
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benchmark ab initio characterization of the abstraction and substitution pathways of the OH + CH4/C2H6 reactions
Balázs
Gruber
and
Gábor
Czakó
 *
*
MTA-SZTE Lendület Computational Reaction Dynamics Research Group, Interdisciplinary Excellence Centre and Department of Physical Chemistry and Materials Science, Institute of Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged H-6720, Hungary. E-mail: gczako@chem.u-szeged.hu
First published on 23rd June 2020
Abstract
We report a comprehensive ab initio investigation of the OH + CH4/C2H6 reactions using a high-level composite approach based on CCSD(T)-F12b/aug-cc-pVTZ geometries and CCSD(T)-F12b/aug-cc-pVnZ n = 5/Q energies augmented with additive corrections of post-CCSD(T) correlation, core correlation, scalar relativity, spin–orbit coupling, and zero-point energy. Besides the hydrogen-abstraction (HA) channel leading to H2O + CH3/C2H5 (ΔH0 = −14.37/−18.19 kcal mol−1), we reveal, for the first time, hydrogen-substitution (HS) and methyl-substitution (MS) pathways resulting in H + CH3OH/C2H5OH (ΔH0 = 13.19/7.12 kcal mol−1) and CH3 + CH3OH (ΔH0 = −2.20 kcal mol−1) products, respectively. The adiabatic barrier heights for HA, MS, and HS in OH + CH4/C2H6 are 4.78/2.18, 39.60, 43.53/41.73(52.48) kcal mol−1, respectively, where substitution proceeds with Walden-inversion or (front-side-attack retention). In the entrance channels van der Waals wells with depths of 0.5–0.8 kcal mol−1 are found and in the exit channels the HOH⋯C2H5, HOH⋯CH3, H3C⋯CH3OH, and H⋯C2H5OH complexes are characterized with De values of 2.4, 1.7, 0.7, and 0.3 kcal mol−1, respectively.
I. Introduction
Moving beyond the reactions of atoms with alkanes,1–16 the reactions of the OH radical with methane (CH4) and ethane (C2H6) have been thoroughly investigated by theory and experiment to uncover the fundamental rules of polyatomic chemical reactivity.16–39 The main product channel of the OH + CH4/C2H6 reactions results in H2O + CH3/C2H5via hydrogen-abstraction (HA). In the early years the kinetics of the OH + CH4/C2H6 reactions were investigated using experimental techniques as well as transition-state theory.17,18,28,30,31 The stationary points along the HA pathway were characterized by various ab initio and density functional methods reporting pre- and post-reaction complexes separated by a transition state for each reaction.19,22,23,27,28,33,35,38 In the case of the OH + CH4 system, the CH4⋯OH and H2O⋯CH3 complexes were also studied by different experimental techniques using stimulated Raman, infrared and electronic excitation,24,36,37 photoelectron–photofragment coincidence,34 and infrared spectroscopy in helium nanodroplets.26,32 Furthermore, detailed reactive scattering experiments were performed for the different isotope-variants of the OH + CH4 → H2O + CH3 reaction by Liu and co-workers.29 For the OH + C2H6 system, the nascent H2O vibrational distributions were measured by infrared chemiluminescence.21 In order to simulate some of the above-mentioned experiments the development of a full-dimensional potential energy surface (PES) was necessary. Following the early work of Espinosa-García and co-workers,22 Li and Guo38 developed such a PES for the OH + CH4 reaction and investigated its dynamics using quasi-classical trajectory and reduced-dimensional quantum methods.39In this study we focus on the high-level ab initio characterization of the PES of the OH + CH4/C2H6 reactions providing benchmark structures and energies for the stationary points, thereby complementing our recent work on atom (F, Cl, Br, I) + CH4/C2H6 systems.13–15 Several aspects of the present study move beyond the previous work: (1) unlike most of the early work except ref. 38, we use the explicitly correlated CCSD(T)-F12 method40 to obtain structures, frequencies, and energies; (2) basis sets as large as aug-cc-pV5Z (OH + CH4) and aug-cc-pVQZ (OH + C2H6) are employed; (3) post-CCSD(T) correlation effects are considered up to the CCSDT(Q) level of theory; (4) the correlation energy contribution of the core electrons is taken into account; (5) scalar relativistic corrections are determined; and (6) spin–orbit effects are computed. Furthermore, besides the above-mentioned quantitative advances, we investigate several new alternative reaction pathways for the OH + CH4/C2H6 systems, namely hydrogen-substitution and methyl-substitution forming H + CH3OH/C2H5OH and CH3 + CH3OH products, respectively. Knowing the energetics of the above channels is essential to develop global PESs for the title reactions and to advance our knowledge on complex reaction mechanisms of multi-channel reactions.
In Section II we describe the computational details of the composite approach used to determine the best technically feasible stationary-point properties. The results are presented and discussed in Section III. The paper ends with summary and conclusions in Section IV.
II. Computational details
Stationary-point search is performed on the basis of previous studies14,38 and chemical intuition using the second-order Møller–Plesset perturbation theory41 (MP2) with the aug-cc-pVDZ basis set.42 The obtained minima and saddle points are further optimized with the explicitly-correlated coupled-cluster singles, doubles, and perturbative triples (CCSD(T)-F12b) method40 using the aug-cc-pVDZ and aug-cc-pVTZ basis sets.42 Harmonic vibrational frequencies are computed for each stationary point using the MP2/aug-cc-pVDZ, CCSD(T)-F12b/aug-cc-pVDZ, and CCSD(T)-F12b/aug-cc-pVTZ (except for some of the OH + C2H6 stationary points) levels of theory.In order to achieve sub-chemical accuracy we perform the following singe-point energy computations at the best geometries obtained at the CCSD(T)-F12b/aug-cc-pVTZ level:
(1) CCSD(T)-F12b computations are carried out using the aug-cc-pVQZ and, in the case of the OH + CH4 system, the aug-cc-pV5Z basis sets.
(2) CCSD(T),43 CCSDT,44 and CCSDT(Q)45 methods are used with the cc-pVDZ and, in the case of the OH + CH4 system, the aug-cc-pVDZ basis sets.
(3) All-electron (AE) and frozen-core (FC) computations are performed at the CCSD(T)-F12b/cc-pCVTZ-F12 level of theory.40,46 In the usual FC treatment only the valence electrons are correlated, whereas the AE computations correlate the 1s2 electrons of the C and O atoms as well.
(4) Second-order Douglas–Kroll47 (DK) relativistic energies are computed at the AE-CCSD(T)-F12b/cc-pCVTZ-F12 level of theory.
(5) Spin–orbit (SO) corrections are determined using the Breit–Pauli Hamiltonian in the interacting-states approach48 using the Davidson-corrected49 multi-reference configuration interaction50 (MRCI+Q) method with the aug-cc-pVDZ and aug-cc-pVTZ basis sets. The MRCI computations utilize an active space of 15/21 electrons in 8/11 spatial orbitals keeping all the 4/6 core electrons frozen for OH + CH4/C2H6. Two doubly-degenerate electronic states are determined, non-SO1 and non-SO2, resulting in a 4 × 4 SO matrix, whose eigenvalues correspond to the two-fold ground (SO1) and excited (SO2) spin–orbit states.
