
Optimized electronic structure and p-band centre control engineering to enhance surface absorption and inherent conductivity for accelerated hydrogen evolution over a wide pH range†
 *a
*a
Abstract
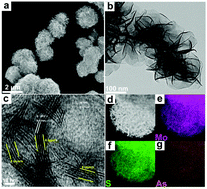
Numerous experiments have demonstrated that an appropriate electronic configuration can effectively activate the electrocatalytic activity. However, systematic studies on the effects of non-metallic elemental doping and its p-orbital center (εp) on electrocatalysis have not yet been carried out. Combining theoretical and experimental methods, we demonstrate an electronic configuration and p-orbital center control engineering for promoting the HER course in both acid and alkaline solutions over group VA elements doped into the inert basal plane of nanoMoS2. In acidic solutions, As-doped MoS2 has the best electrocatalytic activity. Theoretically, the calculated ΔGH of the As atom is only −0.07 eV, indicating that it has excellent catalytic performance. Furthermore, the p-orbital center under and near the Fermi level plays a significant role in the H adsorption course, and the closer the εp value is to the Fermi level, the weaker the H- non-metallic atom bond is. An appropriate εp can insure a proper strength of bond with H and further influence the catalytic activity of the HER. In alkaline solutions, P-doped MoS2 has the best electrocatalytic activity, which is due to the engineering of water dissociation sites by doping P atoms into MoS2 nanosheets. These findings pave the path to develop a rational strategy to trigger the activity of the inert basal plane of MoS2, to enhance the conductivity of inherent MoS2 towards the HER and provide a new idea that can be extended to other layered dichalcogenides.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

