
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of light with a non-covalent zinc porphyrin–graphene oxide nanohybrid†
Ewelina
Gacka
a,
Gotard
Burdzinski
 b,
Bronisław
Marciniak
ac,
Adam
Kubas
*d and
Anna
Lewandowska-Andralojc
*ac
b,
Bronisław
Marciniak
ac,
Adam
Kubas
*d and
Anna
Lewandowska-Andralojc
*ac
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznanskiego 8, 61-614 Poznan, Poland. E-mail: alewand@amu.edu.pl
bFaculty of Physics, Adam Mickiewicz University, Uniwersytetu Poznanskiego 2, 61-614 Poznan, Poland
cCenter for Advanced Technology, Adam Mickiewicz University, Uniwersytetu Poznanskiego 10, 61-614 Poznan, Poland
dInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: akubas@ichf.edu.pl
First published on 4th June 2020
Abstract
The present study explores the influence of graphene oxide (GO) on deactivation pathways of the excited states of zinc 5,10,15,20-tetrakis(4-(hydroxyphenyl))porphyrin (ZnTPPH). The interaction of light with free ZnTPPH molecules and with ZnTPPH molecules adsorbed on graphene oxide sheets was probed via UV-vis spectroscopy, fluorescence spectroscopy, femtosecond pump–probe technique and nanosecond flash photolysis. Formation of the ground-state ZnTPPH–GO complex in solution was monitored by the red-shift of the porphyrin Soret absorption band. It was found that Stern–Volmer fluorescence quenching can be described in terms of two different quenching regimes depending on the GO concentration. In addition, our comprehensive analysis of the steady-state and time-resolved emission experiments led to the conclusion that the observed quenching was entirely attributable to a static mechanism. Laser flash photolysis showed that the triplet lifetime of the ZnTPPH increased in the presence of GO from 174 μs to 292 μs, which is related to the decrease in the rate constant of a radiationless decay mechanism involving rotation of the peripheral hydroxyphenyl rings of the porphyrin. Femtosecond transient absorption spectroscopy demonstrated the presence of a fast photoinduced electron transfer from the singlet excited state of ZnTPPH to the GO sheets, as indicated by the formation of a porphyrin radical cation. Quantum chemical calculations were used to gain deeper insights into the nature of the electronically excited states in the free ZnTPPH as well as in the ZnTPPH–GO complex.
Introduction
Graphene based hybrid nanomaterials have attracted broad research attention due to their potential applications in solar energy conversion (photocatalysis and photovoltaics).1–6 The photoinduced electron transfer (PET) process in those hybrid materials plays a key role in converting solar light into chemical energy or electricity.7 One strategy to fabricate hybrid materials is to functionalize graphene with molecules that are photochemically active. The idea of dye/graphene materials is that graphene acts as electron transporter and supporting matrix for the sensitizer, which in turn expands the light absorption of the nanohybrid into the visible range. However, graphene sheets are hydrophobic and tend to aggregate due to π–π interactions that limit possible functionalization and subsequently applications. On the other hand, graphene oxide (GO), which is a graphene derivative bearing various oxygen-derived functional groups, is hydrophilic and can be applied in photocatalytic systems.8 Although the properties of graphene oxide are inferior to those of pure graphene, the modification with various oxygen-containing groups at the basal planes of graphene sheets allows it to form stable aqueous suspensions. This is important for functionalization and further application.Porphyrins are a well-known group of pigments due to some excellent spectroscopic and electrochemical properties. As planar, electron rich aromatic entities, they possess remarkably high extinction coefficients in the visible region. In addition, their PET ability have caused porphyrins to be widely used as photosensitizers in artificial photosynthetic devices.9 Therefore, porphyrins and graphene oxide are appealing units to prepare new nanohybrid composites, which can find application in solar-energy conversion. For any practical application of those hybrid materials, it is crucial that PET from porphyrin to GO takes place.10–14 In 2010 Wojcik et al. clearly demonstrated for the first time the occurrence of PET in the hybrid material composed of the cationic porphyrin (5,10,15,20-tetra(1-methyl-4-pyridino))porphyrin tetra(p-toluenesulfonate) (TMPyP) and reduced graphene oxide.12 Afterwards, Aly et al. have demonstrated, by applying a series of cationic and neutral porphyrins, that the charge-transfer rate between porphyrins and the graphene carboxylate surface is controlled by both the distance and the electronic coupling between the donor and the acceptor units.13 Interestingly, to date several Zn(II) porphyrin/graphene based materials have been applied in photochemical processes.13–20 Yuan et al. presented a non-noble metal system for photocatalytic H2 generation that combined Zn(II)-5,10,15,20-tetrakis(4-N-methylpyridyl)porphyrin and RGO with decorated MoS2 as the catalyst.15 Yuan et al. have shown that the material that employed Zn(II) porphyrins exhibited much higher hydrogen production efficiency compared to an analogue system where free-base porphyrin was used.15 Guo et al. have shown that zinc 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine nanostructures, assembled onto GO sheets via a surfactant-assisted method, displayed photocatalytic activity for the degradation of rhodamine B.16 It has been also demonstrated that GO sheets can be successfully photoreduced using 5-(4-hydroxyphenyl)-10,15,20-tri(p-tolyl) zinc(II) porphyrin under ambient conditions.19
Taking into account the number of literature examples involving non-covalent hybrids of Zn porphyrin and graphene, it is surprising that to date there are no documented cases of zinc 5,10,15,20-tetrakis(4-(hydroxyphenyl))porphyrin (ZnTPPH) non-covalently anchored to GO sheets. This is especially true considering the Zhu group's reported enhanced photocatalytic activity toward hydrogen generation in a system where the free base 5,10,15,20-tetrakis(4-(hydroxyphenyl))porphyrin (TPPH) molecules played the role of a photosensitizer adsorbed on the surface of RGO sheets with Pt nanoparticles acting as the dispersed co-catalyst.20 Because of the fact that spectroscopic properties of both the ZnTPPH dye itself as well as its hybrids with graphene oxide have not been studied except one report where absorption and emission spectra of ZnTPPH in acetonitrile were measured,21 it is well justified to explore in detail how unbound ZnTPPH interacts with light and how this interaction is affected when the dye is non-covalently anchored to graphene oxide sheets. The main aim of the current study is to describe the nature of interaction between the components as well as to elucidate whether an electron-transfer process occurs between constituents within the nanohybrid material. Herein, we present the preparation of a new non-covalent hybrid of ZnTPPH and graphene oxide (ZnTPPH–GO) (Fig. 1), where the dye molecules are attached to graphene oxide sheets via π–π interactions. This new material was subjected to a detailed photochemical investigation in EtOH–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) as the solvent. In parallel, light-driven properties of the free dye were studied including the excited state lifetimes and transient absorption spectra of the singlet as well as the triplet excited state. We explored in detail the excited-state deactivation processes of ZnTPPH upon addition of GO. The current investigations, based on steady-state and time-resolved fluorescence experiments and femtosecond transient absorption spectroscopy, demonstrated that a fast photoinduced electron transfer from the singlet excited state of the ZnTPPH to the GO moiety occurs in this system. These comprehensive studies coupled to detailed quantum-chemical analysis provide invaluable information that can serve as guidance for fabrication of energy-conversion devices.
:2 v/v) as the solvent. In parallel, light-driven properties of the free dye were studied including the excited state lifetimes and transient absorption spectra of the singlet as well as the triplet excited state. We explored in detail the excited-state deactivation processes of ZnTPPH upon addition of GO. The current investigations, based on steady-state and time-resolved fluorescence experiments and femtosecond transient absorption spectroscopy, demonstrated that a fast photoinduced electron transfer from the singlet excited state of the ZnTPPH to the GO moiety occurs in this system. These comprehensive studies coupled to detailed quantum-chemical analysis provide invaluable information that can serve as guidance for fabrication of energy-conversion devices.