The benchmark classical relative energies are calculated by the following composite energy expression:
| CCSD(T)-F12b/aug-cc-pVnZ + δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO, | (1) |
| δ[CCSDT] = CCSDT/(aug-)cc-pVDZ − CCSD(T)/(aug-)cc-pVDZ, | (2) |
| δ[CCSDT(Q)] = CCSDT(Q)/(aug-)cc-pVDZ − CCSDT/(aug-)cc-pVDZ, | (3) |
| Δcore = AE-CCSD(T)-F12b/cc-pCVTZ-F12 − FC-CCSD(T)-F12b/cc-pCVTZ-F12, | (4) |
| Δrel = DK-AE-CCSD(T)-F12b/cc-pCVTZ-F12 − AE-CCSD(T)-F12b/cc-pCVTZ-F12 | (5) |
| ΔSO = SO1(MRCI+Q/aug-cc-pVTZ) − non-SO1(MRCI+Q/aug-cc-pVTZ). | (6) |
The benchmark adiabatic relative energies are obtained as
| CCSD(T)-F12b/aug-cc-pVnZ + δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO + ΔZPE | (7) |
For open-shell systems the MP2 method is used in a restricted formalism (RMP2),51 unless otherwise noted, whereas all the CCSD(T)-F12b computations utilize the unrestricted UCCSD(T)-F12b52 method based on restricted open-shell Hartree–Fock (ROHF) orbitals. For the determination of the post-CCSD(T) correlation effects we use unrestricted Hartree–Fock (UHF) reference and the unrestricted UCCSD(T), UCCSDT, and UCCSDT(Q) methods. Note that, for the sake of simplicity, we usually omit the reference function as well as the R (restricted) and U (unrestricted) abbreviations in the notations.
All the MP2, CCSD(T)-F12b, MRCI, and SO computations are carried out with the MOLPRO program package,53 whereas the CCSD(T), CCSDT, and CCSDT(Q) energies are obtained with MRCC54 interfaced to MOLPRO.
III. Results and discussion
The topology of the PESs with the benchmark classical and adiabatic energies of the OH + CH4 and OH + C2H6 reactions are shown in Fig. 1 and 2, respectively, and the corresponding stationary-point geometries showing the most important structural parameters at different levels of theory are given in Fig. 3 and 4. The HA pathways are exothermic with benchmark classical(adiabatic) ΔE(ΔH0) values of −13.07(−14.37)/−16.70(−18.19) kcal mol−1 for the OH(2Π3/2) + CH4/C2H6 → H2O + CH3/C2H5 reactions. The HA barrier is significantly higher for OH + CH4, i.e., 6.30(4.78) kcal mol−1 than in the case of OH + C2H6, 3.69(2.18) kcal mol−1. The structures of the hydrogen-abstraction transition-states (HA TSs) are reactant like in both cases as the reactive C–H bonds are only stretched by 0.118/0.087 Å relative to the corresponding bond lengths in CH4/C2H6, whereas the forming O–H bonds are stretched by 0.363/0.428 Å relative to the H2O product as shown in Fig. 3 and 4. Thus, the exothermic HA processes feature early barriers in accord with the Hammond postulate55 and the more exothermic OH + C2H6 reaction has lower and more reactant-like barrier as the above-discussed data show. In the product wells of both systems there are post-reaction minima (HA PostMINs), where H2O binds to the CH3/C2H5 units with a single hydrogen-bond. The De(D0) values of the HA PostMIN complexes are 1.73(0.65)/2.36(1.35) kcal mol−1, in accord with the ratio of the intermolecular C⋯H distances of 2.383/2.312 Å. The greater stability of the latter can be explained by the fact that the C2H5 unit has larger dipole moment than the CH3 fragment.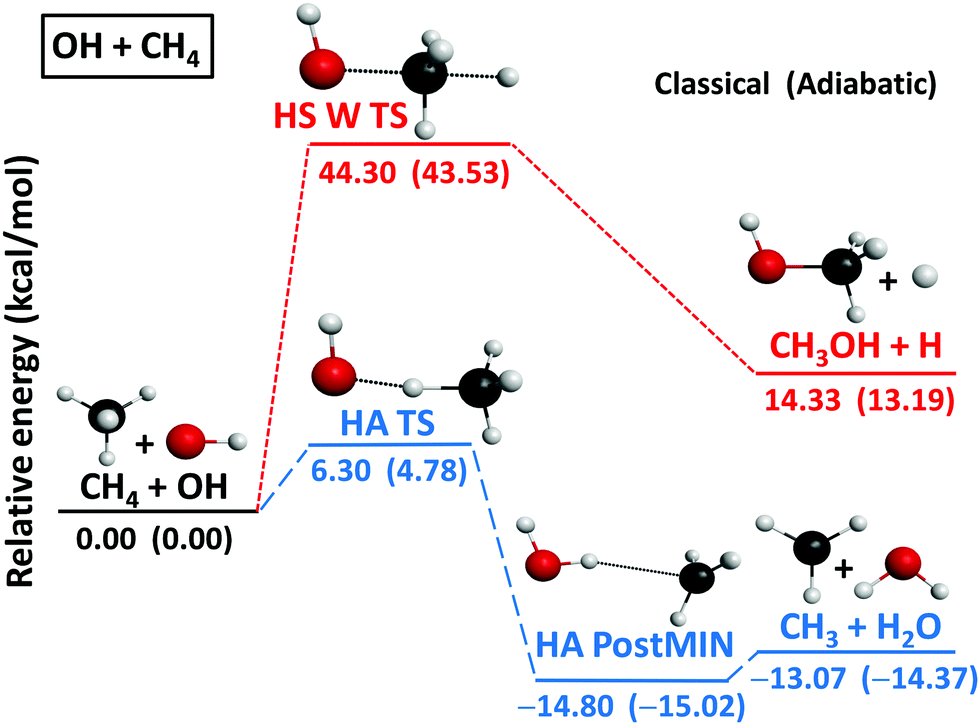 | ||
| Fig. 1 Schematic potential energy surface of the OH(2Π3/2) + CH4 reaction showing the benchmark classical (adiabatic) relative energies of the stationary points along the different reaction pathways. | ||
 | ||
| Fig. 2 Schematic potential energy surface of the OH(2Π3/2) + C2H6 reaction showing the benchmark classical (adiabatic) relative energies of the stationary points along the different reaction pathways. | ||
