
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProperties of gaseous closo-[B6X6]2− dianions (X = Cl, Br, I)†
Markus
Rohdenburg
 a,
Zheng
Yang
b,
Pei
Su
c,
Eduard
Bernhardt
d,
Qinqin
Yuan
b,
Edoardo
Apra
e,
Simon
Grabowsky
f,
Julia
Laskin
c,
Carsten
Jenne
d,
Xue-Bin
Wang
*b and
Jonas
Warneke
*gh
a,
Zheng
Yang
b,
Pei
Su
c,
Eduard
Bernhardt
d,
Qinqin
Yuan
b,
Edoardo
Apra
e,
Simon
Grabowsky
f,
Julia
Laskin
c,
Carsten
Jenne
d,
Xue-Bin
Wang
*b and
Jonas
Warneke
*gh
aInstitut für Angewandte und Physikalische Chemie, Universität Bremen, Fachbereich 2-Biologie/Chemie, 28359 Bremen, Germany
bPhysical Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: xuebin.wang@pnnl.gov
cDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
dAnorganische Chemie, Fakultät für Mathematik und Naturwissenschaften, Bergische Universität Wuppertal, 42119 Wuppertal, Germany
eEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99352, USA
fDepartement für Chemie und Biochemie, Universität Bern, 3012 Bern, Switzerland
gWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, 04103 Leipzig, Germany. E-mail: jonas.warneke@uni-leipzig.de
hLeibniz Institute of Surface Engineering (IOM), Permoserstraße 15, 04318 Leipzig, Germany
First published on 22nd July 2020
Abstract
Electronic structure, collision-induced dissociation (CID) and bond properties of closo-[B6X6]2− (X = Cl–I) are investigated in direct comparison with their closo-[B12X12]2− analogues. Photoelectron spectroscopy (PES) and theoretical investigations reveal that [B6X6]2− dianions are electronically significantly less stable than the corresponding [B12X12]2− species. Although [B6Cl6]2− is slightly electronically unstable, [B6Br6]2− and [B6I6]2− are intrinsically stable dianions. Consistent with the trend in the electron detachment energy, loss of an electron (e− loss) is observed in CID of [B6X6]2− (X = Cl, Br) but not for [B6I6]2−. Halogenide loss (X− loss) is common for [B6X6]2− (X = Br, I) and [B12X12]2− (X = Cl, Br, I). Meanwhile, X˙ loss is only observed for [B12X12]2− (X = Br, I) species. The calculated reaction enthalpies of the three competing dissociation pathways (e−, X− and X˙ loss) indicated a strong influence of kinetic factors on the observed fragmentation patterns. The repulsive Coulomb barrier (RCB) determines the transition state for the e− and X− losses. A significantly lower RCB for X− loss than for e− loss was found in both experimental and theoretical investigations and can be rationalized by the recently introduced concept of electrophilic anions. The positive reaction enthalpies for X− losses are significantly lower for [B6X6]2− than for [B12X12]2−, while enthalpies for X˙ losses are higher. These observations are consistent with a difference in bond character of the B–X bonds in [B6X6]2− and [B12X12]2−. A complementary bonding analysis using QTAIM, NPA and ELI-D based methods suggests that B–X bonds in [B12X12]2− have a stronger covalent character than in [B6X6]2−, in which X has a stronger halide character.
1. Introduction
Polyhedral closo-borate dianions with the general formula [BnXn]2− are among the most famous boron-based molecules. The best explored derivatives are the closo-dodecaborate dianions (n = 12) with exceptional physico-chemical properties. As a weakly coordinating anion, [B12Cl12]2− has been used to stabilize highly reactive cations.1–4 [B12(CN)12]2− is the most electronically stable multiply charged anion (MCA) synthesized so far5–8 and [B12I12]2− belongs to the most chaotropic anions known.9 Understanding interactions of the weakly coordinating [B12X12]2− anions with different counterions,1,2,10–12 solvent molecules,13 coordinating neutral species,14 host molecules in supramolecular complexes,15–17 and biomolecules18,19 is of interest, for example, to boron neutron capture therapy,20,21 stabilization of reactive cations in synthesis and catalysis,1,4 battery science,22 and non-linear optics.21 A systematic study of the molecular properties of closo-[BnXn]2− dianions and their dependence on the substituent X and scaffold size n constitutes the scientific foundation for the rational design of closo-borate anions for specific applications. Several computational studies have investigated the electronic stability of [B12X12]2− ions dependent on the substituents X.6,23–25 Experimentally, the intrinsic molecular properties of different closo-[BnXn]2− anions can be studied in the gas phase, using electrospray ionization, so that possible perturbation by the condensed phase environment is eliminated. Several gas phase studies have examined the reactivity and electronic structure of [B12X12]2− (X = H, F, Cl, Br, I, CN),5,26–29 [B11X11]2− and [B10X10]2− (X = Cl, Br, I).10 Much less is known about smaller closo-borate dianions.30 However, the chemistry of hexaborate dianions has recently received a renewed interest31–33 and their salts have been discussed in the context of hydrogen storage.34,35In this study, we explore the gas-phase ion properties of hexaborate dianions (closo-[B6X6]2−). These ions may be regarded as “half-weight” analogues of the well-explored and understood [B12X12]2− dianions. Both [B6X6]2− and [B12X12]2− are fully symmetric (all B and X atoms are chemically equivalent), see Fig. 1. We investigate how electronic structure, stability, dissociation pathways, and bond properties change when the number of atoms in this dianion is reduced by half. We employ a combination of photoelectron spectroscopy (PES), collision induced dissociation (CID), electronic structure calculations, and complementary bonding analyses to obtain a detailed understanding of the properties of isolated [B6X6]2− anions.
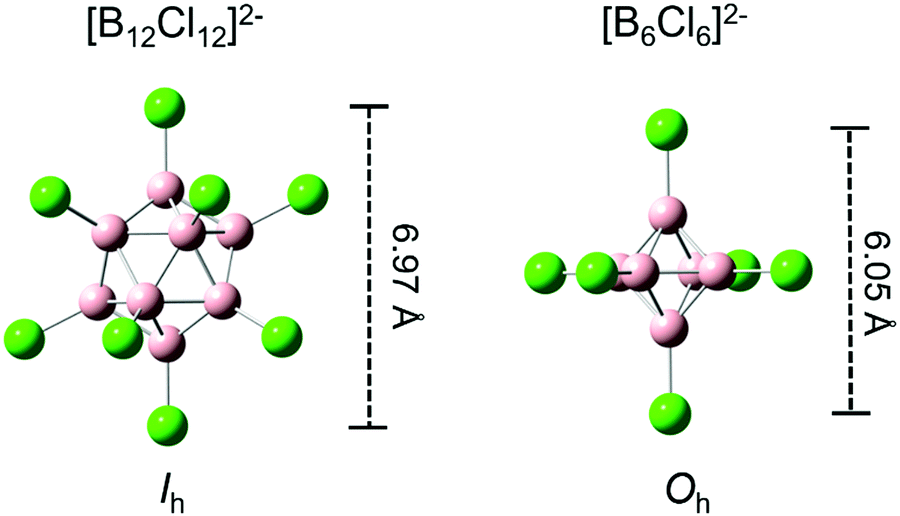 | ||
| Fig. 1 Typical structures of closo-[B12X12]2− (left) and closo-[B6X6]2− anions (right) shown for X = Cl as an example. Distances between opposite substituents and molecular point groups are shown (structural parameters based on geometry optimization at PBE0-GD3BJ/aug-cc-pVTZ). | ||
2. Experimental and theoretical methodologies
2.1. Synthesis of hexaborate anions
Tetrabutylammonium (TBA) salts of the halogenated closo-hexaborates [Bu4N]2[B6X6] (X = Cl, Br, I) were prepared from Na2[B6H6]30 and the elemental halogens according to literature procedures:362.2. Photoelectron spectroscopy (PES) experiments
PES experiments were carried out using an instrument consisting of an electrospray ionization source, a temperature-controlled cryogenic ion trap, and a magnetic-bottle time-of-flight (TOF) photoelectron spectrometer.37 A 1 mmol L−1 acetonitrile solution of each tetrabutylammonium (TBA) salt of [B6X6]2− (X = Cl, Br, I) was used for electrospray ionization to generate gaseous [B6X6]2− ions. The electrospray conditions, i.e., the high voltage applied on the electrospray needle, and the 1st skimmer voltage were tuned and optimized in order to produce strong [B6X6]2− beams. All ions were guided into the ion trap, accumulated and cooled down to temperatures of ∼20 K by collisions with a cold buffer gas (20% hydrogen, 80% helium) for 20–100 ms. The cooling of the anions to 20 K improves the spectral energy resolution and eliminates hot band peaks. The resulting cryogenic anions were then transferred into the extraction zone of the TOF mass spectrometer, mass selected, and decelerated to around 10 eV before the photodetachment process was initiated with a laser beam in the detachment zone. In the current study, three laser photon energies of 7.866 eV (157 nm from a F2 excimer laser), 6.424 eV (193 nm from an ArF excimer laser), and 4.661 eV (266 nm from a Nd:YAG laser) were used. All lasers were operated at a 20 Hz repetition rate with the ion beam turned off at alternating laser shots to afford a shot-by-shot background subtraction. The detached photoelectrons were collected with ∼100% efficiency using the magnetic-bottle and analyzed in a 5.2 m long calibrated electron flight tube. The electron kinetic energy spectrum was converted from the recorded photoelectron TOF spectrum. The electron binding energy (EBE) spectrum was obtained by subtracting the electron kinetic energy from the energy of detaching photons. The energy resolution was about 2%, i.e., ∼20 meV for 1 eV kinetic energy electrons.2.3. Mass spectrometry and collision-induced dissociation (CID)