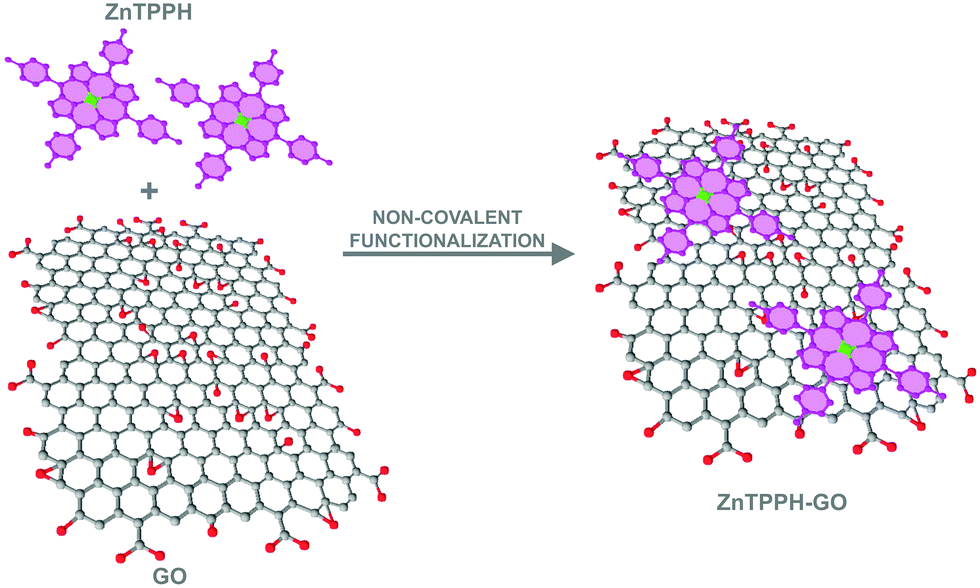 | ||
| Fig. 1 Illustration of formation of the ZnTPPH–GO nanocomposite via noncovalent functionalization. | ||
Experimental
Materials
5,10,15,20-Tetrakis(4-(hydroxyphenyl))porphyrin Zinc(II) (ZnTPPH) and 5,10,15,20-tetrakis(4-(hydroxyphenyl))porphyrin (TPPH) were purchased from Porphyrin Systems, and graphene oxide (GO-powder < 35 mesh. C/O atomic ratio = 2.5–2.6. Ultrasound bath 1/2 h form perfect single layer dispersion) was purchased from Abalonyx, Ethanol (HPLC grade) was bought from J. T. Baker. Solutions were prepared with ultrapure water. For all experiments GO suspensions were prepared by dispersing GO powder in water followed by ultrasonication for 30 min.Experimental apparatus
UV-vis absorption spectra were recorded using a two-beam spectrometer Cary 100 UV-vis scanning from 800 to 200 nm with 1 nm increments. Fluorescence spectra were taken on a LS 50B spectrofluorometer (PerkinElmer) (scan speed 240 nm min−1, excitation and emission slits 5 nm) for solutions with absorbances at the excitation wavelength lower than 0.1. Emission was scanned between 500 and 800 nm for all of the samples. The fluorescence lifetimes were measured on a Fluorescence Lifetime Spectrometer (FluoTime300 from PicoQuant) with a detection system based on time-correlated single-photon counting (TCSPC). The emission decay lifetimes were measured following excitation with 440 nm photodiodes. Samples were placed in 10 mm quartz cells for all of the steady-state and time-resolved emission measurements. The details of the femtosecond transient absorption spectra have been described elsewhere.22 In short, the ultrafast laser system consisted of a short-pulse titanium sapphire oscillator (Mai Tai, Spectra Physics, 70 fs) followed by a high-energy titanium sapphire regenerative amplifier (Spitfire Ace, Spectra Physics, 100 fs). The 800 nm beam was split into two beams to generate: (1) a pump (λexc = 425, 442, or 455 nm) from the optical parametric amplifier (Topas Prime with a NirVis frequency mixer) and (2) probe pulses in the UV-vis range by using sapphire plate (Ultrafast Systems, Helios). The temporal resolution of the setup is about 300 fs. For the transient UV-vis measurements a quartz cell with 2 mm optical path of solution was used with an absorbance of about 0.2 at the excitation wavelength, and the sample solution was stirred by a Teflon-coated bar. The typical pump energy was about 1 μJ. All of the experiments were performed at room temperature. Global analysis of the transient absorption data was made using the Surface Explorer software (Ultrafast Systems). The setup for the nanosecond laser flash photolysis (LFP) experiments and the data acquisition system have been previously described in detail.23 LFP experiments were performed using a Nd:YAG laser (532 nm, 5 mJ) for excitation. The pulse duration is about 8 ns and the shortest meaningful lifetime that can be determined from the time evolution of the signal is about 12–15 ns. Transient kinetic profiles were measured at individual wavelengths by the step-scan method in the range of 400 to 750 nm as the average of 8 pulses. Solutions for LFP were degassed with argon for 15 min prior to the measurements. Experiments were performed in rectangular quartz cells (1 cm × 1 cm).Quantum chemical calculations
The method of construction and geometry optimization of the graphene oxide model was described earlier.10 Briefly, the system is based on the experimental work of Lerf and co-workers24 who investigated a structure of GO prepared according to Hummers procedure.25 The same procedure was used by the supplier of the GO in the present study. In our model, GO is represented as C59O26H26 structure with C/O atomic ratio of 2.3 (experimentally 2.5) and contains all chemically relevant functional groups such as epoxy and carboxy groups. The model was optimized within the density functional theory (DFT) using the BP86 functional26,27 augmented with D3BJ27–29 and gCP30 corrections to improve the description of weak interactions and to decrease basis set superposition error, respectively. The same setup was used to optimize the ZnTPPH geometry as well as the ZnTPPH–GO nanohybrid assembly. All of the structures were subject to numerical second derivative calculations and were confirmed to possess only positive Hessian eigenvalues. Subsequently, single-point energies were evaluated with the BHLYP functional,31 and corresponding D3BJ/gCP corrections were added to the total electronic energy. If not stated otherwise, the def2-TZVP basis set32 was used throughout the study. Interaction energy between ZnTPPH and GO was calculated as the difference between the energy of the nanohybrid and the energy of the monomers obtained at their relaxed geometries. All of the calculations were performed with the ORCA 4.2.1 suite of programs.33Electronic spectra of the GO, ZnTPPH and ZnTPPH–GO systems were obtained with the simplified time-dependent DFT method (sTD-DFT).34 The method allows for efficient calculations of electronic transition energies and transition moments for systems comprising hundreds of atoms and approaches the accuracy of canonical TD-DFT implementations at a fraction of the cost. The method used reference BHLYP orbitals. All of the configuration state functions (CSFs) with energies up to 10 eV above the ground state were considered (EThresh parameter in the ORCA nomenclature). The spectra were simulated by applying Lorenzian broadening with a broadening parameter of 1000 cm−1. In addition, we uniformly red-shifted all of the computed spectra by 950 cm−1 to visually match with the experimental data. In order to analyze the nature of the low-lying excited states, we performed additional multireference calculations using the n-electron valence state perturbation theory (NEVPT2) level.35 Due to the size of the systems studied, the domain based local pair natural orbital approximation was applied with the default settings (DLPNO-NEVPT2).58 Such correlated calculations were based on the state-averaged complete active space self-consistent field (CASSCF)36 wave function which allowed us to rigorously treat orbital near-degeneracy present in the porphyrin-based scaffold. The choices of orbitals that build the active spaces were based on occupation numbers of the BHLYP natural orbitals of the respective species. In the case of ZnTPPH, four Gouterman's orbitals37 entered the active space along with four electrons and formed a space denoted as CAS(4,4). The ZnTPPH–GO nanohybrid active space included an additional two orbitals of GO and two more electrons. Thus, the final active space in this case was CAS(6,6). To estimate the relative intersystem crossing probability, the spin–orbit coupling (SOC) matrix elements were evaluated within the quasi-degenerate perturbation theory38 as implemented in ORCA.
Results and discussion
Absorption properties of ZnTPPH
ZnTPPH is only slightly soluble in water, thus all of the experiments were performed in EtOH–H2O (1:2 v/v) mixtures. The absorption spectra of ZnTPPH exhibited typical bands expected for metalloporphyrins, i.e., the Soret band arising from the transition to the second excited state (S0 → S2) and Q bands corresponding to weak transition to the first excited state (S0 → S1) which are described by the Gouterman four-electron, four orbital model.39,40 As shown in Fig. 2, the UV-vis spectrum of ZnTPPH shows a very strong Soret band with the maximum at ca. 424 nm (ε424 = 5.0 × 105 M−1 cm−1) and two less intense Q-bands at ca. 559 nm and 600 nm. These spectra are similar to those reported earlier for ZnTPPH in acetonitrle.21 The insertion of Zn(II) into the porphyrin ring of TPPH caused a small red-shift of the Soret band by 6 nm together with a decrease in the number of Q-bands.10 The decrease in the number of Q-bands in the UV-vis spectra of ZnTPPH in comparison to TPPH is attributed to the increase of the symmetry of the porphyrin ring upon metalation.39,40
 | ||
| Fig. 2 UV-vis absorption spectra of 3.0 μM solution of ZnTPPH in EtOH–H2O (1:2 v/v) (inset: Q-band region of the same spectra). | ||
Absorption properties of ZnTPPH in the presence of GO
The UV-vis spectroscopy was used to probe the interaction between GO and ZnTPHH in its electronic ground state. UV-vis spectra are illustrated in Fig. 3 for a number of ZnTPPH–GO nanohybrid suspensions measured at a constant ZnTPPH concentration but with a gradual increase of GO. Correction of the UV-vis spectra of ZnTPPH for the GO absorbance revealed that changes in the ZnTPPH absorption are meaningful (inset Fig. 3). Upon adding GO the intensity of the porphyrin's Soret band at 424 nm gradually decreased, and a new Soret band at 440 nm arose. An isosbestic point was observed at 433 nm, indicating that there was a clear change from free ZnTPPH to ZnTPPH adsorbed on the surface of GO. The two Q bands were also red shifted. However no formation of the intense band around 700 nm, reported for TPPH and attributed to a partial charge transfer, was observed upon addition of GO.10 However theoretical calculations predict that the long-wavelength, charge-transfer band could lie beyond our experimental range i.e. 800 nm (Fig. 14). However, it can be noticed that the Soret band of the ZnTPPH–GO exhibited a red-shift of 16 nm. The bathochromic shift observed upon nanohybrid formation could be explained by at least two different mechanisms: (1) J-aggregation of the porphyrin molecules, (2) flattening of the porphyrin molecule. The first possibility is highly unlikely because a clear isosbestic point was observed during titration. In addition the linear dependence of the absorbance at the maximum of the Soret band of the nanohybrid on the GO concentration suggests that the obtained material had a well-defined structure. The most plausible explanation for the observed red shift of the Soret band in the ZnTPPH–GO is the considerable distortion in the structure of ZnTPPH upon adsorption on the GO sheet. This interpretation is supported by theoretical calculations (vide infra). The flattening of the porphyrin molecule was also proposed previously in the case of adsorption of TMPyP onto chemically converted graphene (CCG)41 or TPPH and GO.10 | ||
| Fig. 3 Absorption spectra recorded during the process of titration of 3 mL of 3.0 μM EtOH–H2O (1:2 v/v) solution of ZnTPPH with 3 mg mL−1 of GO dispersion (0–0.09 mg mL−1), the inset is the same spectra presented only in the Soret-band region with the spectra corrected for GO absorption. | ||
The interaction of ZnTPPH with GO was found to be weaker in comparison to GO nanohybrids with positively charged porphyrins TMAP or TMPyP reported earlier.11,12 Even at high GO concentration 0.09 mg mL−1, the Soret band, attributed to free ZnTPPH, unbound to GO, was still present in the UV-vis spectra. It can be explained by weaker π–π interactions in the current case relative to the strong electrostatic attraction present in the interaction of cationic porphyrin and negatively charged GO.
