
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of mono- and poly-dispersed nanoparticles on emulsion droplets: antagonistic vs. synergistic effects as a function of particle size†
Abeer
Khedr
and
Alberto
Striolo
 *
*
Department of Chemical Engineering, University College London, UK. E-mail: a.striolo@ucl.ac.uk
First published on 7th September 2020
Abstract
In this work, using Dissipative Particle Dynamics simulations, we provide fundamental insights into the self-assembly of nanoparticles (NPs) on droplet surfaces in an oil-in-water emulsion. We highlight the effect of particle size on the arrangement of NPs for different interparticle interactions. NPs of two different sizes were considered. In general, when the NP–NP interaction is changed from repulsive to attractive, a transition in the NP arrangement occurs from weekly-connected networks to clusters of NPs separated by particle-free domains. When NP–NP interactions are strongly attractive, NPs yield small 3D aggregates on the droplet surface. These arrangements seem to agree with experimental observations reported in the literature. In addition, our simulations suggest that small NPs are able to diffuse more easily on the droplet surface, which leads to prompt self-organisation, while large NPs are more likely to form metastable structures, perhaps because of slow mobility and strong adsorption to the interface. Our analysis suggests that thermal fluctuations could provide the activation energy for the small NPs to escape local minima in the free energy landscape. The results obtained for systems containing NPs of two sizes provide evidence of size segregation on the droplet surface, which could be useful when NP self-assemblies are used, for example, to template supra-molecular materials. However, analysis of the simulated trajectories suggests that the results depend strongly on the initial configuration, as the larger NPs seem to impose barriers for the small NPs to adsorb and diffuse on the droplet surface.
1. Introduction
The self-assembly of particles at interfaces is a key step in many applications, from materials design to emulsion stabilisation. While adsorbed at fluid–fluid interfaces, particles can arrange to yield structures useful to produce, for example, NP films with unique properties, including reflecting ones.1 On the other hand, assembled particles on droplet surfaces can provide an energetic barrier useful to stabilise dispersed systems in the so-called Pickering emulsions.2,3The adsorption and distribution of particles and nanoparticles (NPs) at interfaces are controlled by the physical/chemical properties such as wettability, size, shape, surface charge, and chemical nature of the particles.4–8 A macroscopic quantity often used to quantify the affinity between particles and the two liquids at the interface is the three-phase contact angle, θC. The desorption energy, e.g., depends on particle size and contact angle, as described by:3
ΔEp = πR2γ(1 ± cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θC)2 θC)2 | (1) |
In eqn (1), R is the particle radius, γ is the fluid–fluid interfacial tension, and negative and positive signs represent the detachment from the interface to one of the two liquids in contact, e.g., water and oil. As shown in eqn (1), because the desorption energy depends on θC, particle adsorption can be manipulated by changing the particle wettability, using, e.g., polymers or surfactants,9–11 or by manufacturing amphiphilic particles.12–14 The importance of particle size should be emphasized as, for example, when adsorbed at fluid–fluid interfaces, NPs are subjected to thermal fluctuations and Brownian motion, which might affect contact angle measurements.4 For example, Isa et al.15 reported a broad distribution of three-phase contact angles for NPs of size ranging from 20 to 500 nm prepared from different materials. The broad distribution of contact angles could be due to the NPs’ heterogeneous surface properties, fluctuations in the direction perpendicular to the interface due to Brownian motion coupled with weak adsorption energy, and the line tension becoming more and more important as the NP size decreases.
The dependency of the adsorption energy on the particle size could lead to competition between particles of different sizes to self-assemble at the interface. Lin et al.,16,17e.g., studied the self-assembly of cadmium selenide NPs of two sizes on water-in-toluene emulsion droplets. The NPs were covered with tri-n-octylphosphine-oxide. The small NPs (2.8 nm) were able to stabilise the emulsion for days.16 These NPs yield monolayers on the droplets in which NPs arranged in liquid-like structures. These NPs were mobile and diffused laterally over the droplet surface. Upon the addition of large NPs (4.6 nm), some small NPs were displaced from the droplet surface. This observation was ascribed to the dependency of the adsorption/desorption energy on particle size [i.e., see eqn (1)]. Lin et al. reported that, over time, the NPs underwent a phase separation on the droplet surface, and an assembly of large NPs surrounded by small ones was observed after 48 hours.17 They concluded that adsorbed NPs of different sizes tend to phase separate, but that for the phase separation to be complete, the NPs need to diffuse, a process that can be slow.18
The in-plane diffusion of NPs at an interface depends on particle size and fluid viscosity. Wang et al.,19e.g., showed that the diffusion coefficient of quantum dots (QDs) of radius 5, 8, and 11 nm at the water/oil interface increases as the particle size decreases. Tarimala and Dai20 investigated the arrangement of polystyrene particles with diameters of 1 and 4 μm on a poly(dimethyl siloxane) droplet dispersed in water. The small NPs formed patches of hexagonally ordered monolayers separated by particle-free domains. When both particles coexisted on the droplet surface, the large NPs compromised the ordered assembly among small NPs. As opposed to Lin et al.,16,17 Tarimala and Dai did not report size-dependent phase separation on the droplet surface, which could be due to slow diffusion.
Reincke et al.,21 among others, demonstrated that the adsorption of NPs at the water/oil interface shows a balance between the NPs’ chemical potential in the bulk aqueous phase and that at the interface. Both van der Waals and electrostatic interactions contribute to these effects. When Reincke et al. compared their theoretical predictions to experiments, they observed that the results were dependent on particle size. They reported a switchable interfacial self-assembly of small carboxylic acid-functionalized gold NPs of size <10 nm when increasing the aqueous solution pH from 2 to 9. They also reported evidence of hysteresis effects, as once the large NPs adsorbed at the interface, changing the solution pH was not sufficient to desorb them.
Others used external stimuli to control NP–NP interactions. Luo et al.,1e.g., considered gold NPs of size 5 and 10 nm at the planar toluene–water interface. The NP surface was modified using ion-pair complexes, so that changing the aqueous solution pH could affect the NPs’ interactions. For both NP sizes, they found that at pH > 10, the NP–NP interactions become so repulsive that the NPs desorbed from the interface. At pH ≤ 10, the NPs assembled at the interface yielding films, probably monolayers, with a mirror-like reflectance. The reflective properties of the film could be manipulated by controlling the interparticle distances, which decrease when the ionic strength increases from 0.1 to 5 mM.
Controlling NP–NP interactions at interfaces could affect the stability of Pickering emulsions as well. For example, in drug delivery applications, pH changes could trigger the release of therapeutics.22 However, effective NP–NP interactions are the result of multiple phenomena, which might occur simultaneously. For example, McClements and co-workers23,24 reported extensive droplet flocculation and aggregation near the isoelectric point of particles adsorbed on droplet surfaces, leading to creaming instability. Other experiments showed an enhancement in the emulsion stability against creaming due to the formation of a 3D network of interconnected particles and emulsion droplets above a certain electrolyte concentration.25
To quantify how NPs adsorbed at interfaces respond to changes in the effective NP–NP interactions, computational studies could be useful, as they allow practitioners to modify systematically individual parameters and test the effect of such changes. While atomistic molecular simulations provide useful information on the properties of individual particles at fluid–fluid interfaces,26 to investigate emergent effects, one needs to implement coarse-grained simulations or other approaches.27–29 For example, Qin and Yong30,31 using a bespoke coarse-grained simulation approach that accounts for electrostatic interactions, were able to describe a disorder-to-order phase transition in the arrangement of functionalized NPs on a planar fluid interface upon increasing the particles’ degree of ionization. In a further study,32 the same group found that it is possible to induce the desorption of NPs from the droplet surface when a high degree of ionisation is combined with high surface coverage. These findings were found to be in agreement with experimental observations. As another example, Cerbelaud et al.,33 using Brownian dynamics, investigated the self-assembly of NPs on octane droplets dispersed in water upon changing the density of carboxylic acid fragments adsorbed on the NPs’ surface. At low carboxylic acid densities, the particles yield a homogeneous layer and remain well dispersed due to repulsive interactions. Higher carboxylic acid densities reduce the repulsive forces between NPs, leading to the formation of closely packed arrays on the surface of the droplets. A further increase in carboxylic acid density results in extensive agglomeration among NPs, both on the droplet surface and in the continuous aqueous phase.
