
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decomposing anharmonicity and mode-coupling from matrix effects in the IR spectra of matrix-isolated carbon dioxide and methane†‡
Dennis F.
Dinu
 abc,
Maren
Podewitz
a,
Hinrich
Grothe
c,
Thomas
Loerting
b and
Klaus R.
Liedl
*a
abc,
Maren
Podewitz
a,
Hinrich
Grothe
c,
Thomas
Loerting
b and
Klaus R.
Liedl
*a
aInstitute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, A-6020 Innsbruck, Austria. E-mail: klaus.liedl@uibk.ac.at
bInstitute of Physical Chemistry, University of Innsbruck, A-6020 Innsbruck, Austria
cInstitute of Materials Chemistry, TU Wien, A-1060 Vienna, Austria
First published on 3rd August 2020
Abstract
Gas-phase IR spectra of carbon dioxide and methane are nowadays well understood, as a consequence of their pivotal roles in atmospheric- and astrochemistry. However, once those molecules are trapped in noble gas matrices, their spectroscopic properties become difficult to conceptualize. Still, such spectra provide valuable insights into the vibrational structure. In this study, we combine new matrix-isolation infrared (MI-IR) spectra at 6 K in argon and neon with in vacuo anharmonic spectra computed by vibrational self-consistent field (VSCF) and vibrational configuration interaction (VCI). The aim is to separate anharmonicity from matrix effects in the mid-infrared spectra of 12C16O2, 12CH4, and 12CD4. The accurate description of anharmonic potential energy surfaces including mode-coupling allows to reproduce gas-phase data with deviations of below 3 cm−1. Consequently, the remaining difference between MI-IR and VSCF/VCI can be attributed to matrix effects. Frequency shifts and splitting patterns turn out to be unsystematic and dependent on the particular combination of analyte and noble gas. While in the case of neon matrices these effects are small, they are pronounced in xenon, krypton, and argon matrices. Our strategy allows us to suggest that methane rotates in neon matrices – in contrast to previous reports.
1. Introduction
Matrix-isolation infrared (MI-IR) spectroscopy, where a guest molecule (or analyte) is trapped in an IR inactive host, can be considered as an ideal technique to study small polyatomic molecules. Trapping at cryo-conditions allows for investigation of single entities by quenching all rotational and translational degrees of freedom. In an ideal MI-IR experiment, very narrow bands are recorded and they are characteristic for the vibrational structure of a single molecule. However, compared to gas-phase experiments, the vibrational frequencies are shifted due to weak interactions between the host and the guest. Such matrix frequency shifts have been extensively evaluated, e.g., by Jacox for diatomics1 and transient species.2,3 In some MI-IR spectra, rotational–vibrational transitions are observed, most prominently in the case of water.4–10 That means, rotation is not always perfectly quenched. Additionally, most MI-IR spectra exhibit band splitting of several wavenumbers that cannot be explained by rotation. Those matrix effects are usually interpreted by a distortion of the analyte from its equilibrium structure as a consequence of the specific geometry of the trapping sites within the matrix.11 Further complications of the spectrum are due to the formation of larger clusters of the analyte itself (oligomerization) or with impurities.Although matrix effects are fairly well studied, their manifestation in spectra of different molecules is not easily conceptualized. In the past, a lot of experimental effort has been devoted to understanding MI-IR and MI-Raman spectra, leaving matrix effects at debate even for such simple molecules as carbon dioxide12–22 and methane.23–36 In order to better understand the particular matrix effects, specialized theoretical models have been derived for carbon dioxide37 and methane.38–41 However, these models are not readily transferable to other systems without further ado. On the other side, approaches for an efficient and accurate calculation of various host–guest combinations rely on approximations that limit the incorporation of different matrix effects and anharmonicity. Some approaches based on molecular dynamics,42 vibrational self-consistent field43 and density functional theory44 have been investigated.
In order to separate matrix effects from anharmonicity, an essential starting point in the spectral assignment is a flexible ab initio computational approach that correctly predicts the anharmonic vibrational structure of the analyte, without explicit consideration of matrix effects. As a matter of fact, the last three decades of algorithm development in computational chemistry have brought up a variety of tools to predict anharmonic vibrational spectra of polyatomic molecules. This field of quantum chemistry is well-reviewed45–51 and the current state of quantum chemistry itself is even dedicated to molecules in motion.48
On these grounds, we see the possibility for thorough studies that combine both high-quality IR experiments and ab initio calculations of anharmonic spectra. From the experimental side, MI-IR spectroscopy is suitable for single-molecule investigations, of course only if the before mentioned matrix effects can be handled. For the calculation, we rely on the vibrational self-consistent field (VSCF) and vibrational configuration interaction (VCI) approaches that are based on multi-mode representations of the potential energy surface (PES),52–54 as implemented by Rauhut et al.47,55–57 This approach allows for accurate calculation of anharmonic spectra and is flexibly applicable for very different polyatomic systems without relying on empirically fitted analytical PESs.
The major breakthroughs in the spectroscopic investigation of carbon dioxide (CO2) are shown on a timeline in the upper part of Fig. 1. Today, CO2 is described as a triatomic molecule, collinear and highly symmetric (D∞h point group), with a vibrational structure dissected into asymmetric stretch ν3, bending ν2 and symmetric stretch ν1. The first elaborate investigations in the 1920s considered a triangular molecular structure as possible.58,59 In 1929, while the discussion on collinear vs. triangular structure was still ongoing,60 the first Raman experiments61 have revealed IR inactive vibrational transitions. A rigorous theoretical model of a collinear, symmetric CO2 was presented by Dennison62 in 1931 and only one year later, Fermi63 interpreted the “Fermi doublet” at 1286 cm−1 und 1389 cm−1 in the Raman spectrum, based on the collinear model, as a resonance between symmetric stretch ν1 and the bending overtone 2ν2. From 1931 to 1933, Dennison and Adel enhanced the theoretical model on the collinear CO264–66 to perfectly interpret gas-phase experiments, which steadily increased in accuracy and revealed fine-splitting that could only be interpreted as rotational–vibrational transitions.67 In the advent of quantum mechanical models for polyatomic molecular systems during the 1930s, the CO2 surely was the most prominently studied linear rotator, as also highlighted in Herzberg's spectroscopy reference in 1945.68 The early theoretical proceedings were extensively reviewed in 1951 by Nielsen69 and consolidated in the seminal book by Wilson, Decius and Cross in 1955.70 One particular cornerstone of this theory is the representation of the molecular force field as a power series around the equilibrium structure. With the success of this theoretical model, the parameters of molecular force fields for the CO2 molecule have been derived from experimental observation,71–74 but also from ab initio calculations.75 This allows for the prediction of further rotational–vibrational transitions to provide all characteristic lines of gaseous CO2. Today, a vast amounts of experimental gas-phase data are evaluated in combination with computational predictions.76,77
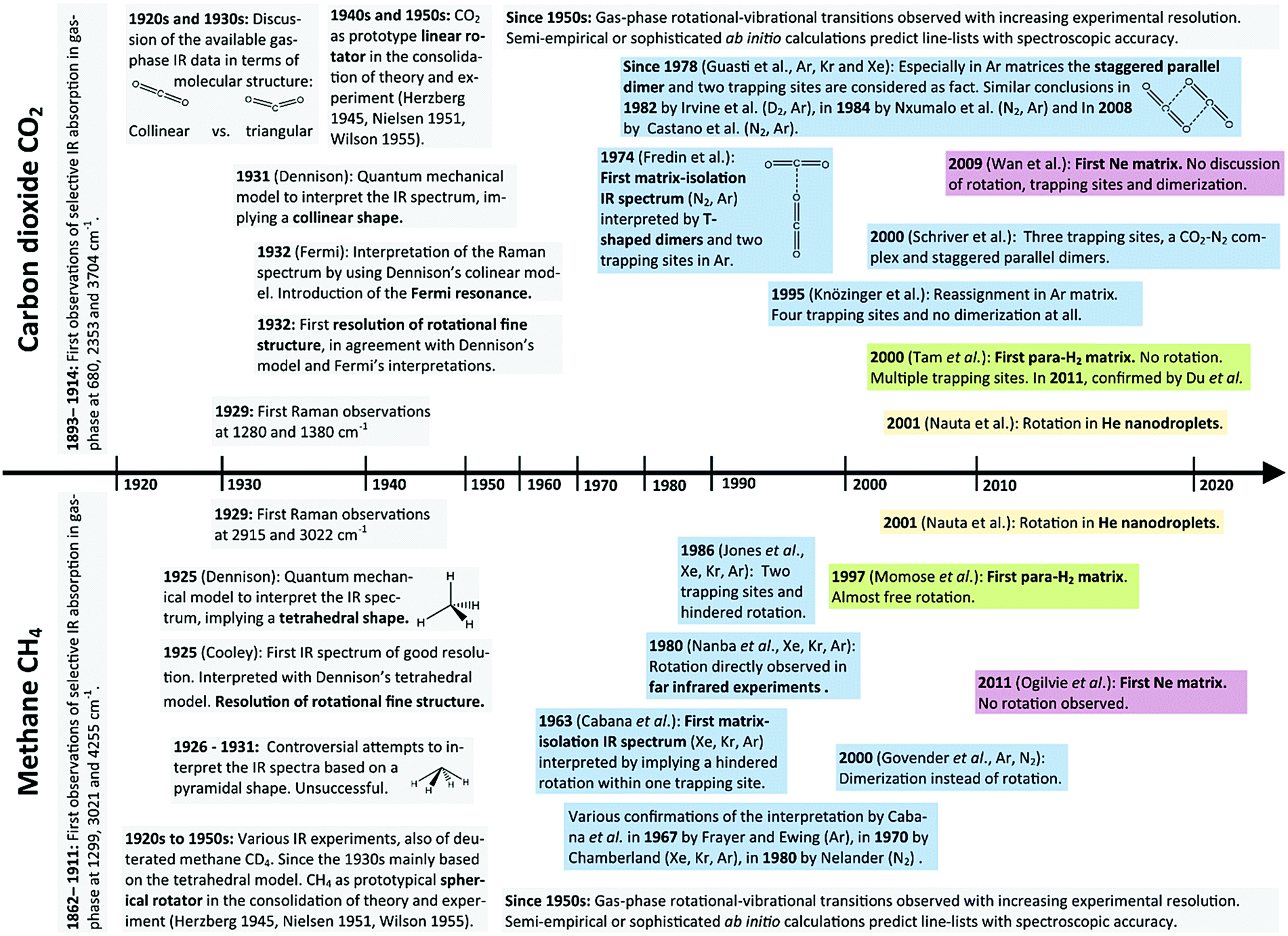 | ||
| Fig. 1 Schematic timeline of spectroscopic progress of gas-phase IR/Raman and matrix-isolation IR of CH4 and CO2, together with the major theoretical foundations to their interpretation. The textboxes are colored by the type of experiment (grey = gas-phase, blue = Kr, Ar, Xe and N2 matrices, red = Ne matrix, green = para-H2 matrix, yellow = He nanodroplets). | ||
In MI-IR spectroscopy, CO2 is a good example for the need of understanding matrix effects. In an ideal MI-IR experiment, CO2 should exhibit the two fundamental vibrational transitions ν2 and ν3 and some overtones and combination bands. In reality, multiple band splitting has been observed. During the last 50 years, different interpretations of this band splitting based on trapping sites, dimerization and complexation have been provided by various authors. The first MI-IR study of CO2 by Fredin et al.12 in 1974 was accompanied by a discussion on the occurrence of the CO2 dimer in Ar matrix to reason the band splitting. They favored a T-shaped dimer (C2v point group) previously identified in phase IR experiments,78 and ruled out a staggered parallel dimer (C2h point group). Furthermore, they found only one trapping site to occur in N2 matrices, yet, at least two trapping sites in Ar matrices. The latter assumption is also made in the Kr MI-IR study by Loewenschuss et al.13 Here, additional MI-Raman experiments also show the Fermi resonance (ν1 and 2ν2). In 1978, the hypothesis of a T-shaped CO2 dimer was challenged by Guasti et al.15 With reference to calculated dimer structures,79 they favored the staggered parallel dimer and verified this by extensive Ar, Kr and Xe MI-IR experiments. For Ar matrices, they confirmed two trapping sites, assumed that also the dimer bands are split due to two trapping sites, assumed that even larger aggregates can occur at higher temperatures, and they even assigned a band to the CO2–H2O cluster. In Kr and Xe matrices, the spectra are reported to be less complicated. Most findings up to the year 1982 were summarized by Irvine et al.,16 who provided further Ar MI-IR17 and D2 MI-IR16 experiments that basically consolidated the picture of CO2 monomers trapped in two substitutional sites and the occurrence of staggered parallel CO2 dimers. While in 1994, Nxumalo et al.18 came to similar conclusions, only one year later in 1995 Knözinger et al.19 came up with a reassignment of the Ar MI-IR spectrum. Based on mass spectroscopy studies of the aggregation of carbon dioxide (CO2)n in gas-phase mixtures with Ar and Kr and MI-IR investigation of the ν3 region of 12CO2 and 13CO2 in those mixtures, they concluded that the four bands in Ar matrices cannot be explained by dimerization but by four trapping sites of the CO2 monomer. In 2000, Schriver et al.21 proposed, aware of the controversies, three trapping sites, a CO2–N2 complex and a tentative assignment of CO2 staggered parallel dimers in Ar matrices. In 2008, however, a reinvestigation of the Ar and N2 MI-IR spectrum of CO2 by Castano et al.22 again revived the “two trapping sites” hypothesis in Ar matrices. Based on computed harmonic frequencies, the authors corroborated the assumption of Schriver et al. on the CO2–N2 complex and reintroduced the CO2 dimer in a similar assignment as Fredin et al. from 1974. They claimed to resolve all controversies, without explicitly disproving the arguments by Knözinger et al. from 1995. Since 2000, further MI-IR experiments in Ne matrix,14 He nanodroplets80 and para-H2 matrix20,81 have been presented. In He nanodroplets the CO2 molecule rotates, whereas in para-H2 matrices it does not, yet, dimerization and up to three trapping sites are discussed.