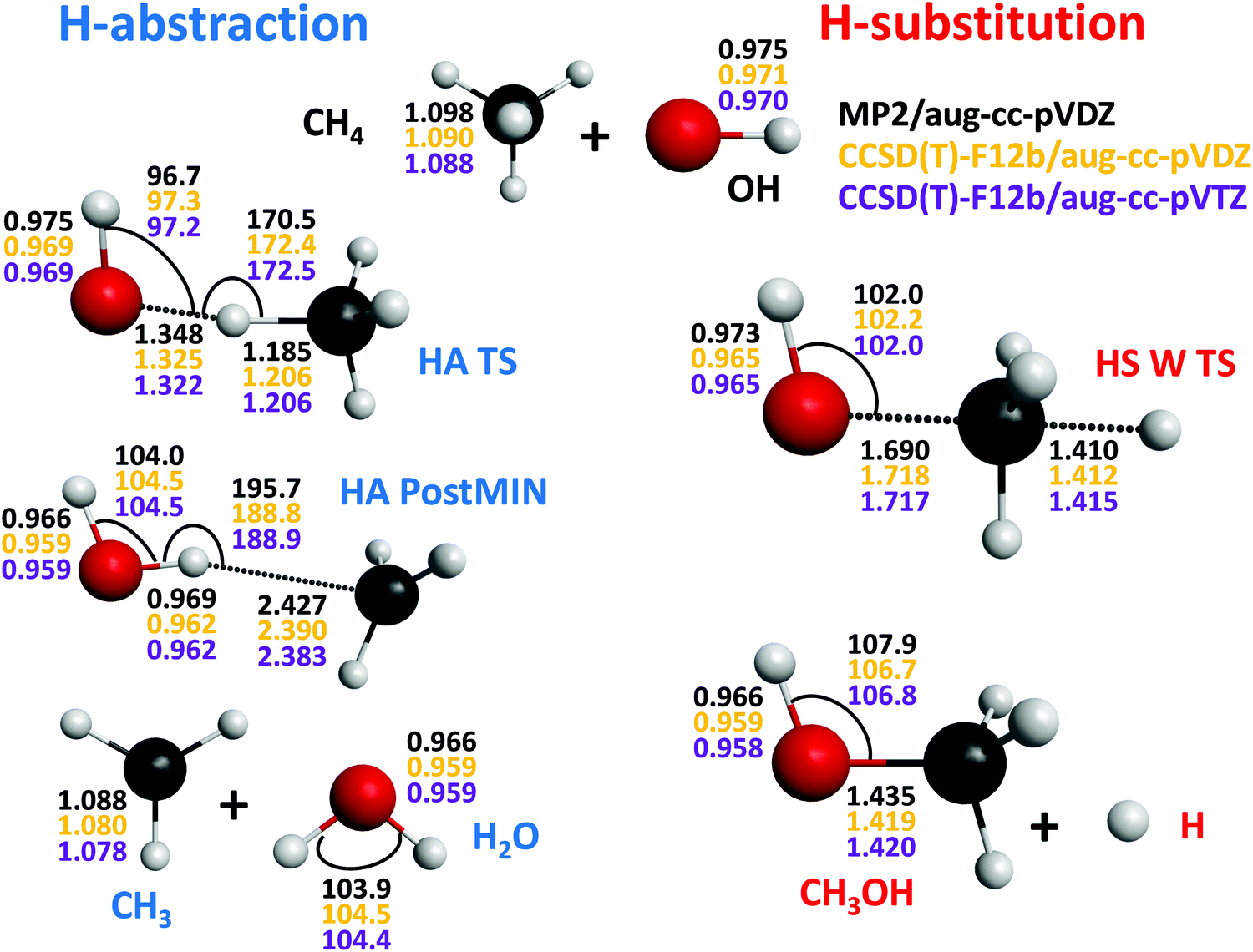 | ||
| Fig. 3 Stationary-point structures of the OH + CH4 system showing the most important distances (Å) and angles (degree) obtained at the MP2/aug-cc-pVDZ, CCSD(T)-F12b/aug-cc-pVDZ, and CCSD(T)-F12b/aug-cc-pVTZ levels of theory. | ||
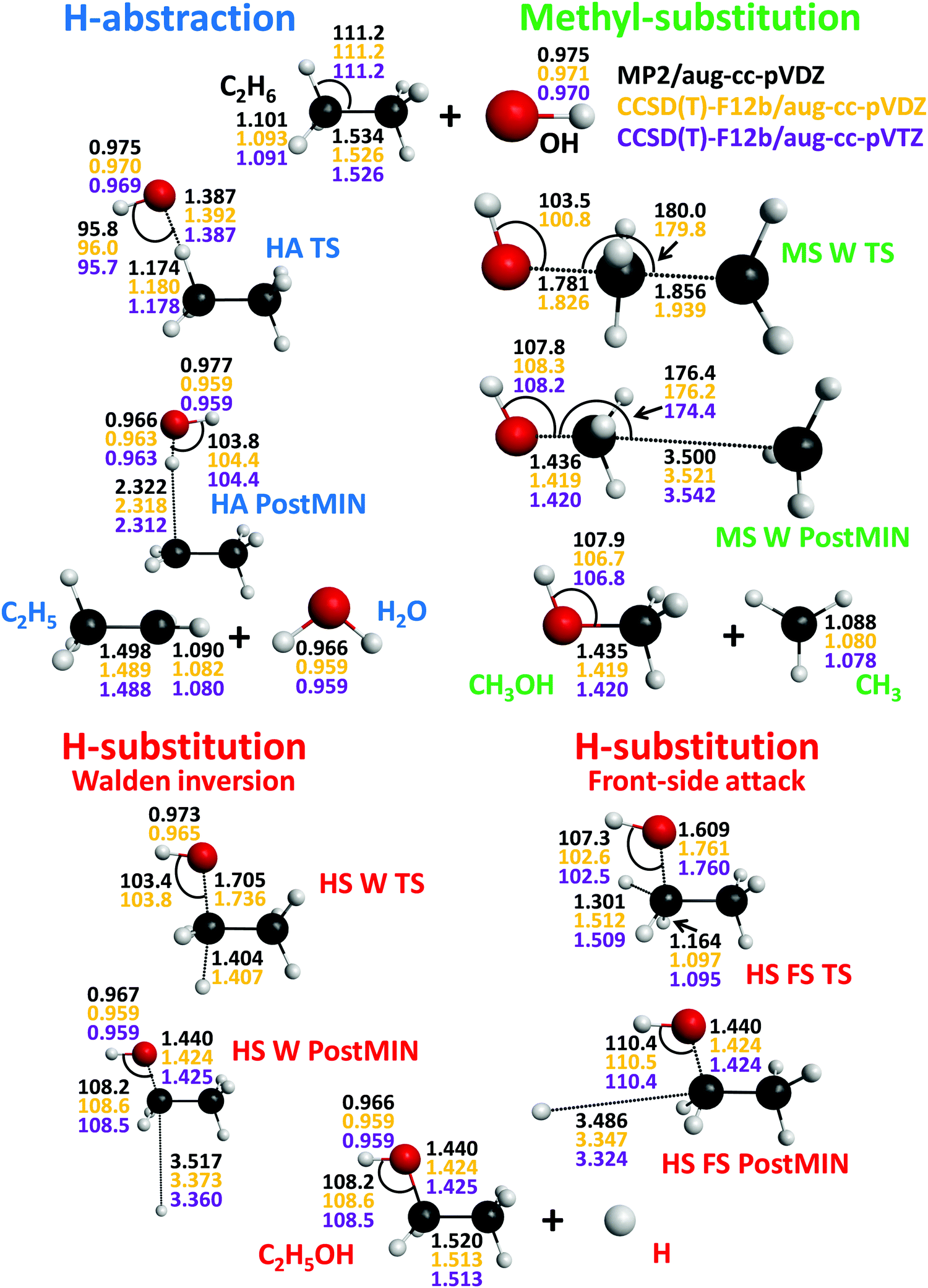 | ||
| Fig. 4 Stationary-point structures of the OH + C2H6 system showing the most important distances (Å) and angles (degree) obtained at the MP2/aug-cc-pVDZ (see footnote k of Table 2), CCSD(T)-F12b/aug-cc-pVDZ, and CCSD(T)-F12b/aug-cc-pVTZ levels of theory. | ||
For the OH + CH4 reaction we have found an endothermic, ΔE(ΔH0) = 14.33(13.19) kcal mol−1, hydrogen-substitution (HS) pathway resulting in H + CH3OH products via a Walden-inversion TS (HS W TS) with a classical(adiabatic) barrier height of 44.30(43.53) kcal mol−1 as shown in Fig. 1. The HS W TS has Cs symmetry with nearly collinear O–C–H arrangement, where the O–C and C–H distances are 1.717 and 1.415 Å, respectively, stretched by 0.297 and 0.327 Å relative the corresponding bonds in CH3OH and CH4 (Fig. 3); thus, HS has a central or slightly-late barrier. The HS pathway of the OH + CH4 reaction is revealed for the first time in the present study, but for atom + methane reactions HS is not unprecedented.4,56–60 For the H and O(3P) + CH4 reactions HS was investigated both experimentally4,56 and theoretically.4,57 For Cl + CH4 we found a Walden-inversion barrier height of 42.08(38.84) kcal mol−1,58 which is similar to that of OH + CH4. Besides Walden inversion, for the O(3P) and Cl + CH4 reactions front-side attack TSs with C2v and C4v symmetry, respectively, were reported.4,60 In the case of OH + CH4 we do not find front-side attack TS; however, reaction dynamics simulations may reveal retention trajectories as we found that the retention pathways of the Cl + CH4 → H + CH3Cl reaction avoid the high-energy C4v TS.60
In the case of the OH + C2H6 reaction HS pathways leading to H + C2H5OH also exist as shown in Fig. 2. The endothermicity of the HS channel, ΔE(ΔH0) = 9.10(7.12) kcal mol−1, is less than that of the OH + CH4 reaction, ΔE(ΔH0) = 14.33(13.19) kcal mol−1, whereas the Walden-inversion classical(adiabatic) barrier heights of the two systems are similar, i.e., 43.32(41.73) kcal mol−1 for OH + C2H6 and 44.30(43.53) kcal mol−1 for OH + CH4. Furthermore, these HS barrier heights are also very similar to that of the Cl + C2H6 system, 41.60(37.66) kcal mol−1.14 For the OH + C2H6 reaction we have found a front-side (FS) attack retention pathway via a higher barrier with classical(adiabatic) height of 54.13(52.48) kcal mol−1.14 At the HS W TS the forming O–C and breaking C–H bonds are nearly collinear (168.3°) and the H2C–C unit is almost planar, whereas at the HS FS TS the O–C–H angle is 58.2° along the reaction coordinate and the H2C–CH3 unit is ethane-like retaining its reactant configuration. Similar HS FS TS was found for the X + C2H6 [X = F, Cl, Br, I] reactions, comparing the barrier heights again the X = Cl case has similar, though slightly larger, value of 59.14(54.97) kcal mol−1. For the OH + C2H6 reaction we have found weakly-bound H⋯C2H5OH complexes in the exit channels of the HS inversion and retention pathways with small De values of 0.26 and 0.32 kcal mol−1, respectively. However, these shallow HS product-channel wells may not support a bound vibrational state, because the ZPE-corrected energies of the HS W PostMIN and HS FS PostMIN complexes are slightly above the vibrationally ground-state product asymptote by 0.05 and 0.04 kcal mol−1 (Fig. 2).