Tetrabutylammonium salts of [B6X6]2− (X = Cl, Br, I) were dissolved in acetonitrile to a final concentration of ∼10−6 mol L−1. Each solution was injected through a fused silica capillary (50 μm ID, 150 μm OD) using a syringe pump into the inlet of a mass spectrometer at a flow rate of ∼0.5 μL min−1. Low-energy ion-trap CID experiments were conducted on a Thermo LTQ XL (Thermo Fisher Scientific GmbH, Bremen, Germany) ion trap mass spectrometer under the following instrument conditions: electrospray voltage: −3 kV, capillary temperature: 200 °C, capillary voltage: −10 V, tube lens: −20 V, scan range: m/z 100–2000. Ion-trap CID experiments were performed by isolating the ion of interest and subsequently fragmenting it (MS2 experiment) by applying a dipolar AC voltage to one pair of the rods of the ion trap using helium as a collision gas at a typical activation time of 30 ms. Ions with a specific m/z value were isolated using: (1) a wide isolation window of 10 m/z to include the natural broad isotopic pattern of the precursor ion and identify the corresponding product ions; (2) a narrow isolation window of 1 m/z to unambiguously determine the mass of a neutral loss in a CID spectrum. Higher-energy CID experiments were conducted on an Agilent 6560 IM Q-TOF (Santa Clara, CA, USA) mass spectrometer under the following instrument conditions: electrospray voltage: −4 kV, capillary temperature: 325 °C, m/z range: 100–3200. Higher-energy CID spectra were acquired using nitrogen as the collision gas at a typical collision energy of 10 V.2.4. Quantum chemical calculations
DFT modeling with the B3LYP38–40 and PBE1PBE41 (equivalent to PBE0 and therefore called PBE0 in the following) hybrid functionals and calculation of natural population analysis (NPA)42 charges was conducted with the Gaussian09, rev. E.01,43 and Gaussian16, rev. C.01,44 software packages. DFT modeling with PBE0 for the vertical and adiabatic detachment energy, CAM-B3LYP45 and Hatree Fock (HF) calculations and post-Hartree Fock calculations (see Table S1 in the ESI†) were performed using the NWChem software.46 Geometry optimizations on DFT level were carried out employing the B3LYP/def2-TZVPP47 and PBE0/aug-cc-pVTZ48 methods with additional dispersion corrections according to Grimme's GD3 method involving Becke–Johnson damping (GD3BJ).49,50 ECPs were used to describe the core electrons of iodine. Subsequent frequency analyses ensured that minima on the potential energy surface were obtained by the absence of imaginary frequencies. To confirm that the observed trends are not an artifact of the use of ECPs for X = I, we additionally employed an all-electron basis set (B3LYP-GD3BJ/6-311G**) for X = Cl–I (Table S2, ESI†).The theoretical Vertical Detachment Energies (VDEs) were calculated as energy differences between the singly charged anion and the corresponding dianion, both at the dianion's optimized geometry. The theoretical Adiabatic Detachment Energies (ADE) were determined by computing the energy difference between the singly charged anion at its own optimized geometry and the dianion at the dianion's optimized geometry. Zero-Point Energy (ZPE) corrections were computed using the harmonic approximation.
Dissociation enthalpies for ionic or radical loss of a substituent X from [B6X6]2− and [B12X12]2− were calculated by subtracting the 0 K molecular enthalpy (i.e., zero-point vibrational energy corrected electronic energy) of the dissociation products from that of the intact parent ion which was additionally BSSE-corrected using the counterpoise method.51,52 In some cases, additional calculations were performed on a MP2, SCS-MP253 and SOS-MP254 level to confirm that trends derived from DFT results are reproduced independent of the calculational method.
The electrostatic potential (ESP) was evaluated from the PBE0-GD3BJ/aug-cc-pVTZ wavefunctions using either the cubegen utility of the Gaussian software package43,44 to generate three-dimensional cube files or the Multiwfn software package, version 3.755 for evaluation along a certain direction. The electric field was derived from ESP cube files by differentiating the data points numerically employing difference quotients. The procedure is described in more detail in ref. 5 and 56. The electric field is a vector quantity. Herein, we evaluated the magnitude of the field with respect to a fixed position, which was chosen to be the center of the boron scaffolds of the investigated species.
We performed a complementary bonding analysis57 of the B–X bonds of the [B6X6]2− anions in direct comparison to their [B12X12]2− analogues based on the PBE0-GD3BJ/aug-cc-pVTZ wavefunctions. Quantum Theory of Atoms in Molecules (QTAIM)58 analyses and subsequent evaluation of electron-density-related properties (e.g. Laplacian of the electron density, total energy density H(r)) at the QTAIM bond critical points as well as calculation of AIM charges were performed with the Multiwfn software package version 3.7.55 The electron localizability indicator (ELI-D)59 was calculated using DGrid version 5.1.60 Grid files of electron density and ELI-D were calculated with a step size of 0.1 Bohr. The ELI-D was topologically analyzed yielding electron-pair basins. These ELI-D basins were cropped at an electron-density iso-contour of 0.001 a.u. Integration of the electron density was performed within the resulting basins to obtain the electron population of the ELI-D basins N(ELI). The Raub-Jansen Index RJI61 was as well calculated using DGrid version 5.160 by determining the contribution to the electron density in the intersection region of atomic QTAIM basins and ELI-D bond basins yielding a measure for bond polarity.61
3. Results and discussion
3.1. Electronic structure and stability
PES experiments provide insights into the electronic structure and stability of dianions. Fig. 2 displays the molecular structures and photoelectron spectra of [B6Cl6]2−, [B6Br6]2− and [B6I6]2− anions. The vertical detachment energy (VDE) of each dianion is determined from the maximum of the first spectral band indicated by a dashed blue line. The adiabatic detachment energy (ADE) is determined from the onset of the first spectral band in each spectrum, as indicated by a dotted green line in Fig. 2. Table 1 compares these experimental values with the theoretical VDEs and ADEs calculated using the two DFT functionals B3LYP and PBE0 both employing the aug-cc-pVTZ basis set. We note that more cost-demanding methods like MP2 and CCSD(T) do not improve the accuracy of the predicted VDE and ADE values (see Table S1, ESI†). HF orbital energy levels (eigenvalues) were calculated for the optimized geometry obtained in PBE0 calculations. These energy levels from HOMO to HOMO−5 are all triply degenerate and are denoted with dashed black lines below the photoelectron spectra in Fig. 2. All values are shifted by a constant (∼+0.7 eV) so that the HOMO matches the experimental VDE to compensate for the deviation from Koopmans’ theorem.62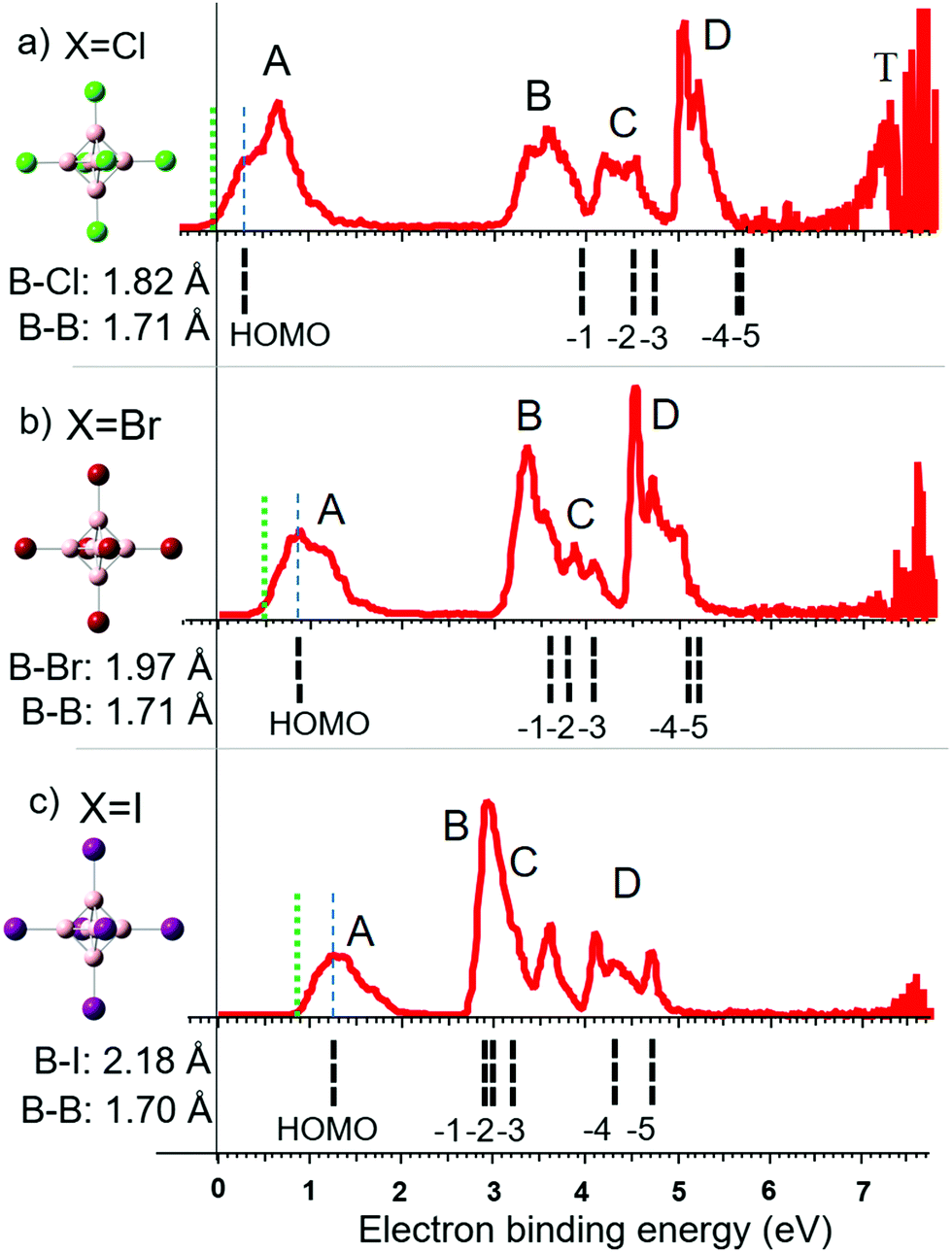 | ||