Striking parallels can be found in the history of the spectroscopy of the methane (CH4) molecule (cf. lower part of Fig. 1). The molecular structure of CH4 was identified as a tetrahedron based on its stereochemistry,82 way before vibrational spectroscopy became available. The vibrational structure of the highly symmetric (Td point group) tetrahedral CH4 molecule is described by the asymmetric deformation ν4, asymmetric stretch ν3, symmetric deformation ν2, and the symmetric stretch ν1, since the first gas-phase IR experiments of good resolution by Cooley83 and a first theoretical model by Dennison84 in 1925 were reported. However, in 1927 a pyramidal model of CH4 was considered,85,86 as it is capable of explaining the rotational fine structure in Cooley's spectrum, which implied at least two different moments of inertia in the molecule. At about the same time it was discussed that both models actually could occur as “dynamical isomers”.87 It is perplexing that the tetrahedral CH4 model was only gradually accepted in the 1930s, based on further IR spectra of CH4 and CD4 by the use of improved spectrometers.88–93 Even Herzberg dedicated this puzzling discussion a rather lengthy part in his seminal compendium.68 This should be warning enough that spectra can be misinterpreted even for very simple systems and especially when adequate theoretical models have not been developed. The CH4 molecule played a prominent role as a spherical rotator in the theoretical foundation for polyatomic molecular systems.94–96 Similar to the CO2 molecule, the molecular force field parameters have been of central interest and were derived from spectroscopy94,97–100 and from ab initio electronic structure calculations.101–104 Today extensive line-lists for CH4 are provided,105,106 and computations of rotational–vibrational frequencies on highly accurate ab initio potential energy surfaces reach spectroscopic accuracy.107–109
Considering MI-IR spectroscopy, the first Xe, Kr, and Ar MI-IR experiments of CH4 and CD4 by Cabana et al.23 in 1963 have revealed the protruding complexity of the spectra. The authors considered dimerization, various trapping sites, symmetry breakdown, and rotation of the molecule within the matrix. Based on thorough theoretical grounds,38 they concluded that methane exhibits a hindered rotation in their MI-IR experiments, in contrast to its free rotation in gas-phase. They confirmed this conclusion by Kr MI-Raman experiments.24 Later, Frayer and Ewing29,30 studied the rotational–vibrational transitions of the methane molecule in Ar MI-IR with a focus on its nuclear spin, as it was previously described by Wilson.96 They observed time-dependent changes in the IR absorption intensity, which served as evidence for nuclear spin interconversion from ortho- to meta-methane. In 1970, as a continuation of the work by Cabana et al., the MI-IR temperature change studies of Chamberland et al.31 have further consolidated the hypothesis of hindered rotation in Ar, Kr and Xe matrices. In the 1980s, two pure rotational transitions of CH4 in Ar, Kr, and Xe matrices were observed by Nanba et al.32 in the far-IR region, proving the assumption of methane's rotation in the matrix environment. Kr and Xe experiments by Sagara et al.33 resolved the fine structure also in the Raman spectrum of CH4 and CD4, which was previously not observed. Nelander110 confirmed the hindered rotation of CH4 and CD4 in N2 MI-IR experiments. Further Ar, Kr, Xe and Ne experiments of high-resolution by Jones et al.34,35 led to the conclusion that methane is trapped in two substitutional trapping sites, where it can undergo a hindered rotation in only one of those sites, in all matrix materials except neon. In the late 1990s, Momose36 and Hoshina27 have provided the first para-H2 MI-IR experiments, where their interpretation and assignment rests on an almost free rotation of methane. At the same time, Govender et al.111 explained the observed fine structure by dimerization of methane rather than its rotation. Their assignment based on in vacuo harmonic calculations is somewhat controversial, because the lack of anharmonicity and mode-coupling leaves significant discrepancies between theory and experiment while the matrix environment introduce further discrepancies on top of that. Howsoever, the rotation of CH4 remained commonly accepted. Almost simultaneously with the dimerization studies by Govender et al., Nauta et al.112 have provided the first IR experiments in He nanodroplets. They have observed a rotational fine structure similar to the gas-phase and argue that methane is essentially a free rotor in He nanodroplets, too. On the other hand, Ogilvie et al.28 provided Ar and Ne MI-IR spectra in 2011, making the case that methane is not rotating in Ne matrices.
Despite this extensive work, controversies remain. In MI-IR spectroscopy, deviations of theory from experiment are due to matrix effects as well as anharmonicity. While some theoretical investigations have fostered the conceptualization of specific matrix effects, the inability to accurately account for anharmonicity and mode-coupling limited the applicability. By minimizing the error due to anharmonicity, calculated in vacuo spectra provide a reliable starting point for the subsequent identification of matrix effects. Based on this combined experimental and theoretical methodology, we revisit central concepts in MI-IR spectroscopy related to the impact of the matrix on the vibrational spectra of carbon dioxide (CO2) and methane (CH4, CD4) trapped in various noble gas matrices.
2. Combined methodology of experimental and theoretical spectroscopy
Matrix isolation infrared spectroscopy (MI-IR)
The matrix-isolation (MI) setup used in this study has been previously described by Galvez et al.,113 and a detailed depiction of the apparatus is found in the supplementary information of ref. 114. The highlights of the setup are the 180° rotatable high-vacuum cryostat, which can be cooled to 5.8 K to allow for stable solid Ne matrices, and the Bruker spectrometer Vertex 80v, which is operating under 2 mbar with a liquid nitrogen cooled MCT detector to eliminate all atmospheric noise in the IR region. This is especially necessary when studying the IR spectrum of the omnipresent carbon dioxide molecule. The spectrometer itself is vibration-cushioned and uncoupled from the MI apparatus to avoid vibration-induction. The arising gap between the spectrometer and the MI apparatus is constantly flushed with pure N2 gas. In all experiments, the atmospheric carbon dioxide is not observed with this setup. Analyte-noble gas mixtures are prepared by barometric monitoring in a volume of about 2 liters. A mixture of 2 mbar of the analyte in dilution with about 990–1010 mbar of noble gas, is denoted as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500. It is injected with a constant flow of 4 mbar min−1 from a volume of about 200 ml and a pressure of 900–980 mbar. The gas is deposited as a matrix onto a gold plate in the high-vacuum cryostat chamber at 10−7 mbar and a temperature of 5.8 K. The deposition time is about 45 min per layer. Background spectra of the gold plate are taken before each sample measurement. The spectra shown in the figures are corrected for this background. To record spectra 512 scans are accumulated at a resolution of 0.3 cm−1. We measure a sample of 12C16O2 gas (99.9995%, Messer Austria) at dilutions of 1:200, 1:1000 and 1:5000, both in Ar and Ne. The sample of 12CH4 (99.995%, Messer Austria) is measured at dilutions of 1:500, 1:1000, 1:2000 in Ar and at dilutions of 1:500, 1:1000, 1:2000, 1:4000, 1:8000 and 1:32000 in Ne. Spectra of the 1:500 mixtures, both in Ar and Ne, are repeatedly recorded over a time of 2 h. The sample of 12CD4 (99.99%, Euriso-top France) is measured at dilutions of 1:1000 and 1:2000 in Ar and at 1:2000 and 1:32000 in Ne. In Ar matrix, temperature change experiments (5.8 K, 7.8 K, 10.4 K, 5.8 K) are carried out for the 1:1000 and 1:2000 mixtures of both the 12CH4 and the 12CD4 species. The CO2 IR spectra are recorded in the MIR region from 7500–500 cm−1, whereas the CH4 and CD4 spectra are recorded from 4000–500 cm−1.
:500. It is injected with a constant flow of 4 mbar min−1 from a volume of about 200 ml and a pressure of 900–980 mbar. The gas is deposited as a matrix onto a gold plate in the high-vacuum cryostat chamber at 10−7 mbar and a temperature of 5.8 K. The deposition time is about 45 min per layer. Background spectra of the gold plate are taken before each sample measurement. The spectra shown in the figures are corrected for this background. To record spectra 512 scans are accumulated at a resolution of 0.3 cm−1. We measure a sample of 12C16O2 gas (99.9995%, Messer Austria) at dilutions of 1:200, 1:1000 and 1:5000, both in Ar and Ne. The sample of 12CH4 (99.995%, Messer Austria) is measured at dilutions of 1:500, 1:1000, 1:2000 in Ar and at dilutions of 1:500, 1:1000, 1:2000, 1:4000, 1:8000 and 1:32000 in Ne. Spectra of the 1:500 mixtures, both in Ar and Ne, are repeatedly recorded over a time of 2 h. The sample of 12CD4 (99.99%, Euriso-top France) is measured at dilutions of 1:1000 and 1:2000 in Ar and at 1:2000 and 1:32000 in Ne. In Ar matrix, temperature change experiments (5.8 K, 7.8 K, 10.4 K, 5.8 K) are carried out for the 1:1000 and 1:2000 mixtures of both the 12CH4 and the 12CD4 species. The CO2 IR spectra are recorded in the MIR region from 7500–500 cm−1, whereas the CH4 and CD4 spectra are recorded from 4000–500 cm−1.