Unlike for OH + CH4, for the OH + C2H6 reaction a third product channel is possible resulting in CH3OH + CH3via methyl substitution (MS). As Fig. 2 shows, MS is slightly exothermic ΔE(ΔH0) = −0.94(−2.20) kcal mol−1 and goes over a classical(adiabatic) Walden-inversion barrier of 39.89(39.60) kcal mol−1 height. Thus, similarly to the halogen + C2H6 reactions,14 MS is both thermodynamically and kinetically favored over HS. At the central MS W TS the O–C–C atoms are nearly collinear and the forming O–C and breaking C–C bonds are stretched by 0.407 and 0.413 Å, respectively, relative to their corresponding equilibrium values in the product and reactant (Fig. 4). For the halogen + C2H6 systems we reported a higher-energy front-side attack MS pathway as well,14 however, for OH + C2H6 MS FS TS is not found, though its existence cannot be ruled out. Nevertheless, we have found a H3C⋯CH3OH complex in the product channel of MS with De(D0) values of 0.69(0.38) kcal mol−1. The stability trend, De = {2.4, 0.7, 0.3} kcal mol−1, of the {HA, MS, HS} PostMIN complexes, {HOH⋯C2H5, H3C⋯CH3OH, H⋯C2H5OH}, reflects the facts that HA PostMIN is stabilized by dipole–dipole interaction and CH3 is more polarizable than the H atom.
The above-discussed stationary-point properties correspond to our new benchmark values obtained by the composite ab initio approach described in Section II. Now let us discuss the accuracy of the computed results. The most important structural parameters of the stationary points obtained by the MP2/aug-cc-pVDZ, CCSD(T)-F12b/aug-cc-pVDZ, and CCSD(T)-F12b/aug-cc-pVTZ levels of theory are shown in Fig. 3 and 4. As seen, the MP2 and CCSD(T)-F12b results may differ by about 0.01 Å, whereas the CCSD(T)-F12b distances with the aug-cc-pVDZ and aug-cc-pVTZ basis sets usually agree within 0.001 Å, except for large intermolecular distances where an order of magnitude larger uncertainties are found due to the flatness of the potential along the dissociation coordinate.
The convergence of the relative energies are shown in Tables 1 and 2 for the OH + CH4 and OH + C2H6 systems, respectively, and the corresponding auxiliary corrections are detailed in Tables 3 and 4. The MP2 and CCSD(T)-F12b relative energies differ by 1–6 kcal mol−1, for example, MP2 overestimates the HA barrier heights by 2.4–2.8 kcal mol−1 and underestimates the endoergicity of the HS channels by 5.8–6.0 kcal mol−1 as seen in Tables 1 and 2. Thus, it is clear that the use of the CCSD(T)-F12b method is needed to achieve chemical accuracy (uncertainty less than 1 kcal mol−1). For the OH + CH4 system we have CCSD(T)-F12b energies up to the large aug-cc-pV5Z basis and on the basis of Table 1 we find that the average absolute deviations of the aug-cc-pVDZ, aug-cc-pVTZ, and aug-cc-pVQZ relative energies from the aug-cc-pV5Z data decrease as 0.25, 0.06, 0.04 kcal mol−1, respectively, showing the excellent basis convergence of the explicitly-correlated CCSD(T)-F12b method. Thus, we expect that even the CCSD(T)-F12b/aug-cc-pVQZ result is basis converged within 0.1 kcal mol−1. This finding for OH + CH4 is useful for the larger OH + C2H6 system, where we do not perform the CCSD(T)-F12b/aug-cc-pV5Z computations. For OH + C2H6, the aug-cc-pVDZ and aug-cc-pVTZ CCSD(T)-F12b relative energies are converged with average absolute deviations of 0.27 and 0.04 kcal mol−1, respectively, with respect to the CCSD(T)-F12b/aug-cc-pVQZ results.
