sulfonate headgroup of approximately 6.5 if each water provides only 1 H-bond and does not bridge neighbouring oxygens. This is only slightly lower than the value reported by Shao et al.22 for SB zwitterions where the sulfonate group had a hydration number of 7.08 ± 0.01. This effect has been observed in phosphocholines25 where the hydration of the anionic sites of ammonium-based zwitterions is significantly less than half that of the positively-charged sites, and is thus a secondary component in defining the hydration characteristics for the molecules. Interestingly, this is slightly higher than the number of hydrogen-bonds per oxygen group seen in pure water,41 so the sulfonate group oxygens are relatively ‘oversaturated’ and experience a stronger local hydration than bulk water.
From the Cn–OW and Cn–HW pRDFs shown in Fig. 5, it is also possible to examine how water molecules respond to changes in the size of the alkyl-chain spacer between the two charged zwitterions in the three DSBs. Cn–HW interactions (m–p) are weaker and more diffuse than the corresponding Cn–OW interactions (e–h), showing the expected water dipole alignment with HW directed away from the central zwitterion around alkyl groups. At the same time, the Cn–OW pRDFs are most sensitive to change as a function of alkyl spacer length. With a few exceptions these interactions are relatively weak, only slightly above the probability of unity observed at infinite length. The most prominent CN–OW interactions are those which neighbour the hydrophilic groups, including the previously discussed C5–OW ammonium ion hydration, and C4–OW, followed by C2–OW.
This indicates that interaction of the alkyl groups with water is a secondary effect that arises from the dominant ammonium–water, sulfonate–water and water–water interactions, and which only add a slight additional stability through the donation of weak H-bonds from CH2 units. The C3–OW interactions show an interesting multimodal form with 3 distinct peaks up to 8 Å, which represents the highly ordered hydration shells of the neighbouring ammonium and sulfonate groups. The C6–HW and C6–OW pRDFs change most markedly in form as the alkyl chain length is increased from DSB-2 to DSB-6, with the secondary OW shell and primary HW shell changing from bimodal to a single feature, with slightly closer onset for the C6 alkyl spacer. Similarly, C2–OW and C2–HW see a slightly shorter onset in DSB-6 than in DSB-2. DSB-6 is the only alkyl chain long enough to see any hydrophobic hydration of the alkyl core, shown in C7–11–OW pRDFs in Fig. 8. However, even this is relatively minor due to the size and strength of the hydration of the conjoined zwitterionic groups, and as seen with the alkyl spacer between the positively and negatively charged unit of a solitary zwitterion, it is likely that far longer alkyl spacers (>n = 12) are required to see significant hydrophobic behaviour.