Schwenke et al.34 combined Monte Carlo and molecular dynamics simulations with experiments to investigate the self-assembly of NPs at planar water/oil interfaces. They considered both mono- and poly-dispersed NPs. They studied the kinetics of NP adsorption at the interface by measuring the surface coverage as a function of time. When starting with a high NPs concentration in the bulk, the rate of adsorption was initially fast, but then it slowed down once a plateau in the estimated surface coverage was approached. However, once the adsorbed NPs organised, more NPs adsorbed from the bulk, filling in available defects on the interfacial NP film.
To complement the previous investigations, we implement here Dissipative Particle Dynamics (DPD) simulations to (1) quantify the effect of particle size on the NP contact angle and in-plane diffusion at planar interfaces and compare our results to available experimental data;15,19 (2) describe the effect of NP size on the arrangement of mono-dispersed NPs on a curved oil/water interface as a function of the effective NP–NP interactions, with emphasis on the transition of NP–NP interactions from repulsive to attractive and on available experimental observations;20,23–25 and (3) investigate the adsorption and the arrangement of poly-dispersed NPs on droplet surfaces, with emphasis on possible competing effects and on the comparison against available experimental observations.16,17,20
The remainder of the manuscript is organized as follows: in Section 2, we briefly summarise the parameters and computational details. In Section 3, we present our main simulation results and we discuss how they favourably compare to experimental and theoretical observations from the literature. We then conclude by highlighting the main outcomes of this work.
2. Methods and algorithms
In this work, the Dissipative Particle Dynamics (DPD)35–38 technique was employed. The methodology and the parameterization details are fully described in our prior work.39,40 All simulations presented here were conducted using the simulation package LAMMPS,41 with the isothermal pair style DPD force fields set at a scaled temperature of kBT = 1. All simulations were conducted in the NPH ensemble, with a constant number of particles, N, constant pressure, P, and constant enthalpy, H. The temperature is maintained constant because of the use of the DPD pair-style. The volume of the box was adjusted to maintain a pressure similar to the one obtained when only water beads are present in a simulation box with a density of 3 beads per rc3.The degree of coarse graining (Nm) was chosen to be 5, which means that each DPD water bead represents 5 water molecules. Distances are measured in terms of the cut-off distance (rc), which in reduced units is 1. According to the relation  , rc ∼ 7.66 Å. The radius of one DPD bead is equal to 0.43rc, which is equivalent to 3.3 Å. As explained elsewhere,39,40 in our model, the simulation time scale τ = 136.3 ps was obtained by fitting the self-diffusion coefficient of water in DPD simulations to the experimental one. All the DPD interaction parameters implemented here are listed in Table 1. The interaction potential between water and oil beads is taken from our previous study,39 where we showed that our parameters are able to reproduce the water–benzene interfacial tension. As we intend to model hydrophilic NPs, the repulsion parameter between water and NP beads (aNP–Water) is chosen equal to the self-repulsion parameter (131.5kBT/rc), whereas the interaction between the NP and oil beads is relatively more repulsive, with aNP–Oil = 150kBT/rc, as shown in Table 1. aNP–NP was set equal to 1, 50, 131.5 and 200kBT/rc to mimic the changes in the NP–NP interactions observed in experiments.
, rc ∼ 7.66 Å. The radius of one DPD bead is equal to 0.43rc, which is equivalent to 3.3 Å. As explained elsewhere,39,40 in our model, the simulation time scale τ = 136.3 ps was obtained by fitting the self-diffusion coefficient of water in DPD simulations to the experimental one. All the DPD interaction parameters implemented here are listed in Table 1. The interaction potential between water and oil beads is taken from our previous study,39 where we showed that our parameters are able to reproduce the water–benzene interfacial tension. As we intend to model hydrophilic NPs, the repulsion parameter between water and NP beads (aNP–Water) is chosen equal to the self-repulsion parameter (131.5kBT/rc), whereas the interaction between the NP and oil beads is relatively more repulsive, with aNP–Oil = 150kBT/rc, as shown in Table 1. aNP–NP was set equal to 1, 50, 131.5 and 200kBT/rc to mimic the changes in the NP–NP interactions observed in experiments.
| Water | Oil | Nanoparticle | |
|---|---|---|---|
| Water | 131.5 | 171.43 | 131.5 |
| Oil | 131.5 | 150 | |
| Nanoparticle | 1/50/131.5/200 |
For the simulations conducted here, we consider hollow spherical NPs of two diameters. The DPD beads are distributed over surfaces with diameter 1rc and 2rc for small and large NPs, respectively. As shown in Fig. 1, the resultant diameter of the NPs is 1.86 and 2.86rc, respectively, which correspond to ∼1.4 nm (small NP) and ∼2.2 nm (large NP). To cover small and large NPs, 50 and 200 beads are required, respectively, yielding a surface density of ∼16 beads per rc2 on the NP surface. This is sufficient to prevent other beads (e.g., solvent beads) from entering the hollow NPs, which would be unphysical, as documented elsewhere.18 One additional bead is located at the centre of each NP; its position is used for estimating, for example, the NP diffusion coefficients.