| Fig. 2 Photoelectron spectra of (a) closo-[B6Cl6]2−, (b) closo-[B6Br6]2− and (c) closo-[B6I6]2−. Calculated orbital energy levels (HF/aug-cc-pVTZ) are shown as black lines below the spectra. The calculated HOMO is shifted in energy to the measured VDE, indicated by a dashed blue line. The ADE is indicated by a dotted green line. Distinct spectral bands are denoted with the labels A–D and T. On the left side of the spectra, molecular structures are shown, and the B–X and B–B bond length are given. HOMO isosurfaces can be found in Fig. S1 (ESI†). | ||
| X | ADE | VDE | ||||
|---|---|---|---|---|---|---|
| Expt. | PBE0 | B3LYP | Expt. | PBE0 | B3LYP | |
| F | n.a. | −1.88 | −1.83 | n.a. | −1.29 | −1.22 |
| Cl | −0.05 ± 0.05 | −0.11 | −0.09 | 0.25 ± 0.10 | 0.28 | 0.32 |
| Br | 0.45 ± 0.05 | 0.34 | 0.34 | 0.75 ± 0.08 | 0.67 | 0.69 |
| I | 0.85 ± 0.05 | 0.83 | 0.79 | 1.15 ± 0.07 | 1.09 | 1.04 |
Along the investigated halogen series, the theoretically predicted electronic structures show a reasonable correlation with the observed spectral bands. The first spectral band (band A) is associated with the HOMO orbital. With increasing halogen size, this orbital becomes lower in energy (more stable) so that [B6I6]2− is the most electronically stable dianion in the series. For X = Cl, three clearly distinct spectral features labeled B, C and D were observed at higher electron binding energies. Band B may be associated with the HOMO−1. The HOMO−2 and −3 are lying closer in energy and account together for band C. In the case of X = Cl, HOMO−4 and −5 have nearly the same energy, resulting in an intense sharp band (band D). In contrast to the HOMO energy, which becomes more negative from X = Cl to I, the orbitals HOMO−1 to −5 increase in energy (less stable) along the halogen series. This trend is in agreement with the observed shift in the positions of B, C and D. We note that comparison of the PES bands with HF energy levels can be used to rationalize general trends in band positions across a series of ions but cannot explain the fine structure and intensity. This requires more sophisticated models, which take into account the Jahn–Teller distortion, excited electronic states, and outgoing electron intensity wave function matrix.26,63,64
The observed trend is well reflected in the contribution of boron and halogen atomic orbitals (AOs) to the molecular orbitals (MOs). The contributions of boron and halogen AOs to the HOMO are: B: 64%, Cl: 36%; B: 56%, Br: 44%; B: 35%, I: 65%. For small halogens, the HOMO has a large boron atomic orbital contribution. The overlap with halogen orbitals increases with increase in the halogen size resulting in a stabilization of the HOMO. In contrast to the HOMO, the energy development of HOMO−1 to HOMO−5 is almost exclusively determined by the halogen-free electron pairs, which become more loosely bound along the halogen series.
The observed stabilization of the HOMO on going from the lighter to the heavier halogens accompanied by a destabilization of the lower lying orbitals (HOMO−1 to −5) is similar to the trend reported for [BnXn]2−n = 10–12.10,26 The only exception from this trend was [B12I12]2− which was found to be less electronically stable than [B12Br12]2−. The HOMOs in [B12Cl12]2− and [B12Br12]2− are very stable and only a small energy gap exists to the orbitals of the halogen's free electron pairs. Further stabilization of this orbital in the case of [B12I12]2− results in an orbital energy below the highest lying orbitals of the iodine free electron pairs. In the case of [B6X6]2−, the energy gap between the HOMO and the orbitals of the halogen free electron pairs is much larger and is still present for X = I. Therefore, electronic stability increases for [B6X6]2− from X = Br to X = I in contrast to [B12X12]2−.
[B6F6]2− is experimentally unknown. In agreement with previous computational investigations,65,66 we calculated this dianion to be electronically unstable, see Table 1. To the best of our knowledge, gas phase electronic stabilities of [B6Br6]2− and [B6I6]2− have never been predicted in the literature, but these ions are clearly shown here to be electronically stable. [B6Cl6]2− represents a particularly interesting case because it is very slightly electronically unstable (positive VDE but slightly negative ADE). The ability to observe the electronically unstable [B6Cl6]2− experimentally must result from the kinetic trapping of the excess electron (similar behavior has been observed for [C60]2−)67 due to the repulsive Coulomb barrier (RCB).68
An additional band (T) near the photon energy limit is observed exclusively in the PES spectrum of [B6Cl6]2− (Fig. 2). Band T is the result of extremely low energy electrons which do not originate from a direct photodetachment process. This can be evidenced by measurements using different laser energies.69 The energy of direct photoelectrons is dependent on the laser energy, while the kinetic energy of electrons originating from indirect detachment processes are usually not affected. Fig. S2 (ESI†) shows spectra for three different laser energies and additional information is given. We assume that the electronic excitation due to broad UV absorption bands (which can occur as a competing process to direct photodetachment) populates the electronically excited states of the dianion. These excited states have finite lifetimes allowing either coupling with the ionization continuum or internal conversion leading to emission from the hot ground state.69,70
3.2. Collision-induced dissociation
CID experiments provide insights into the stability of dianions towards fragmentation, which is correlated to their electronic stability discussed earlier. Three different dissociation pathways of dianions in the gas phase are observed. These include (i) electron detachment, (ii) fragmentation into two singly-charged anions, and (iii) loss of a neutral fragment.71 For [BnXn]2− (n = 6, 12, X = Cl, Br, I), reaction pathway (ii) is exclusively observed as a X− loss and pathway (iii) as a radical loss of X˙, see Scheme 1.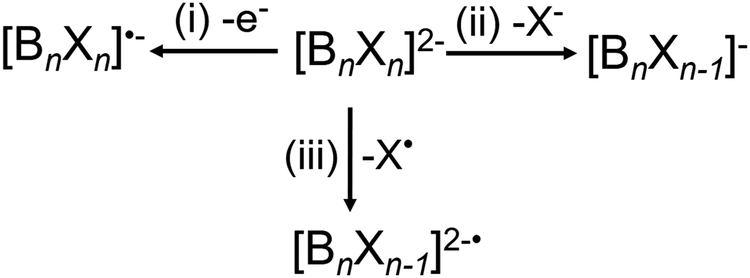 | ||
| Scheme 1 The three competing reaction pathways of closo-[BnXn]2−. | ||
Fig. 3(a) shows low-energy CID spectra of [B6X6]2− (X = Cl, Br, I) obtained on an ion trap mass spectrometer. The corresponding low-energy CID spectra of [B12X12]2− ions are shown in Fig. 3(b). The competition between the dissociation pathways of both [B6X6]2− and [B12X12]2− dianions is strongly dependent on the halogen atom. Specifically, we observe an exclusive loss of electron (e− loss) for [B6Cl6]2−, both e− loss and Br− loss for [B6Br6]2−, and exclusive I− loss for [B6I6]2−. Although e− loss is a dominant pathway for [B6X6]2−, it is not observed for [B12X12]2− (X = Cl–I) dianions. However, it was observed for X = F.27 In contrast, X− loss is observed for all [B12X12]2− species. In addition, a competing X˙ loss is observed for [B12Br12]2− and becomes the dominant reaction channel for [B12I12]2−.