In order to evaluate the amount of the ZnTPPH that can be adsorbed on 1 mg of GO surfaces, the absorption spectra of GO were measured with the gradual addition of concentrated ZnTPPH solution (Fig. S1, ESI†). After exceeding the concentration of ZnTPPH above 1.65 μM, the absorbance of the Soret band at 424 nm attributed to free porphyrin started to be visible. Based on this experiment, the maximum amount of ZnTPPH adsorbed onto the GO sheets was estimated to be ca. 0.012 mg per mg of GO. This amount of ZnTPPH adsorbed on the GO surface is almost three times smaller than the analogous value for TPPH, indicating that the introduction of the Zn(II) into the porphyrin core decreased the strength of the interaction with GO.10
Spectroscopic results clearly showed that ZnTPPH interacted with GO in its ground state. The possibility to isolate the newly prepared porphyrin–GO material was examined by a centrifugation experiment controlled by UV-vis spectroscopy. 350 mL of ZnTPPH solution (4.6 × 10−6 M) (EtOH–H2O, 1:2 v/v) were mixed with 39 mL of aqueous GO suspension (3 mg mL−1). The mixture was then stirred at room temperature for 2 h, resulting in ZnTPPH–GO hybrids. Subsequently the obtained suspension was centrifuged at 12000 rpm for 90 minutes. The obtained supernatant was light yellow, and the precipitate was dark brown. UV-vis spectra of the suspension of the nanohybrid ZnTPPH–GO were recorded before and after centrifugation (Fig. S2, ESI†). Based on the minor peak attributed to the porphyrin detected in the suspension, it was estimated that ca. 90% of the nanohybrid was successfully isolated as the precipitate. Based on that value, it was calculated that the weight content of ZnTPPH in the nanohybrids was 1.0%, which is in good agreement with the value obtained in the UV-vis titration experiment (Fig. S1, ESI†). Interestingly, upon suspending again this precipitate, the resulting spectrum was a reconstruction of the UV-vis spectra of the nanohybrid. This experiment confirms that GO can be successfully non-covalently functionalized with ZnTPPH.
Emission studies
The studied free porphyrin displayed two emission peaks at 612 nm and 663 nm (Fig. 4). This spectral pattern is characteristic for the monomeric porphyrin molecules. The quantum yield of the emission of free ZnTPPH fluorescence was determined to be 5.3%. A decrease in the ZnTPPH fluorescence intensity was observed with increasing GO concentration (Fig. 4). However, one has to be aware that although fluorescence spectroscopy is a great technique, getting valid data can often be demanding.42
 | ||
| Fig. 4 Quenching of the fluorescence of 0.7 μM ZnTPPH recorded during addition of an aqueous suspension of 0.4 mg mL−1 GO (0–0.01 mg mL−1), λexc = 433 nm. | ||
Absorption of light by GO sheets can be taken into account by introducing the equation for the inner filter effect.42 In our study the correction factor for GO absorption of the excitation light was calculated to be as much as 9.5% for 0.01 mg mL−1 of GO. A decrease in the ZnTPPH fluorescence intensity was observed with increasing GO concentration (Fig. 5). The quenching efficiency for the GO concentration 0.01 mg mL−1 was calculated to be 61%. The most interesting observation from the steady-state emission experiment was that the Stern–Volmer plot I0/I versus the concentration of graphene is not linear. It can be noticed that fitting of the experimental points matches better for with the presence of two different quenching regimes (Fig. 5).
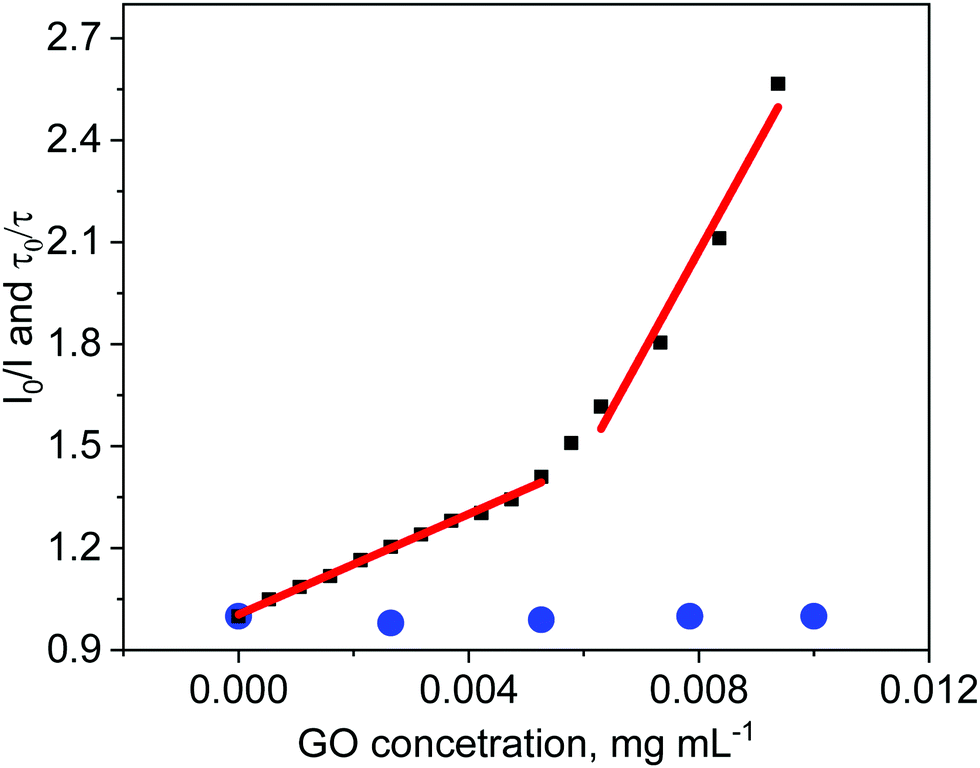 | ||
| Fig. 5 The relationship between fluorescence intensity I0/I (black symbols) (I0 − If without GO, I − If after addition of GO) and GO concentration and the relationship between fluorescence lifetime τ0/τ (blue symbols) (τ0 – fluorescence lifetime without GO, τ – fluorescence lifetime after addition of GO) and GO concentration for ZnTPPH. Data corrected for inner filter effect. | ||
The same quenching behavior was observed for methylene blue, acridine orange and meso-tetra(sulfonatophenyl)porphine in the presence of graphene.43 De Miquel et al. rationalized this remarkable behavior as related to the graphene surface area availability to each dye molecule.43 The monitored reducing of fluorescence intensity after introducing GO to the ZnTPPH solution (Fig. 5) might be related to the dynamic quenching of an singlet excited state of free ZnTPPH by GO (Fig. 6). The possibility of a dynamic mechanism was probed by recording the emission decay profiles of ZnTPPH in the presence of GO. The fluorescence lifetime was calculated based on a monoexponential fit of the fluorescence decay of ZnTPPH in the absence of GO and was found to be ca. 1.39 ns (Fig. S3, ESI†). With an increasing amount of GO in the sample, no shortening in the decay profiles of the excited state was noticed. One would normally have expected a shortening of the singlet excited state of unbound ZnTPPH if the quenching were the result of a dynamic process induced by GO. There might also be the chance to see the appearance of a second decay associated with any fluorescence of the nanohybrid. Neither of these decays was detected during the time correlated single photon counting experiment. No measurable change in the singlet excited state lifetime of the free porphyrins excludes the possibility of dynamic quenching by GO (Fig. 5). Our detailed analysis of fluorescence quenching (steady-state and time-resolved) provided clear evidence that the observed quenching was entirely attributable to static quenching of the porphyrin by GO.