Vibrational self consistent field (VSCF) and vibrational configuration interaction (VCI)
All calculations are performed with the MOLPRO software package.115,116 The Born–Oppenheimer equilibrium structures of the CO2 and CH4 monomers are optimized at coupled cluster level with singles, doubles and perturbative triples excitations and the explicit correlation of interelectronic distances, i.e., the CCSD(T)-F12 ansatz.117,118 This ansatz allows for faster basis-set convergence than conventional coupled-cluster approaches with a little additional computational cost. The accuracy of this method using triple-ζ (VTZ-F12) basis sets119,120 corresponds roughly to a conventional calculation using quintuple-ζ basis sets, which is sufficiently close to the complete basis set limit. Harmonic vibrational energies are calculated at the same level of theory by numerical second derivatives and diagonalization of the Hessian matrix, which also yields normal coordinates. Although the term normal mode actually describes the change along a normal coordinate in time, we shall use it interchangeably for the term normal coordinate. The normal modes qi are used as a coordinate system for the subsequent construction of a multi-mode representation of the potential energy surface (PES), a sum of one-mode (1D) potentials, two-mode (2D) potentials, etc., as depicted in Fig. 2. The MOLPRO SURF algorithm constructs the PES on a grid, where each grid point is generated by elongation along the normal modes, with the 1D sub-potentials along one normal mode V(qi), the 2D sub-potentials along two normal modes V(qi,qj), etc. Symmetry considerations, iterative interpolation, and prescreening techniques significantly lower the amount of ab initio electronic structure single point energy calculations needed, with negligible loss of accuracy in the final PES, as pioneered by Rauhut.55 The grid form of the multi-mode PES representation is transformed into an analytical form by a polynomial fitting procedure.121 Based on the multi-mode representation of the PES, the nuclear Schrödinger equation using the Watson operator is solved variationally within the VSCF,52–54 where the wave function is approximated as a Hartree product of one-mode functions. This approximated solution is subsequently correlation-corrected by the configuration-selective state-specific VCI.47,56,122 It yields the VCI wave function as a linear combination of various excited Hartree products, with the anharmonic vibrational energies corresponding to specific VCI states. An analysis of the dominant configurations that contribute to a specific energy allows identification of resonances. The nuclear wave function also allows for the calculation of IR intensities by employing a multi-mode representation of the dipole moment surface. The latter is calculated here at HF/VTZ-F12 level of theory. Furthermore, the VSCF/VCI approach based on a multi-mode PES inherits the normal mode notation in the VCI states, which in further consequence can be directly compared to the spectroscopist's notation of vibrational transitions that has evolved from normal mode analyses. The whole approach is ab initio and is analogously employed for both the CO2 and the CH4 molecules. The CO2 normal modes qi are derived within the Abelian D2h subgroup as q4 asymmetric stretch, q3 symmetric stretch and q2/q1 bend. Considering the actual D∞h point group and reordering by the irreducible representations, those normal modes give rise to the spectroscopic labels for the fundamental vibrations as ν3 asym. str., ν1 sym. str., and ν2 bend. The CH4 normal modes qi are derived within the Abelian C2v subgroup, as q9/q8/q7 asymmetric stretch, q6 symmetric stretch, q5/q4 symmetric deformation and q3/q2/q1 asymmetric deformation. In the actual Td point group, and after reordering by the irreducible representations, those normal modes give rise to the spectroscopic labels as ν3 asym. str., ν1 sym. str., ν2 sym. def., ν4 asym. def. For both CO2 and CH4 the PES is calculated as a four-mode representation within the respective qi coordinate system, where 1D to 4D sub-potentials are calculated using CCSD(T)-F12 single points with frozen-core (fc) approximation and the cc-pVTZ-F12 basis119,120,123,124 in the case of CO2 and with all-electron (ae) correlation and the cc-pCVTZ-F12123–126 basis in the case of CH4. In Fig. 2, the grid form of the one-mode (1D) and two-mode (2D) sub-potentials are depicted for the normal modes q4, q3, and q1 in the case of CO2 (Fig. 2A) and for the normal modes q9, q6, q5 and q1 in the case of CH4 (Fig. 2B). As one may comprehend from the graphs in the CO2 case, the 1D sub-potentials V(q4) and V(q1) constitute a rather harmonic shape (quadratic and quartic potentials), while the V(q3) is definitely of anharmonic shape. The latter propagates into the 2D sub-potentials V(q3,q1) and V(q4,q3), which also illustrates the occurrence of mode-coupling. Depending on the magnitude of anharmonicity and mode-coupling, the calculated VSCF/VCI vibrational frequencies can be expected to deviate from the results of the harmonic approximation. Similar statements can be made in the case of CH4.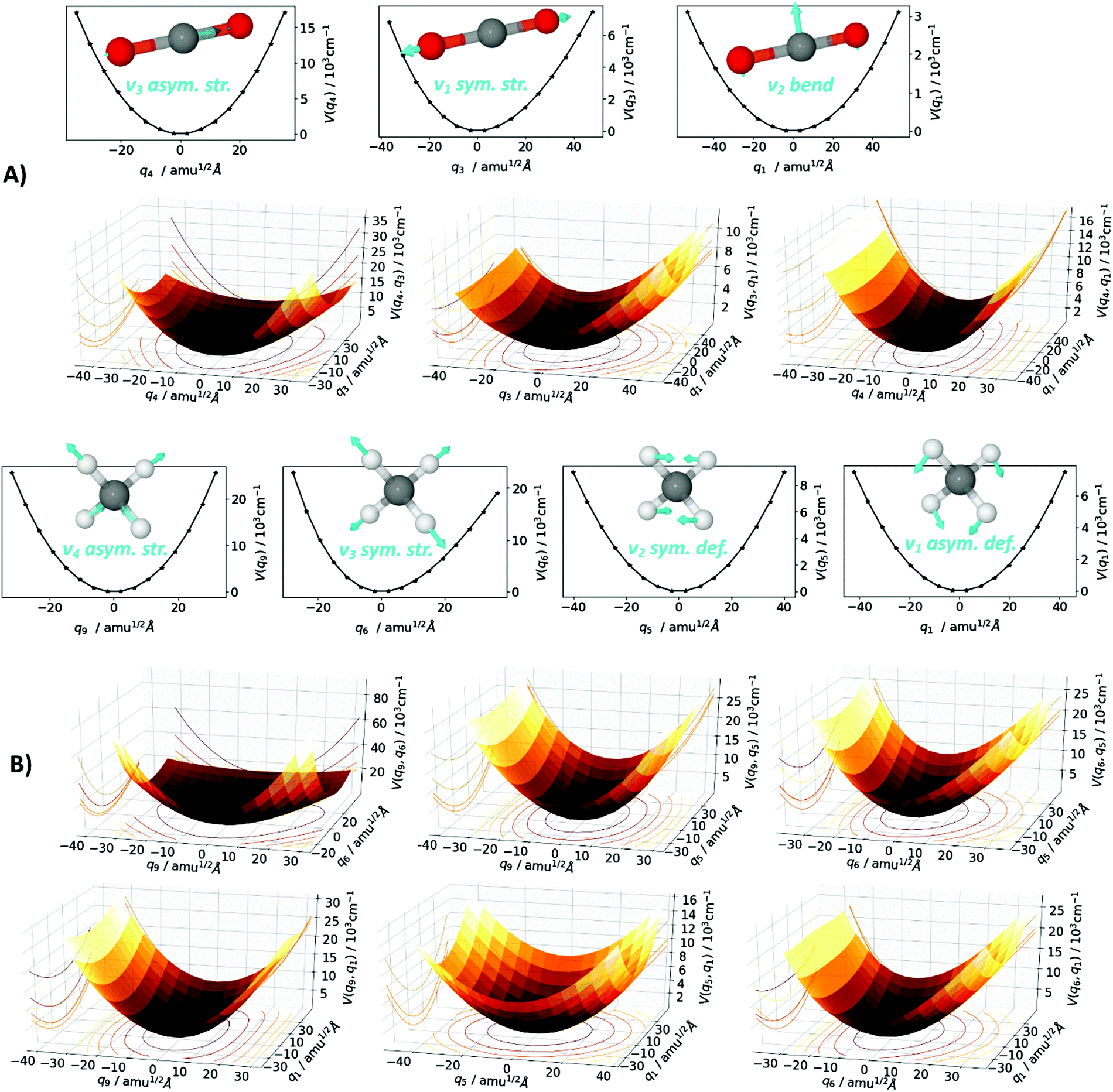 | ||
| Fig. 2 Depiction of the one-mode V(qi) and two-mode V(qi,qj) potentials within the multi-mode representation of the potential energy surface of (A) carbon dioxide, using fc-CCSD(T)-F12/cc-pVTZ-F12 single points, and of (B) methane, using ae-CCSD(T)-F12/cc-pCVTZ-F12 single points. Within the graphs of the one-mode potentials, the corresponding normal mode vectors are depicted. The spectroscopic labels ν should not be confused with the normal mode labels q. | ||
3. Results
Carbon dioxide (12C16O2)
All observed features of carbon dioxide 12C16O2 in the Ar and Ne MI-IR spectra are shown in Fig. 3. The Ar MI-IR spectrum reveals four distinct regions, each comprising a regular splitting pattern of two bands. In the Ne MI-IR spectrum, the same four regions can be identified. The energies are slightly blue-shifted compared to the Ar MI-IR spectrum and the occurrence of band splitting is less regular. The spectra were recorded up to 7500 cm−1, yet, no features were observed above 4000 cm−1. Changes in the sample dilution have a more pronounced influence on the Ne MI-IR spectrum than on the Ar MI-IR spectrum.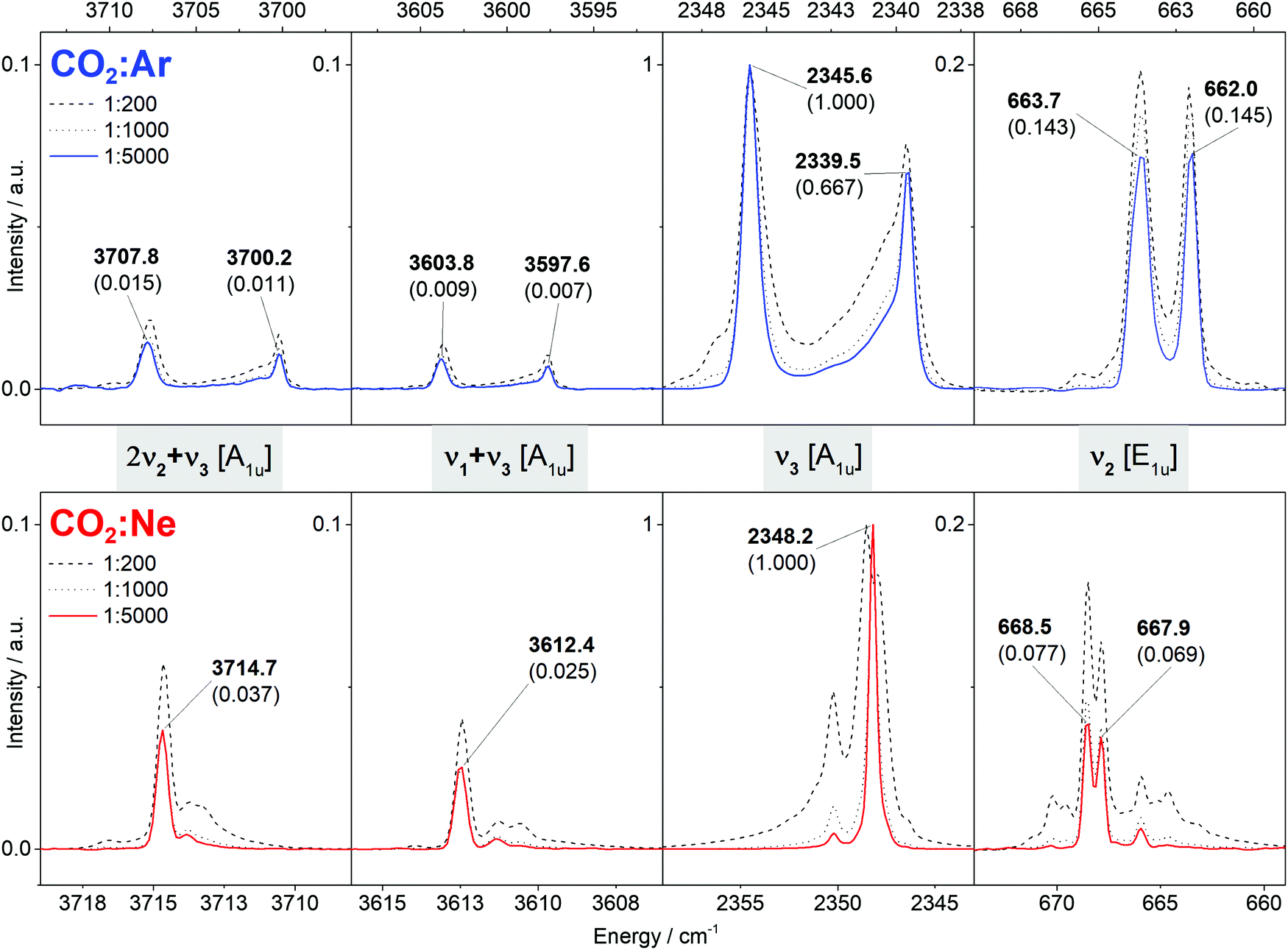 | ||
| Fig. 3 Mid-infrared spectrum of carbon dioxide (12C16O2) isolated in argon (top, blue) and neon (bottom, red) matrices at 5.8 K. The spectra depicted as colored solid lines are considered in the assignment. Frequencies assigned to the monomer are labeled with their relative intensity in brackets. In between two panels, the notation of the vibrational states are given in grey boxes. Unlabeled spectral features are due to dimers or 13C and 18O isotopomers (cf. ESI‡). | ||
Both spectra can be uniquely assigned, purely by the comparison with VCI calculation. The assignment is summarized in Table 1 and discussed in Section 4. In general, the VCI calculated vibrational transitions are in very good agreement with the observation in gas-phase IR (MAD: 1.5 cm−1). Similarly, the transitions observed in Ne MI-IR agree very well with gas-phase IR (MAD: 1.1 cm−1). For other host systems, the frequencies are systematically shifted towards lower energies (MAD: 6.7 cm−1 for Ar, 6.3 cm−1 for Kr, 12.1 cm−1 for Xe).