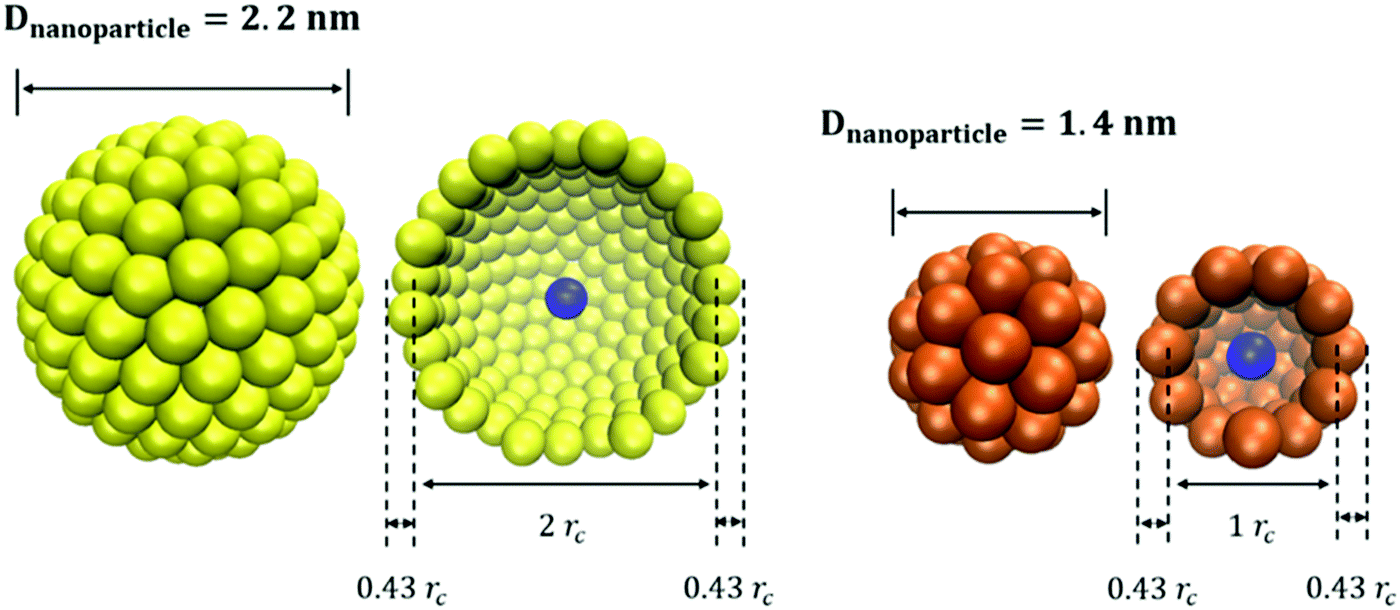 | ||
| Fig. 1 Schematic representation of small and large NPs with diameter 1.86rc (orange) and 2.86rc (yellow), respectively. The DPD beads are placed on the NP surface, achieving a surface density of ∼16 beads per rc2. | ||
The three-phase contact angles of the NPs are measured in rectangular simulation boxes of dimension 20 × 20 × 40 (Lx × Ly × Lz) with the NP placed at the interface between water and oil, as shown in Fig. 2. To estimate the three-phase contact angle (θC), we calculated the fraction of the spherical NP surface area that is wetted by water according to:42,43
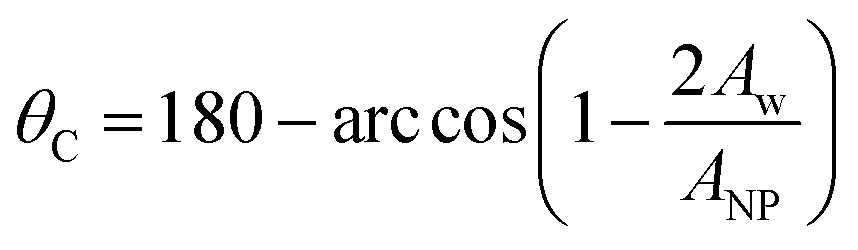 | (2) |
 | ||
| Fig. 2 Small (left) and large (right) NPs adsorbed at the planar oil (grey)/water (blue) interface. | ||
In eqn (2), Aw is the area of the NP surface that is immersed in the water phase. A schematic representation of the algorithm employed to compute the three-phase contact angle is provided in Fig. S1 in the ESI.† The ratio Aw/ANP is obtained by dividing the number of NP surface beads immersed in the water phase by the total number of beads on the NP surface. One surface bead is considered to be immersed in water if a water bead is the solvent bead nearest to it. The contact angle was estimated over 1 × 106 simulation steps (equivalent to 5.45 μs), during which one frame was collected every 1000 steps, yielding 1000 frames for our analysis. The contact angle was averaged over these 1000 frames.
The diffusion of NPs at the water/oil interface was also studied. For these calculations, we estimated the mean squared displacement (MSD) of a single NP adsorbed at a planar water/oil interface parallel to the x–y plane. The simulation box dimensions are 20 × 20 × 40 (Lx × Ly × Lz). For each particle size, the MSD is an average over 30 simulations conducted for 5.45 μs. The simulated diffusion coefficient is estimated according to:
 | (3) |
To study the arrangement of monodispersed NPs on a curved surface, we considered an oil droplet with diameter 26.15rc, which corresponds to ∼20 nm. At the beginning of each simulation, the NPs were randomly placed at the droplet surface. For the small NPs, we simulated systems with either 200 or 430 NPs on the droplet. For the large NPs, we simulated three systems with 128, 200, and 250 NPs on the droplet surface, respectively, as shown in Fig. 3. The dimensions of the simulation boxes used for each system are listed in Table 2. The simulation box size was adjusted to prevent unphysical interactions between NPs adsorbed on the same droplet under the periodic boundary conditions. These simulations were conducted for up to 65 μs with no obvious change in the NPs’ arrangement over the last 44 μs.
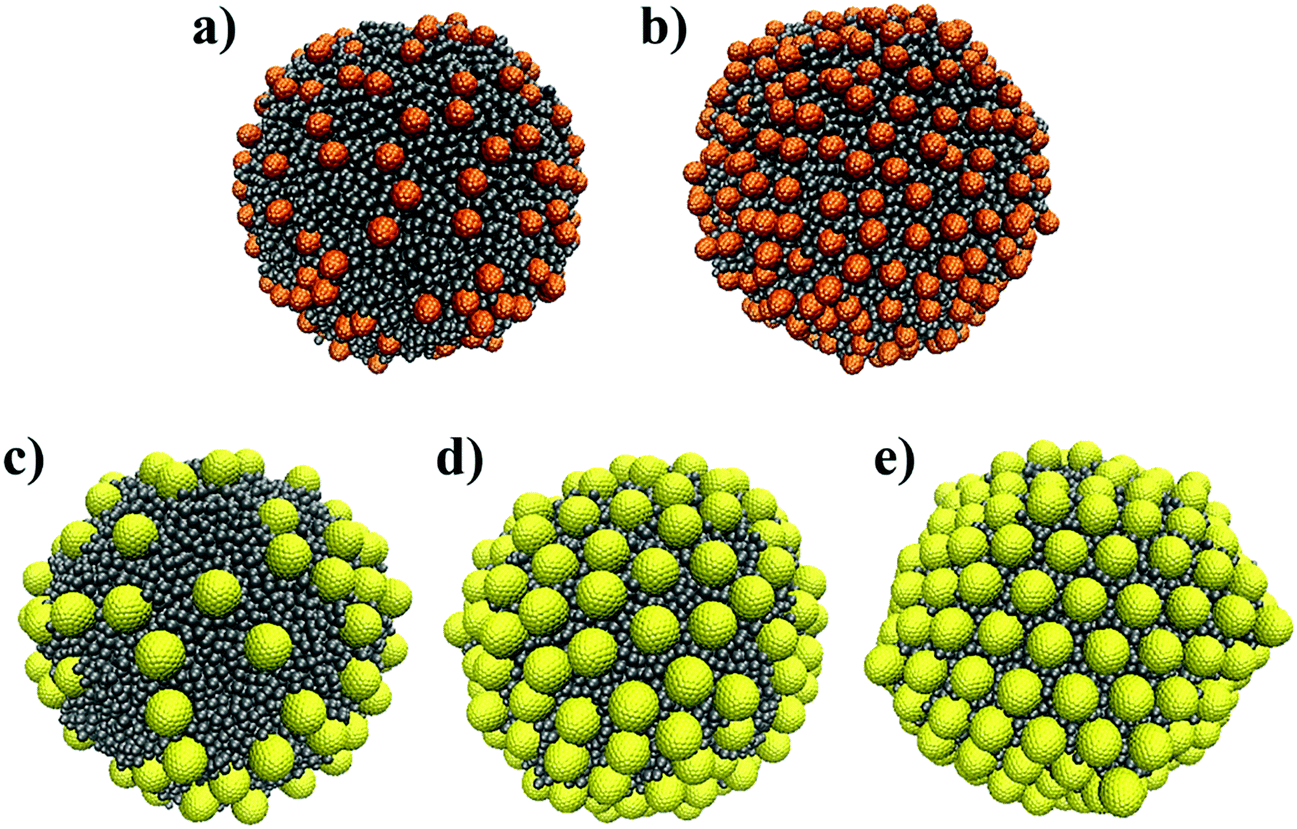 | ||
| Fig. 3 Initial configurations of 200 (a) and 430 (b) small NPs and 128 (c), 200 (d) and 250 large NPs arranged on an oil droplet (grey) immersed in water. Water beads are not shown for clarity. | ||
| Number of small NPs | Number of large NPs | Number of oil beads | Number of water beads | Dimensions of simulation box (Lx × Ly × Lz) |
|---|---|---|---|---|
| 200 | — | 27988 |
111980 |
39 × 36 × 39 |
| 430 | — | 27988 |
111980 |
38 × 36 × 40 |
| — | 128 | 27988 |
111980 |
36 × 36 × 40 |
| — | 200 | 27988 |
111980 |
39 × 36 × 39 |
| — | 250 | 27988 |
245387 |
45 × 45 × 45 |
| 200 | 200 | 27988 |
111980 |
39 × 37 × 39 |
Finally, we studied the adsorption and structure of poly-dispersed NPs on the droplet surface. For this study, aNP–NP was set to 131.5kBT/rc, because larger values lead to strong repulsion between NPs, while smaller values lead to strong attraction between NPs, as discussed in the results section. We considered systems of 200 small NPs and 200 large NPs. The simulation box dimensions are listed in Table 2. Three different initial configurations were considered, in all of which one oil droplet was immersed in water, as shown in Fig. 4. In the first initial configuration, all the NPs were dispersed in bulk water. In the second initial configuration, the small NPs were adsorbed on the droplet while the large NPs were dispersed in bulk water. In the third initial configuration, the large NPs were adsorbed on the droplet while the small NPs were dispersed in bulk water. Each of these systems was simulated for 403 μs.