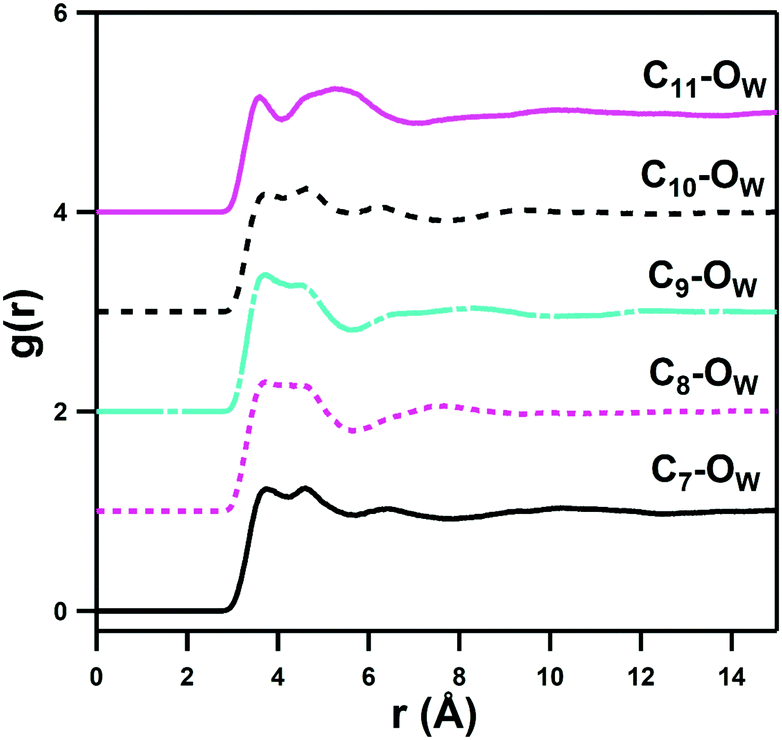 | ||
| Fig. 8 pRDFs showing interactions between the hexyl spacer in the DSB-6 zwitterion with the water oxygen atom. Some slight hydration is seen, though the intensity of the primary peak at around 3.8 Å is directly related to the distance of the atom site from an ammonium group, which also helps to explain the bimodality observed in some of the features. The alkyl group may contribute some weak H-bonds to help stabilise the oriented primary hydration shell of the zwitterion. | ||
Effect on solvent structure
pRDFs for the three water–water interactions, OW–OW, OW–HW and HW–HW are shown in Fig. 9 for the three disulfobetaines, and corresponding Ncoord values are given in Table 3, alongside data collated by Soper41 on bulk water for comparison.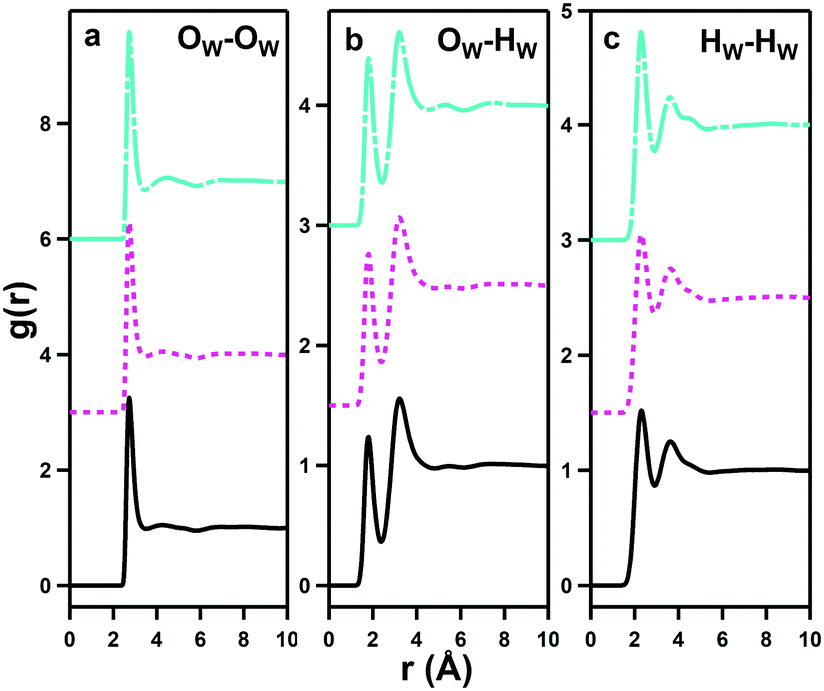 | ||
| Fig. 9 Water–water pRDFs showing interactions between water oxygen sites (a), OW–HW interactions (b), and proton–proton interactions (c) for DSB-2 (black solid lines, bottom), DSB-3 (magenta dotted lines, middle), and DSB-6 (cyan dashed lines, top). | ||
| A | B | R min/Å | R max/Å | DSB-2 | DSB-3 | DSB-6 | Water | TMABr | TPABr |
|---|---|---|---|---|---|---|---|---|---|
| OW | OW | 1.00 | 3.36 | 3.75 ± 1.21 | 3.67 ± 1.19 | 3.49 ± 1.11 | 4.67 | 4.30 ± 1.20 | 4.40 ± 1.20 |
| OW | OW | 3.36 | 5.76 | 14.69 ± 3.56 | 14.00 ± 3.50 | 13.15 ± 3.33 | — | — | — |
| OW | HW | 1.00 | 2.40 | 2.06 ± 1.02 | 2.05 ± 1.01 | 2.04 ± 0.98 | 1.88 | 2.00 ± 0.87 | 1.98 ± 0.88 |
| OW | HW | 2.40 | 4.74 | 19.53 ± 4.80 | 19.08 ± 4.75 | 17.36 ± 4.43 | — | — | — |
| HW | HW | 1.00 | 2.95 | 5.11 ± 1.46 | 5.04 ± 1.44 | 4.82 ± 1.37 | 5.27 | 6.30 ± 1.60 | 6.44 ± 1.70 |
| HW | HW | 2.95 | 5.25 | 24.38 ± 5.78 | 23.81 ± 5.71 | 21.84 ± 5.37 | — | — |
All of the water–water pRDFs show the same form as bulk water, however the integrated Ncoord values from Table 3 reveal significant differences from those of bulk water. In particular, the number of water molecules in the first water coordination shell, derived from the OW–OW correlations, is 3.75 ± 1.21 for DSB-2, 3.67 ± 1.19 for DSB-3 and 3.49 ± 1.11 for DSB-6 compared to 4.67 for bulk water. This shows a reduction of ca. 20–25% in the number of water molecules and this becomes more pronounced with the increase in the length of the alkyl chain spacer in the DSB. HW–HW coordination numbers are also reduced from 5.27 in bulk H2O to 5.11 ± 1.46, 5.04 ± 1.44 and 4.82 ± 1.37 respectively for DSB-2, DSB-3 and DSB-6. Similarly, the second solvation shells for all three interactions (OW–OW, OW–HW and HW–HW) show a large reduction, and greater disruption as the alkyl chain length increases. However, this is accompanied by a slight increase in the OW–HW coordination number which increases from 1.88 in bulk water to 2.05 on average for the three zwitterions.