| Transition [irrep]a | Experimentb | Calculationc | |||||
|---|---|---|---|---|---|---|---|
| Xe | Kr | Ar | Ne | Gas | VCI | Harm. | |
|
a Fermi resonant transitions are marked with FR.
b Data from this work are in bold letters (CO2:Ar 1:5000 at 5.8 K, CO2:Ne 1:5000 at 5.8 K), data from the literature in italics.
c VSCF/VCI calculation with up to quintuple excitations on a 4-mode PES at CCSD(T)-F12A/cc-pVTZ-F12. Harmonic approximation (Harm.) at CCSD(T)-F12A/cc-pVTZ-F12.
d Taken from ref. 17 (Xe/CO2 ratio between 100 and 23000, at 6–8 K).
e Taken from ref. 13 (Kr/CO2 ratio between 500 and 30000, at 20 K).
f Taken from ref. 77.
g Taken from ref. 127.
h Mean absolute deviation (MAD) and maximum deviation (MAX) with reference to gas-phase IR. For Ar and Kr the higher-lying energies of the split bands have been considered.
|
|||||||
| 2ν2 + ν3 FR [A1u] | 3707.8/3700.2 | 3714.7 | 3715.6 | 3713.8 | |||
| ν 1 + ν3 FR [A1u] | 3603.8/3597.6 | 3612.4 | 3613.7 | 3610.6 | |||
| ν 3 [A1u] | 2334.7 | 2344.0/2342.2 | 2345.1/2339.6 | 2348.2 | 2349.9 | 2347.4 | 2394.9 |
| 2ν2 [A1g] | 1382.0 | 1388.2 | 1388.1 | ||||
| ν 1 [A1g] | 1277.9 | 1285.5 | 1284.6 | 1353.0 | |||
| ν 2 [E1u] | 659.3 | 662.6/660.2 | 663.7/662.1 | 668.5/667.9 | 668.2 | 667.8 | 673.1 |
| MAD (MAX)h | 12.1 (15.3) | 6.3 (7.6) | 6.7 (9.9) | 1.1 (1.8) | 0 | 1.5 (3.1) | 39.1 (67.5) |
In both Ar and Ne MI-IR, some transitions with rather low intensity are observed. Some arise from aggregation and vanish with increasing dilution. Others are due to the isotopomers 13C16O2 and 12C18O16O. VCI calculations for those species show the same good agreement with the experiment as mentioned for 12C16O2. Those results are provided in the ESI‡ (cf. Table S1).
Methane (12CH4 and 12CD4)
In Fig. 4 and 5, the MI-IR spectra of both the 12CH4 and the 12CD4 isotopologue are depicted. We observe temperature-dependent band shapes in the experiments performed with Ar matrices (Fig. 4). In both, Ar and Ne matrices, dilution-dependent band shapes can be observed (Fig. 5).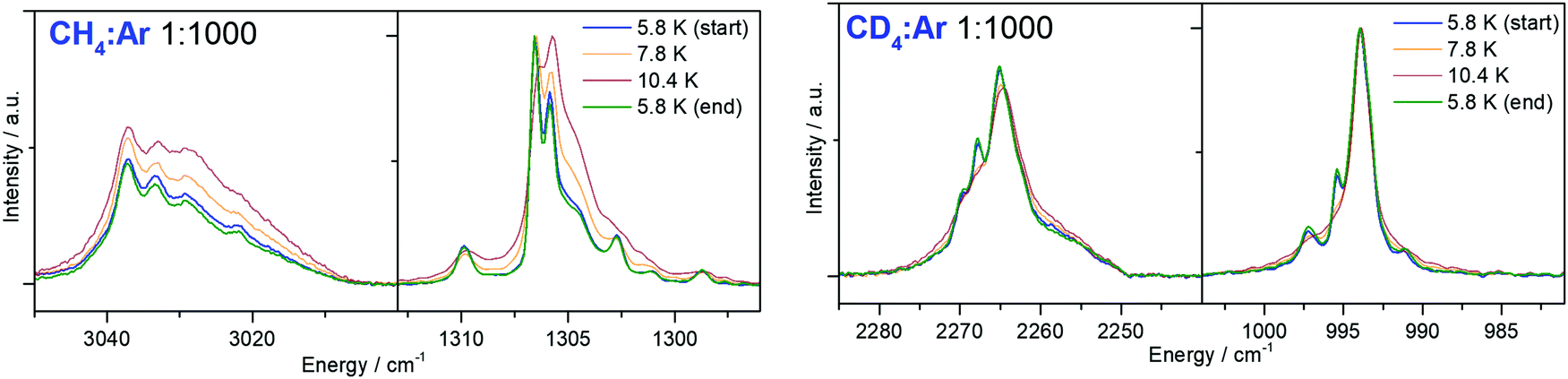 | ||
| Fig. 4 Impact of temperature changes in the mid-infrared spectrum of a methane–argon matrix (left: 12CH4, right: 12CD4), initially deposited at 5.8 K. | ||
 | ||
| Fig. 5 Mid-infrared spectra of methane (left: 12CH4, right: 12CD4) isolated in argon (top, blue) and neon (bottom, red) matrices at 5.8 K and various dilutions. The spectra depicted as colored solid lines are considered in the assignment. Frequencies assigned to the monomer are labeled with their relative intensity in brackets. In between two panels, the notation of the vibrational states are given in grey boxes. | ||
Temperature-change experiments in Ar matrix (Fig. 4) show that already a slight increase in temperature strongly influences the shape of the observed bands. For the 12CH4 isotopoloque (cf.Fig. 4, left), the width of the bands significantly increases upon heating from 5.8 to 10.4 K. In the region of 3050–3000 cm−1, the intensities of the observed bands equalize and the weakest sideband even disappears at 10.4 K. In the region of 1310–1290 cm−1, the originally most intense band weakens, while the intensity of its nearest neighbor increases. At 10.4 K, there are only two out of six bands left. However, when cooling back to 5.8 K, the initial band shapes in both mentioned regions perfectly recover. In temperature-change experiments of 12CD4 in Ar matrices (cf.Fig. 4, right), heating to 10.4 K leads to dissolution of multiple bands into one band, each in the region of 2280–2250 cm−1 and 1010–980 cm−1. By cooling back to 5.8 K, again, the original band shapes perfectly recover.
For the 12CH4 isotopoloque in Ar at 5.8 K and a dilution of 1:500 (cf.Fig. 5, top left), we observe four strongly overlapping bands with similar intensity in the region of 3050–3000 cm−1, and seven bands with very different intensities in the region of 1310–1290 cm−1. Those two patterns do not significantly change with increasing dilution. They are basically the same for 1:500, 1:1000, and 1:2000. In contrast to that, the Ne MI-IR experiments tend to be more sensitive toward increasing dilution (cf.Fig. 4, bottom left). In each of the two mentioned spectral regions, one band is observed at a dilution of 1:500. With increasing dilution to 1:1000 and 1:2000, the band at 3020 cm−1 loses intensity, while the band at 1307 cm−1 slightly blue-shifts. At a dilution of 1:4000 the bands begin to split, while this splitting is best observed at a dilution of 1:8000. At a dilution of 1:32000, the splitting of the band at 3020 cm−1 becomes difficult to be distinguished from spectral noise. In contrast to that, the intensity of the bands around 1307 cm−1 is high enough so that the splitting pattern may also be analyzed in the 1:32000 experiment. We here rely on the 1:8000 experiment for subsequent assignment. In this experiment, the shape of the band at 3020 cm−1 is somewhat mirrored to the corresponding band in Ar matrix. The band shape at 1307 cm−1 is similar to the corresponding band in the Ar MI-IR spectrum. For both regions, the bands in the Ne spectrum are much closer to each other than the corresponding bands in the Ar spectrum.
Considering the 12CD4 isotopologue, similar observations are made. The Ar MI-IR spectra (cf.Fig. 5, top right) are rather insensitive to dilution, exhibiting two or three strongly overlapping bands in the region of 2280–2250 cm−1 and four partly overlapping bands in the region of 1010–980 cm−1. The Ne MI-IR spectra (cf.Fig. 5, bottom right) drastically change with dilution. Here, one broad band is observed each in the regions of 2270–2250 cm−1 and 1010–980 cm−1. Upon dilution of 1:32000, those bands transform to very narrow bands comprising also some band splitting pattern. The intensities in the 12CD4 experiments are stronger than in the 12CH4 experiments. Thus, for the 12CD4 experiments, we can consider the high dilution experiment of 1:32000 in our assignment, as this spectrum is not complicated by spectral noise.