 | ||
| Fig. 6 Pathways of deactivation of excited states of porphyrin ZnTPPH in the presence of graphene oxide. | ||
Numerous papers, dealing with the photophysics of graphene–dye hybrids, interpreted emission data as proof of an electron-transfer process occurring between a dye and graphene, however one cannot identify definitely what is the quenching mechanisms based solely on the steady-state emission experiments.20,44–46 Since ZnTPPH–GO has a different electronic structure as found by the changes in its UV-vis absorption spectra compared to the absorption spectrum of unbound ZnTPPH, any emission from the hybrid material would also be expected to be red shifted compared to that of free ZnTPPH. It is important in this case to remark that upon addition of a GO suspension to a ZnTPPH solution, no change in the shape, as well as, in the position of the peaks in the emission spectra was noticed. In addition, as depicted in Fig. 7, the fluorescence excitation spectrum measured for the ZnTPPH solution, after addition of 0.01 mg mL−1 of GO, coincided perfectly with the absorption spectrum of free ZnTPPH with a maximum at 424 nm. The results presented above, clearly show that ZnTPPH–GO did not exhibit any measurable emission and that the observed fluorescence in Fig. 4 arose entirely from the free ZnTPPH in the suspension.
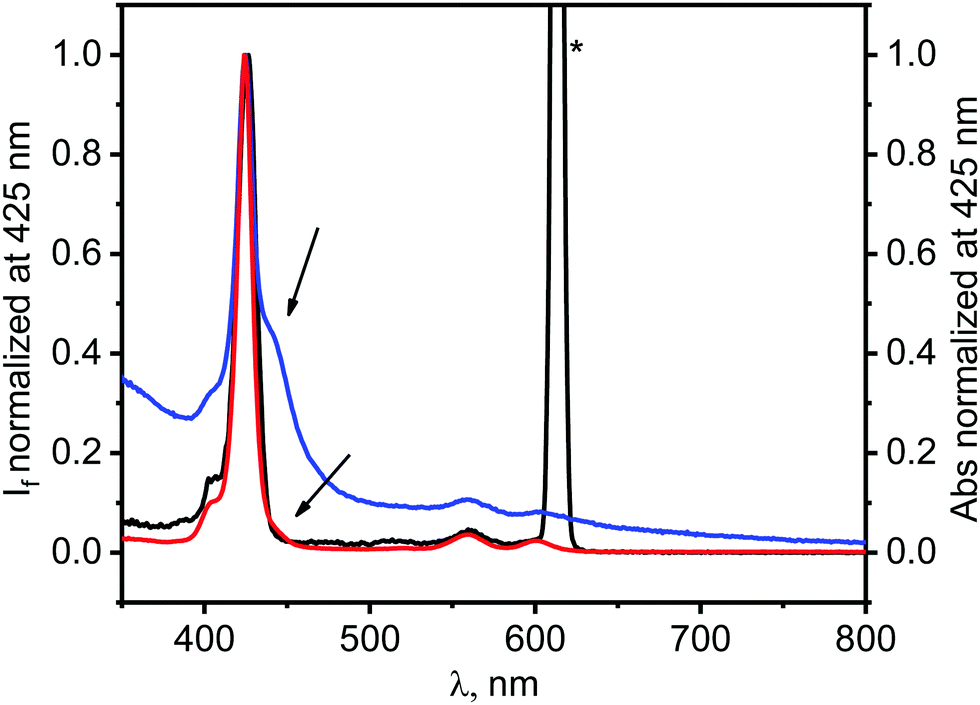 | ||
| Fig. 7 Normalized fluorescence excitation spectrum measured at 612 nm of the mixture of ZnTPPH (0.7 μM) and GO (0.01 mg mL−1) (black), absorption spectrum of this mixture (blue) and normalized absorption spectrum of ZnTPPH (0.7 μM) in the absence of GO (red). Asterisk denotes scattered excitation light reaching to the detector. | ||
The plausible reason for ZnTPPH–GO complex to be non-emissive is the presence of other fast processes that deactivate quickly the singlet excited state.
 | ||
| Fig. 8 Transient absorption spectra registered at various time delays for ZnTPPH (10.0 μM) in EtOH–H2O (1:2 v/v) following the 425 nm laser excitation. | ||
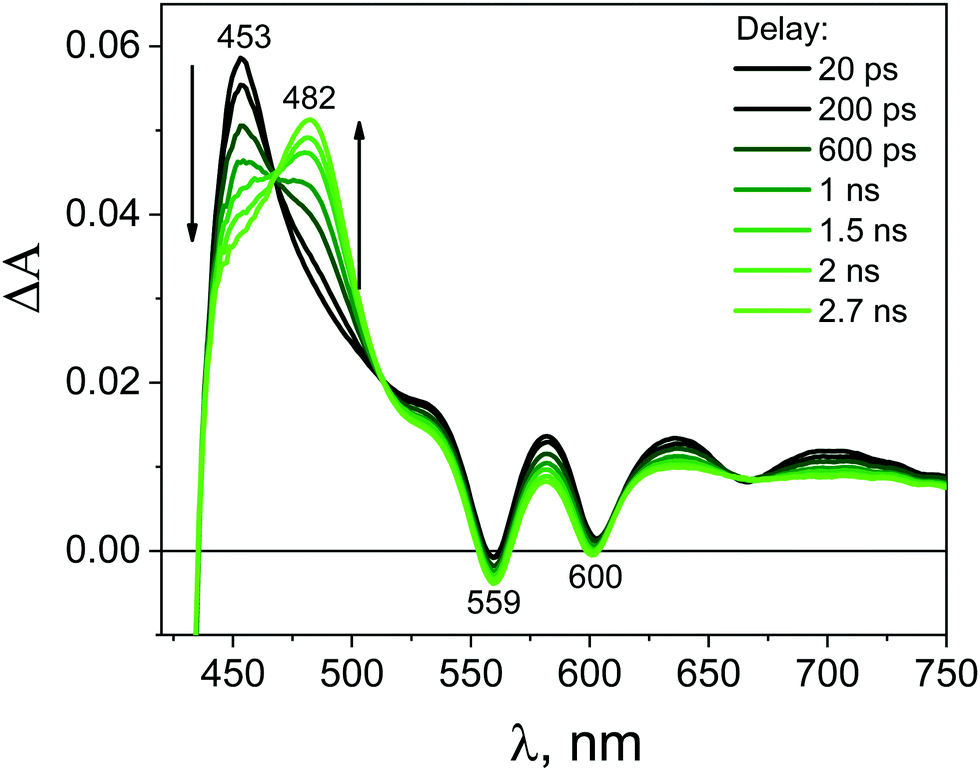
After mixing the GO and ZnTPPH solutions in such ratio that all ZnTPPH molecules are adsorbed on the GO surface, the recorded transient absorption (TA) spectra (Fig. 9) varied in comparison to the TA spectra (Fig. 8) of free ZnTPPH. At first, before drawing any conclusions concerning the interaction between ZnTPPH in the singlet excited state and GO, it was of great importance to correct the obtained spectra for the transient absorbance of the GO itself (Fig. S7, ESI†).50,51 Fig. S8 (ESI†) presents the TA recorded before the correction for the GO transient absorption.