| Stationary points | MP2 | CCSD(T)-F12b | ∑Δf | Classicalg | Δ ZPE | Adiabatici | |||
|---|---|---|---|---|---|---|---|---|---|
| aVDZa | aVDZb | aVTZc | aVQZd | aV5Ze | |||||
| a MP2/aug-cc-pVDZ relative energies obtained at MP2/aug-cc-pVDZ geometries. b CCSD(T)-F12b/aug-cc-pVDZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVDZ geometries. c CCSD(T)-F12b/aug-cc-pVTZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. d CCSD(T)-F12b/aug-cc-pVQZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. e CCSD(T)-F12b/aug-cc-pV5Z relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. f ∑Δ = δ[CCSDT/aVDZ] + δ[CCSDT(Q)/aVDZ] + Δcore + Δrel + ΔSO(aVTZ). g Benchmark classical relative energy obtained as CCSD(T)-F12b/aV5Z + ∑Δ. h ZPE correction obtained at CCSD(T)-F12b/aug-cc-pVTZ. i Benchmark adiabatic relative energy obtained as classical + ΔZPE. | |||||||||
| HA TS | 8.50 | 6.08 | 6.27 | 6.30 | 6.38 | −0.08 | 6.30 | −1.52 | 4.78 |
| HA PostMIN | −18.22 | −15.15 | −14.99 | −15.00 | −14.96 | +0.16 | −14.80 | −0.22 | −15.02 |
| HS W TS | 41.07 | 44.52 | 44.40 | 44.38 | 44.42 | −0.12 | 44.30 | −0.77 | 43.53 |
| CH3OH + H | 7.62 | 13.39 | 14.00 | 13.95 | 13.98 | +0.35 | 14.33 | −1.14 | 13.19 |
| CH3 + H2O | −16.28 | −13.19 | −13.15 | −13.25 | −13.25 | +0.18 | −13.07 | −1.30 | −14.37 |
| Stationary points | MP2 | CCSD(T)-F12b | ∑Δe | Classicalf | Δ ZPE | Adiabatich | ||
|---|---|---|---|---|---|---|---|---|
| aVDZa | aVDZb | aVTZc | aVQZd | |||||
| a MP2/aug-cc-pVDZ relative energies obtained at MP2/aug-cc-pVDZ geometries. b CCSD(T)-F12b/aug-cc-pVDZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVDZ geometries. c CCSD(T)-F12b/aug-cc-pVTZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. d CCSD(T)-F12b/aug-cc-pVQZ relative energies obtained at CCSD(T)-F12b/aug-cc-pVTZ geometries. e ∑Δ = δ[CCSDT] + δ[CCSDT(Q)] + Δcore + Δrel + ΔSO(aVTZ). f Benchmark classical relative energy obtained as CCSD(T)-F12b/aVQZ + ∑Δ. g ZPE correction obtained at CCSD(T)-F12b/aug-cc-pVTZ (for exemptions see j). h Benchmark adiabatic relative energy obtained as classical + ΔZPE. i Obtained at CCSD(T)-F12b/aug-cc-pVDZ geometries. j ZPE correction obtained at CCSD(T)-F12b/aug-cc-pVDZ. k Obtained with UMP2. | ||||||||
| HA TS | 6.21 | 3.45 | 3.65 | 3.70 | −0.01 | 3.69 | −1.51 | 2.18 |
| HA PostMIN | −21.78 | −19.28 | −19.07 | −19.09 | +0.03 | −19.06 | −0.48 | −19.54 |
| HS W TS | 39.89 | 43.57 | 43.49i | 43.51i | −0.19 | 43.32 | −1.59j | 41.73 |
| HS W PostMIN | 2.11 | 7.96 | 8.62 | 8.63 | +0.21 | 8.84 | −1.67 | 7.17 |
| HS FS TS | 55.68k | 54.33 | 54.47 | 54.53 | −0.40 | 54.13 | −1.65j | 52.48 |
| HS FS PostMIN | 2.00k | 7.90 | 8.56 | 8.57 | +0.21 | 8.78 | −1.62j | 7.16 |
| MS W TS | 44.80k | 40.29 | 40.21i | 40.21i | −0.32 | 39.89 | −0.29j | 39.60 |
| MS W PostMIN | −3.14k | −2.02 | −1.85 | −1.89 | +0.26 | −1.63 | −0.95j | −2.58 |
| C2H5OH + H | 2.29 | 8.29 | 8.91 | 8.89 | +0.21 | 9.10 | −1.98 | 7.12 |
| CH3OH + CH3 | −2.24 | −1.16 | −1.13 | −1.21 | +0.27 | −0.94 | −1.26 | −2.20 |
| C2H5 + H2O | −18.96 | −16.64 | −16.61 | −16.75 | +0.05 | −16.70 | −1.49 | −18.19 |
| Stationary points | δ[CCSDT]a | δ[CCSDT(Q)]b | Δ core | Δ rel | Δ SO | Δ ZPE | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VDZ | aVDZ | VDZ | aVDZ | aVDZ | aVTZ | MP2/DZ | CC/DZ | CC/TZ | |||
| a CCSDT – CCSD(T) obtained with the cc-pVDZ (VDZ) and aug-cc-pVDZ (aVDZ) basis sets at CCSD(T)-F12b/aug-cc-pVTZ geometries. b CCSDT(Q) – CCSDT obtained with the cc-pVDZ (VDZ) and aug-cc-pVDZ (aVDZ) basis sets at CCSD(T)-F12b/aug-cc-pVTZ geometries. c Core-correlation correction obtained as the difference between all-electron and frozen-core CCSD(T)-F12b/cc-pCVTZ-F12 relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. d Scalar relativistic effect obtained as the difference between Douglas–Kroll and non-relativistic all-electron CCSD(T)-F12b/cc-pCVTZ-F12 relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. e Spin–orbit (SO) corrections obtained as the difference between the SO and non-SO ground-state MRCI+Q/aug-cc-pVDZ (aVDZ) or MRCI+Q/aug-cc-pVTZ (aVTZ) relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. f ZPE corrections obtained at MP2/aug-cc-pVDZ (MP2/DZ), CCSD(T)-F12b/aug-cc-pVDZ (CC/DZ), and CCSD(T)-F12b/aug-cc-pVTZ (CC/TZ). | |||||||||||
| HA TS | −0.11 | −0.12 | −0.15 | −0.23 | 0.06 | 0.02 | 0.18 | 0.19 | −1.05 | −1.67 | −1.52 |
| HA PostMIN | 0.00 | 0.04 | −0.13 | −0.15 | −0.04 | 0.12 | 0.18 | 0.19 | −0.06 | −0.01 | −0.22 |
| HS W TS | −0.26 | −0.18 | −0.35 | −0.43 | 0.25 | 0.05 | 0.17 | 0.19 | −0.75 | −0.84 | −0.77 |
| CH3OH + H | 0.09 | 0.18 | −0.20 | −0.19 | 0.02 | 0.15 | 0.18 | 0.19 | −1.24 | −1.12 | −1.14 |
| CH3 + H2O | 0.01 | 0.05 | −0.13 | −0.14 | −0.04 | 0.12 | 0.18 | 0.19 | −1.26 | −1.26 | −1.30 |
| Stationary points | δ[CCSDT]a | δ[CCSDT(Q)]b | Δ core | Δ rel | Δ SO | Δ ZPE | |||
|---|---|---|---|---|---|---|---|---|---|
| aVDZ | aVTZ | MP2/DZ | CC/DZ | CC/TZ | |||||
| a CCSDT – CCSD(T) obtained with the cc-pVDZ basis set at CCSD(T)-F12b/aug-cc-pVTZ geometries. b CCSDT(Q) – CCSDT obtained with the cc-pVDZ basis set at CCSD(T)-F12b/aug-cc-pVTZ geometries. c Core-correlation correction obtained as the difference between all-electron and frozen-core CCSD(T)-F12b/cc-pCVTZ-F12 relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. d Scalar relativistic effect obtained as the difference between Douglas–Kroll and non-relativistic all-electron CCSD(T)-F12b/cc-pCVTZ-F12 relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. e Spin–orbit (SO) corrections obtained as the difference between the SO and non-SO ground-state MRCI+Q/aug-cc-pVDZ (aVDZ) or MRCI+Q/aug-cc-pVTZ (aVTZ) relative energies at CCSD(T)-F12b/aug-cc-pVTZ geometries. f ZPE corrections obtained at MP2/aug-cc-pVDZ (MP2/DZ), CCSD(T)-F12b/aug-cc-pVDZ (CC/DZ), and CCSD(T)-F12b/aug-cc-pVTZ (CC/TZ). g Obtained at CCSD(T)-F12b/aug-cc-pVDZ geometries. h Obtained with UMP2. | |||||||||