 | ||
| Fig. 4 The three initial configurations for systems with poly-dispersed NPs adsorbing on an oil droplet dispersed in water: (a) all the NPs were dispersed in bulk water; (b) small NPs were on the droplet, while the large NPs were in bulk water; and (c) large NPs were on the droplet, and the small ones were in bulk water. | ||
3. Results
3.1. Effect of NP size on the structure and dynamics at water–oil interfaces
The three-phase contact angle (θC) values of the NPs are calculated at the planar oil/water interface, starting from the configurations shown in Fig. 2. In Fig. 5, we report the estimated contact angle for both NPs over 5.45 μs. We observe a fluctuation in the estimated contact angle values over the simulation time. These fluctuations reflect the perpendicular movement of NPs at the interface, due to the thermal fluctuations, in agreement with experimental observations.15 The resultant contact angles are ∼80° ± 11° and 81° ± 8° for small and large NPs, respectively. From the error bars observed, we can see that the small NPs are more sensitive to thermal fluctuations at the interface compared to the large ones. This is in agreement with eqn (1), which shows that the adsorption energy increases with the particle radius.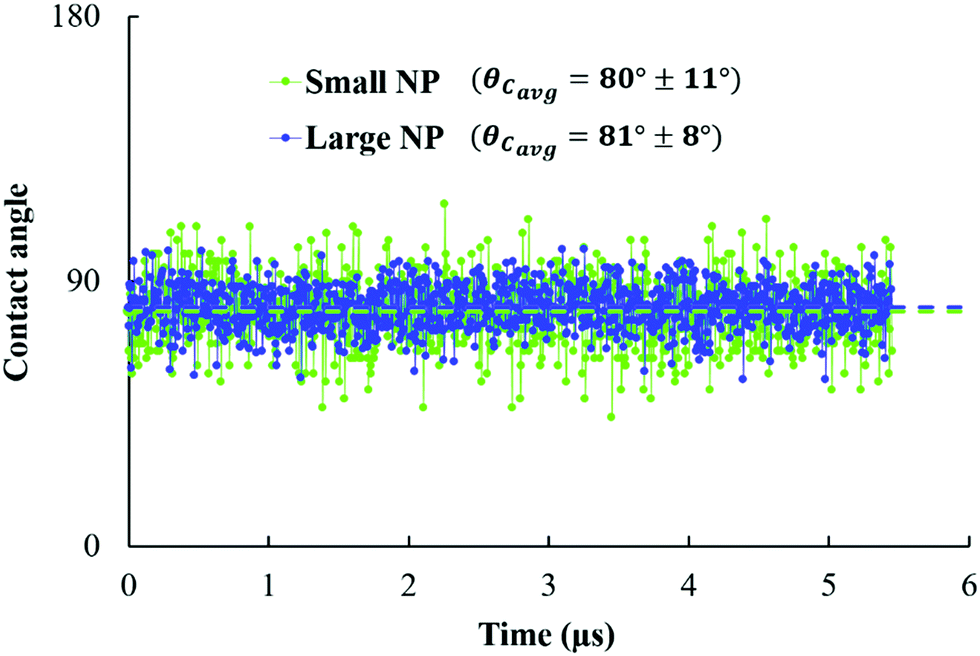 | ||
| Fig. 5 Contact angle estimated over 1000 frames (equivalent to 5.45 μs) for small and large NPs (green and blue, respectively) adsorbed at planar water/oil interfaces. | ||
In Fig. 6, we present the calculated mean squared displacement (MSD) as a function of time for small and large NPs at planar interfaces, averaged over 30 simulations. From these results, the diffusion coefficient of small and large NPs equals 9 ± 6 × 10−7 cm2 s−1 and 5.5 ± 3.75 × 10−7 cm2 s−1, respectively. As expected, large NPs are less diffusive than smaller ones, in qualitative agreement with experimental observations.19 It should be noted that the change in the diffusion coefficient with the NP radius as predicted by our simulations is consistent with the Stokes–Einstein relation for the diffusion of spherical particles in uniform mediums.