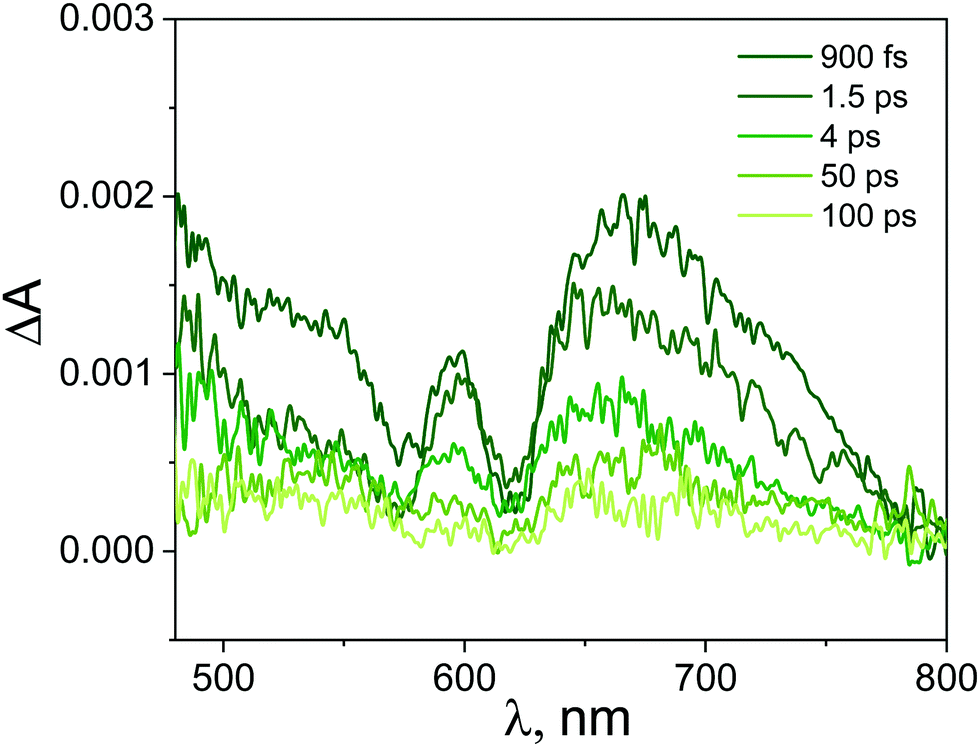 | ||
| Fig. 9 Transient absorption spectra registered at various time delays for ZnTPPH–GO (ZnTPPH concentration 10.0 μM, GO concentration 0.4 mg mL−1) in water following the 442 nm laser excitation. Transient absorption spectra were corrected for the contribution from GO itself. | ||
Importantly, the intense signal in the region 450–550 nm assigned to the singlet excited state absorption of ZnTPPH disappeared. As shown in Fig. 9, a new band in the range of 650–800 nm was observed. In addition, the small bleach of the two Q bands could be detected. It seems reasonable to attribute the band 650–800 nm to the porphyrin cation radical based on the similarity to the reported spectra for the ZnTPPH radical cation52 and the predicted shape of the spectra in the theoretical calculations (vide infra). Neta et al. obtained the spectra of the ZnTPPH radical cation in a pulse radiolysis experiment, and it was found that in neutral solution, the radical cation showed an absorption spectra characterized by a fairly broad band centered at ca. 650–690 nm and a weak shoulder at 760 nm.52
The detection of the radical cation of ZnTPPH provides evidence for a photoinduced electron transfer from the porphyrin to the GO. Interestingly the radical cation of ZnTPPH in our studied system was already detected within the temporal resolution of the instrument (300 fs), which indicates two things: first, electron transfer from the zinc porphyrin to GO is very fast and second that the observed spectra of ZnTPPH radical cation corresponds to vertical electron transfer without relaxation (see theoretical calculations section).
Kinetic profiles recorded for ZnTPPH in the absence and presence of GO are depicted in Fig. 10. The decay of the transient absorption was registered at 700 nm. It is clear that within the same time observation window (50 ps), a significant difference can be seen between the two investigated samples.
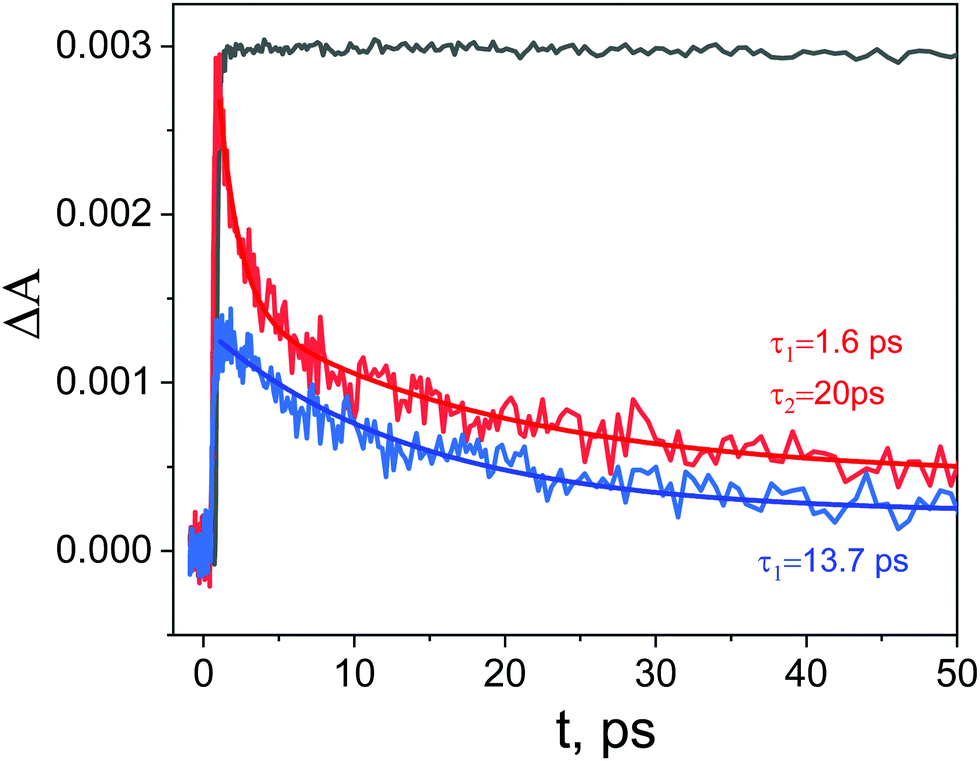 | ||
| Fig. 10 Transient absorption decays at 700 nm in EtOH–H2O (1:2 v/v) registered for the ZnTPPH (black curve) and ZnTPPH–GO (red curve) and GO (blue) following the 425 nm laser excitation for ZnTPPH and 442 nm for ZnTPPH–GO and GO. Two exponential decay fit for ZnTPPH–GO (red line) and monoexponential decay fit for GO (blue line). | ||
The analysis of the kinetic profile obtained for the nanohybrid ZnTPPH–GO is complex due to its similarity to the kinetic profile measured for the GO itself (blue curve in Fig. 10). However, undoubtedly the kinetic profile at 700 nm for ZnTPPH–GO showed a very fast decay dynamics (two exponential decay) compared with the absence of any decay of the excited state of free porphyrin on the same time scale. The time constants extracted from the decay of the ZnTPPH radical cation are 1.6 ps and 20 ps. The slower component is convoluted with the signal from the GO itself. However, the very fast decay of the signal of the ZnTPPH radical cation can be assigned to efficient back electron transfer and recovery of the ground state complex. This is confirmed in transient spectra in Fig. 9, since at delay of 4 ps practically no characteristic ZnTPPH ground state S0 depletion is observed.
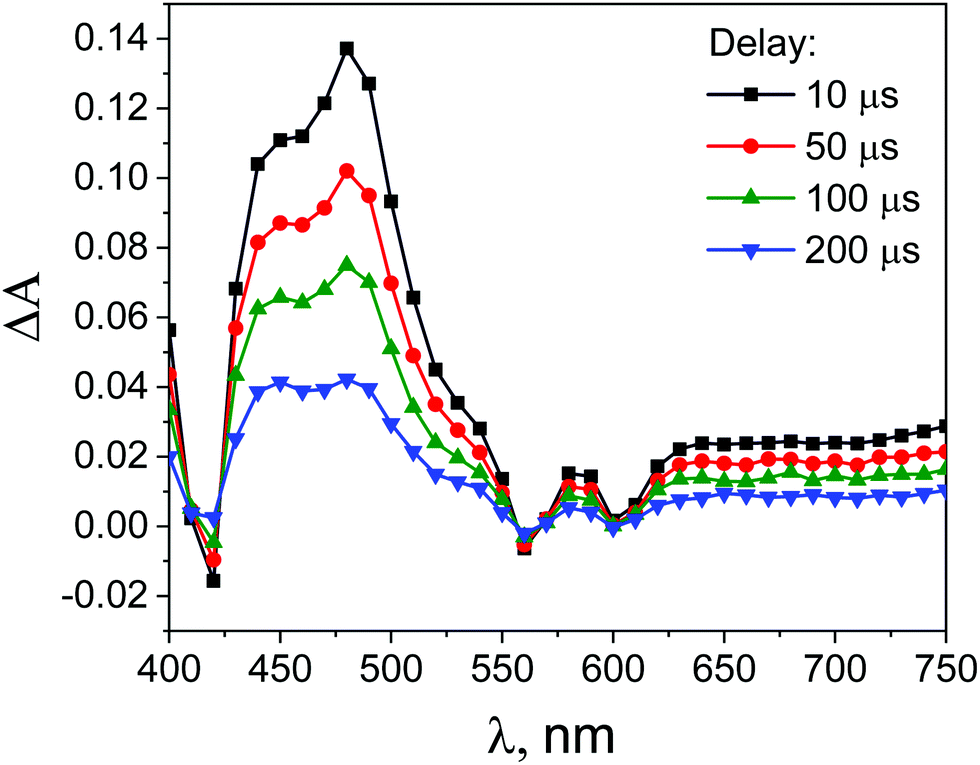 | ||
| Fig. 11 Transient absorption spectra obtained during laser flash photolysis (with excitation at 532 nm) of deoxygenated solutions of ZnTPPH (35 μM) in EtOH–H2O (1:2 v/v) after different time delays. | ||
Surprisingly, the addition of the GO led to the increase of the ZnTPPH triplet lifetime (Fig. S9, ESI†). The triplet lifetime obtained from the monoexponential fit to the 480 nm decay profile was found to be as much as 292 μs. This interesting observation of the increase of the porphyrin triplet lifetime upon addition of graphene has been reported once before in the case of meso-tetra(sulfonatophenyl)porphine.43 To explain this somehow unexpected ZnTPPH triplet excited state lifetime increase upon addition of graphene oxide, we propose that in this case the lifetime enhancement is related to conformational restrictions arising when porphyrin molecule in its excited state is adsorbed on the GO surface. Several reports indicate that that triplet excited states decay to the ground state via radiationless mechanisms requires flipping and twisting of conformational free meso-phenyl rings around the core of the porphyrin in which the electronic density of the triplet excited state is mainly located.53,54 It was observed that for flexible molecules the triplet lifetimes increase when the molecules are immobilized on a solid surface of cucurbit[n]urils55 or in the porous channels of zeolite.56 In our case, once the ZnTPPH molecule in the triplet state is adsorbed on the GO sheet, the possibility for the free rotation of the hydroxyphenyl rings, the substituents to the porphyrin core, is largely suppressed. Thus the radiationless deactivation pathway is impeded leading to the observed elongation of the excited state lifetime. The attribution of the increase of the triplet excited state lifetime of ZnTPPH upon adsorption on GO surface to the decrease of the radiationless rate constant were further discussed based on theoretical calculations (vide infra).