| HA TS | −0.11 | −0.14 | 0.03 | 0.02 | 0.18 | 0.19 | −2.08 | −1.74 | −1.51 |
| HA PostMIN | −0.04 | −0.13 | −0.11 | 0.12 | 0.18 | 0.19 | −0.08 | −0.31 | −0.48 |
| HS W TS | −0.27g | −0.40g | 0.24g | 0.05g | 0.18g | 0.19g | −1.55 | −1.59 | |
| HS W PostMIN | 0.10 | −0.20 | −0.02 | 0.14 | 0.18 | 0.19 | −1.82 | −1.69 | −1.67 |
| HS FS TS | −0.38 | −0.50 | 0.26 | 0.03 | 0.18 | 0.19 | −1.88h | −1.65 | |
| HS FS PostMIN | 0.10 | −0.20 | −0.02 | 0.14 | 0.18 | 0.19 | −1.80h | −1.62 | |
| MS W TS | −0.48g | −0.43g | 0.39g | 0.01g | 0.18g | 0.19g | 1.19h | −0.29 | |
| MS W PostMIN | −0.02 | −0.12 | 0.09 | 0.12 | 0.18 | 0.19 | −0.84h | −0.95 | |
| C2H5OH + H | 0.10 | −0.20 | −0.02 | 0.14 | 0.18 | 0.19 | −2.08 | −1.92 | −1.98 |
| CH3OH + CH3 | −0.02 | −0.11 | 0.09 | 0.12 | 0.18 | 0.19 | −1.15 | −1.21 | −1.26 |
| C2H5 + H2O | −0.03 | −0.12 | −0.11 | 0.12 | 0.18 | 0.19 | −1.51 | −1.47 | −1.49 |
The large-basis CCSD(T)-F12b computations provide a very good estimate of the complete-basis-set limit of the CCSD(T) relative energies. If one aims to approach the “exact” energies additional small corrections, such as post-CCSD(T) and core electron correlation as well as scalar and SO relativistic effects should be considered. Furthermore, to get experimentally observable quantities the ZPE corrections have to be taken into account. These so-called auxiliary corrections are given in Tables 3 and 4 for the OH + CH4 and OH + C2H6 systems, respectively. For OH + CH4 we have determined the δ[CCSDT] and δ[CCSDT(Q)] corrections with the cc-pVDZ and aug-cc-pVDZ basis sets. As Table 3 shows even the small cc-pVDZ basis provides good estimates for these post-CCSD(T) correlation effects suggesting that it is sufficient to perform only the CCSDT(Q)/cc-pVDZ computations for the larger OH + C2H6 system. In the case of the products and HA PostMIN of the OH + CH4 reaction the δ[CCSDT] and δ[CCSDT(Q)] absolute corrections are between 0–0.2 kcal mol−1 and partially cancel each other. However, for the HA TS and HS W TS the aug-cc-pVDZ(cc-pVDZ) corrections add up to −0.35(−0.26) and −0.61(−0.61) kcal mol−1, respectively, showing the good performance of the smaller basis set. For the OH + C2H6 system most of the post-CCSD(T) corrections have the same negative sign, except the HS products and PostMINs (Table 4). The largest cumulative post-CCSD(T) corrections are found for the TSs, namely −0.25, −0.67, −0.88, and −0.91 kcal mol−1, for HA TS, HS W TS, HS FS TS, and MS W TS, respectively, which are clearly not negligible if sub-chemical accuracy is desired.
For both reactions the core correlation corrections are in the range from −0.11 to +0.39 kcal mol−1, whereas the scalar relativistic effects on the relative energies are always small positive values between 0.01 and 0.15 kcal mol−1. The largest core corrections are obtained for the HS TSs (0.24–0.26 kcal mol−1) and for the MS W TS (0.39 kcal mol−1) as seen in Tables 3 and 4.
SO interaction almost fully quenches at all the stationary points, while lowers the reactant asymptote, thereby increasing all the relative energies by 0.19 kcal mol−1. As seen in Tables 3 and 4 we have performed SO computations with the aug-cc-pVDZ and aug-cc-pVTZ basis sets, which result in almost the same corrections of 0.18 (once 0.17) and 0.19 kcal mol−1, respectively. These values are in good agreement with the experimental data of 0.20 kcal mol−1 deduced from the measured SO splitting (ε = 0.40 kcal mol−1) of the OH radical as ε/2. To get deeper insight, we have computed SO and non-SO potential energy curves along the intermolecular coordinates of the CH4⋯OH and C2H6⋯OH systems while the OH approaches the CH4 and C2H6 molecules from different directions as shown in Fig. 5 and 6. The ground electronic state of the OH radical is the 2 × 2-fold degenerate 2Π, which splits to a 2-fold SO ground (2Π3/2) and a 2-fold SO excited state (2Π1/2). As OH approaches CH4 the two doublet non-SO state remains quasi-degenerate and the SO ground and excited states are below and above the non-SO states by ε/2 as shown in Fig. 5. There is a van der Waals well in the entrance channel whose depth is 0.5 and 0.7 kcal mol−1 with HCH3⋯OH and H3CH⋯OH C3v orientations, respectively. The well depths and positions are not affected by the SO interaction, because the wells are at C⋯O distances of 3–4 Å, whereas the SO correction is almost constant until the C⋯O distance decreases to about 2 Å, where the difference between the SO and non-SO ground state energies rapidly drops to zero (see insets in Fig. 5). This fast quenching of SO interaction occurs at high relative energies above 100 kcal mol−1, where the quasi-degeneracy of the non-SO states starts lifting. Interestingly, in the case of the halogen (X) + CH4 reactions, the HCH3⋯X minimum is the deeper with depth of 0.6(0.9) kcal mol−1 and the H3CH⋯X well is the shallower, 0.3(0.3) kcal mol−1, with(without) SO correction.13,58 In the case of the H3CCH3⋯OH (C3v) and H3CH2CH⋯OH (Cs) arrangements in the entrance well of OH + C2H6 reaction, the conclusions are qualitatively the same as for OH + CH4, i.e., the former minimum is 0.6 kcal mol−1 deep, whereas the latter is deeper, 0.7 kcal mol−1, and these are not affected by the SO interactions. If OH approaches C2H6 perpendicularly to the C–C bond, the well is the deepest, 0.8 kcal mol−1, and slightly affected by the SO interaction as the quenching and the departure of the two non-SO states occur in the 2.5–3.5 Å range of the CC⋯O distance as seen in Fig. 6. For the X + C2H6 systems, the H3CCH3⋯X (C3v) minimum was found to be the deepest, slightly below the perpendicular well, and the H3CH2CH⋯X (Cs) arrangement gave the shallowest minimum.14 These entrance-channel wells may play an important role in the dynamics of the title reactions at low collision energies, especially for OH + C2H6, which has a low barrier for HA.