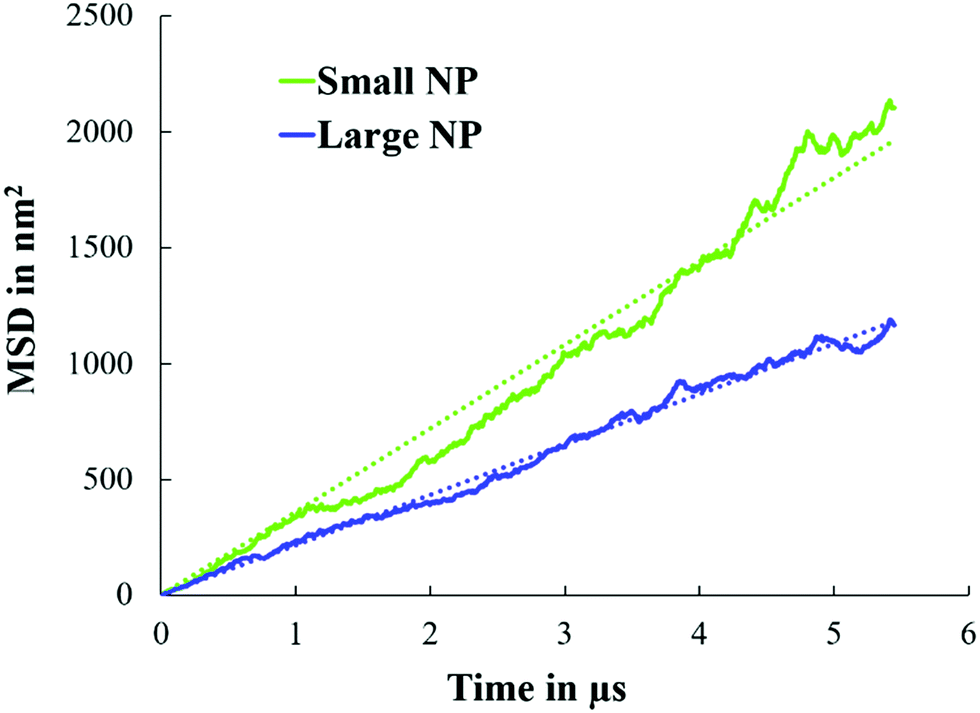 | ||
| Fig. 6 MSD as a function of simulation time for small and large NPs at water/oil planar interfaces. For these simulations, single NPs were considered (see Fig. 2). | ||
3.2. NP aggregates on oil droplets: effects of NP size and NP–NP interactions
To quantify the structure of NP aggregates on oil droplets, we prepared several initial configurations, as well as several NP densities on the droplet surfaces. Starting from the initial configurations shown in Fig. 3, we conducted equilibrium simulations. In addition to varying the surface density, we also changed the effective NP–NP interactions. We considered the aNP–NP values reported in Table 1 (i.e., from 1 to 200 kBT/rc). These values reflect conditions at which the effective NP–NP interactions range from strongly attractive to strongly repulsive. We discuss the results first for the small NPs, and then for the large ones. | ||
| Fig. 7 Simulation snapshots of the arrangement of 430 small NPs on the surface of the oil droplet, when changing aNP–NP to 200, 131.5, 50 and 1kBT/rc from left to right, respectively. Snapshots were taken after 65 μs. | ||
When aNP–NP is reduced to 131.5kBT/rc, the small NPs form patches on the droplet surface separated by particle-free domains, as shown in Fig. 7. Hexagonal order is observed in some of the patches, although such an order is not pervasive. The structure just described becomes less evident when the NP surface coverage decreases to 200 NPs, as shown in Fig. S2 in the ESI.† Similar monolayers were observed in several experiments.20,44,45
When aNP–NP is reduced to 50kBT/rc, the small NPs yield very dense clusters. In some of these clusters, a bilayer is formed by the NPs, while in others, hexagonally ordered structures are observed. A representative snapshot is provided in Fig. 8.
 | ||
| Fig. 8 Formation of a second NP layer on the oil droplet surface when aNP–NP is equal to 50kBT/rc. Snapshots were taken after 65 μs. | ||
When aNP–NP is further reduced to 1kBT/rc, the very attractive NPs yield 3D clusters (flocs) on the droplet surface. Snapshots of a few such flocs are provided in Fig. 9 and in Fig. S3 in the ESI.† Each floc is formed by a different number of NPs. In Fig. 10, we present the number distribution of NPs in the flocs when 200 or 430 NPs are present on the droplet. Under the conditions chosen, it could have been expected that all the NPs would agglomerate into a single aggregate, adsorbed at the oil–water interface. However, the formation of multiple flocs dispersed on the droplet interface contributes to lowering the interfacial energy due to the water–oil interactions (i.e., see eqn (1)). Thus, a balance between the two driving forces may give rise to the formation of flocs of different sizes dispersed on the droplet surface. Further, because the size distribution of the flocs is rather wide, it is possible that the floc size is also affected by the diffusion of the particles, as well as that of the flocs, on the droplet surface.
 | ||
| Fig. 9 3D flocs created by small NPs at the droplet surface when aNP–NP = 1kBT/rc. Snapshots were taken after 65 μs. The insets show top views of each floc. | ||
 | ||
| Fig. 10 Number distribution of small NPs in flocs in the presence of 200 (left) and 430 (right) NPs on the droplet surface when aNP–NP is equal to 1kBT/rc. The flocs obtained during the last 20 μs of the simulations were used for analysis. | ||
From the findings just summarised, we conclude that when aNP–NP is high, the NPs repel each other and uniformly cover the droplet, while when aNP–NP is low, the NPs attract each other and form 3D flocs. When aNP–NP = 131.5kBT/rc, an intermediate behaviour is observed; atb the corresponding conditions, the NPs begin to assemble. The transition in the NPs’ arrangement from a dispersed structure, to patches, and then to 3D agglomerates resembles the simulation results reported by Cerbelaud et al.,33 when the NP interaction changed from repulsive to attractive with the increase in the carboxylic acid density on the NP surfaces.
 | ||
| Fig. 11 Simulation snapshots of the arrangement of 250 large NPs on the surface of the oil droplet, changing aNP–NP from 200 to 131.5, 50 and 1kBT/rc from left to right, respectively. All results were obtained at the same temperature but following two simulation protocols for snapshots in panels (a) and (b), respectively. See narrative for details. | ||
In detail, when aNP–NP equals 200kBT/rc, the large NPs distribute rather uniformly on the droplet (see also results in Fig. S4 of the ESI†). This result does not seem to depend on the simulation procedure nor the initial configuration. At the other extreme, when aNP–NP equals 1kBT/rc, the NP–NP interactions are very attractive, and the NPs yield 3D flocs on the droplet surface. This result does not depend on initial configuration nor simulation protocol. Snapshots of a few 3D flocs are provided in Fig. 12 and in Fig. S5 of the ESI.† Each floc can contain up to 14 NPs when there are 250 NPs on the droplet surface. At a lower surface density, the flocs are still present, but they contain, for the most part, 4 NPs. In some cases, they contain 7 large NPs. The number distribution of NPs in the flocs is shown in Fig. 13. It is evident that the flocs formed by the large NPs are smaller, overall, than those formed by the small NPs. This could be due to the fact that for large NPs, the driving force to form small flocs dispersed on the droplet is stronger than that leading lo large aggregates. The results in Fig. 13 suggest that the large NPs tend to form flocs of ∼4 NPs. Sometimes, these flocs merge, yielding larger aggregates. Perhaps this is indicative of the mechanism by which the flocs form, although much deeper statistical analysis should be conducted to verify this possibility.
 | ||
| Fig. 12 3D flocs formed by large NPs at the droplet surface when aNP–NP = 1kBT/rc. Snapshots were taken after 65 μs. The insets show top views of each floc. | ||
 | ||
| Fig. 13 Number distribution of 200 (left) and 250 (right) large NPs in flocs on the droplet surface when aNP–NP is equal to 1kBT/rc, at a scaled temperature kBT = 1. The analyses were performed on the flocs obtained during the last 20 μs of the simulations. | ||
For the simulations conducted at intermediate values of the aNP–NP parameter (131.5 and 50kBT/rc), the structures formed by the large NPs depend on the simulation sequence. Some small patches or networks are visible, especially at aNP–NP = 50 kBT/rc, and some NPs desorb from the interface, with the effect being more pronounced as both the surface density and the repulsive parameter increase (more details are shown in Fig. S4 of the ESI†). In some cases, specifically when the NPs are provided with increased thermal energy for a short period of time at the beginning of the simulations (bottom panels in Fig. 11), patches of NPs are obtained, which are similar to those observed for the small NPs. Analysis of the simulation trajectories suggests that once NP aggregates are formed, they persist in our simulations.