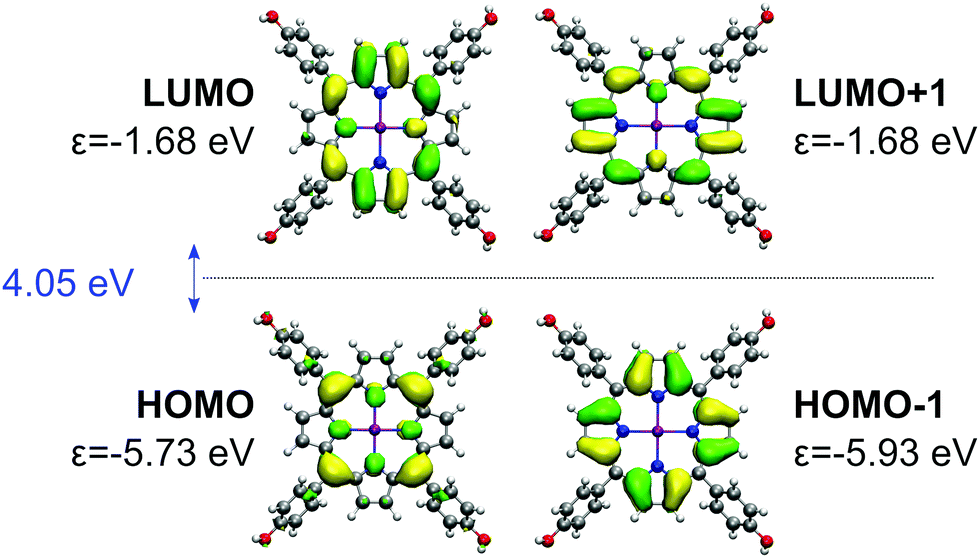 | ||
| Fig. 12 Gouterman's four orbitals of the ZnTPPH. For each orbital, BHLYP one-electron energy is denoted with ε. HOMO–LUMO gap is given in blue. | ||
The electronic interaction energy of ZnTPPH with the GO model is relatively large and yields −22.8 kcal mol−1, comparable to one calculated for TPPH (−22.4 kcal mol−1).10 The optimal distance for this interaction is 4.4 Å, marginally shorter than in the case of the TPPH–GO interaction (4.6 Å). The interaction of ZnTPPH with graphene oxide leads to a twisting of the side rings relative to the porphyrin core from about 60° to 45°, and therefore ZnTPPH gets more flat upon complexation. Analysis of the frontier molecular orbitals of the ZnTPPH–GO nanohybrid, depicted in Fig. 13, reveals a HOMO–LUMO gap of 2.62 eV, the value exactly the same like in the case of TPPH. The HOMO and HOMO−1 orbitals are located within the ZnTPPH moiety while the first three LUMO orbitals are GO-centered. The LUMO+3 and LUMO+4 orbitals correspond to two of Gouterman's π* orbitals of the free ZnTPPH.
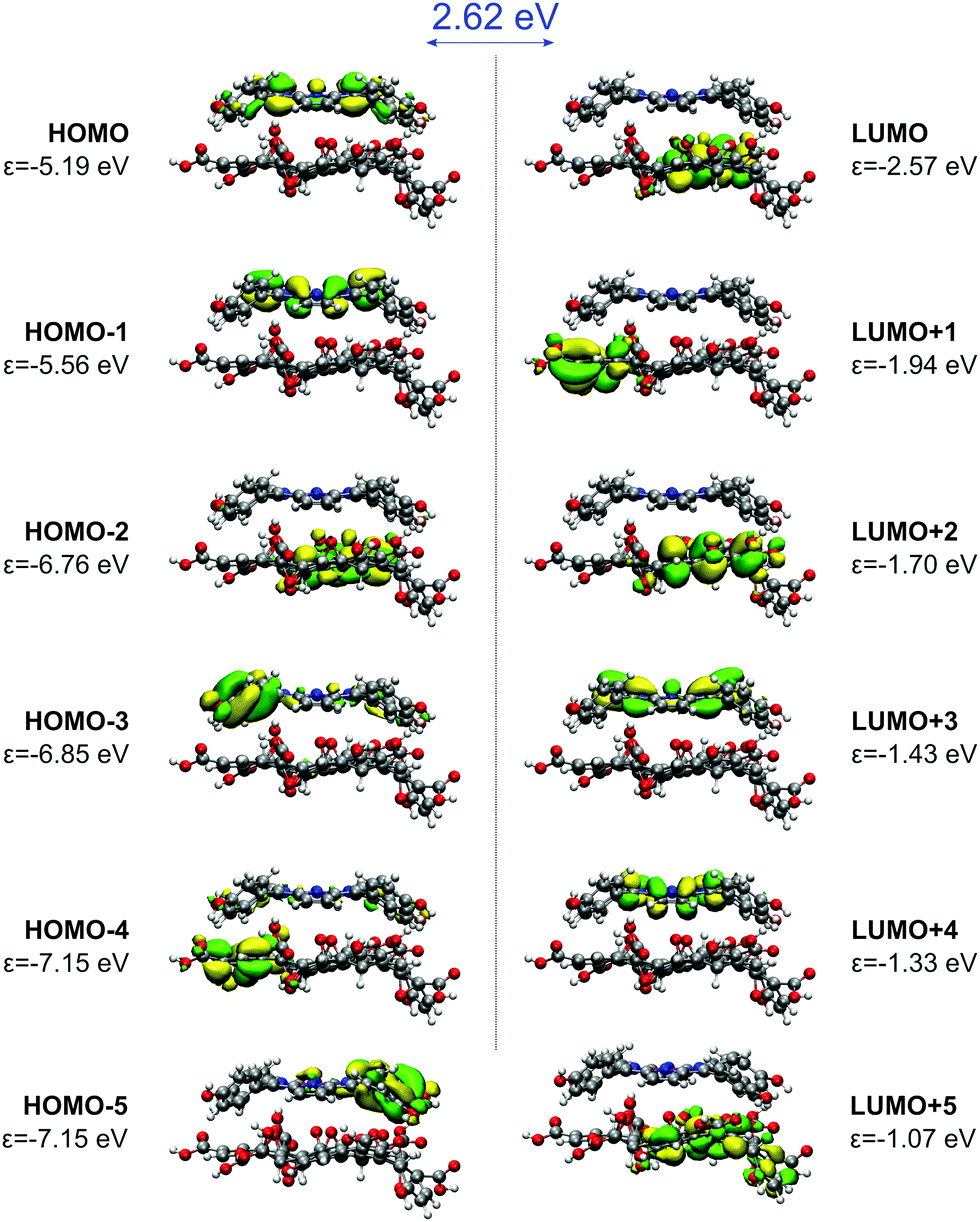 | ||
| Fig. 13 Frontier molecular orbitals of the ZnTPPH–GO nanohybrid. HOMO–LUMO gap is given in blue. | ||
The presented orbital picture constitutes the basis for the absorption spectra interpretation. Fig. 14 provides a comparison between experimental and sTD-DFT computed spectral lines. The calculations reproduce essentially all non-vibronic transitions. Moreover, the experimentally observed Soret band red-shift of 16 nm is well reproduced in theory (36 nm). Interestingly, the experimentally broad Soret band has contributions from multiple transitions. As expected, the most intense are the HOMO−1 → LUMO+3/+4 transitions that are within the Gouterman's orbitals system. Interestingly, a lower energy satellite about 490 nm is of charge-transfer (CT) character from ZnTPPH to GO and involves primarily the HOMO → LUMO+5 orbitals.