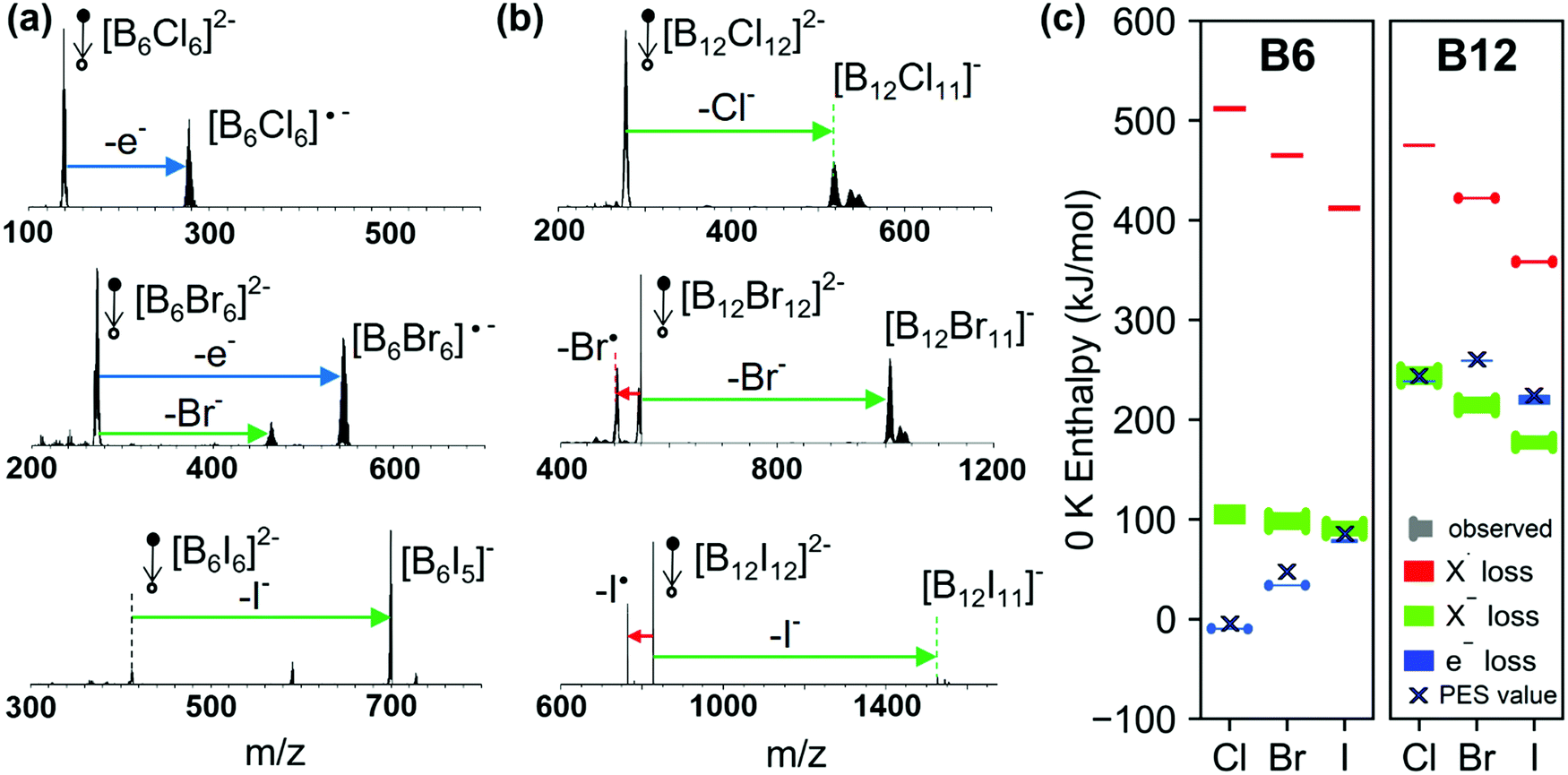 | ||
| Fig. 3 Low-energy CID spectra (MS2) of mass-selected (a) [B6X6]2− and (b) [B12X12]2− (X = Cl, Br, and I) dianions. Ions detected at higher masses than the singly charged [BnXn-1]− are attributed to reactions with residual gases H2O and N2.27 (c) Calculated 0 K enthalpies (PBE0-GD3BJ/aug-cc-pVTZ and B3LYP-GD3BJ/aug-cc-pVTZ) for the three competing pathways shown in Scheme 1 for all the dianions examined in this study. Calculated energy ranges determined with the two methods are either shown as bars with (reaction observed) or without (reaction not observed) end caps. Black crosses mark the ADE results derived from PES (see Section 3.1). The values of the calculated enthalpies are provided in Table S3 (ESI†). Note that to enhance visibility, enthalpy ranges are shown with a minimum width of 2 kJ mol−1. | ||
Fig. 3(c) shows a comparison of the calculated 0 K enthalpies of the three reaction pathways for [B6X6]2− and [B12X12]2−: e−, X− and X˙ loss. To visualize the uncertainties of the computational method, we show ranges derived from B3LYP and PBE0 calculations both employing the aug-cc-pVTZ basis set. The calculated ranges of experimentally observed reactions are denoted with end-capped markers. Ranges of non-observed reactions are denoted with markers without endcaps. ADEs derived from PES (see Section 3.1) are included as crosses. In the following, we will rationalize the competition between the three pathways in the context of the calculated enthalpies and estimated heights of reverse activation barriers.
We start by discussing the competition between the two charge separation pathways: e− loss and X− loss. Electron detachment is by far more thermochemically favorable for [B6X6]2− than for [B12X12]2−, which is consistent with the lower electronic stability of [B6X6]2− as discussed in Section 3.1. For [B6X6]2− dianions, the enthalpy of the e− loss increases (becomes less favorable) with an increase in the size of the halogen atom. The relative yield of the e− loss pathway observed in CID is consistent with the calculated increase in enthalpy. However, it is remarkable that [B6I6]2− and [B12Cl12]2− show exclusive X− loss although the 0 K enthalpy for e− loss is calculated to be very similar using DFT (see Fig. 3(c)). We confirmed with MP2, SCS-MP2 and SOS-MP2 calculations that the enthalpy for X− loss is not more favorable than for e− loss (see Table S4, ESI†). Furthermore, although the reaction enthalpy of the e− loss from [B6Br6]2− is more favorable than for X− loss by 41–75 kJ mol−1 (estimated based on PBE0-GD3BJ/aug-cc-pVTZ and B3LYP-GD3BJ/aug-cc-pVTZ), both channels are observed for this precursor ion. Thermochemistry appears not to be the only factor that determines the competition between the dissociation pathways. Dissociation of multiply charged anions in the gas phase is usually influenced by the RCB, which determines the barrier for both X− and e− loss. These results indicate that X− loss from [BnXn]2− ions may be a kinetically favored pathway (smaller RCB) in comparison with e− loss.
Comparison of low- and higher-energy CID of [B6Br6]2− shown in Fig. 4(a) and (b) provides interesting insights into the competition between the Br− loss and e− loss, respectively. The branching ratio of the e− loss to Br− loss pathways changes from ∼5 in the low-energy CID spectrum (Fig. 4(a)) to ∼0.1 in the higher-energy CID spectrum (Fig. 4(b)). Fig. 4(c) shows a schematic drawing of the potential energy surface, which rationalizes this observation. It is well-established that low-energy CID favors fragmentation pathways with energetically low-lying transition states (TS). The higher abundance of the e− loss product [B6Br6]˙− in comparison with [B6Br5]− in the low-energy CID spectrum (Fig. 4(a)) indicates that the TS for the e− loss (TS1) is lower in energy than the TS for the Br− loss (TS2). However, considering that Br− loss is still observed in the spectrum, we expect the difference between TS1 and TS2 to be small (within several kJ mol−1) and considerably smaller than the difference between the reaction enthalpies (41–75 kJ mol−1). Therefore, the reverse activation barrier, which is dominated by the RCB for charge separation reactions, appears to be substantially smaller for Br− loss than for e− loss (RCB2 < RCB1).
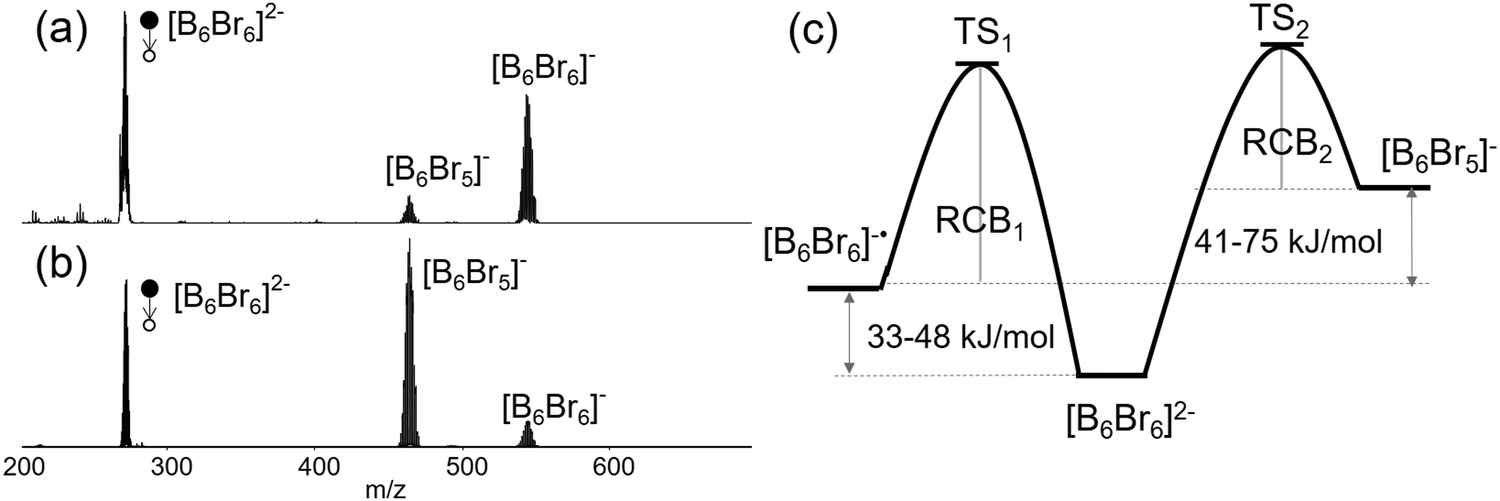 | ||
| Fig. 4 MS2 spectra of [B6Br6]2− produced from (a) low-energy CID on an ion trap and (b) higher-energy CID on a Q-TOF instrument. (c) A schematic drawing of the potential energy surface, which is consistent with the CID spectra, showing the relative energies of the products and transition states for the competing e− and Br− losses from [B6Br6]2−. | ||
This assertion is further supported by higher-energy CID experiments shown in Fig. 4(b), in which [B6Br5]− is the dominant fragment and [B6Br6]˙− is a minor product. Kinetically favored fragmentation pathways often become dominant in higher-energy CID experiments due to the relatively high internal energies accessible in these experiments. The drastic difference in the [B6Br6]−/[B6Br5]− branching ratio observed in low-energy and higher-energy CID suggests that although the energy of TS1 is lower than that of TS2, the formation of the [B6Br5]− fragment is entropically more favorable than the e− loss pathway. An entropically-preferred TS is usually associated with a smaller reverse activation barrier (here smaller RCB). As a result, at higher internal energies, Br− loss becomes the dominant dissociation channel. Based on these experimental findings, we propose that the Br− loss has a considerably smaller RCB than the e− loss channel. The difference between the heights of the RCBs is expected to be close to the enthalpy difference of e− and X− loss (41–75 kJ mol−1) resulting in comparable energies of TS1 and TS2. In the following, we rationalize this finding using PES and different computational methods.