Comparing the results obtained for small and large NPs, we conclude that large NPs are more likely to form metastable structures on a droplet surface. This could be due to the lower mobility of the large NPs (e.g., see Fig. 6), stronger adsorption at the interface (e.g., see eqn (1) and Fig. 5), and stronger NP–NP adhesion forces.
To quantify the results discussed so far, in Table S1 in the ESI,† we report the difference in the total energy of selected simulated systems due to the adsorption of either small or large NPs on an oil droplet dispersed in water. The results confirm that NP adsorption lowers the total system energy because, as shown in eqn (1), it reduces the contact area between the droplet surface and the bulk water. The results in Table S1 (ESI†) suggest that large NPs have a more pronounced effect than small NPs.
3.3. Poly-dispersed NPs
In this section, we investigate poly-dispersed NP systems. We introduce 200 ‘small’ and 200 ‘large’ NPs near the surface of the oil droplet in water. The initial configurations shown in Fig. 4 are used for comparison. These simulations were conducted using aNP–NP = 131.5kBT/rc. This value was chosen because larger values lead to strong repulsion between NPs, while smaller values lead to strong attractions between NPs, especially for small NPs. As shown in Section 3.2.1, this interaction parameter value is intermediate between effectively repulsive and attractive interactions between the NPs. However, the corresponding results for the large NPs suggest that using this parameter value can yield metastable results. In fact, several NP aggregates, as obtained from different initial configurations, are shown in Fig. S6–S8 of the ESI.† These snapshots suggest that the results depend on the initial configurations.When all NPs are initially in the bulk, or when the small NPs are initially on the droplet surface, the small NPs yield aggregates reminiscent of those observed when only small NPs are present on the droplet (see Fig. 7), while the large NPs occupy the empty spaces separating the clusters of small NPs. In Fig. 14 (bottom left panel), we show the final structure, after 403 μs of simulations, which suggests some sort of size segregation between the NPs on the droplet surface. At the end of the simulations, some NPs of both sizes are found in the bulk water. Assuming that the contact angle shown in Fig. 5 does not depend on the curvature of the interface, nor on NP–NP interactions, we estimate that the final surface coverage obtained from these initial configurations (left and middle in Fig. 4) is ∼74%.
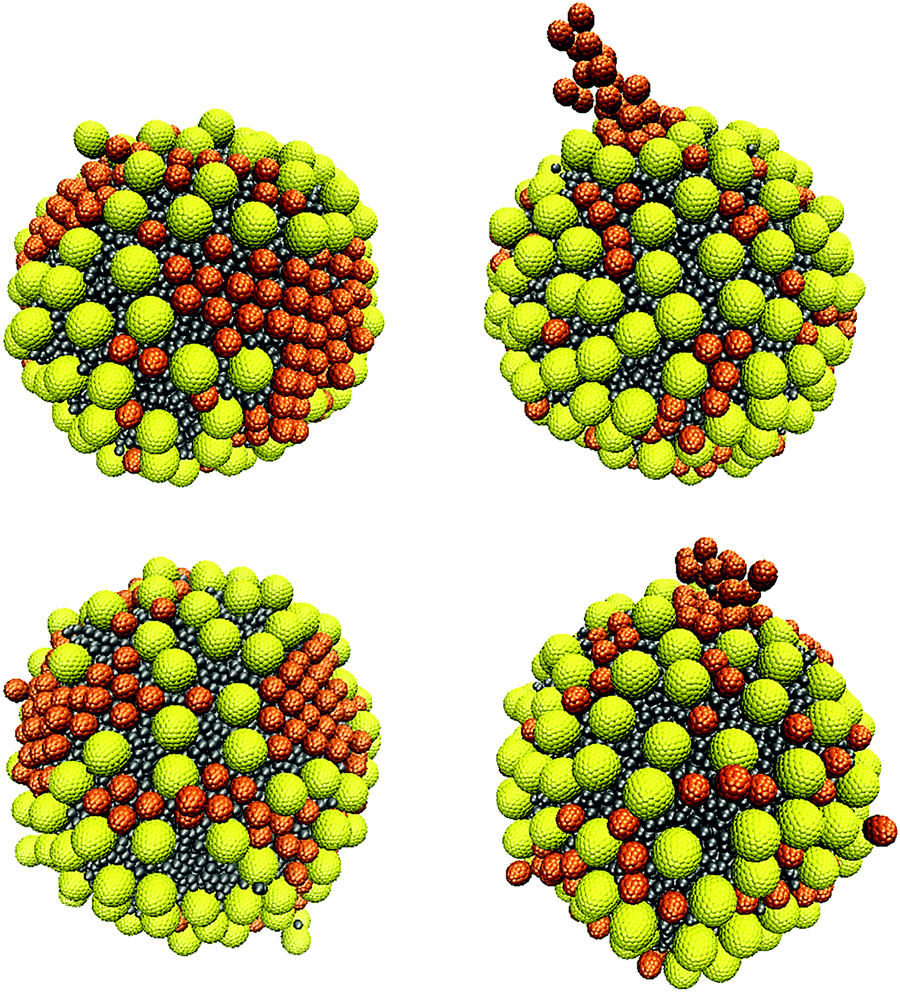 | ||
| Fig. 14 Simulation snapshots of the arrangement of a mixture of small and large NPs, when starting with the initial configuration shown in Fig. 4(a) and (c) from left to right, respectively. Snapshots were taken after 164 μs (top) and 403 μs (bottom). The simulations were conducted using the parameter aNP–NP = 131.5kBT/rc. | ||
Experimental results reported by Lin et al.16,17 are consistent with the possibility that NPs of different sizes segregate on a droplet interface. In our simulations, a complete phase separation is not observed, which might be due to the relatively short simulation times compared to experiments, the small size of the droplets simulated as well as that of the simulated system, the possibility that the system is trapped in local minima in the free energy landscape, and the possibility that the interaction parameters implemented in our models do not precisely match the experimental conditions.