The band assignment of methane is summarized in Table 2 and discussed in Section 4. Considering the CH4 isotopologue, the VCI vibrational transitions are in good agreement with the observation in gas-phase (MAD: 3.7 cm−1), and the Ne MI-IR experiments are very close to the gas-phase IR data (MAD: 0.4 cm−1). For the other host systems, the frequencies are shifted towards lower energies (MAD: 6.7 cm−1 for Ar, 3.4 cm−1 for Kr, 11.8 cm−1 for Xe). It should be seen that these shifts are not systematically decreasing from xenon to neon. Similar results are obtained for CD4.
| Assignmenta | Experimentb | Calculationc | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mol. | Vib. [irrep] | Rot. | Xe | Kr | Ar | Ar | Ne | Gas | VCI | Harm. |
|
a Rotational transitions (J′ ← J′′) are labeled as R(J′′) for ΔJ = J’ − J′′ = +1, as Q(J′′) for ΔJ = 0 and as P(J′′) for ΔJ = −1.
b Data from this work in bold letters (CH4:Ar 1:8000 at 5.8 K, CH4:Ne 1:8000 resp. 1:32000 at 5.8 K). Data from literature in italics.
c VSCF/VCI calculation with up to quintuple excitations on a 4-mode PES at ae-CCSD(T)-F12A/cc-pCVTZ-F12. Harmonic approximation (Harm.) at ae-CCSD(T)-F12A/cc-pCVTZ-F12.
d Taken from ref. 31 (0.06% CH4 in Xe at 8 K, 0.10% CH4 in Kr at 8 K, 0.06% CD4 in Kr at 8 K).
e Taken from ref. 23 (0.75% CH4 in Kr at 5 K, 2% CD4 in Xe at 5 K). We here reassigned the ν4 region of CD4 in Xe.
f Taken from ref. 28 (CH4 and CD4 in Ne and Ar at molar fractions of 0.0001–0.005 at 3–21 K).
g Taken from ref. 33 (1% CH4 in Kr at 10 K, 1.3% CH4 in Xe at 10 K, 2.5% CD4 in Kr at 10 K).
h Taken from ref. 34 (Xe/CH4 = 5000 at 5 K, Kr/CH4 = 10000 at 5 K).
i Taken from ref. 128.
j Taken from ref. 129.
k Taken from ref. 130.
l Taken from ref. 93. The P-Branch therein has been reassigned.
m Mean absolute deviation (MAD) and maximum deviation (MAX) with reference to gas-phase.
|
||||||||||
| CH4 | ν 3 [F2] | R(1) | 3018.3 | 3030.0 | 3041.2 | 3040.9 | 3027.1 | 3038.5 | ||
| R(0) | 3013.3 | 3026.5 | 3037.3 | 3037.3 | 3021.4 | 3028.8 | ||||
| R(0)′ | 3022.2 | 3033.5 | 3033.5 | 3020.3 | ||||||
| Q | 3005.3 | 3018.8 | 3027.6 | 3029.2 | 3018.6 | 3018.8 | 3024.5 | 3163.7 | ||
| P(1) | 2995.6 | 3010.1 | 3022.2 | 3022.2 | 3014.9 | 3009.0 | ||||
| ν 1 [A1] | Q | 2905.3 | 2915.7 | 2917 | 2919.6 | 3040.8 | ||||
| ν 2 [E] | Q | 1534 | 1533.9 | 1572.3 | ||||||
| ν 4 [F2] | R(1) | 1304.8 | 1306.9 | 1309.6 | 1309.8 | 1309.5 | 1316.8 | |||
| R(0) | 1300.7 | 1303.4 | 1306.3 | 1306.6 | 1308.4 | 1311.4 | ||||
| R(0)’ | 1305.6 | 1305.9 | 1307.3 | |||||||
| Q | 1295.7 | 1299.1 | 1302.5 | 1302.7 | 1306.3 | 1305.8 | 1312.1 | 1346.8 | ||
| P(1) | 1290.9 | 1294.8 | 1298.4 | 1298.7 | 1303.9 | 1300.3 | ||||
| MAD (MAX) for Q-branchm | 11.8 (13.5) | 3.4 (6.7) | 6.0 (8.8) | 6.7 (10.4) | 0.4 (0.5) | 0 | 3.7 (6.3) | 87.0 (144.9) | ||
| CD4 | ν 3 [F2] | R(2) | 2263.2 | 2273.5 | ||||||
| R(1) | 2253.2 | 2260.8 | 2271.5 | 2269.7 | 2261.7 | 2268.7 | ||||
| R(0) | 2250.5 | 2258.9 | 2265.5 | 2267.9 | 2259.1 | 2264.2 | ||||
| Q | 2248.1 | 2255.4 | 2261.9 | 2265.1 | 2258.2 | 2259.0 | 2257.2 | 2341.8 | ||
| P(1) | 2244.0 | 2252.0 | 2261.9 | 2251.2 | ||||||
| P(2) | 2248.1 | 2246.9 | ||||||||
| ν 1 [A1] | Q | 2099.5 | 2109 | 2102.9 | 2151.0 | |||||
| ν 2 [E] | Q | 1092 | 1094.1 | 1112.2 | ||||||
| ν 4 [F2] | R(2) | 993.8 | 997.3 | 997.2 | 996.9 | 1006.8 | ||||
| R(1) | 991.2 | 994.9 | 995.4 | 996.3 | 1003.3 | |||||
| R(0) | 990.2 | 993.3 | 995.3 | 994.0 | 995.6 | 1000.1 | ||||
| Q | 988.8 | 990.8 | 992.7 | 991.2 | 993.7 | 995.9 | 999.1 | 1018.3 | ||
| P(1) | 987.4 | 988.3 | 990.2 | |||||||
| P(2) | 982.9 | 985.8 | 986.6 | |||||||
| MAD (MAX) for Q-branchm | 9.0 (10.9) | 4.3 (5.1) | 3.1 (3.2) | 5.4 (6.1) | 1.5 (2.2) | 0 | 3.3 (6.1) | 41.9 (82.8) | ||
An important observation is made for the line spacing of the rotational–vibrational transitions. Those are in general smaller in the MI-IR experiments than their counterparts in the gas-phase spectra, as can be seen in Table 3. For example, in the gas-phase spectra of the CH4 isotopologue, the line spacing between P(1) and Q(1) for the ν3 fundamental is 19.7 cm−1. In matrix-isolation spectra, this particular spacing is 13.0 cm−1 in Xe, 11.7 cm−1 in Kr, 11.7 cm−1 in Ar and 8.5 cm−1 in Ne. Considering the ν4 fundamental vibration, the same line spacing between P(1) and Q(1) is narrower, with 11.0 cm−1 in gas-phase, 9.1 cm−1 in Xe, 7.8 cm−1 in Kr, 7.1 cm−1 in Ar and 3.2 cm−1 in Ne. Similar results are observed for other line spacings (cf.Table 3). For the CD4 isotopologue, the line spacings are in general smaller than for the CH4 isotopologue. Also here, the spacings show a systematical decrease when going from Xe to Ne, whereas they are generally lower than observed in the gas-phase. For example, the line spacing between P(1) and Q(1) considering the ν4 fundamental vibration are 9.7 cm−1 in gas-phase, 5.1 cm−1 in Xe, 5.4 cm−1 in Kr, 4.6 cm−1 in Ar and 3.5 cm−1 in Ne.
| Assignment | Experiment | ||||||
|---|---|---|---|---|---|---|---|
| Mol. | Vib. [irrep] | Rot. | Xe | Kr | Ar | Ne | Gas |
| CH4 | ν 3 [F2] | R(1) | 13.0 | 11.7 | 11.7 | 8.5 | 19.7 |
| R(0) | 8.0 | 8.3 | 8.1 | 2.8 | 10.0 | ||
| Q | 0 | 0 | 0 | 0 | 0 | ||
| P(1) | −9.7 | −8.2 | −7.0 | −3.7 | −9.8 | ||
| ν 4 [F2] | R(1) | 9.1 | 7.8 | 7.1 | 3.2 | 11.0 | |
| R(0) | 4.9 | 4.3 | 3.9 | 2.1 | 5.6 | ||
| Q | 0 | 0 | 0 | 0 | 0 | ||
| P(1) | −4.9 | −4.3 | −4.0 | −2.4 | −5.5 | ||
| CD4 | ν 3 [F2] | R(1) | 5.1 | 5.4 | 4.6 | 3.5 | 9.7 |
| R(0) | 2.4 | 3.5 | 2.9 | 0.9 | 5.2 | ||
| Q | 0 | 0 | 0 | 0 | 0 | ||
| P(1) | −4.1 | −3.5 | −3.2 | −7.8 | |||
| ν 4 [F2] | R(1) | 2.4 | 4.1 | 4.2 | 1.3 | 7.4 | |
| R(0) | 1.4 | 2.5 | 2.8 | 0.7 | 4.2 | ||
| Q | 0 | 0 | 0 | 0 | 0 | ||
| P(1) | −1.4 | −2.5 | −1.9 | −5.7 | |||
In the here observed spectral range, two non-fundamental transitions occur with detectable intensity. According to the VCI calculations, there should be the 2ν4 overtone at 2615.0 cm−1 for CH4 (resp. at 1992.8 cm−1 for CD4) and the ν2 + ν4 combination band at 2834.2 cm−1 (resp. at 2092.0 cm−1 for CD4). With very weak intensities, we observe in the Ar MI-IR spectra the 2ν4 transition at 2616.2 cm−1 for CH4 (resp. at 1981.9 cm−1 for CD4) and the ν2 + ν4 transition at 2834.3 cm−1 for CH4 (not observed for CD4). In Ne matrix, no overtones and combination bands are observed.
4. Discussion
In the introduction we presented a historical retrospect of CO2 and CH4 infrared spectroscopy. This retrospect shows that well-resolved gas-phase IR spectra need the help of a generalizable theoretical model for conclusive interpretation. It also highlights that the assignment of gas-phase IR spectra was inherently linked to controversial discussions of molecular structure of CO2 and CH4. In the 1960s, roughly 30 years after the gas-phase IR spectra have been successfully interpreted by quantum theoretical models, the novel matrix-isolation technique led to new insights but also new difficulties in describing the vibrational structure of the trapped molecules. The unambiguous interpretation of MI-IR spectra requires not only knowledge of the molecular structure of the analyte but also of the structure of a host–guest system. Therefore, the assignment of MI-IR spectra is inherently linked to a controversial discussion of matrix effects that arise from the peculiarities of the host–guest structure.First of all, the laboratory preparation of a matrix is an intricate process depending on various experimental parameters. The phase transition of the analyte-noble gas mixture to a solid matrix depends on, inter alia, the deposition speed, the pressure and temperature during deposition, the deposition technique (pulsed or continuous) and the mixing behaviour of the analyte and the noble gas, which may not be the same for different mixing ratios. Changing these parameters leads to different properties of the solid matrix. For example, it is often not known whether the matrix is in an amorphous or crystalline phase. In most studies, however, the local environment around the analyte is assumed to resemble the crystal-like ordering of the noble gas that serves as host.
Although assumptions on the local host–guest structure may be valid, it is actually not possible to directly derive the host–guest structure from an MI-IR spectrum. However, the matrix effects impacting the spectra can be very well investigated and rationalized by some basic concepts of the local host–guest structure. Such matrix effects are, among others, band-splitting due to differences in trapping sites, frequency shifts due to the interaction of the host with the guest, and analyte rotation within the matrix trapping site. In the following, we discuss these matrix effects and their conceptualization in a systematic series of host–guest combinations and evaluate the transferability of successful concepts from one host–guest system to another.
Matrix trapping sites
CO2 in Ar matrices exhibits a band-splitting pattern that has been extensively and controversially discussed in the literature.12,17,19,21,22 Two equally intense bands are observed in each spectral window (cf.Fig. 3). This doublet can be reasonably explained by assuming that CO2 monomers are trapped in two different trapping sites. As the band-splitting pattern is not changing with increasing dilution, dimerization cannot be the reason for this doublet. The shoulders observed for the bands at 2345.6 cm−1 and 2339.5 cm−1 may be reasoned by further trapping sites, as claimed by Knözinger et al.19 or as dimers, as claimed originally by Fredin et al.12 As these shoulders are barely existent in our experiments, it is sufficient to explain our spectra by the hypothesis of two trapping sites. In literature, similar band-splitting has been observed in Kr matrices, yet, not in Xe matrices.15 However, as experimental literature data are rather sparse for Xe MI-IR, it can be assumed that the band-splitting was simply not reported.In a previous study on CO2 in Ne matrices, no discussion on band splitting and matrix trapping sites has been provided,14 perhaps because the band splitting in Ne matrices is much simpler than in corresponding Ar matrices. Only for the region around 668 cm−1, a variety of bands occur, yet, their intensity is heavily dependent on the dilution. It is reasonable to assume that a great part of those bands is due to dimerization as most of them disappear upon dilution. However, the doublet remains in this region even with the highest dilution (1:5000). It is possible to explain this by two different trapping sites for Ne matrices. However, this is problematic to justify because these two trapping sites should lead to a similar splitting pattern in the other spectral regions, too, which is not the case (cf.Fig. 3). The difference in the Ar and Ne MI-IR spectra of carbon dioxide is a vivid example on how band-splitting patterns are dictated by the chosen matrix material.