Although it is not possible for us to determine the RCB for the Br− loss experimentally, the magnitude of the RCB for e− loss can be estimated by comparing photoelectron spectra measured at different laser energies (EL). Photoelectron spectra of closo-[B6Br6]2− measured at 193 nm (EL = 6.4eV) and 157 nm (EL = 7.9eV) are shown in Fig. 5. It is established that the presence of an RCB suppresses spectral bands at binding energies larger than (EL − RCB).72 Note that a higher binding energy (x-axis) correlates with a lower kinetic energy of the detected electrons. Electrons with extremely low kinetic energy cannot overcome the RCB which leads to signal suppression at high binding energies. Electrons with kinetic energies only slightly below the barrier height may tunnel through the RCB, which results in partial signal suppression. Three areas are marked in the 193 nm spectrum in Fig. 5. A complete signal suppression occurs for bands between 4.9 and 6.5 eV binding energy (region I). The intense signal at 4.5 eV in the 157 nm spectrum is visible in the 193 nm spectrum but is partially suppressed, meaning that the kinetic energy of electrons in this region of the PES spectrum is close to but still below the RCB height (region II). No considerable change in the signal is observed below 4.2 eV (region IV), meaning that the corresponding photoelectrons are not affected by the RCB. Since no significant band between 4.2 and 4.5 eV is present (region III, corresponding to 2.0–2.3 eV kinetic energy at 193 nm), this region determines the uncertainty of the experimental value. Therefore, based on the signal suppression in the PE spectra, we estimate that the height of the RCB for electron loss from [B6Br6]2− is in the range of 2.0 eV to 2.3 eV. A list of the heights of RCBs estimated based on the observed suppression of the electron loss from [B6X6]2− and [B12X12]2− (X = Cl, Br, I) at different laser wavelengths is shown in Table S5 (ESI†) and the corresponding spectra for [B6X6]2− are shown in Fig. S3 (ESI†).
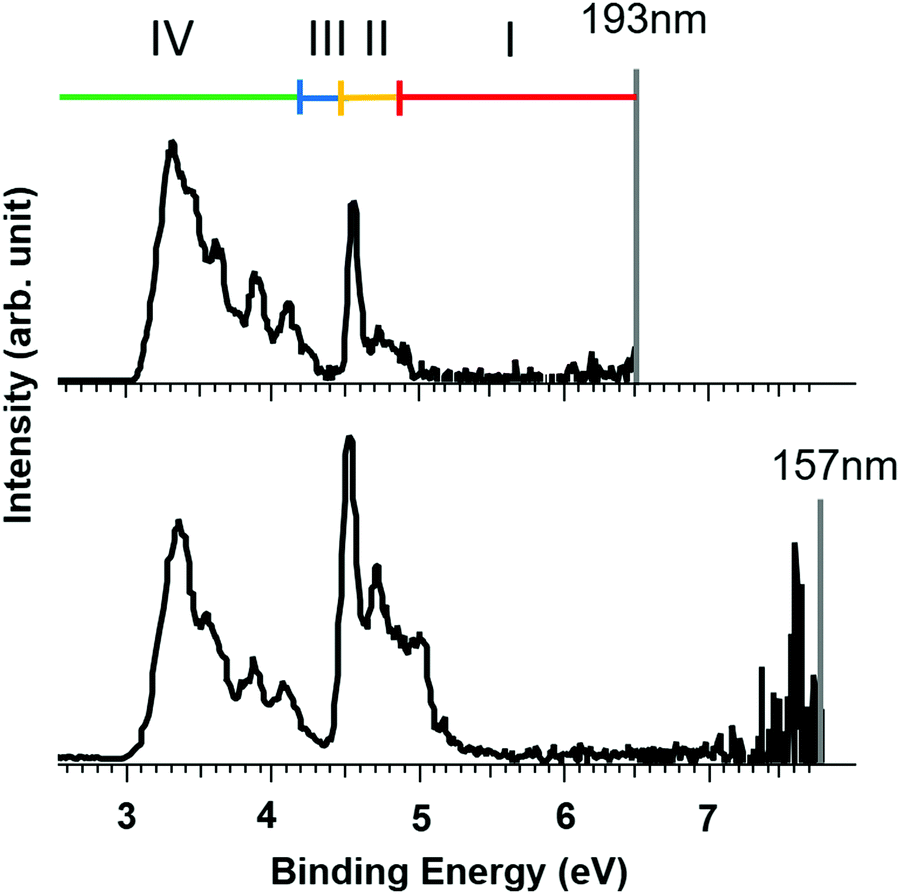 | ||
| Fig. 5 Photoelectron spectra of closo-[B6Br6]2− at two different laser wavelengths (193 nm and 157 nm). The comparison exemplifies suppression effects of spectral bands due to the RCB. For details, see text. | ||
For a theoretical evaluation of the RCB height, we calculated the potential energy of a negative test charge based on the electrostatic potential (ESP) of [B6Br6]−. The ESP was calculated along three different possible detaching directions: through the middle of the B–B bond, through the middle of the B-triangle and along the B–X bond. The results for the three different pathways are visualized in Fig. S4 (ESI†). The lowest barrier for electron detachment of 2.5 eV is found along the B–X bond. The calculated barrier is slightly higher than the RCB estimated from suppression effects in photoelectron spectra (2.0–2.3 eV). Because the polarizability of the ion is not taken into account, RCB calculations based on the ESP typically overestimate the barrier height.
For comparison, we calculated the ESP of [B6Br5]− along the direction of the Br− detachment. The energies of the negative test charge along the detachment pathways for [B6Br5]− and [B6Br6]− are shown in Fig. 6(a). Although [B6Br5]− and [B6Br6]− have the same total charge, the electrostatic barrier is substantially smaller for [B6Br5]− than for [B6Br6]−. The origin of this effect lies in the unusual charge distribution in the [B6Br5]− ions. A [BnXn−1]− ion possesses a vacant boron atom. Although the total charge of the ion is negative, this vacant boron exhibits a significant positive partial charge. NPA indicates that the atomic charge on the vacant boron atom is +0.4 e for [B6Br5]−. This unusual phenomenon has been previously reported for [B12X11]− ions which are highly reactive and have been classified as “superelectrophilic anions”.5,8,56
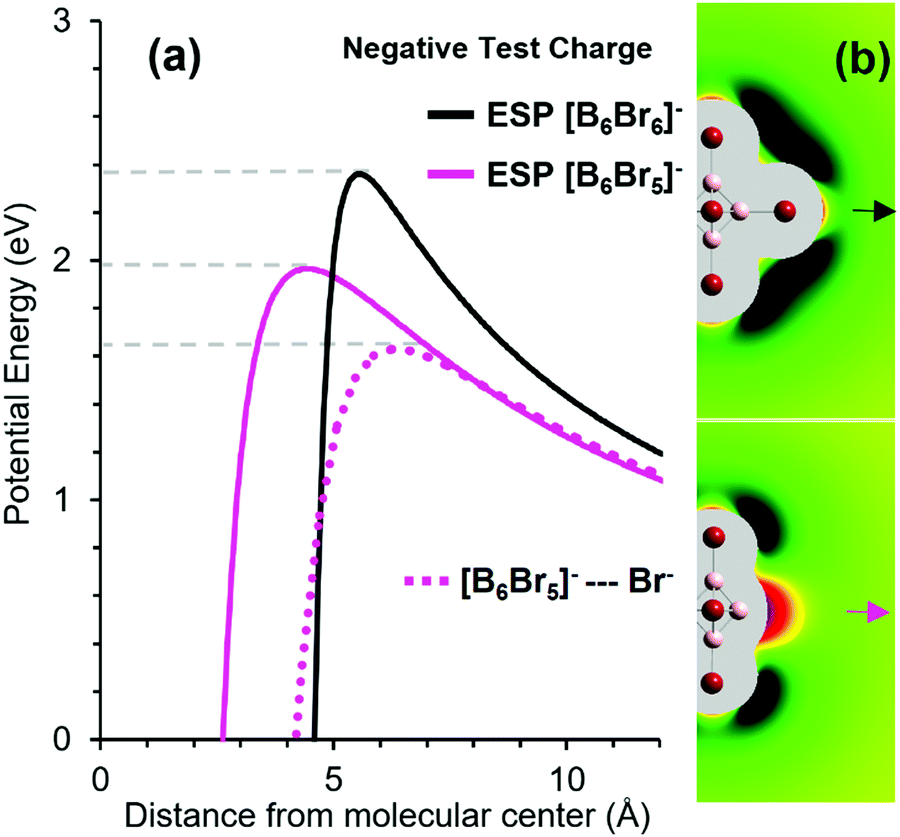 | ||
| Fig. 6 (a) Potential energy of a negative test charge positioned at different distances from [B6Br6]− along the B–Br bond direction (black) and from [B6Br5]− along the C4v symmetry axis (magenta). Distances are given with respect to the boron scaffold center defined by the middle point between the four equivalent boron atoms. The dotted magenta line shows the potential energy of the [B6Br5]− + Br− system as a function of the center-Br− distance. (b) The electric field close to the ions [B6Br6]− (top) and [B6Br5]− (bottom). Green areas mark regions of repulsion (black: strong repulsion) from the ion on a negative test charge, while red areas mark regions of attraction. Quantitative details of the color coding can be found in Fig. S5 (ESI†). The arrows mark the direction of e−/Br− loss. | ||
In order to examine differences in the charge distributions of [B6Br6]− and [B6Br5]−, we calculated the electric fields outside the molecular surfaces of these ions by taking the first derivative of the ESP. The results of these calculations are shown in Fig. 6(b). Green areas represent repulsion of a negative particle, while red areas show attraction. Similar to [B12X11]− ions, [B6Br5]− is an electrophilic anion, in which a partial positive charge is localized on the vacant boron atom. In contrast, the electric field around the [B6Br6]− ion is purely repulsive. The positive charge at the binding site of [B6Br5]− substantially lowers the barrier for a negative particle.