 | ||
| Fig. 5 Potential energy curves along the C3 axis of the CH4⋯OH system obtained at the MRCI+Q/aug-cc-pVDZ level of theory while the structures of the CH4 and OH units are kept frozen at their equilibrium geometries. SO1 and SO2 denote the spin–orbit ground and excited states, whereas non-SO1 and non-SO2 are the non-relativistic ground and excited electronic states, respectively. The insets show the distance dependence of the spin–orbit corrections obtained as difference between the SO1 and non-SO1 energies. | ||
 | ||
| Fig. 6 Potential energy curves along the C3 (left), CH (middle), and CC-perpendicular (right) axes of the C2H6⋯OH system obtained at the MRCI+Q/aug-cc-pVDZ level of theory while the structures of the C2H6 and OH units are kept frozen at their equilibrium geometries. SO1 and SO2 denote the spin–orbit ground and excited states, whereas non-SO1 and non-SO2 are the non-relativistic ground and excited electronic states, respectively. The insets show the distance dependence of the spin–orbit corrections obtained as difference between the SO1 and non-SO1 energies. | ||
The ZPE effects on the relative energies are always negative and in the most cases the absolute corrections are in the range of 1–2 kcal mol−1 as shown in Tables 1 and 2. The mean absolute deviation of the MP2 ZPE corrections from the CCSD(T)-F12b results is about 0.2 kcal mol−1, whereas the CCSD(T)-F12b/aug-cc-pVTZ data are converged within 0.1 kcal mol−1 as Tables 3 and 4 show. These ZPE effects are clearly not negligible if we aim to compute chemically accurate, measurable adiabatic relative energies.
For the reaction enthalpies comparison between the present computed adiabatic relative energies and the “experimental” data deduced from 0 K enthalpies of formation taken from the Active Thermochemical Tables (ATcT)61 is possible. Note that ATcT collects the best measured and computed thermochemical data, thereby providing the best available predictions for enthalpies of formation and their uncertainties of several chemical species. We call these ATcT data as “experimental” even if they rely on both theory and experiment. As Table 5 shows the agreement between the present ab initio 0 K reaction enthalpies and experiment is excellent; the mean absolute deviation between theory and experiment is only 0.07 kcal mol−1 and, in most cases, the theoretical predictions are within the error bars of the experiment. This comparison demonstrates that one needs to consider the auxiliary corrections (post-CCSD(T), core, scalar relativistic, SO) to achieve this outstanding accuracy, because the cumulative effect of these corrections averaged for the five reaction channels is 0.21 kcal mol−1, which is significantly larger than the above-mentioned mean deviation between theory and experiment. Furthermore, the excellent agreement for these measurable quantities confirms the accuracy of the present theoretical predictions for the experimentally hardly accessible properties, such as barrier heights.
| Reaction | Theorya | Experimentb |
|---|---|---|
| a Benchmark theoretical reaction enthalpies (this work). b Data obtained from the latest version (1.122 g) of the Active Thermochemical Tables (ATcT).61 Uncertainties are derived from the uncertainties of each 0 K enthalpy of formation given in ATcT using the Gaussian error-propagation law. | ||
| OH + CH4 → CH3OH + H | 13.19 | 13.22 ± 0.04 |
| OH + CH4 → CH3 + H2O | −14.37 | −14.28 ± 0.02 |
| OH + C2H6 → C2H5OH + H | 7.12 | 7.13 ± 0.06 |
| OH + C2H6 → CH3OH + CH3 | −2.20 | −2.16 ± 0.05 |
| OH + C2H6 → C2H5 + H2O | −18.19 | −18.35 ± 0.07 |
IV. Summary and conclusions
The reactions of the OH radical with methane and ethane have become benchmark systems to understand the dynamics and mechanisms of polyatomic reactions. Despite many previous experimental and theoretical studies16–39 focusing on the exothermic (ΔH0 = −14.37/−18.19 kcal mol−1) hydrogen-abstraction reaction resulting in H2O + CH3/C2H5via low adiabatic barriers of 4.78/2.18 kcal mol−1, other product channels and their energetic requirements were unknown until the present study. Here, we show that hydrogen-substitution leading to H + CH3OH/C2H5OH is endothermic with ΔH0 = 13.19/7.12 kcal mol−1 and can proceed via a Walden-inversion barrier with adiabatic height of 43.53/41.73 kcal mol−1 or for the latter we have also found a front-side attack pathways via an adiabatic barrier of 52.48 kcal mol−1. For OH + C2H6 a methyl-substitution channel forming CH3 + CH3OH products is also possible, which is exothermic, ΔH0 = −2.20 kcal mol−1, but has a large adiabatic barrier of 39.60 kcal mol−1. For the product channel several complexes have been revealed and characterized, showing a stability order of HOH⋯C2H5, HOH⋯CH3, H3C⋯CH3OH, and H⋯C2H5OH with De values of 2.4, 1.7, 0.7, and 0.3 kcal mol−1, respectively. For the first time, we have performed SO computations for the entrance channel, thereby revealing van der Waals wells with depths of 0.5–0.8 kcal mol−1 depending on the relative orientation of the reactants. Unlike for the halogen + CH4/C2H6 systems,13,14 the well is the deepest for perpendicular C–C⋯OH approach (0.8 kcal mol−1), followed by the H3CH/H3CH2CH⋯OH (0.7 kcal mol−1) and HCH3/H3CCH3⋯OH (0.5–0.6 kcal mol−1) arrangements. These pre- and post-reaction wells may play significant roles in the dynamics of the hydrogen-abstraction processes, especially at low collision energies, by steering the reactants in the entrance channels and affecting product rotation in the exit channels, respectively.The stationary-point properties have been computed using an accurate composite ab initio approach which goes beyond the widely-used standard quantum chemistry. The complete-basis-set limit of CCSD(T) is approached well within 0.1 kcal mol−1 with explicitly-correlated CCSD(T)-F12b/aug-cc-pVnZ computations with n = 5 and 4(Q) for OH + CH4 and OH + C2H6, respectively. Post-CCSD(T) correlation (−0.9 to −0.1), core correlation (−0.1 to +0.4), scalar relativistic (+0.0 to +0.2), SO (+0.2), and ZPE (−2 to −1) effects are determined resulting in typical values for the title reactions as indicated in parentheses in kcal mol−1. We conclude that the present theoretical predictions provide sub-chemically accurate relative energies with an estimated uncertainty of around 0.1 kcal mol−1, which is about an order of magnitude better than that of the standard quantum chemistry studies. The outstanding accuracy of the present results can be confirmed by the comparison of the computed reaction enthalpies with the corresponding experimental data61 showing only 0.07 kcal mol−1 mean absolute deviation.
The new insights into the mechanisms and alternative reaction pathways of the title reactions are essential to develop global PESs for the OH + CH4/C2H6 systems, allowing dynamical investigations over a large collision energy range. Furthermore, the relative energies of the stationary points may guide future experimental investigations showing the thermodynamical and kinetical controls of the different processes. Finally, the present composite approach may be utilized in several similar ab initio investigations if high accuracy is desired.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Research, Development and Innovation Office-NKFIH, K-125317, the Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT, and the Momentum (Lendület) Program of the Hungarian Academy of Sciences for financial support.References
- C. Murray and A. J. Orr-Ewing, Int. Rev. Phys. Chem., 2004, 23, 435 Search PubMed
.