It should, however, be noted that when we start the simulations from a different initial configuration, i.e., with the large NPs adsorbed on the oil droplet surface (panel (c) in Fig. 4), the final configuration observed differs somewhat from the one just described. The small NPs reach the droplet surface either particle-by-particle or as clusters. Single particles tend to occupy the interparticle spaces between large NPs on the droplet surface, while a small NP cluster adsorbing on the droplet can gradually push the large NPs until all the small NPs in the cluster adsorb, as shown in Fig. S9 of the ESI.† When clusters of small NPs adsorb on the droplet, it is possible that they remain ‘dangling’ towards the bulk, as shown in Fig. 14 (right), as well as in Fig. S8 of the ESI.† As observed for the other two initial configurations, even in this case, some NPs remain in the bulk, and the estimated surface coverage is ∼77.8% at the end of the simulations, which is somewhat larger than what was mentioned above. In some regions, high NP packing on the droplet surface leads to changes in contact angle, which is probably due to crowding effects.46 It is possible that the right panel in Fig. 14 differs from the left one because the large NPs distributed over the droplet surface delay the mobility of the small NPs on the droplet surface via, e.g., caging, which would prevent size segregation within the length of our simulations. Dai et al.20 also reported that large NPs could prevent the formation of ordered structures of small NPs. Such effects could become more pronounced as the surface coverage increases, as suggested by prior simulation results.18
Noting that the final surface coverages for the structures observed in Fig. 14 are similar, we monitored the adsorption kinetics of small and large NPs on the droplet surface, starting from the initial configurations shown in Fig. 4. As shown in Fig. 15(a), when both NPs are initially in the bulk, the small NPs reach the droplet surface first. When the small NPs are initially on the droplet surface, the results shown in Fig. 15(b) suggest that the fast initial adsorption of the large NPs causes the displacement of some small NPs during the first few μs of simulations. After that, the large NPs continue to adsorb, slowly (see, e.g., plateaus such as the one from 207 to 240 μs of simulations), showing evidence of bursts of additional adsorption. During the latter events, a few small NPs desorbed, suggesting that large NPs are preferentially adsorbed under those conditions. However, when the large NPs are initially on the droplet, and the small ones are in the bulk, the results in Fig. 15(c) show that as the small NPs adsorb, some of the large ones desorb, suggesting that a competition between the two NPs for adsorption indeed takes place in the investigated system. This competition, combined with the slow mobility of NPs within a crowded interface, is probably the reason why the systems considered seem to sample a free energy landscape characterised by deep local minima, from which the system is not always able to escape. It is, however, interesting to observe that the rate of adsorption of the small NPs on the droplet covered by large NPs is much slower than that observed for all the other systems (compare Fig. 15(c) to (a) and (b)), especially at the beginning of the simulations, suggesting that the large NPs are more strongly adsorbed at the interface, as expected based on eqn (1), and that perhaps they also prevent the mobility of both small and large NPs on the droplet interface. The general trend of the results shown in Fig. 15, namely a fast initial adsorption rate followed by a much slower adsorption rate, is consistent with the simulation results reported by Schwenke et al.34 In particular, this group showed that when the adsorption rate is fast (i.e., when the NP concentration in the bulk is high), a pseudo plateau is observed in the amount of NPs adsorbed, which is followed by additional adsorption after some time, presumably because the adsorbed NPs require some time to rearrange on the surface. Our results suggest that a few such events can occur during a long simulation, because of successive relaxations of the adsorbed NPs (see, e.g., Fig. S9 in the ESI†).
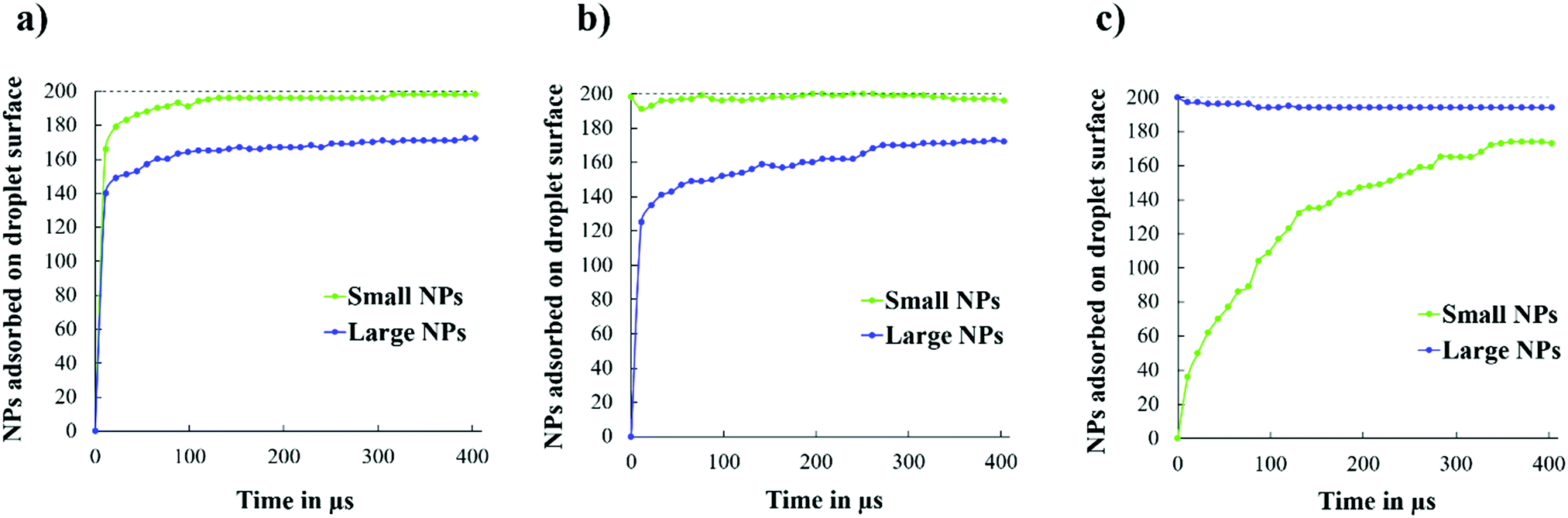 | ||
| Fig. 15 Number of small and large NPs adsorbed on the droplet surface as a function of simulation time, starting from the three initial configurations shown in Fig. 4. | ||
Certainly, the simulation results strongly depend on the interaction parameters. For example, to quantify the possibility that size segregation is important in determining the final aggregates for poly-dispersed NPs on a droplet, one could choose interaction parameters that lead to strong repulsions between the NPs. We conducted sample simulations for systems similar to those shown in Fig. 14 but imposing aNP–NP = 200kBT/rc. We conducted 40 μs-long simulations starting from the initial configuration shown in Fig. 4(a). The results are shown in Fig. 16. In agreement with previous results, as well as with expectations, size segregation was observed.
 | ||
| Fig. 16 Simulation snapshots illustrating the arrangement of a mixture of small and large NPs, taken from different angles, when starting from the initial configuration shown in Fig. 4(a). Snapshots were taken after 40 μs. The simulations were conducted using the parameter aNP–NP = 200kBT/rc. | ||
4. Conclusions
Nanoparticles (NPs) of two sizes were simulated on oil droplets dispersed in water. The coarse-grained dissipative particle dynamics formalism was implemented, which allowed us to simulate the systems considered for up to several hundreds of micro-seconds. This allows for semi-quantitative comparison to experimental observations available in the literature, although the simulated systems are smaller than the experimental ones, and the force fields represent significant simplifications of the reality.When mono-dispersed NPs were considered, the simulations showed larger fluctuations in the contact angle for small NPs than for large NPs, which is consistent with the adsorption energy increasing with the square of the particle radius. The simulations also showed that small NPs diffuse faster at the oil–water interface than the large NPs, consistent with expectations based on the Stokes–Einstein relation for particles diffusing in homogeneous fluids. When the NPs are adsorbed on curved interfaces, the NP–NP interactions determine the structure of the resultant aggregates. Small NPs yield uniform distributions on the droplet surface, interconnected patches of NPs with some evidence of order, incipient multi-layered aggregates, and eventually small 3-dimensional clusters of NPs as the effective NP–NP interactions transition from strongly repulsive to strongly attractive. The aggregates formed by large NPs seem to follow the same trend, although evidence of multi-layered structures was not observed, and analysis of the simulation results suggests that large NPs could in some cases be trapped in local minima of the free-energy landscape.
When equimolar mixtures of NPs with different sizes were simulated, the simulation results suggest that size segregation is likely to occur. The simulation results are dependent on the initial configurations, possibly because of the strong adsorption energy of large vs. small NPs at the oil–water interface. Differences in diffusion coefficients both in the bulk and at the interface yield differences in the kinetics of adsorption of the two NPs, which can be linked with the structure of the final NP aggregates obtained on the droplet surface.