The DSB:water ratio in these samples, 1:30, corresponds to 15 water molecules per zwitterionic unit and so, in the solutions, all the water molecules contribute to primary solvation of the DSB molecules. This is reflected in the reduction in the water–water Ncoord that show a compressed structure of bound water, similar to that recorded for liquid metal salt hydrates,42 rather than that of bulk water usually observed because few solutes produce significant perturbation of the water structure.18,40,41,43
Therefore, the solubilised zwitterions cause a clear rearrangement in the water present in these solutions. This effect is likely to arise from the strongly localised and directional ordering of water molecules in the primary hydration shell of the zwitterions. Greater disruption is seen as the alkyl spacer length is increased, with the introduction of additional hydration about the hydrophobic moieties.
It is interesting to examine the differences between the composite set of effects on water caused by DSBs with those from solutions of tetramethylammonium bromide and tetrapropylammonium bromide (TPABr),39 shown in Table 3. Despite polarity and the presence of charge, water does not hydrate the cations of tetramethylammonium bromide or chloride with strongly oriented dipoles across a wide concentration range.44 Rather, hydration occurs in a diffuse and apolar regime,45 whereas the zwitterions here are clearly hydrated in a hydrophilic regime with strongly oriented water dipoles.
Self-association of zwitterions
pRDFs that show auto-associative modes of the DSB dizwitterions are given in Fig. 10. These interactions are of interest, in part because it has been proposed that clustering of aqueous hydrophilic solutes, such as urea, plays a role in the denaturation of protein molecules.41 Moreover, it is interesting to determine whether any aggregation in solution is induced by alignment and charge matching of sulfobetaine headgroup dipoles,15 particularly as the spacer chain length increases,46 to form aggregates resembling those formed by bolaform surfactants.47 The aggregation of bolaforms leads to micelles with different shapes, sizes and structures to conventionally-oriented or gemini amphiphiles, and are potentially useful for material templating.48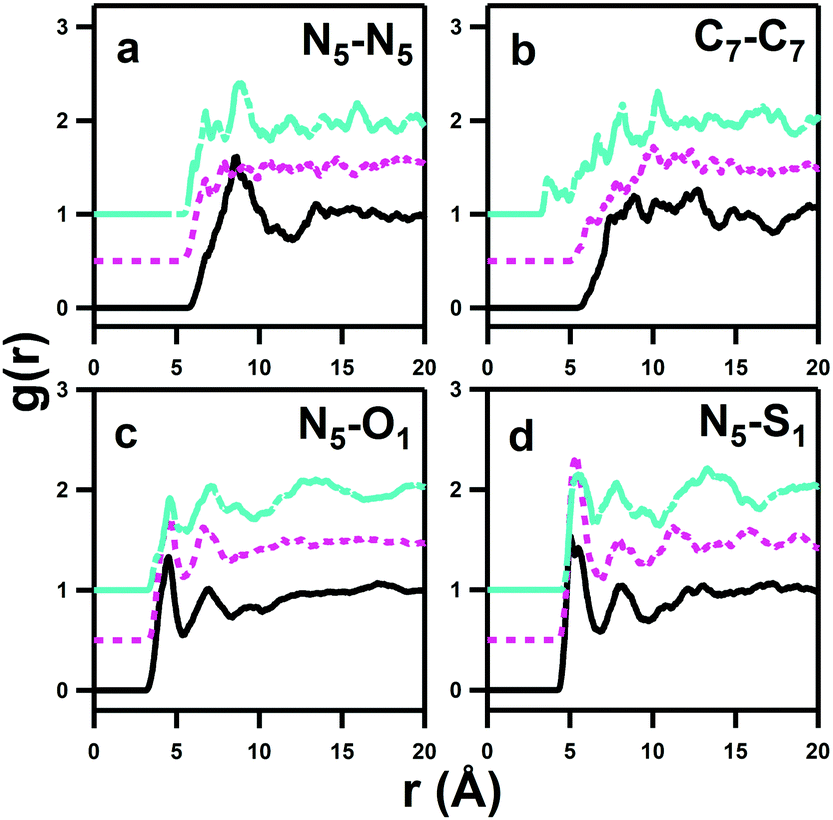 | ||
| Fig. 10 pRDFs showing the limits of self-association interactions between like zwitterions. DSB–DSB interactions are then shown between nitrogen sites (a), the furthest chemically-similar carbon from the origin of the molecules (b), and nitrogen–sulfonate interactions (c and d). Data are offset to aid viewing. | ||