The simple electrostatic model based on ESPs suggests that RCB2 as introduced in Fig. 4 is by 0.4 eV (= 39 kJ mol−1) smaller than RCB1 (Fig. 6(a)). For a more sophisticated calculation of RCB2, we performed a “relaxed scan” (optimization of the [B6Br5]−–Br− geometry for a fixed B–Br bond length). As shown in Fig. 6(a), the height of RCB2 was calculated to be 1.6 eV by this method. Therefore, including structural relaxation and polarization effects in the calculation lowers the calculated barrier height by additional 0.4 eV compared to the simple ESP-based model. Note that the experimentally determined RCB1 (Fig. 5(a)) was 2.0–2.3 eV. Therefore, we can estimate RCB1–RCB2 to lie in the range of 0.4–0.7 eV (= 39–67 kJ mol−1), which is consistent with the assertion that TS2 and TS1 are lying close in energy (compare Fig. 4(c)).
Collectively, both the experimental and theoretical results discussed earlier confirm that the competition between the charge separation pathways (e−vs. X− loss) is adequately described by the potential energy surface shown in Fig. 4(c). Furthermore, theoretical calculations rationalize the observed kinetic preference for the X− loss from [BnXn]2− over the e− loss. Although the reverse activation barrier for the charge separation pathways is dominated by electrostatics and both fragments ([BnXn]˙− and [BnXn−1]−) are singly charged, the positive site within [BnXn−1]− is responsible for a substantially smaller RCB. Furthermore, our results show for the first time that similar to [B12X12]2−, smaller [B6X6]2− species are precursors for “electrophilic anions” generated by abstraction of a negative substituent, X−, from the dianion.
According to the calculated reaction enthalpies shown in Fig. 3(c), loss of X˙ observed in competition with the charge separation pathways is thermodynamically the least favorable reaction channel. However, for [B12I12]2−, I˙ loss is the dominant pathway observed experimentally. Furthermore, although the enthalpy for Br− loss from [B12Br12]2− is 200–215 kJ mol−1 lower than the enthalpy for Br˙ loss, the products of Br˙ and Br− loss are both observed in comparable abundances. While fragmentation into two singly-charged ions is hindered by the RCB, fragmentation of [BnXn]2− into [BnXn−1]2−˙ and X˙ is the reverse of a radical–radical recombination, which usually has no or a very small barrier. Efficient competition of the thermochemically unfavorable X˙ loss with the more favorable charge separation pathways becomes possible if the enthalpy difference between X˙ and X− losses is comparable to the height of the RCB. For all [B6X6]2− ions, 0 K enthalpy differences between X˙ and X− loss are 4.1–4.3 eV (X = Cl) to 3.3–3.4 eV (X = I). These values are much larger than the estimated RCBs (see Fig. 6(a) and Table S5, ESI†). As a result, X˙ loss from [B6X6]2− cannot compete with charge separation pathways. The larger difference between X˙ and X− loss is a result of both an enthalpically preferred X− loss and a less preferred X˙ loss compared to [B12X12]2−, see Fig. 3(c). Structural relaxation of the fragment ions generated by X˙ and X− loss may contribute to the reaction enthalpies. However, the calculated relaxation energies (Table S6, ESI†) are very small indicating that structural relaxation is not a dominant factor in determining the enthalpies of X˙ and X− losses. An intrinsic difference in the nature of the B–X bond in [B6X6]2− and [B12X12]2− may play a role, which we evaluate in Section 3.3.
3.3. Complementary bonding analysis of the B–X bond
Table 2 summarizes the calculated properties of the B–X bonds in [B6X6]2− and [B12X12]2− for X = Cl–I. The B–Cl bond in [B6Cl6]2− is slightly longer than in [B12Cl12]2−. However, with increasing halogen size, the increase in the B–X bond length is larger for [B12X12]2−. The B–I bond lengths of [B12I12]2− and [B6I6]2− anions are almost equal. This may be rationalized by steric effects between the substituents X which may be more pronounced in [B12X12]2− than in [B6X6]2−. For more information on the bond character, we employed different methods of bonding analysis based on PBE0-GD3BJ/aug-cc-pVTZ wavefunctions. Table 2 lists the values which we consider most relevant to the comparison of the efficiency of X− and X˙ loss from the dianions. A comprehensive table showing additional parameters related to bond critical point (bcp) properties can be found in Table S7 (ESI†). We also show that the observed trends are largely independent of the level of theory by showing the results for the same descriptors but obtained using B3LYP-GD3BJ/def2-TZVPP wavefunctions (Tables S8 and S9, ESI†).| Property | [B6Cl6]2− | [B12Cl12]2− | [B6Br6]2− | [B12Br12]2− | [B6I6]2− | [B12I12]2− |
|---|---|---|---|---|---|---|
| d/Å | 1.818 | 1.793 | 1.967 | 1.949 | 2.178 | 2.176 |
| δ | 0.66 | 0.66 | 0.72 | 0.73 | 0.86 | 0.93 |
| Q AIM(X)/e | −0.80 | −0.72 | −0.72 | −0.62 | −0.51 | −0.30 |
| Q NPA(X)/e | −0.25 | −0.14 | −0.19 | −0.06 | −0.08 | +0.08 |
| N (ELI)/e | 1.55 | 1.55 | 1.55 | 1.61 | 1.33 | 1.40 |
| RJI/% | 88.5 | 85.7 | 84.1 | 79.6 | 62.7 | 51.0 |
Quantum theory of atoms in molecules (QTAIM) describes the properties of chemical bonds by means of a topological analysis of the electron density. The delocalization index δ is derived by integration over the exchange density in the two analyzed atomic QTAIM basins and is usually interpreted as the number of electron pairs delocalized between two atoms, i.e. as a covalent bond order.73 With increasing halogen size the value of δ increases, the negative atomic charges QAIM of the halogens decrease. This points to an increase in the covalent bond character and a decrease in the halide character from X = Cl to I. For a given halogen, δ of [B12X12]2− has a larger or the same value than [B6X6]2− and QAIM of the halogens is more negative for [B6X6]2−. This analysis suggests that the B–X bonds have more halide character in the [B6X6]2− clusters and a stronger covalent character in [B12X12]2−.
To confirm this assertion, we use methods complementary to the QTAIM analysis. NPA charges are often considered to be the most chemically meaningful calculated atomic charges.42 Although absolute numbers of QNPA differ considerably from the determined QAIM values, the general trend along the series is consistent and it can be concluded that the halogens in [B6X6]2− carry a larger negative charge than in [B12X12]2−. This difference is more pronounced for larger halogens. As a complementary measure of the covalent bond strength, we integrated the electron density over the electron localizability indicator (ELI-D) bond basins (N (ELI)).59 Similar to the δ values, slightly higher values for [B12X12]2− than for [B6X6]2− confirm a stronger covalent bond character in the former case. The Raub-Jansen index (RJI) uses the intersections of ELI-D bond basins and QTAIM based electron density basins to quantify bond polarity.61 A value larger than 50% shows a polarization toward the halogen atom of the B–X bond. The calculated RJIs for B–X bonds in [B12X12]2− and [B6X6]2− (Table 2) are in full agreement with the discussed trends. They show that the electron density of the bonding electrons is heavily shifted towards the halogen atom. The value decreases from X = Cl to I and for a given halogen, and it is always larger for [B6X6]2−.
The absolute values for δ, N (ELI) and RJI are very similar for a given halogen, as it would be expected for B–X bonds in very similar environments. However, the slight differences shown in Table 2 consistently show that the electron density in the B–X bond is more polarized towards the halogen in [B6X6]2− than in [B12X12]2−. The higher intrinsic halide character of X in [B6X6]2− rationalizes advantages for a heterolytic cleavage (X− loss) and disadvantages for homolytic cleavage (X˙ loss) in comparison to a less polar and more covalent B–X bonds in [B12X12]2−.