In our CH4 and CD4 experiments, we observe characteristic band splitting patterns, both for CH4 and CD4 in Ar and Ne matrices (cf.Fig. 5). However, these patterns are not as systematic as one may expect from the occurrence of distinct trapping sites, e.g., as seen for the doublet pattern in CO2:Ar (cf.Fig. 3). For both CH4 and CD4, the splitting in the v4 region is very different to the splitting in the v3 region. In the pioneering studies of CH4 and CD4 trapped in noble gas matrices, no direct evidence for distinct trapping sites was given.23,31 Based on high-resolution experiments in Xe, Kr, and Ar matrices, Jones et al.34 demonstrated later for the v4 region that CH4 may enter two different trapping sites. However, they showed that only one of these trapping sites is predominant and that this predominant trapping site is linked to the characteristic band splitting pattern observed in most other studies. We will discuss the assignment of this pattern as rotational–vibrational transitions in the following.
Rotation in the matrix
The ν3 and ν2 region of the CO2 spectrum have been previously studied by MI-IR with all types of noble gas matrices.13,15,17,18,21 With reference to those studies, our observations agree with the interpretation that CO2 does not rotate in solid noble gas matrices. When the main band splitting pattern is assigned by the occurrence of two trapping sites, there remain basically no bands that one could assign as rotational–vibrational transitions.In various MI-IR studies of CH4, the characteristic band splitting patterns (cf.Fig. 5) are commonly interpreted by the occurrence of a hindered rotation.23–31,34,35,110 As the temperature is very low in MI-IR experiments, only a handful of rotational–vibrational transitions can be observed in the spectra. Considering the isolation of CH4 in Ar matrices, our observations in various dilution and temperature change experiments directly support this interpretation. High dilution experiments help to rule out oligomerization and complexation as the origin of these band splitting patterns. At high dilutions, the methane molecules are well separated and diffusion within the Ar solid at 5.8 K is hardly possible, thus, oligomerization or complexation is considered as inhibited in our matrices. Oligomerization and complexation may be induced by so-called annealing experiments (e.g. in ref. 23), where diffusion is increased by heating and partial melting of the matrix. However, the chance of complexation and oligomerization in our temperature-change experiments is negligible, as we performed those experiments at a high dilution of 1:1000, where the amount of methane molecules within the solid is very low. Consequently, the here observed temperature-change induced alternation of the spectral pattern in Ar matrices is due to nuclear-spin interconversion, as shown by Frayer et al.,30 and is an evidence for rotation within the matrix. Most authors agree on the assignment of rotational–vibrational transitions, particularly R(1), R(0), Q(1) and P(1), for the characteristic band splitting pattern. We adopt this notation in our Ar and Ne MI-IR experiments and update previous literature assignments for the sake of consistency (cf.Table 2).
While it is established in literature to assign rotational–vibrational transitions in the spectra of CH4 and CD4 in Xe, Kr, and Ar matrices, this is not the case for Ne matrices. However, our new data strongly suggest an assignment of rotational–vibrational transitions for CH4 and CD4 in the Ne MI-IR spectra (cf.Fig. 5, bottom). Our assignment is rationalized based on following reasoning: (1) Assuming rotation of CH4 in noble gas matrices, the rotational barrier should increase from Xe to Ne, resulting in decreasing rotational–vibrational line spacings. This trend is confirmed in Table 3, individually for the ν3 and ν4 fundamentals of both CH4 and CD4. (2) When going from CH4 to CD4, the rotational constants should decrease by a factor of 2, and with that the rotational–vibrational line spacings should halve approximately. This is observed for all noble gases, also for Ne (cf.Table 3).
Matrix frequency shifts
Combining our results for CO2 in Ar and Ne matrices with the results in other noble gases from previous studies,13,15,17,18,21 we observe an anticipated trend when going from Xe to Ne: The frequencies are successively blue-shifted toward the values obtained from gas-phase experiments (cf.Table 1). This holds, at least, for the ν3 and ν2 fundamental vibrations. For CO2 in noble gas matrices, the following widely-accepted concept holds: The number of electrons decreases from Xe to Ne, which leads to a systematic decrease in the non-bonding interactions of the host with the analyte. Thus, the host's influence on the vibrational structure of the analyte is successively diminished, i.e., the matrix shift is minimized. In the Ne matrix, CO2 behaves like single molecules in highly diluted gas-phase. Consequently, a very good agreement between Ne MI-IR spectra, gas-phase IR spectra and the VCI calculations based on the model of collinear CO2in vacuo was found.From Raman experiments of CO2, the overtone 2ν2 is expected to be in resonance with the ν1 fundamental.63 This has been observed in Kr MI-Raman experiments,13 where the frequencies are red-shifted by approximately 6 cm−1 compared to gas-phase. Another such resonance is present for the IR active ν1 + ν3 and 2ν2 + ν3 combination bands. In accordance with previous studies,17,18,21 we observe this resonance in Ar MI-IR. Here, each of the two resonant transitions exhibits a onefold band-splitting (cf.Fig. 3), which can be reasoned with two trapping sites. The centres of those singly split bands are each red-shifted by approximately 11 cm−1 compared to gas-phase. In Ne MI-IR, we do not observe an additional band-splitting of the resonance, which is in accordance with previous experiments.14 Here, the frequencies of the resonant transitions are only red-shifted by approximately 1 cm−1 compared to gas-phase. To the best of our knowledge, there are no Kr and Xe MI-IR experiments published considering the ν1 + ν3 and 2ν2 + ν3 combination bands for spectral interpretation. Yet, recent He nanodroplet experiments80 found those resonating states at 3612.42 cm−1 and 3714.44 cm−1, very close to our observation in Ne MI-IR (3612.4 cm−1 and 3714.7 cm−1). From these observations, one may deduce that the matrix frequency shifts of the Fermi resonance of ν1 + ν3 and 2ν2 + ν3 show a similar systematic trend as for the fundamental transitions. However, further experiments are needed to prove this.
Comparing the non-rotating Q(1) transition of CH4 in matrix-isolation with the fundamental vibrational transition in gas-phase, the absolute matrix shift is about 12 cm−1 for Xe and 6 cm−1 for Ar, while it is only about 3 cm−1 for Kr and 1 cm−1 for Ne. In other words, for CH4 and CD4 the matrix frequency shift is not systematically decreasing when going from Xe to Ne. This is in central contradiction to the observation made for CO2 and undermines the concept that the matrix frequency shifts systematically change with the electronic structure of the host system. The concept may be extended by incorporation of other factors that have an impact on the matrix frequency shifts in order to interpret the case of CH4 and CD4, e.g., by considering the crystal structure of the host system. However, such ideas cannot be transferred from one host–guest system to the other, because they are intrinsic to the specific size distribution and electronic interaction between analyte and noble gas. This will be discussed in the up-following paragraph.
Matrix effects in various host–guest combinations
At first glance, the MI-IR spectra of CO2 and CH4 (resp. CD4) are very simple compared to the vast number of bands in the gas-phase IR spectra. At a closer look, matrix frequency shifts and splitting patterns complicate the spectra and its interpretation is not straight-forward. While oligomerization can be identified by probing various dilutions of the analyte, further spectral features are rather unsystematic for both species in different matrix materials. The linear molecule CO2 is best described as a non-rotating monomer in all noble gas matrices. In Ar matrices, it has at least two different matrix trapping sites, whereas (potentially) only one in Ne. The spectra of the spherical rotator CH4 (resp. CD4) may best be explained by a hindered rotation in all matrices. Different matrix trapping sites can mostly be ruled out in both Ar and Ne. Although matrix frequency shifts systematically decrease from Xe to Ne for CO2, this systematic trend is not observed for CH4 (resp. CD4). In other words, the impact of the matrix on the vibrational structure depends on the molecular symmetry of the analyte, the properties of the host material and the intricate interplay between the two. Table 4 shows that this statement holds true also in a bigger context when we consider our previous Ne and Ar MI-IR studies on water10 and fluoroethane.131 The matrix effects depend on the specific host–guest combination.| H2O [C2v] a | CH4 [Td] | CO2 [D∞h] | CH3CH2Fb [Cs] | |
|---|---|---|---|---|
| a Taken from ref. 10. b Taken from ref. 131. c Rotation in Ne based on the new data presented in this study. d Mean absolute deviation w.r.t. gas-phase considering the non-rotating fundamentals of the most abundant isotopoloques. | ||||
| Rotation within the matrix | ||||
| Ar/Ne | Yes | Yes (hindered)c | No | No |
| Number of matrix trapping sites (at least) | ||||
| Ar | 1 | 1 | 2 | 2 |
| Ne | 1 | 1 | 1 | 2 |
| Matrix frequency shifts /cm −1 | ||||
| Ar | 14.7 | 6.7 | 6.4 | 3.8 |
| Ne | 2.6 | 0.4 | 0.9 | 1.1 |
These findings demonstrate the challenges for a theoretical model to accurately calculate MI-IR spectra. The fundamental question is, whether matrix effects can be rationalized by a host–guest structure localized around the molecule under study. If so, one may ask how this host–guest structure can be obtained and how it is most accurately described in a theoretical model.
Matrix frequency shifts can be computed as the difference between the vibrational spectrum of the host–guest structure and the vibrational spectrum of the molecule in vacuo. Thus, several distinct host–guest structures may have to be tested to identify the one that best matches to the experimental spectrum. Several approaches can be invoked. For example, the host can be modelled with an implicit force field or by placement of host atoms. With a VSCF/VCI approach based on multi-mode PES representations, an extension of the molecular framework by the addition of noble gas atoms was successfully demonstrated for iodine in argon.43
The computation of matrix splitting patterns is more tedious, because observation of band splitting in experimental spectra typically implies the occurrence of different trapping sites. Consequently, a variety of host–guest structures have to be evaluated to reproduce these experimental findings. This approach has recently been applied in a theoretical study of CO2 in argon within the harmonic approximation, showing promising results.44 The agreement with experimental spectra would further profit from incorporation of anharmonicity and mode-coupling in the calculations. At this level, most matrix effects should be predictable from theory with high accuracy.
5. Conclusion
The correct account of anharmonicity and mode-coupling in computations of molecular vibration, opens a promising route towards the identification of matrix effects in a MI-IR spectrum. In this study, we demonstrate that in vacuo VSCF/VCI calculations based on one-the-fly calculated multi-mode PES representations, i.e., a theoretical model derived for the description of gas-phase spectra, provide a suitable starting point for the interpretation of MI-IR spectra. Especially when Ne matrices are used, the discrepancy between theory and experiment is within 1–3 cm−1, considering the here presented results of methane and carbon dioxide. With the access to accurate anharmonic molecular spectra, it remains to address matrix effects. Those are observed in all kind of noble gas matrices. Incorporating also observations from our previous MI-IR and VCI studies on water and fluoroethane, we see that matrix effects cannot systematically be accounted for but intricately depend on the host–guest combinations. Historically, the conclusive interpretation of gas-phase IR spectra was ultimately linked to the development of a general and transferable theoretical description of molecular vibration. Similarly, we suppose that the unambiguous interpretation of matrix effects in MI-IR spectra requires general and transferable concepts to model the host–guest structure. Such a universally applicable model deems necessary to ultimately solve controversies, such as the occurrence of trapping sites or rotation within the matrix.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Prof. Dr Erich Knözinger. His experimental studies on the matrix-isolation technique in the 1990s are a central pillar on which our work today rests. We gratefully acknowledge support by the Austrian Science Fund FWF (project I1392 and project P30565) and the Austria Research Promotion Agency FFG (bridge project EARLYSNOW, 850689). M. P. would like to thank the FWF for a Lise Meitner Postdoctoral Fellowship (M-2005). Special thanks to C. Rameshan for providing the CO2 sample.Notes and references
- M. E. Jacox, Comparison of the ground state vibrational fundamentals of diatomic molecules in the gas phase and in inert solid matrices, J. Mol. Spectrosc., 1985, 113, 286–301 CrossRef CAS.