It should be noted that the pRDFs in Fig. 10 are of longer length scales than those between the DSBs and water shown in Fig. 5 and that the interactions are themselves significantly less favourable, despite the high concentration and absence of significant ‘bulk’ water in the solutions. This is reflected in the large intrinsic uncertainty and ‘noise’ in the self-association correlations, despite long (>10000 cycle) accumulation.
A broad first correlation shell up to ca. 12 Å is evident in the N5–N5 pRDF for DSB-2, but this disappears for DSB-3 and DSB-6. Meanwhile, the C7–C7 interaction (Fig. 10b) that would respond to association and aggregation of the hydrophobic alkyl-spacers between the zwitterionic groups shows a progressive shortening of onset distance of the correlation as the spacer length increases from 2 to 6, with the first feature occurring at just 4 Å for DSB-6. However, this correlation peak is exceptionally broad, and largely featureless, indicating that clustering is therefore essentially negligible, and a much longer alkyl chain is likely necessary to see any true hydrophobic domain and thus hydrotropic or surfactant behaviour. The N5–O1 and N5–S1 pRDFs (Fig. 10c and d) do however show non-negligible ordering, meaning that configurations are possible where a primary molecule oriented in the direction N–S–O can align with a molecule whose dipole is opposite, i.e. O–S–N, or indeed a perpendicular interaction is possible. However, the Ncoord of the N5–S1 interaction in the first solvation sphere is only 1.71 ± 0.66, 1.76 ± 0.90 and 1.55 ± 0.63 for DSB-2, DSB-3 and DSB-6 respectively. This is indicative of low probability statistical association rather than formation of thermodynamic aggregates. The interdigitation of opposite dipoles of zwitterions in this manner has been shown previously by Gong et al.15 who observed absorption of PC lipids on SB-functionalised surfaces and vice versa using AFM and QCM.
4 Conclusions
Neutron scattering, isotopic substitution and EPSR modeling were used to characterise the hydration of a series of disulfobetaine bolaform zwitterions in aqueous solution at 1:30 molar dilution. Interrogation of the solvation structures derived from the EPSR models, fitted to experimental data, show only minor changes in the hydration behaviour of the zwitterionic moieties as the alkyl spacer between zwitterion pairs is increased from C2 to C3 to C6, while the N–S spacer was held constant. The DSB molecules were highly hydrated and the hydration is entirely local, with no persistent long-range interactions observed. Water molecules are directionally oriented around the ammonium and sulfonate groups with the water dipoles preferentially orienting with HW towards anionic, and OW towards cationic moieties in the DSBs. The sulfonate groups see lower local hydration than the cationic moieties but experience stronger hydration than bulk water molecules due to being relatively coordinatively oversaturated.