4. Conclusion
We investigated the properties of isolated closo-[B6X6]2− dianions using PES, CID and a variety of computational methods. Our results clarify that [B6Cl6]2− is unstable against electron autodetachment, while [B6Br6]2− and [B6I6]2− are intrinsically stable dianions with low second electron binding energies in the range of 0.4–0.5 eV (X = Br) and 0.8–0.9 eV (X = I), respectively. The low electronic stability is reflected in the observed fragmentation pathways of [B6X6]2− with e− loss being the dominant dissociation channel for X = Cl, Br. Meanwhile, X− loss is observed for X = Br, I. In contrast, the electronically more stable [B12X12]2− shows X− loss (X = Cl, Br, I) and X˙ loss (X = Br, I). Although the enthalpies of X− and e− losses are calculated to be similar for [B6Br6]2−, [B6I6]2− and [B12Cl12]2−, a preferred loss of X− from these ions was observed experimentally. The RCB (= the reverse activation barrier) for X− loss is considerably lower than for e− loss indicating that X− loss is a kinetically preferred pathway. The origin of this difference in the magnitude of the RCB is attributed to the charge distribution within [BnXn−1]− ions. Although the ion has a net negative charge, the vacant boron atom possesses a strong positive partial charge. The presence of such strongly electrophilic centers is known for [B12X11]− but evidenced here for the first time for a smaller closo-borate anion. Observation of an enthalpically unfavorable X˙ loss from [B12Br12]2− and [B12I12]2− can be explained by the absence of a significant barrier for this reaction channel. In general, 0 K enthalpies for X− loss were found to be smaller for [B6X6]2− than for [B12X12]2−. In contrast, X˙ losses are more favorable for [B12X12]2− than for [B6X6]2−. This is rationalized by a stronger polarization of the B–X bond, which results in a smaller covalent bond order and a larger halide character of X in [B6X6]2− in comparison with [B12X12]2−. Future studies will examine closo-halogenohexaborates containing a mixture of halogen ligands74 to gain additional insights into the effect of heteroleptic ligands attached to the B6 boron scaffold on the electronic stability and fragmentation of the dianion.The results reported in this study deepen our understanding of the chemical and physical properties of [BnXn]2− anions as a function of size and halogen atom. In addition, we introduce the smallest known electrophilic anion, [B6X5]−, confirming that the concept of electrophilic anions extends beyond the [B12X11]− species described in previous studies. The formation of the electrophilic anion lowers the RCB for the X− loss making this reaction pathway kinetically favored over the more thermochemically favorable e− loss pathway.
Author contribution
MR: performed the major part of the computational work, prepared figures and co-wrote the manuscript; ZY: performed photoelectron spectroscopy experiments; PS: performed collision induced dissociation experiments; EB: Synthesized the closo-hexaborate samples; QY: performed photoelectron spectroscopy experiments; EA: Performed part of the computational work; SG: designed theoretical investigations (complementary bonding analysis); JL: designed CID experiments and co-wrote the manuscript; CJ: initiated the work together with JW and co-wrote the manuscript; X. B. W.: designed photoelectron spectroscopy experiments and co-wrote the manuscript; JW: initiated, designed and coordinated the study and wrote the major part of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of the computations for this work were done with resources of Leipzig University Computing Centre (M. R. & J. W.). Photoelectron spectroscopy work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences at Pacific Northwest National Laboratory (X.-B. W.). A portion of this research (E. A.) was performed in Environmental Molecular Sciences Laboratory (EMSL), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at PNNL. J. W. is grateful to the Volkswagen foundation for a Freigeist Fellowship.References
- C. Bolli, J. Derendorf, M. Keßler, C. Knapp, H. Scherer, C. Schulz and J. Warneke, Synthesis, Crystal Structure, and Reactivity of the Strong Methylating Agent Me2B12Cl12, Angew. Chem., Int. Ed., 2010, 49, 3536–3538 CrossRef CAS PubMed.
- M. Kessler, C. Knapp, V. Sagawe, H. Scherer and R. Uzun, Synthesis, Characterization, and Crystal Structures of Silylium Compounds of the Weakly Coordinating Dianion [B12Cl12]2−, Inorg. Chem., 2010, 49, 5223–5230 CrossRef CAS PubMed.
- M. Kessler, C. Knapp and A. Zogaj, Cationic Dialkyl Metal Compounds of Group 13 Elements (E = Al, Ga, In) Stabilized by the Weakly Coordinating Dianion [B12Cl12]2−, Organometallics, 2011, 30, 3786–3792 CrossRef CAS.
- C. Knapp, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 651–679 Search PubMed.
- M. Mayer, V. van Lessen, M. Rohdenburg, G.-L. Hou, Z. Yang, R. M. Exner, E. Aprà, V. A. Azov, S. Grabowsky, S. S. Xantheas, K. R. Asmis, X.-B. Wang, C. Jenne and J. Warneke, Rational design of an argon-binding superelectrophilic anion, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8167–8172 CrossRef CAS PubMed.
- H. Zhao, J. Zhou and P. Jena, Stability of B12(CN)122−: Implications for Lithium and Magnesium Ion Batteries, Angew. Chem., Int. Ed., 2016, 55, 3704–3708 CrossRef CAS PubMed.
- A. A. Kamin and M. A. Juhasz, Exhaustive Cyanation of the Dodecaborate Dianion: Synthesis, Characterization, and X-ray Crystal Structure of [B12(CN)12]2−, Inorg. Chem., 2020, 59, 189–192 CrossRef CAS PubMed.
- M. Mayer, M. Rohdenburg, V. van Lessen, M. C. Nierstenhöfer, E. Aprà, S. Grabowsky, K. R. Asmis, C. Jenne and J. Warneke, First steps towards a stable neon compound: Observation and bonding analysis of [B12(CN)11Ne]−, Chem. Commun., 2020, 56, 4591–4594 RSC.
- K. I. Assaf and W. M. Nau, The Chaotropic Effect as an Assembly Motif in Chemistry, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- J. Warneke, S. Z. Konieczka, G.-L. Hou, E. Aprà, C. Kerpen, F. Keppner, T. C. Schäfer, M. Deckert, Z. Yang, E. J. Bylaska, G. E. Johnson, J. Laskin, S. S. Xantheas, X.-B. Wang and M. Finze, Properties of perhalogenated {closo-B10} and {closo-B11} multiply charged anions and a critical comparison with {closo-B12} in the gas and the condensed phase, Phys. Chem. Chem. Phys., 2019, 21, 5903–5915 RSC.
- W. S. Hopkins, P. J. J. Carr, D. Huang, K. P. Bishop, M. Burt, T. B. McMahon, V. Steinmetz and E. Fillion, Infrared-Driven Charge Transfer in Transition Metal B12F12 Clusters, J. Phys. Chem. A, 2015, 119, 8469–8475 CrossRef CAS PubMed.
- I. J. S. De Vlugt, M. J. Lecours, P. J. J. Carr, A. Anwar, R. A. Marta, E. Fillion, V. Steinmetz and W. S. Hopkins, Infrared-Driven Charge-Transfer in Transition Metal-Containing B12X122− (X = H, F) Clusters, J. Phys. Chem. A, 2018, 122, 7051–7061 CrossRef CAS PubMed.
- P. Fan, J. Neumann, S. Stolte, J. Arning, D. Ferreira, K. Edwards and D. Gabel, Interaction of dodecaborate cluster compounds on hydrophilic column materials in water, J. Chromatogr. A, 2012, 1256, 98–104 CrossRef CAS PubMed.
- M. J. Lecours, R. A. Marta, V. Steinmetz, N. Keddie, E. Fillion, D. O’Hagan, T. B. McMahon and W. S. Hopkins, Interaction of B12F122− with All-cis 1,2,3,4,5,6 Hexafluorocyclohexane in the Gas Phase, J. Phys. Chem. Lett., 2017, 8, 109–113 CrossRef CAS PubMed.
- J. Warneke, C. Jenne, J. Bernarding, V. A. Azov and M. Plaumann, Evidence for an intrinsic binding force between dodecaborate dianions and receptors with hydrophobic binding pockets, Chem. Commun., 2016, 52, 6300–6303 RSC.
- K. I. Assaf, A. Hennig, S. Peng, D.-S. Guo, D. Gabel and W. M. Nau, Hierarchical host–guest assemblies formed on dodecaborate-coated gold nanoparticles, Chem. Commun., 2017, 53, 4616–4619 RSC.
- K. I. Assaf, M. S. Ural, F. Pan, T. Georgiev, S. Simova, K. Rissanen, D. Gabel and W. M. Nau, Water structure recovery in chaotropic anion recognition: High-affinity binding of dodecaborate clusters to γ-cyclodextrin, Angew. Chem., Int. Ed., 2015, 54, 6852–6856 CrossRef CAS PubMed.
- A. V. Efremenko, A. A. Ignatova, A. A. Borsheva, M. A. Grin, V. I. Bregadze, I. B. Sivaev, A. F. Mironov and A. V. Feofanov, Cobalt bis(dicarbollide) versus closo-dodecaborate in boronated chlorin e6 conjugates: implications for photodynamic and boron-neutron capture therapy, Photochem. Photobiol. Sci., 2012, 11, 645 RSC.
- D. Awad, M. Bartok, F. Mostaghimi, I. Schrader, N. Sudumbrekar, T. Schaffran, C. Jenne, J. Eriksson, M. Winterhalter, J. Fritz, K. Edwards and D. Gabel, Halogenated Dodecaborate Clusters as Agents to Trigger Release of Liposomal Contents, ChemPlusChem, 2015, 80, 656–664 CrossRef CAS PubMed.
- A. H. Soloway, W. Tjarks, B. A. Barnum, F. G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, The chemistry of neutron capture therapy, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed.
- Boron Science, ed. N. S. Hosmane, CRC Press, 2016 Search PubMed.