- M. E. Jacox, The vibrational energy levels of small transient molecules isolated in neon and argon matrices, Chem. Phys., 1994, 189, 149–170 CrossRef CAS.
- M. E. Jacox, The spectroscopy of molecular reaction intermediates trapped in the solid rare gases, Chem. Soc. Rev., 2002, 31, 108–115 RSC.
- E. Catalano and D. E. Milligan, Infrared spectra of H2O, D2O, and HDO in solid argon, krypton, and xenon, J. Chem. Phys., 1959, 30, 45–47 CrossRef CAS.
- R. L. Redington and D. E. Milligan, Infrared Spectroscopic Evidence for the Rotation of the Water Molecule in Solid Argon, J. Chem. Phys., 1962, 37, 2162 CrossRef CAS.
- R. L. Redington and D. E. Milligan, Molecular rotation and ortho–para nuclear spin conversion of water suspended in solid Ar, Kr, and Xe, J. Chem. Phys., 1963, 39, 1276–1284 CrossRef CAS.
- G. P. Ayers and A. D. E. Pullin, Reassignment of the vibrational spectra of matrix isolated H2O and HDO, Chem. Phys. Lett., 1974, 29, 609–615 CrossRef CAS.
- D. Forney, M. E. Jacox and W. E. Thompson, The Mid- and Near-Infrared Spectra of Water and Water Dimer Isolated in Solid Neon, J. Mol. Spectrosc., 1993, 157, 479–493 CrossRef CAS.
- J. Ceponkus, P. Uvdal and B. Nelander, The coupling between translation and rotation for monomeric water in noble gas matrices, J. Chem. Phys., 2013, 138, 244305 CrossRef CAS PubMed.
- D. F. Dinu, M. Podewitz, H. Grothe, K. R. Liedl and T. Loerting, Toward Elimination of Discrepancies between Theory and Experiment: Anharmonic rotational–vibrational Spectrum of Water in Solid Noble Gas Matrices, J. Phys. Chem. A, 2019, 123, 8234–8242 CrossRef CAS PubMed.
- G. C. Pimentel and S. W. Charles, Infrared spectral perturbations in matrix experiments, Pure Appl. Chem., 1963, 7, 111–124 CAS.
- L. Fredin, B. Nelander and G. Ribbegård, On the dimerization of carbon dioxide in nitrogen and argon matrices, J. Mol. Spectrosc., 1974, 53, 410–416 CrossRef CAS.
- A. Loewenschuss and A. Givan, Raman and Infrared Spectra of Matrix Isolated CO2 and CS2, Spectrosc. Lett., 1977, 10, 551–558 CrossRef CAS.
- L. Wan, L. Wu, A.-W. Liu and S.-M. Hu, Neon matrix isolation spectroscopy of CO2 isotopologues, J. Mol. Spectrosc., 2009, 257, 217–219 CrossRef CAS.
- R. Guasti, V. Schettino and N. Brigot, The structure of carbon dioxide dimers trapped in solid rare gas matrices, Chem. Phys., 1978, 34, 391–398 CrossRef CAS.
- M. J. Irvine and A. E. P. Pullin, Spectra of Carbon Dioxide. I Deuterium Matrices: Identification of Bands due to Carbon Dioxide Dimers, Aust. J. Chem., 1982, 35, 1961 CrossRef CAS.
- M. J. Irvine, J. G. Mathieson and A. D. E. Pullin, The infrared matrix isolation spectra of carbon dioxide. II. Argon matrices: the CO2 monomer bands, Aust. J. Chem., 1982, 35, 1971–1977 CrossRef CAS.
- L. M. Nxumalo and T. A. Ford, IR spectra of the dimers of carbon dioxide and nitrous oxide in cryogenic matrices, J. Mol. Struct., 1994, 327, 145–159 CrossRef CAS.
- E. Knoezinger and P. Beichert, Matrix isolation studies of CO2 clusters emerging from adiabatic expansion, J. Phys. Chem., 1995, 99, 4906–4911 CrossRef CAS.
- S. Tam and M. E. Fajardo, Observation of the high-resolution infrared absorption spectrum of CO2 molecules isolated in solid parahydrogen, Low Temp. Phys., 2000, 26, 653–660 CrossRef CAS.
- A. Schriver, L. Schriver-Mazzuoli and A. A. Vigasin, Matrix isolation spectra of the carbon dioxide monomer and dimer revisited, Vib. Spectrosc., 2000, 23, 83–94 CrossRef CAS.
- J. A. Gómez Castaño, A. Fantoni and R. M. Romano, Matrix-isolation FTIR study of carbon dioxide: Reinvestigation of the CO2 dimer and CO2⋯N2 complex, J. Mol. Struct., 2008, 881, 68–75 CrossRef.
- A. Cabana, G. B. Savitsky and D. F. Hornig, Vibration–Rotation Spectra of CH4 and CD4 Impurities in Xenon, Krypton, and Argon Crystals, J. Chem. Phys., 1963, 39, 2942–2950 CrossRef CAS.
- A. Cabana, A. Anderson and R. Savoie, Raman spectrum of CH4 trapped in solid krypton. Evidence for molecular rotation, J. Chem. Phys., 1965, 42, 1122–1123 CrossRef.
- T. Momose and T. Shida, Matrix-Isolation Spectroscopy Using Solid Parahydrogen as the Matrix: Application to High-Resolution Spectroscopy, Photochemistry, and Cryochemistry, Bull. Chem. Soc. Jpn., 1998, 71, 1–15 CrossRef CAS.
- S. Tam, M. E. Fajardo, H. Katsuki, H. Hoshina, T. Wakabayashi and T. Momose, High resolution infrared absorption spectra of methane molecules isolated in solid parahydrogen matrices, J. Chem. Phys., 1999, 111, 4191–4198 CrossRef CAS.
- H. Hoshina, T. Wakabayashi, T. Momose and T. Shida, Infrared spectroscopic study of rovibrational states of perdeuterated methane (CD4) trapped in parahydrogen crystal, J. Chem. Phys., 1999, 110, 5728–5733 CrossRef CAS.
- J. F. Ogilvie, S. L. Chou, M. Y. Lin and B. M. Cheng, Mid-infrared spectra of methane dispersed in solid neon and argon, Vib. Spectrosc., 2011, 57, 196–206 CrossRef CAS.
- F. H. Frayer and G. E. Ewing, Spectroscopic Observation of Nuclear Spin Conversion in Methane, J. Chem. Phys., 1967, 46, 1994–1995 CrossRef CAS.
- F. H. Frayer and G. E. Ewing, Nuclear-Spin Conversion and Vibration-Rotation Spectra of Methane in Solid Argon, J. Chem. Phys., 1968, 48, 781–792 CrossRef CAS.
- A. Chamberland, R. Belzile and A. Cabana, Infrared spectra and structure of methane – noble gas mixed crystals: the influence of temperature and methane concentration on the v3 vibration band of methane, Can. J. Chem., 1970, 48, 1129–1139 CrossRef CAS.
- T. Nanba, M. Sagara and M. Ikezawa, Optical Excitation of the Rotational Transition of Methane in Condensed Rare Gas Matrices, J. Phys. Soc. Jpn., 1980, 48, 228–234 CrossRef CAS.
- M. Sagara and M. Ikezawa, Raman Scattering Spectra of Methane in Solid Rare Gases, J. Phys. Soc. Jpn., 1983, 52, 327–332 CrossRef CAS.
- L. H. Jones, S. A. Ekberg and B. I. Swanson, Hindered rotation and site structure of methane trapped in rare gas solids, J. Chem. Phys., 1986, 85, 3203–3210 CrossRef CAS.
- L. H. Jones and S. A. Ekberg, Hindered rotation and site structure of CD4 trapped in rare gas solids, J. Chem. Phys., 1987, 87, 4368 CrossRef CAS.
- T. Momose, M. Miki, T. Wakabayashi, T. Shida, M.-C. Chan, S. Lee. and T. Oka, Infrared spectroscopic study of rovibrational states of perdeuterated methane trapped in parahydrogen crystal, J. Chem. Phys., 1997, 107, 5728–5733 Search PubMed.
- P. R. Dahoo, A. Lakhlifi, H. Chabbi and J. M. Coanga, Matrix effect on triatomic CO2 molecule: Comparison between krypton and xenon, J. Mol. Struct., 2006, 786, 157–167 CrossRef CAS.
- H. F. King and D. F. Hornig, Rotational states of a tetrahedron in a cubic crystal field, J. Chem. Phys., 1966, 44, 4520–4531 CrossRef CAS.
- K. Nishiyama and T. Yamamoto, Theory of phase transitions in solid methanes. IX. The infrared absorption of methane in rare gas matrices, J. Chem. Phys., 1973, 58, 1001 CrossRef CAS.
- Y. Ozaki, Calculation of Rotational Energy Levels of CH4 in Solid Ar, J. Phys. Soc. Jpn., 1990, 59, 1277–1284 CrossRef CAS.
- T. Sugimoto, I. Arakawa and K. Yamakawa, Nuclear spin relaxation of methane in solid xenon, Eur. Phys. J. D, 2018, 72, 2–7 CrossRef.
- R. Fraenkel and Y. Haas, Trapping of guests in a rare gas matrix: A molecular dynamics simulation, J. Chem. Phys., 1994, 100, 4324–4328 CrossRef CAS.
- Z. Bihary, R. B. Gerber and V. A. Apkarian, Vibrational self-consistent field approach to anharmonic spectroscopy of molecules in solids: Application to iodine in argon matrix, J. Chem. Phys., 2001, 115, 2695–2701 CrossRef CAS.
- F. Bader, T. Lindic and B. Paulus, A Validation of Cluster Modeling in the Description of Matrix Isolation Spectroscopy, J. Comput. Chem., 2020, 41, 751–758 CrossRef CAS PubMed.
- O. Christiansen, Vibrational structure theory: new vibrational wave function methods for calculation of anharmonic vibrational energies and vibrational contributions to molecular properties, Phys. Chem. Chem. Phys., 2007, 9, 2942 RSC.
- O. Christiansen, Selected new developments in vibrational structure theory: potential construction and vibrational wave function calculations, Phys. Chem. Chem. Phys., 2012, 14, 6672 RSC.
- D. Oschetzki, M. Neff, P. Meier, F. Pfeiffer and G. Rauhut, Selected Aspects Concerning the Efficient Calculation of Vibrational Spectra beyond the Harmonic Approximation, Croat. Chem. Acta, 2012, 85, 379–390 CrossRef CAS.
- A. G. Császár, C. Fábri, T. Szidarovszky, E. Mátyus, T. Furtenbacher and G. Czakó, The fourth age of quantum chemistry: Molecules in motion, Phys. Chem. Chem. Phys., 2012, 14, 1085–1106 RSC.
- J. Tennyson, Perspective: Accurate ro-vibrational calculations on small molecules, J. Chem. Phys., 2016, 145(12), 120901 CrossRef PubMed.