Hydration numbers of 18–19 around each ammonium group and 6–7 around each sulfonate were calculated, giving an approximate total hydration number of 48–52. This averaged first shell hydration number is larger than the water:DSB ratio (1:30) in the experimental compositions, and reflects the role of water molecules as bridging species between molecules that show minimal evidence of self-aggregation. These hydration spheres are partially reinforced by weak H-bond donation from the neighbouring alkyl chains, but there is only weak secondary hydration of the alkyl spacer groups for the lengths measured; longer chain lengths are required to observe true apolar hydration in molecules containing this many charged sites. This leads to a significant and ordered local hydration shell which offers potential to disrupt attachment of protein residues and helps to explain the high lubricity of zwitterionic polymer brush surfaces.
This is different to the behaviour of other solutes studied in the literature and provides some basis for the strong local ordering and suggested kosmotropic behaviour of zwitterions, as they influence the bulk structure due to interaction through a strongly ordered primary solvation shell. The introduction of a longer alkyl chain slightly exacerbates these effects, by inducing slight increases in the disruption to the solvent structure and marginal reduction of water coordination around DSB-6, though within the error. Therefore, it is shown that despite the presence of the alkyl chains and geminal methyl groups of the quaternary ammonium centres, the solvation of the zwitterions is in a hydrophilic regime rather than the apolar hydration exhibited by simple tetraalkylammonium salts. Additionally, it is suggested that the effect of the hydrated zwitterion on the bulk solvent structure may affect the conformation of solubilised proteins, which may affect attachment. The interaction of the sulfobetaine headgroup was also assessed, showing a slight possibility of alignment with opposite dipoles and perpendicular arrangements, but this was not a major contributor to structure, even at the high molar concentrations of the solutions measured. Overall, the structural findings here are of broad relevance to those working with zwitterionic species such as those introduced into polymers and protein-resistant materials for biomedical and maritime applications and lubrication, as it provides structural confirmation of the suspected hydration layer formed by these zwitterionic materials. Additionally, the structural rearrangement of bulk water may be of interest to groups using such materials to induce and investigate salting in/out phenomena.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank STFC for access to the SANDALS instrument at ISIS for neutron diffraction measurements under beamtime allocation RB1820154, the EPSRC for funding through grant EP/S002669/1 (MB, OH, GL) and the Royal Academy of Engineering (LM).References
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41, 5599 RSC.
- (a) J. A. Callow and M. E. Callow, Nat. Commun., 2011, 2, 244 CrossRef PubMed; (b) S.-B. Yeh, C.-S. Chen, W.-Y. Chen and C.-J. Huang, Langmuir, 2014, 30, 11386–11393 CrossRef CAS PubMed; (c) L. D. Blackman, P. A. Gunatillake, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2019, 48, 757–770 RSC.
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS.
- (a) R. S. Kane, P. Deschatelets and G. M. Whitesides, Langmuir, 2003, 19, 2388–2391 CrossRef CAS; (b) S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS; (c) J. Drelich, E. Chibowski, D. D. Meng and K. Terpilowski, Soft Matter, 2011, 7, 9804 RSC.
- L. Li, S. Chen, J. Zheng, B. D. Ratner and S. Jiang, J. Phys. Chem. B, 2005, 109, 2934–2941 CrossRef CAS PubMed.
- K. W. Bewig and W. A. Zisman, J. Phys. Chem., 1965, 69, 4238–4242 CrossRef CAS.
- D. Wang, Biomaterials, 2003, 24, 3969–3980 CrossRef CAS.
- (a) C.-M. Chan, T.-M. Ko and H. Hiraoka, Surf. Sci. Rep., 1996, 24, 1–54 CrossRef CAS; (b) M. Strobel, V. Jones, C. S. Lyons, M. Ulsh, M. J. Kushner, R. Dorai and M. C. Branch, Plasmas Polym., 2003, 8, 61–95 CrossRef CAS; (c) K. Tadanaga, J. Morinaga, A. Matsuda and T. Minami, Chem. Mater., 2000, 12, 590–592 CrossRef CAS.
- N. Kumar, M. N. Ravikumar and A. Domb, Adv. Drug Delivery Rev., 2001, 53, 23–44 CrossRef CAS.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed.