- M. F. Roll, Ionic borohydride clusters for the next generation of boron thin-films: Nano-building blocks for electrochemical and refractory materials, J. Mater. Res., 2016, 31, 2736–2748 CrossRef CAS.
- J. Moon, H. Baek and J. Kim, Unusually high stability of B12(BO)122− achieved by boronyl ligand manipulation: Theoretical investigation, Chem. Phys. Lett., 2018, 698, 72–76 CrossRef CAS.
- H. Fang and P. Jena, B12(SCN)12−: An Ultrastable Weakly Coordinating Dianion, J. Phys. Chem. C, 2017, 121, 7697–7702 CrossRef.
- M. M. Zhong, H. Fang and P. Jena, Record-high stability and compactness of multiply-charged clusters aided by selected terminal groups, Phys. Chem. Chem. Phys., 2020, 22, 4880–4883 RSC.
- J. Warneke, G.-L. Hou, E. Aprà, C. Jenne, Z. Yang, Z. Qin, K. Kowalski, X.-B. Wang and S. S. Xantheas, Electronic Structure and Stability of [B12X12]2− (X = F–At): A Combined Photoelectron Spectroscopic and Theoretical Study, J. Am. Chem. Soc., 2017, 139, 14749–14756 CrossRef CAS PubMed.
- J. Warneke, T. Dülcks, C. Knapp and D. Gabel, Collision-induced gas-phase reactions of perhalogenated closo-dodecaborate clusters – a comparative study, Phys. Chem. Chem. Phys., 2011, 13, 5712–5721 RSC.
- E. Aprà, J. Warneke, S. S. Xantheas and X.-B. Wang, A benchmark photoelectron spectroscopic and theoretical study of the electronic stability of [B12H12]2−, J. Chem. Phys., 2019, 150, 164306 CrossRef PubMed.
- P. Farràs, N. Vankova, L. L. Zeonjuk, J. Warneke, T. Dülcks, T. Heine, C. Viñas, F. Teixidor and D. Gabel, From an Icosahedron to a Plane: Flattening Dodecaiodo-dodecaborate by Successive Stripping of Iodine, Chem. – A Eur. J., 2012, 18, 13208–13212 CrossRef PubMed.
- W. Preetz and G. Peters, The Hexahydro-closo-hexaborate Dianion [B6H6]2− and Its Derivatives, Eur. J. Inorg. Chem., 1999, 1831–1846 CrossRef CAS.
- J. C. Axtell, K. O. Kirlikovali, D. Jung, R. M. Dziedzic, A. L. Rheingold and A. M. Spokoyny, Metal-Free Peralkylation of the closo-Hexaborate Anion, Organometallics, 2017, 36, 1204–1210 CrossRef CAS.
- J. C. Axtell, L. M. A. Saleh, E. A. Qian, A. I. Wixtrom and A. M. Spokoyny, Synthesis and Applications of Perfunctionalized Boron Clusters, Inorg. Chem., 2018, 57, 2333–2350 CrossRef CAS PubMed.
- X. Mu, J. C. Axtell, N. A. Bernier, K. O. Kirlikovali, D. Jung, A. Umanzor, K. Qian, X. Chen, K. L. Bay, M. Kirollos, A. L. Rheingold, K. N. Houk and A. M. Spokoyny, Sterically Unprotected Nucleophilic Boron Cluster Reagents, Chem, 2019, 5, 2461–2469 CAS.
- A. Rahmani, A. Zabardasti and A. Kakanejadifard, Intermolecular complexes of [B6H6]2− with nH2 (n = 1–8) molecules: a theoretical study, Struct. Chem., 2019, 30, 669–680 CrossRef CAS.
- Q. L. Lu, J. W. Meng, W. J. Song and J. G. Wan, High capacity hydrogen storage in closo-hexaborate dianion, Int. J. Hydrogen Energy, 2013, 38, 13328–13334 CrossRef CAS.
- W. Preetz and J. Fritze, Darstellung, 11B-NMR- und Schwingungsspektren der oktaedrischen closo-Boratanionen B6X62−; X = H, Cl, Br, I, Z. Naturforsch. B: Chem. Sci., 1984, 39, 1472–1477 Search PubMed.
- X.-B. Wang and L.-S. Wang, Development of a low-temperature photoelectron spectroscopy instrument using an electrospray ion source and a cryogenically controlled ion trap, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09 Revision E.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian16 Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers?, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- S. Grimme, Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- Y. Jung, R. C. Lochan, A. D. Dutoi and M. Head-Gordon, Scaled opposite-spin second order Møller–Plesset correlation energy: An economical electronic structure method, J. Chem. Phys., 2004, 121, 9793–9802 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. Rohdenburg, M. Mayer, M. Grellmann, C. Jenne, T. Borrmann, F. Kleemiss, V. A. Azov, K. R. Asmis, S. Grabowsky and J. Warneke, Superelectrophilic Behavior of an Anion Demonstrated by the Spontaneous Binding of Noble Gases to [B12Cl11]−, Angew. Chem., Int. Ed., 2017, 56, 7980–7985 CrossRef CAS PubMed.
- M. Fugel, J. Beckmann, D. Jayatilaka, G. V. Gibbs and S. Grabowsky, A Variety of Bond Analysis Methods, One Answer? An Investigation of the Element–Oxygen Bond of Hydroxides HnXOH, Chem. – A Eur. J., 2018, 24, 6248–6261 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1990 Search PubMed.
- F. R. Wagner, M. Kohout and Y. Grin, Direct Space Decomposition of ELI-D: Interplay of Charge Density and Pair-Volume Function for Different Bonding Situations, J. Phys. Chem. A, 2008, 112, 9814–9828 CrossRef CAS PubMed.
- M. Kohout, DGrid, version 5.1, Dresden, 2017 Search PubMed.
- S. Raub and G. Jansen, A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules, Theor. Chem. Acc., 2001, 106, 223–232 Search PubMed.
- H. Häkkinen, B. Yoon, U. Landman, X. Li, H.-J. Zhai and L.-S. Wang, On the Electronic and Atomic Structures of Small AuN− (N = 4–14) Clusters: A Photoelectron Spectroscopy and Density-Functional Study, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef.
- Z. Qin, G.-L. Hou, Z. Yang, M. Valiev and X.-B. Wang, Negative ion photoelectron spectra of ISO3−, IS2O3−, and IS2O4− intermediates formed in interfacial reactions of ozone and iodide/sulfite aqueous microdroplets, J. Chem. Phys., 2016, 145, 214310 CrossRef PubMed.
- E. Hossain, S. M. Deng, S. Gozem, A. I. Krylov, X.-B. Wang and P. G. Wenthold, Photoelectron Spectroscopy Study of Quinonimides, J. Am. Chem. Soc., 2017, 139, 11138–11148 CrossRef CAS PubMed.
- N. Zint, A. Dreuw and L. S. Cederbaum, Gas-Phase Stability of Derivatives of the closo-Hexaborate Dianion B6H62−, J. Am. Chem. Soc., 2002, 124, 4910–4917 CrossRef CAS PubMed.
- M. Zhong, J. Zhou, H. Fang and P. Jena, Role of ligands in the stability of BnXn and CBn−1Xn (n = 5–10; X = H, F, CN) and their potential as building blocks of electrolytes in lithium ion batteries, Phys. Chem. Chem. Phys., 2017, 19, 17937–17943 RSC.
- R. L. Hettich, R. N. Compton and R. H. Ritchie, Doubly charged negative ions of carbon-60, Phys. Rev. Lett., 1991, 67, 1242–1245 CrossRef CAS PubMed.
- X.-B. Wang, C.-F. Ding and L.-S. Wang, Photodetachment Spectroscopy of a Doubly Charged Anion: Direct Observation of the Repulsive Coulomb Barrier, Phys. Rev. Lett., 1998, 81, 3351–3354 CrossRef CAS.
- A. Sen, E. M. Matthews, G.-L. Hou, X.-B. Wang and C. E. H. Dessent, Photoelectron spectroscopy of hexachloroplatinate-nucleobase complexes: Nucleobase excited state decay observed via delayed electron emission, J. Chem. Phys., 2015, 143, 184307 CrossRef PubMed.
- K. Matheis, L. Joly, R. Antoine, F. Lépine, C. Bordas, O. T. Ehrler, A.-R. Allouche, M. M. Kappes and P. Dugourd, Photoelectron Spectroscopy of Gramicidin Polyanions: Competition between Delayed and Direct Emission, J. Am. Chem. Soc., 2008, 130, 15903–15906 CrossRef CAS PubMed.
- W. E. Boxford and C. E. H. Dessent, Probing the intrinsic features and environmental stabilization of multiply charged anions, Phys. Chem. Chem. Phys., 2006, 8, 5151–5165 RSC.
- L.-S. Wang and X.-B. Wang, Probing Free Multiply Charged Anions Using Photodetachment Photoelectron Spectroscopy, J. Phys. Chem. A, 2000, 104, 1978–1990 CrossRef CAS.
- R. F. W. Bader and M. E. Stephens, Spatial localization of the electronic pair and number distributions in molecules, J. Am. Chem. Soc., 1975, 97, 7391–7399 CrossRef CAS.
- J. Thesing, M. Stallbaum and W. Preetz, Vibrational Spectra and Normal Coordinate Analysis of the Heteroleptic Halogenohexaborates B6XnY6−n2−, n = 1–5; X ≠ Y = Cl, Br, I, Z. Naturforsch. B: Chem. Sci., 1991, 46, 602–608 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02581j |
| This journal is © the Owner Societies 2020 |