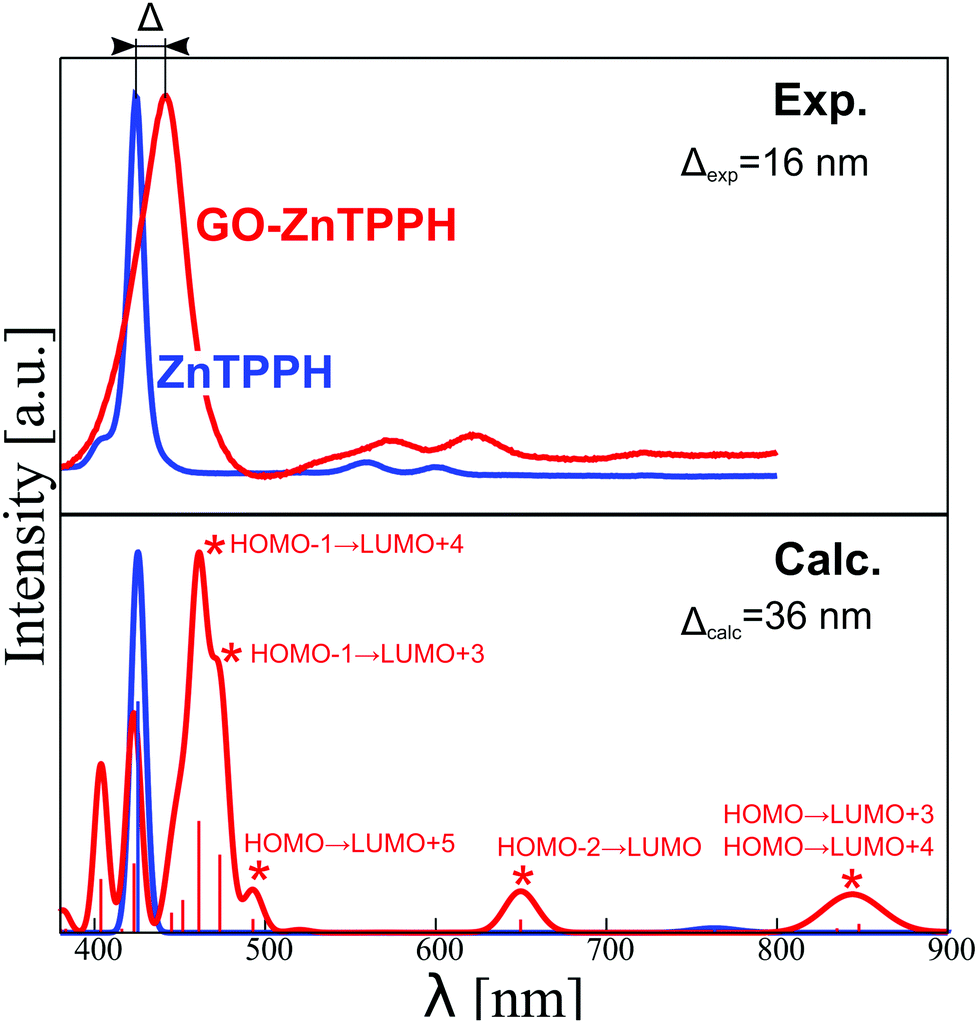 | ||
| Fig. 14 Comparison of normalized experimental (top) and calculated (bottom) absorption spectra of ZnTPPH (blue) and ZnTPPH–GO complex (red). For the latter, assignment of the observed absorption bands is provided. | ||
This CT excited state is presumably responsible for changes in transient absorption spectra recorded with excitation wavelength of 442 nm in the femtosecond transient absorption spectroscopy. To further confirm the origin of this spectral feature (experiment in Fig. 9), we computed the spectrum of the ZnTPPH radical cation at two geometries: the geometry of the neutral ZnTPPH and the relaxed, optimal geometry of the radical cation (Fig. S11, ESI†). The former corresponds to a vertical electron detachment (electron transfer to GO) while the latter involves slow molecular scaffold relaxation. Comparison of simulated and experimental spectra shown in Fig. 9 and Fig. S11 (ESI†), respectively, undoubtedly proves that the transient absorption bands correspond to ZnTPPH radical cation that did not relaxed to the optimal geometry. This is reasonable taking into account that the ZnTPPH radical cation was detected within the temporal resolution of the instrument (300 fs).
To get further insight into the nature of low-lying excited states of the ZnTPPH–GO nanoassembly, we performed high-level DLPNO-NEVPT2/CASSCF calculations. The results of these calculations are presented in a form of state-energy diagrams in Fig. 15. As expected, for free ZnTPPH, two degenerated pairs of singlet-excited states (S1/S2 and S3/S4) are found 1.98 eV and 2.63 eV, respectively, above the ground state. The same holds for T1/T2 and T3/T4 triplet states (1.70 and 2.02 eV higher in energy than the ground state, respectively). The computed spin–orbit coupling (SOC) matrix element between T1 and S0 states is 0.8 cm−1. Once the nanohybrid with GO is formed, the orbital quasi-degeneracy is lifted. Interestingly, the nature of first excited singlet and triplet states changes from porphyrin-centered to charge transfer (Fig. S12, ESI†). Such an S1 CT state was found 1.23 eV above the ground state, but the corresponding S1 ← S0 transition will gain only marginal intensity (fosc ≈ 10−4 a.u.) as compared to the S2 ← S0 transition (fosc ≈ 1.6 a.u.). The higher-lying CT state (S4) has a markedly larger fosc of 0.03 a.u. Thus, by tuning the laser excitation to 442 nm, the S4 excited state will gain significant population, and the charge-transfer state would be observed as confirmed experimentally (vide supra).
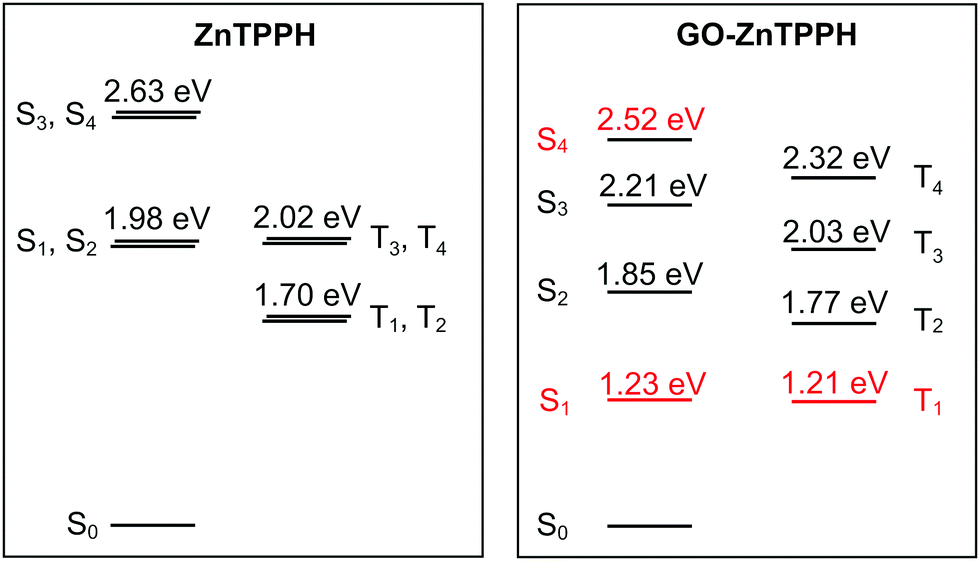 | ||
| Fig. 15 State-energy diagrams for free ZnTPPH (a) and ZnTPPH–GO nanohybrid (b) computed at the DLPNO-NEVPT2/CASSCF(4,4) and DLPNO-NEVPT2/CASSCF(6,6) levels, respectively. CT states are marked in red. Analysis of dominant configurations for relevant states in the case of GO–ZnTPPH is provided in the ESI,† Fig. S12. | ||
Flash photolysis experiments showed that the lifetime of the triplet state of the ZnTPPH increased upon complexation with the GO. The SOC matrix elements between S0 and all of the triplet states are larger than in the case of free porphyrin (>0.8 cm−1, largest element 2.3 cm−1 for 〈S0|Hsoc|T2〉). The rate constant for decay of the triplet state is proportional to the square of SOC and inversely proportional to energy difference between the involved states at the crossing geometry.57 To first approximation, the crossing geometry can be taken as the ground state geometry. The calculated small increase in SOC is thus compensated by the larger difference in energies between T2 and ground S0 states (∼0.07 eV) that further underlines the role of relaxation effects that contribute to the elongated lifetime of the triplet state.