- H. Zhu, L. Li and J. Shen, Chem. Phys., 2019, 532, 110599 CrossRef.
- Z. Zhang, T. Chao, S. Chen and S. Jiang, Langmuir, 2006, 22, 10072–10077 CrossRef CAS PubMed.
- A. L. Lewis, Colloids Surf., B, 2000, 18, 261–275 CrossRef CAS.
- K. Ishihara, H. Nomura, T. Mihara, K. Kurita, Y. Iwasaki and N. Nakabayashi, J. Biomed. Mater. Res., 1998, 39, 323–330 CrossRef CAS PubMed.
- L. Gong, L. Xiang, J. Zhang, L. Han, J. Wang, X. Wang, J. Liu, B. Yan and H. Zeng, Langmuir, 2019, 35, 2842–2853 CrossRef CAS.
- J. C. Hower, M. T. Bernards, S. Chen, H.-K. Tsao, Y.-J. Sheng and S. Jiang, J. Phys. Chem. B, 2009, 113, 197–201 CrossRef CAS PubMed.
- (a) J. H. Jensen and M. S. Gordon, J. Am. Chem. Soc., 1995, 117, 8159–8170 CrossRef CAS; (b) R. A. Jockusch, A. S. Lemoff and E. R. Williams, J. Phys. Chem. A, 2001, 105, 10929–10942 CrossRef CAS; (c) E. Tajkhorshid, K. J. Jalkanen and S. Suhai, J. Phys. Chem. B, 1998, 102, 5899–5913 CrossRef CAS; (d) S. Xu, J. M. Nilles and K. H. Bowen, J. Chem. Phys., 2003, 119, 10696–10701 CrossRef CAS.
- S. E. McLain, A. K. Soper, A. E. Terry and A. Watts, J. Phys. Chem. B, 2007, 111, 4568–4580 CrossRef CAS.
- A. Sanchez-Fernandez, G. L. Moody, L. C. Murfin, T. Arnold, A. J. Jackson, S. M. King, S. E. Lewis and K. J. Edler, Soft Matter, 2018, 14, 5525–5536 RSC.
- M. L. Berkowitz, D. L. Bostick and S. Pandit, Chem. Rev., 2006, 106, 1527–1539 CrossRef CAS.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698–1701 CrossRef CAS PubMed.
- Q. Shao, Y. He, A. D. White and S. Jiang, J. Phys. Chem. B, 2010, 114, 16625–16631 CrossRef CAS PubMed.
- (a) Y. Higaki, Y. Inutsuka, T. Sakamaki, Y. Terayama, A. Takenaka, K. Higaki, N. L. Yamada, T. Moriwaki, Y. Ikemoto and A. Takahara, Langmuir, 2017, 33, 8404–8412 CrossRef CAS PubMed; (b) H. Du and X. Qian, J. Comput. Chem., 2015, 37, 877–885 CrossRef PubMed.
- Q. Shao and S. Jiang, J. Phys. Chem. B, 2013, 117, 1357–1366 CrossRef CAS PubMed.
- F. Foglia, M. J. Lawrence, C. D. Lorenz and S. E. McLain, J. Chem. Phys., 2010, 133, 145103 CrossRef PubMed.
- R. J. Gillams, C. D. Lorenz and S. E. McLain, J. Chem. Phys., 2016, 144, 225101 CrossRef.
- G. Level, J. Zhang, J. Brown, O. Hammond, B. Hannigan, L. Stella, P. Nockemann and M. Blesic, J. Colloid Interface Sci., 2020, 562, 391–399 CrossRef CAS.
- (a) D. Fang, D.-Z. Zhang and Z.-L. Liu, Monatsh. Chem., 2010, 141, 419–423 CrossRef CAS; (b) D. Fang, J. Yang and C. Jiao, ACS Catal., 2011, 1, 42–47 CrossRef CAS; (c) H.-F. Liu, F.-X. Zeng, L. Deng, B. Liao, H. Pang and Q.-X. Guo, Green Chem., 2013, 15, 81–84 RSC; (d) F.-X. Zeng, H.-F. Liu, L. Deng, B. Liao, H. Pang and Q.-X. Guo, ChemSusChem, 2013, 6, 600–603 CrossRef CAS PubMed; (e) D.-J. Tao, Y. Dong, Z.-J. Cao, F.-F. Chen, X.-S. Chen and K. Huang, J. Ind. Eng. Chem., 2016, 41, 122–129 CrossRef CAS; (f) G. Zheng and X. Li, Synth. Commun., 2019, 49, 933–941 CrossRef CAS.