The present results are in general agreement with experimental observations reported in the literature, which suggests that coarse-grained simulations within the formalism implemented here could be useful for interpreting experiments as well as for designing practical approaches to advance applications in materials sciences.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous allocations of computing time were provided by the University College London (UCL) Research Computing Platforms Support (LEGION), and the National Energy Research Scientific Computing Center (NERSC) at the Lawrence Berkeley National Laboratory. The NERSC is supported by the DOE Office of Science under Contract DE-AC0205CH11231. The authors acknowledge financial support from the Department of Chemical Engineering at UCL.References
- M. X. Luo, G. K. Olivier and J. Frechette, Soft Matter, 2012, 8, 11923–11932 RSC.
- R. J. G. Lopetinsky, J. H. Masliyah and Z. Xu, in Colloidal Particles at Liquid Interfaces, ed. P. B. Bernard and S. H. Tommy, Cambridge University Press, Cambridge, 2006, ch. 6, pp. 186–224 Search PubMed.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- A. Maestro, E. Santini, D. Zabiegaj, S. Llamas, F. Ravera, L. Liggieri, F. Ortega, R. G. Rubio and E. Guzman, Adv. Condens. Matter Phys., 2015, 917516 Search PubMed.
- B. Madivala, J. Fransaer and J. Vermant, Langmuir, 2009, 25, 2718–2728 CrossRef CAS.
- E. P. Lewandowski, M. Cavallaro, L. Botto, J. C. Bernate, V. Garbin and K. J. Stebe, Langmuir, 2010, 26, 15142–15154 CrossRef CAS.
- X. C. Luu and A. Striolo, J. Phys. Chem. B, 2014, 118, 13737–13743 CrossRef CAS.
- T. S. Horozov, R. Aveyard, J. H. Clint and B. P. Binks, Langmuir, 2003, 19, 2822–2829 CrossRef CAS.
- A. Maestro, E. Guzman, E. Santini, F. Ravera, L. Liggieri, F. Ortega and R. G. Rubio, Soft Matter, 2012, 8, 837–843 RSC.
- Y. Zhu, J. Z. Jiang, K. H. Liu, Z. G. Cui and B. P. Binks, Langmuir, 2015, 31, 3301–3307 CrossRef CAS.
- C. P. Whitby and E. J. Wanless, Materials, 2016, 9, 626–647 CrossRef.
- F. Tu, B. J. Park and D. Lee, Langmuir, 2013, 29, 12679–12687 CrossRef CAS.
- B. P. Binks and P. D. I. Fletcher, Langmuir, 2001, 17, 4708–4710 CrossRef CAS.
- A. Boker, J. He, T. Emrick and T. P. Russell, Soft Matter, 2007, 3, 1231–1248 RSC.
- L. Isa, F. Lucas, R. Wepf and E. Reimhult, Nat. Commun., 2011, 2, 438 CrossRef.
- Y. Lin, H. Skaff, T. Emrick, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226–229 CrossRef CAS.
- Y. Lin, A. Boker, H. Skaff, D. Cookson, A. D. Dinsmore, T. Emrick and T. P. Russell, Langmuir, 2005, 21, 191–194 CrossRef CAS.
- X.-C. Luu, J. Yu and A. Striolo, Langmuir, 2013, 29, 7221–7228 CrossRef CAS.
- D. P. Wang, S. Yordanov, H. M. Paroor, A. Mukhopadhyay, C. Y. Li, H. J. Butt and K. Koynov, Small, 2011, 7, 3502–3507 CrossRef CAS.
- S. Tarimala and L. L. Dai, Langmuir, 2004, 20, 3492–3494 CrossRef CAS.
- F. Reincke, W. K. Kegel, H. Zhang, M. Nolte, D. Y. Wang, D. Vanmaekelbergh and H. Mohwald, Phys. Chem. Chem. Phys., 2006, 8, 3828–3835 RSC.
- C. L. G. Harman, M. A. Patel, S. Guldin and G. L. Davies, Curr. Opin. Colloid Interface Sci., 2019, 39, 173–189 CrossRef CAS.
- K. Demetriades, J. N. Coupland and D. J. McClements, J. Food Sci., 1997, 62, 342–347 CrossRef CAS.
- A. Kulmyrzaev, R. Chanamai and D. J. McClements, Food Res. Int., 2000, 33, 15–20 CrossRef CAS.
- T. S. Horozov, B. P. Binks and T. Gottschalk-Gaudig, Phys. Chem. Chem. Phys., 2007, 9, 6398–6404 RSC.
- H. Fan, D. E. Resasco and A. Striolo, Langmuir, 2011, 27, 5264–5274 CrossRef CAS.
- N. Wu, D. Lee and A. Striolo, Anisotropic Particle Assemblies: Synthesis, Assembly, Modeling, and Applications, Elsevier, Amsterdam, 2018 Search PubMed.
- X. B. Dai, P. Y. Chen, G. L. Zhu, Z. Y. Xu, X. H. Zhang and L. T. Yan, J. Phys. Chem. Lett., 2019, 10, 7970–7979 CrossRef CAS.
- G. L. Zhu, Z. H. Huang, Z. Y. Xu and L. T. Yan, Acc. Chem. Res., 2018, 51, 900–909 CrossRef CAS.
- S. Y. Qin, J. Kang and X. Yong, Langmuir, 2018, 34, 5581–5591 CrossRef CAS.
- S. Y. Qin and X. Yong, Soft Matter, 2017, 13, 5137–5149 RSC.
- S. Y. Qin and X. Yong, Soft Matter, 2019, 15, 3291–3300 RSC.
- M. Cerbelaud, A. Videcoq, L. Alison, E. Tervoort and A. R. Studart, Langmuir, 2017, 33, 14347–14357 CrossRef CAS.
- K. Schwenke, L. Isa and E. Del Gado, Langmuir, 2014, 30, 3069–3074 CrossRef CAS.
- P. J. Hoogerbrugge and J. M. V. A. Koelman, Europhys. Lett., 1992, 19, 155 CrossRef.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, Inc., Orlando, Florida, 2002 Search PubMed.
- R. D. Groot and K. L. Rabone, Biophys. J., 2001, 81, 725–736 CrossRef CAS.
- A. Khedr and A. Striolo, J. Chem. Theory Comput., 2018, 14, 6460–6471 CrossRef CAS.
- A. Khedr and A. Striolo, J. Chem. Theory Comput., 2019, 15, 5058–5068 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- F. Sicard and A. Striolo, Faraday Discuss., 2016, 191, 287–304 RSC.
- H. Fan and A. Striolo, Soft Matter, 2012, 8, 9533–9538 RSC.
- F. Gautier, M. Destribats, R. Perrier-Cornet, J. F. Dechezelles, J. Giermanska, V. Heroguez, S. Ravaine, F. Leal-Calderon and V. Schmitt, Phys. Chem. Chem. Phys., 2007, 9, 6455–6462 RSC.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2001, 17, 4540–4547 CrossRef CAS.
- F. Sicard and A. Striolo, Nanoscale, 2017, 9, 8567–8572 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation snapshots illustrating the arrangement of NPs on the droplet surface for different surface coverages when changing the NP–NP interaction; 3D flocs of small and large NPs with different NP densities when aNP–NP = 1kBT/rc; simulation snapshots illustrating aggregates of small and large NPs on the droplet surface, after 403 μs, as well as a possible mechanism of NP adsorption connected with rearrangement on the droplet surface. See DOI: 10.1039/d0cp02588g |
| This journal is © the Owner Societies 2020 |