Conclusions
First, in the presented work, we have characterized in detail singlet and triplet excited state properties of ZnTPPH in EtOH–H2O (1:2 v/v). It was determined that the singlet and triplet excited states lifetimes are 1.39 ns and 174 μs, respectively. Afterwards, we explored the interaction of GO with the ground state as well as both triplet excited states of ZnTPPH. Formation of the ground-state ZnTPPH–GO complex in solution was confirmed by the red-shift of the porphyrin Soret absorption band as well as the successful isolation of the ZnTPPH–GO nanohybrid material. The computed interaction energy is about 22 kcal mol−1. It was found that Stern–Volmer fluorescence quenching can be described in terms of two different quenching regimes depending on the GO concentration. In addition, our comprehensive analysis of the steady-state and time-resolved emission experiments led to the conclusion that fluorescence quenching was solely attributed to static quenching of the porphyrin by GO. Laser flash photolysis showed that the triplet lifetime of the ZnTPPH was increased in the presence of GO from 174 μs to 292 μs. This lifetime enhancement can be rationalized by the restriction in the rotation of the hydroxyphenyl rings, a motion which is required in the radiationless deactivation of the triplet excited-state mechanism. Surprisingly, fluorescence was not detected for the nanohybrid which indicates that a very fast deactivation process must take place. Ultrafast time-resolved transient absorption spectroscopy demonstrated the occurrence of fast electron transfer from the photoexcited ZnTPPH singlet state to GO sheets, as indicated by the formation of a porphyrin radical cation. The presence of charge-transfer states was also confirmed by our comprehensive theoretical study that also provided detailed insights into the nature of the electronically excited states involved in ZnTPPH–GO photochemistry. These results are relevant to the use of such systems in developing energy-conversion assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Centre (project no. 2015/19/D/ST5/00682). A. K. acknowledges support from the National Science Centre, Poland, grant no. 2018/30/E/ST4/00004. Access to high-performance computing resources was provided by the Interdisciplinary Centre for Mathematical and Computational Modelling in Warsaw, Poland, under grants no. GB77-11 and GB79-5. We would like to express our gratitude to Dr G. L. Hug for critically reading the manuscript.References
- J. Albero, D. Mateo and H. García, Molecules, 2019, 24, 906 CrossRef PubMed.
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242–251 CrossRef CAS.
- D. Kiessling, R. D. Costa, G. Katsukis, J. Malig, F. Lodermeyer, S. Feihl, A. Roth, L. Wibmer, M. Kehrer, M. Volland, P. Wagner, G. G. Wallace, D. L. Officer and D. M. Guldi, Chem. Sci., 2013, 4, 3085–3098 RSC.
- I. V. Lightcap and P. V. Kamat, Acc. Chem. Res., 2013, 46, 2235–2243 CrossRef CAS PubMed.
- Q. Xiang, B. Cheng and J. Yu, Angew. Chem., Int. Ed., 2015, 54, 11350–11366 CrossRef CAS PubMed.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820–3839 CrossRef CAS PubMed.
- A. Arrigo, A. Santoro, M. T. Indelli, M. Natali, F. Scandola and S. Campagna, Phys. Chem. Chem. Phys., 2014, 16, 818–826 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304–305, 38–54 CrossRef CAS.
- E. Gacka, A. Wojcik, M. Mazurkiewicz-Pawlicka, A. Malolepszy, L. Stobiński, A. Kubas, G. L. Hug, B. Marciniak and A. Lewandowska-Andralojc, J. Phys. Chem. C, 2019, 123, 3368–3380 CrossRef CAS.
- D. Larowska, A. Wojcik, M. Mazurkiewicz-Pawlicka, A. Malolepszy, L. Stobiński, B. Marciniak and A. Lewandowska-Andralojc, ChemPhysChem, 2019, 20, 1054–1066 CrossRef CAS PubMed.
- A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697–6706 CrossRef CAS PubMed.
- S. M. Aly, M. R. Parida, E. Alarousu and O. F. Mohammed, Chem. Commun., 2014, 50, 10452–10455 RSC.
- D. Masih, S. M. Aly, A. Usman, E. Alarousu and O. F. Mohammed, Phys. Chem. Chem. Phys., 2015, 17, 9015–9019 RSC.
- Y.-J. Yuan, D. Chen, J. Zhong, L.-X. Yang, J.-J. Wang, Z.-T. Yu and Z.-G. Zou, J. Phys. Chem. C, 2017, 121, 24452–24462 CrossRef CAS.
- P. Guo, P. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 5336–5345 CrossRef CAS PubMed.
- G. Rotas, M. B. Thomas, R. Canton-Vitoria, F. D’Souza and N. Tagmatarchis, Chem. – Eur. J., 2020, 26, 6652–6661 CrossRef CAS PubMed.
- M. B. M. Krishna, V. P. Kumar, N. Venkatramaiah, R. Venkatesan and D. N. Rao, Appl. Phys. Lett., 2011, 98, 081106 CrossRef.
- M. A. Khaderbad, V. Tjoa, T. Z. Oo, J. Wei, M. Sheri, R. Mangalampalli, V. R. Rao, S. G. Mhaisalkar and N. Mathews, RSC Adv., 2012, 2, 4120–4124 RSC.
- M. Zhu, Z. Li, B. Xiao, Y. Lu, Y. Du, P. Yang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1732–1740 CrossRef CAS PubMed.
- Y. B. Ivanova, A. S. Parfenov and N. Z. Mamardashvili, Russ. J. Phys. Chem. A, 2017, 91, 94–99 CrossRef CAS.
- M. Wendel, S. Nizinski, D. Tuwalska, K. Starzak, D. Szot, D. Prukala, M. Sikorski, S. Wybraniec and G. Burdzinski, Phys. Chem. Chem. Phys., 2015, 17, 18152–18158 RSC.
- T. Pedzinski, A. Markiewicz and B. Marciniak, Res. Chem. Intermed., 2009, 35, 497–506 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- C. Bannwarth and S. Grimme, Comput. Theor. Chem., 2014, 1040–1041, 45–53 CrossRef CAS.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS.
- M. Gouterman, J. Chem. Phys., 1959, 30, 1139–1161 CrossRef CAS.
- D. Ganyushin and F. Neese, J. Chem. Phys., 2006, 125, 024103 CrossRef PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- M. Gouterman, G. H. Wagnière and L. C. Snyder, J. Mol. Spectrosc., 1963, 11, 108–127 CrossRef CAS.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490–13497 CrossRef CAS PubMed.
- A. Lewandowska-Andralojc and B. Marciniak, ACS Energy Lett., 2019, 4, 1898–1901 CrossRef CAS.
- M. de Miguel, M. Álvaro and H. García, Langmuir, 2012, 28, 2849–2857 CrossRef CAS PubMed.
- Z. Mou, Y. Dong, S. Li, Y. Du, X. Wang, P. Yang and S. Wang, Int. J. Hydrog. Energy, 2011, 36, 8885–8893 CrossRef CAS.
- W. Zhang, Y. Li and S. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 15187–15195 CrossRef CAS PubMed.
- S. Min and G. Lu, Int. J. Hydrog. Energy, 2012, 37, 10564–10574 CrossRef CAS.
- G. G. Gurzadyan, T.-H. Tran-Thi and T. Gustavsson, J. Chem. Phys., 1998, 108, 385–388 CrossRef CAS.
- N. Mataga, Y. Shibata, H. Chosrowjan, N. Yoshida and A. Osuka, J. Phys. Chem. B, 2000, 104, 4001–4004 CrossRef CAS.
- C.-K. Min, T. Joo, M.-C. Yoon, C. M. Kim, Y. N. Hwang, D. Kim, N. Aratani, N. Yoshida and A. Osuka, J. Chem. Phys., 2001, 114, 6750–6758 CrossRef CAS.
- S. Kaniyankandy, S. N. Achary, S. Rawalekar and H. N. Ghosh, J. Phys. Chem. C, 2011, 115, 19110–19116 CrossRef CAS.
- X. Wen, P. Yu, Y.-R. Toh, Y.-C. Lee, K.-Y. Huang, S. Huang, S. Shrestha, G. Conibeer and J. Tang, J. Mater. Chem. C, 2014, 2, 3826–3834 RSC.
- P. Neta, M.-C. Richoux, A. Harriman and L. R. Milgrom, J. Chem. Soc., Faraday Trans., 1986, 82, 209–217 RSC.
- A. Lukaszewicz, J. Karolczak, D. Kowalska, A. Maciejewski, M. Ziolek and R. P. Steer, Chem. Phys., 2007, 331, 359–372 CrossRef CAS.
- U. Tripathy, D. Kowalska, X. Liu, S. Velate and R. P. Steer, J. Phys. Chem. A, 2008, 112, 5824–5833 CrossRef CAS PubMed.
- C. G. Silva, M. de Miguel, B. Ferrer, M. Álvaro and H. García, Photochem. Photobiol. Sci., 2009, 8, 1650–1654 RSC.
- M. Álvaro, A. Corma, B. Ferrer, H. García and E. Palomares, Phys. Chem. Chem. Phys., 2004, 6, 1345–1349 RSC.
- A. O. Lykhin and S. A. Varganov, Phys. Chem. Chem. Phys., 2020, 22, 5500–5508 RSC.
- Y. Guo, K. Sivalingam, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 114, 94111 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02545c |
| This journal is © the Owner Societies 2020 |