- (a) M. Blesic, B. F. Gilmore, J. D. Holbrey, J. Jacquemin, G. Level, P. Nockemann and L. Stella, Green Chem., 2017, 19, 4007–4011 RSC; (b) G. Level, M. VieiraFadul and M. Blesic, ChemPhysChem, 2018, 19, 575–580 CrossRef CAS PubMed.
- A. Soper, Chem. Phys., 1996, 202, 295–306 CrossRef CAS.
- A. K. Soper, GudrunN and GudrunX: Programs for correcting raw neutron and X-ray diffraction data to differential scattering cross section, Rutherford Appleton Laboratory Technical Report, 2011 Search PubMed.
- A. K. Soper, Mol. Phys., 2001, 99, 1503–1516 CrossRef CAS.
- A. K. Soper, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104204 CrossRef.
- A. Soper, Chem. Phys., 2000, 258, 121–137 CrossRef CAS.
- (a) W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS; (b) P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS; (c) J. N. C. Lopes, A. A. H. Pádua and K. Shimizu, J. Phys. Chem. B, 2008, 112, 5039–5046 CrossRef PubMed.
- A. Soper, Mol. Phys., 2009, 107, 1667–1684 CrossRef CAS.
- S. Zhang, Y. Chen, R. X.-F. Ren, Y. Zhang, J. Zhang and X. Zhang, J. Chem. Eng. Data, 2005, 50, 230–233 CrossRef CAS.
- T. Iwashita, B. Wu, W.-R. Chen, S. Tsutsui, A. Q. R. Baron and T. Egami, Sci. Adv., 2017, 3, e1603079 CrossRef PubMed.
- E. J. Nilsson, V. Alfredsson, D. T. Bowron and K. J. Edler, Phys. Chem. Chem. Phys., 2016, 18, 11193–11201 RSC.
- E. Hulme, A. Soper, S. McLain and J. Finney, Biophys. J., 2006, 91, 2371–2380 CrossRef CAS.
- A. K. Soper, ISRN Phys. Chem., 2013, 1–67 CAS.
- S. Lenton, N. H. Rhys, J. J. Towey, A. K. Soper and L. Dougan, Nat. Commun., 2017, 8, 919 CrossRef PubMed.
- (a) D. T. Bowron, J. Phys.: Conf. Ser., 2009, 190, 012022 CrossRef; (b) J. L. Finney, D. T. Bowron and A. K. Soper, J. Phys.: Condens. Matter, 2000, 12, A123–A128 CrossRef CAS; (c) S. Busch, C. D. Lorenz, J. Taylor, L. C. Pardo and S. E. McLain, J. Phys. Chem. B, 2014, 118, 14267–14277 CrossRef CAS PubMed; (d) R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2012, 51, 7468–7471 CrossRef CAS PubMed; (e) O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- (a) J. Turner, A. Soper and J. Finney, Mol. Phys., 1990, 70, 679–700 CrossRef CAS; (b) J. Turner, A. Soper and J. Finney, Mol. Phys., 1992, 77, 411–429 CrossRef CAS; (c) A. Soper, J. Turner and J. Finney, Mol. Phys., 1992, 77, 431–437 CrossRef CAS.
- J. Z. Turner, A. K. Soper and J. L. Finney, J. Chem. Phys., 1995, 102, 5438–5443 CrossRef CAS.
- H. Hirata, N. Hattori, M. Ishida, H. Okabayashi, M. Frusaka and R. Zana, J. Phys. Chem., 1995, 99, 17778–17784 CrossRef CAS.
- T. W. Davey, W. A. Ducker and A. R. Hayman, Langmuir, 2000, 16, 2430–2435 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, J. Kim, Y. Han and G. D. Stucky, Chem. Mater., 1999, 11, 2668–2672 CrossRef CAS.
Footnote |
| † Present address: Laboratoire de Chimie, École Normale Supérieure de Lyon, Lyon, France. |
| This journal is © the Owner Societies 2020 |