- E. S. Whitney, A. M. Zolot, A. B. McCoy, J. S. Francisco and D. J. Nesbitt, J. Chem. Phys., 2005, 122, 124310 CrossRef PubMed
- W. Li, C. S. Huang, M. Patel, D. Wilson and A. Suits, J. Chem. Phys., 2006, 124, 011102 CrossRef PubMed
- D. Troya, G. C. Schatz, D. J. Garton, A. L. Brunsvold and T. K. Minton, J. Chem. Phys., 2004, 120, 731 CrossRef CAS PubMed
- R. J. Holiday, C. H. Kwon, C. J. Annesley and F. F. Crim, J. Chem. Phys., 2006, 125, 133101 CrossRef PubMed
- H. A. Bechtel, Z. H. Kim, J. P. Camden and R. N. Zare, J. Chem. Phys., 2004, 120, 791 CrossRef CAS PubMed
- K. Liu, J. Chem. Phys., 2015, 142, 080901 CrossRef PubMed
- B. Fu, X. Shan, D. H. Zhang and D. C. Clary, Chem. Soc. Rev., 2017, 46, 7625 RSC
- J. Espinosa-Garcia, J. C. Corchado, M. Garcia-Chamorro and C. Rangel, Phys. Chem. Chem. Phys., 2018, 20, 19860 RSC
- F. Meng, W. Yan and D. Wang, Phys. Chem. Chem. Phys., 2012, 14, 13656 RSC
- Z. Chen, J. Chen, R. Chen, T. Xie, X. Wang, S. Liu, G. Wu, D. Dai, X. Yang and D. H. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9202 CrossRef CAS PubMed
- R. Welsch and U. Manthe, J. Chem. Phys., 2015, 142, 064309 CrossRef PubMed
- G. Czakó and J. M. Bowman, J. Phys. Chem. A, 2014, 118, 2839 CrossRef PubMed
- D. Papp, B. Gruber and G. Czakó, Phys. Chem. Chem. Phys., 2019, 21, 396 RSC
- G. Czakó, T. Győri, B. Olasz, D. Papp, I. Szabó, V. Tajti and D. A. Tasi, Phys. Chem. Chem. Phys., 2020, 22, 4298 RSC
- B. Jiang and H. Guo, J. Chin. Chem. Soc., 2014, 61, 847 CrossRef CAS
- D. L. Baulch, R. J. B. Craven, M. Din, D. D. Drysdale, S. Grant, D. J. Richardson, A. Walker and G. Watling, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 689 RSC
- F. P. Tully, A. T. Droege, M. L. Koszykowski and C. F. Melius, J. Phys. Chem., 1986, 90, 691 CrossRef CAS
- K. D. Dobbs, D. A. Dixon and A. Komornicki, J. Chem. Phys., 1993, 98, 8852 CrossRef CAS
- L. N. Krasnoperov and J. V. Michael, J. Phys. Chem. A, 2004, 108, 5643 CrossRef CAS
- N. I. Butkovskaya and D. W. Setser, Int. Rev. Phys. Chem., 2003, 22, 1 Search PubMed
- J. Espinosa-Garcia and J. C. Corchado, Theor. Chem. Acc., 2015, 134, 6 Search PubMed
- F. Khaled, B. R. Giri, M. Szőri, B. Viskolcz and A. Farooq, Chem. Phys. Lett., 2015, 641, 158 CrossRef CAS
- M. Tsiouris, M. D. Wheeler and M. I. Lester, Chem. Phys. Lett., 1999, 302, 192 CrossRef CAS
- L. Bonnet and J. Espinosa-Garcia, Chem. Phys. Lett., 2018, 711, 184 CrossRef CAS
- S. Rudić, J. M. Merritt and R. E. Miller, Phys. Chem. Chem. Phys., 2009, 11, 5345 RSC
- T. Hashimoto and S. Iwata, J. Phys. Chem. A, 2002, 106, 2652 CrossRef CAS
- J. P. Senosiain, C. B. Musgrave and D. M. Golden, J. Phys. Chem. A, 2001, 105, 1669 CrossRef CAS
- B. Zhang, W. Shiu and K. Liu, J. Phys. Chem. A, 2005, 109, 8983 CrossRef CAS PubMed
- J. Korchowiec, S. Kawahara, K. Matsumura, T. Uchimaru and M. Sugie, J. Phys. Chem. A, 1999, 103, 3548 CrossRef CAS
- H. Lin, Y. Zhao, B. A. Ellingson, J. Pu and D. G. Truhlar, J. Am. Chem. Soc., 2005, 127, 2830 CrossRef CAS PubMed
- P. L. Raston, E. I. Obi and G. E. Douberly, J. Phys. Chem. A, 2017, 121, 7597 CrossRef CAS PubMed
- B. A. Ellingson, J. Pu, H. Lin, Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2007, 111, 11706 CrossRef CAS PubMed
- Y. Benitez, D. Lu, K. G. Lunny, J. Li, H. Guo and R. E. Continetti, J. Phys. Chem. A, 2019, 123, 4825 CrossRef CAS PubMed
- L. Masgrau, Á. González-Lafont and J. M. Lluch, J. Chem. Phys., 2001, 115, 4515 CrossRef CAS
- M. Tsiouris, M. D. Wheeler and M. I. Lester, J. Chem. Phys., 2001, 114, 187 CrossRef CAS
- M. D. Wheeler, M. Tsiouris, M. I. Lester and G. Lendvay, J. Chem. Phys., 2000, 112, 6590 CrossRef CAS
- J. Li and H. Guo, J. Chem. Phys., 2015, 143, 221103 CrossRef PubMed
- H. Song, Y. Lu, J. Li, M. Yang and H. Guo, J. Chem. Phys., 2016, 144, 164303 CrossRef PubMed
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS
- J. Noga and R. J. Bartlett, J. Chem. Phys., 1987, 86, 7041 CrossRef CAS
- M. Kállay and J. Gauss, J. Chem. Phys., 2005, 123, 214105 CrossRef PubMed
- J. G. Hill, S. Mazumder and K. A. Peterson, J. Chem. Phys., 2010, 132, 054108 CrossRef PubMed
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89 CAS
- A. Berning, M. Schweizer, H.-J. Werner, P. J. Knowles and P. Palmieri, Mol. Phys., 2000, 98, 1823 CrossRef CAS
- S. R. Langhoff and E. R. Davidson, Int. J. Quantum Chem., 1974, 8, 61 CrossRef CAS
- K. R. Shamasundar, G. Knizia and H.-J. Werner, J. Chem. Phys., 2011, 135, 054101 CrossRef CAS PubMed
- R. D. Amos, J. S. Andrews, N. C. Handy and P. J. Knowles, Chem. Phys. Lett., 1991, 185, 256 CrossRef CAS
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, et al., MOLPRO, version 2015.1, a package of ab initio programs, see http://www.molpro.net.
- MRCC, a quantum chemical program suite written by M. Kállay, Z. Rolik, I. Ladjánszki, L. Szegedy, B. Ladóczki, J. Csontos and B. Kornis, see also Z. Rolik and M. Kállay, J. Chem. Phys., 2011, 135, 104111, as well as: http://www.mrcc.hu.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334 CrossRef CAS
- A. Chattopadhyay, S. Tasaki, R. Bersohn and M. Kawasaki, J. Chem. Phys., 1991, 95, 1033 CrossRef CAS
- Z. Zhao, Z. Zhang, S. Liu and D. H. Zhang, Nat. Commun., 2017, 8, 14506 CrossRef PubMed
- G. Czakó and J. M. Bowman, J. Chem. Phys., 2012, 136, 044307 CrossRef PubMed
- G. Czakó, J. Phys. Chem. A, 2012, 116, 7467 CrossRef PubMed
- L. Krotos and G. Czakó, J. Phys. Chem. A, 2017, 121, 9415 CrossRef CAS PubMed
-
B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.122g of the Thermochemical Network, Argonne National Laboratory, 2019, available at http://ATcT.anl.gov Search PubMed
| This journal is © the Owner Societies 2020 |