- T. Carrington, Perspective: Computing (ro-)vibrational spectra of molecules with more than four atoms, J. Chem. Phys., 2017, 146(12), 120902 CrossRef PubMed.
- C. Qu and J. M. Bowman, Quantum approaches to vibrational dynamics and spectroscopy: is ease of interpretation sacrificed as rigor increases?, Phys. Chem. Chem. Phys., 2018, 3397–3413 Search PubMed.
- G. D. Carney, L. L. Sprandel and C. W. Kern, Adv. Chem. Phys., 1978, XXXVII, 305–379 Search PubMed.
- J. M. Bowman, The Self-Consistent-Field Approach to Polyatomic Vibrations, Acc. Chem. Res., 1986, 19, 202–208 CrossRef CAS.
- M. A. Ratner and R. B. Gerber, Excited vibrational states of polyatomic molecules: The semiclassical self-consistent field approach, J. Phys. Chem., 1986, 90, 20–30 CrossRef CAS.
- G. Rauhut, Efficient calculation of potential energy surfaces for the generation of vibrational wave functions, J. Chem. Phys., 2004, 121, 9313–9322 CrossRef CAS PubMed.
- G. Rauhut, Configuration selection as a route towards efficient vibrational configuration interaction calculations, J. Chem. Phys., 2007, 127, 184109 CrossRef PubMed.
- G. Rauhut, G. Knizia and H.-J. Werner, Accurate calculation of vibrational frequencies using explicitly correlated coupled-cluster theory, J. Chem. Phys., 2009, 130, 054105 CrossRef PubMed.
- J. W. Ellis, Emmision and Absorption Bands of Carbon Dioxide in the Infrared, Phys. Rev., 1925, 26, 469 CrossRef CAS.
- C. Schaefer and B. Philipps, Das Absorptionsspektrum der Kohlensäure und die Gestalt der CO2-Molekel, Z. Phys., 1926, 36, 641–656 CrossRef CAS.
- F. I. G. Rawlins, The Form of the Molecule of Carbon Dioxide, Trans. Faraday Soc., 1929, 25, 925 RSC.
- R. G. Dickinson, R. T. Dillon and F. Rasetti, Raman Spectra of Polyatomic Gases, Phys. Rev., 1929, 34, 582 CrossRef CAS.
- D. M. Dennison, The infrared spectra of polyatomic molecules Part I, Rev. Mod. Phys., 1931, 3, 280–345 CrossRef CAS.
- E. Fermi, Über den Ramaneffekt des CO2, Z. Phys., 1932, 79, 492–494 CrossRef.
- D. M. Dennison, The vibrational levels of linear symmetrical triatomic molecules, Phys. Rev., 1932, 41, 304–312 CrossRef CAS.
- A. Adel and D. M. Dennison, The Infrared Spectrum of Carbon Dioxide. Part I, Phys. Rev., 1933, 43, 716–723 CrossRef CAS.
- A. Adel and D. M. Dennison, The Infrared Spectrum of Carbon Dioxide. Part II, Phys. Rev., 1933, 44, 99–104 CrossRef CAS.
- P. E. Martin and E. F. Barker, The infrared absorption spectrum of carbon dioxide, Phys. Rev., 1932, 41, 291–303 CrossRef CAS.
- G. Herzberg, Molecular spectra and molecular structure. Vol. 2: Infrared and Raman spectra of polyatomic molecules, D. Van Nostrand Company, Inc., New York, 1945 Search PubMed.
- H. H. Nielsen, The vibration-rotation energies of molecules, Rev. Mod. Phys., 1951, 23, 90–136 CrossRef CAS.
- E. Bright Wilson, J. Decius and P. Cross, Molecular Vibrations. The Theory of Infrared and Raman Vibrational Spectra, Dover Publications Inc., 1980 Search PubMed.
- I. Suzuki, General anharmonic force constants of carbon dioxide, J. Mol. Spectrosc., 1968, 25, 479–500 CrossRef.
- A. Chedin, The carbon dioxide molecule, J. Mol. Spectrosc., 1979, 76, 430–491 CrossRef CAS.
- M. Lacy, The anharmonic force field of carbon dioxide, Mol. Phys., 1982, 45, 253–258 CrossRef CAS.
- A. Chedin and J. L. Teffo, The carbon dioxide molecule: A new derivation of the potential, spectroscopic, and molecular constants, J. Mol. Spectrosc., 1984, 107, 333–342 CrossRef CAS.
- A. G. Csaszar, Anharmonic force field of carbon dioxide, J. Phys. Chem., 1992, 96, 7898–7904 CrossRef CAS.
- X. Huang, D. W. Schwenke, R. S. Freedman and T. J. Lee, Ames-2016 line lists for 13 isotopologues of CO2: Updates, consistency, and remaining issues, J. Quant. Spectrosc. Radiat. Transfer, 2017, 203, 224–241 CrossRef CAS.
- E. Zak, J. Tennyson, O. L. Polyansky, L. Lodi, N. F. Zobov, S. A. Tashkun and V. I. Perevalov, A room temperature CO2 line list with ab initio computed intensities, J. Quant. Spectrosc. Radiat. Transfer, 2016, 177, 31–42 CrossRef CAS.
- L. Mannik, J. C. Stryland and L. Welsh, An Infrared Spectrum of CO2 Dimers in the ‘Locked’ Configuration, Can. J. Phys., 1971, 49, 3056–3057 CrossRef CAS.
- N. Brigot, S. Odiot and S. H. Walmsley, The structure of the carbon dioxide dimer, Chem. Phys. Lett., 1977, 49(1), 157–159 CrossRef CAS.
- K. Nauta and R. E. Miller, Rotational and vibrational dynamics of CO2 and N2O in helium nanodroplets, J. Chem. Phys., 2001, 115, 10254–10260 CrossRef CAS.
- J. H. Du, L. Wan, L. Wu, G. Xu, W. P. Deng, A. W. Liu, Y. Chen and S. M. Hu, CO2 in solid para-hydrogen: Spectral splitting and the CO2-(o-H2)n clusters, J. Phys. Chem. A, 2011, 115, 1040–1046 CrossRef CAS PubMed.
- G. M. Richardson, L. Pasteur, J. H. van’t Hoff, J.-A. Le Bel and J. Wislicenus, Scientific Memoirs – The Foundations of Stereo Chemistry, American Book Company, 1901 Search PubMed.
- J. P. Cooley, The Infra-red absorbtion bands of methane, Astrophys. J., 1925, 62, 73 CrossRef CAS.
- D. M. Dennison, The molecular structure and infra-red spectrum of methane, Astrophys. J., 1925, 62, 84 CrossRef CAS.
- V. Guillemin, Zur Molekülstruktur des Methan, Ann. Phys., 1926, 4, 12 Search PubMed.
- V. Henri, The structure of the methane molecule, Chem. Rev., 1927, 4, 189–201 CrossRef CAS.
- J. Morse, The Molecular Structures of Methane, Proc. Natl. Acad. Sci. U. S. A., 1928, 14, 166–171 CrossRef CAS PubMed.
- J. W. Ellis, New Infra-red Absorption Bands of Methane, Proc. Natl. Acad. Sci. U. S. A., 1927, 13, 202–207 CrossRef CAS PubMed.
- D. M. Dennison and S. B. Ingram, A new band in the absorption spectrum of methane gas, Phys. Rev., 1930, 36, 1451–1459 CrossRef CAS.
- J. Moorhead, The Near Infrared Absorption Spectrum of Methane, Phys. Rev., 1931, 39, 83 CrossRef.
- W. V. Norris and H. J. Unger, Infrared absorption bands of methane, Phys. Rev., 1933, 43, 467–472 CrossRef CAS.
- A. H. Nielsen and H. H. Nielsen, The infrared absorption bands of methane, Phys. Rev., 1935, 48, 864–867 CrossRef CAS.
- A. Nielsen and H. Nielsen, The Infra-Red Absorbtion Spectrum of CD4, Phys. Rev., 1938, 54, 118 CrossRef CAS.
- D. M. Dennison, The infrared spectra of polyatomic molecules Part II, Rev. Mod. Phys., 1940, 12, 280–345 CrossRef.
- W. H. Shaffer, H. H. Nielsen and L. H. Thomas, The Rotation-Vibration Energies of Tetrahedrally Symmetric Pentatomic Molecules. II, Phys. Rev., 1939, 56, 1051–1059 CrossRef CAS.
- E. B. Wilson, The Statistical Weights of the Rotational Levels of Polyatomic Molecules, Including Methane, Ammonia, Benzene, Cyclopropane and Ethylene, J. Chem. Phys., 1935, 3, 276–285 CrossRef CAS.
- H. Kaylor and A. Nielsen, Infrared spectrum and molecular constants of CD4, J. Chem. Phys., 1955, 23, 2139 CrossRef CAS.
- I. M. Mills, The calculations of accurate normal co-ordinates – I General theory and application to methane, Spectrochim. Acta, 1959, 16, 35–48 CrossRef.
- L. H. Jones and R. S. McDowell, Force constants of methane. Infrared spectra and thermodynamic functions of isotopic methanes, J. Mol. Spectrosc., 1959, 3, 632–653 CrossRef CAS.
- E. Venuti, L. Halonen and R. G. Della Valle, High dimensional anharmonic potential energy surfaces: The case of methane, J. Chem. Phys., 1999, 110, 7339–7347 CrossRef CAS.
- P. Pulay, W. Meyer and J. E. Boggs, Cubic force constants and equilibrium geometry of methane from Hartree-Fock and correlated wavefunctions, J. Chem. Phys., 1978, 68, 5077–5085 CrossRef CAS.
- T. J. Lee, J. M. L. Martin and P. R. Taylor, An accurate ab initio quartic force field and vibrational frequencies for CH4 and isotopomers, J. Chem. Phys., 1995, 102, 254–261 CrossRef CAS.
- D. W. Schwenke and H. Partridge, Vibrational energy levels for CH4 from an ab initio potential, Spectrochim. Acta, Part A, 2001, 57, 887–895 CrossRef CAS.
- R. Marquardt and M. Quack, Global analytical potential hypersurfaces for large amplitude nuclear motion and reactions in methane. I. Formulation of the potentials and adjustment of parameters to ab initio data and experimental constraints, J. Chem. Phys., 1998, 109, 10628–10643 CrossRef CAS.
- L. R. Brown, K. Sung, D. C. Benner, V. M. Devi, V. Boudon, T. Gabard, C. Wenger, A. Campargue, O. Leshchishina, S. Kassi, D. Mondelain, L. Wang, L. Daumont, L. Régalia, M. Rey, X. Thomas, V. G. Tyuterev, O. M. Lyulin, A. V. Nikitin, H. M. Niederer, S. Albert, S. Bauerecker, M. Quack, J. J. O’Brien, I. E. Gordon, L. S. Rothman, H. Sasada, A. Coustenis, M. A. H. Smith, T. Carrington, X. G. Wang, A. W. Mantz and P. T. Spickler, Methane line parameters in the HITRAN2012 database, J. Quant. Spectrosc. Radiat. Transfer, 2013, 130, 201–219 CrossRef CAS.
- S. N. Yurchenko and J. Tennyson, ExoMol line lists-IV. The rotation-vibration spectrum of methane up to 1500 K, Mon. Not. R. Astron. Soc., 2014, 440, 1649–1661 CrossRef CAS.
- A. V. Nikitin, M. Rey and V. G. Tyuterev, Rotational and vibrational energy levels of methane calculated from a new potential energy surface, Chem. Phys. Lett., 2010, 501, 179–186 CrossRef.
- X.-G. Wang and T. Carrington, Computing rovibrational levels of methane with curvilinear internal vibrational coordinates and an Eckart frame, J. Chem. Phys., 2013, 138, 104106 CrossRef PubMed.
- A. V. Nikitin, M. Rey and V. G. Tyuterev, First fully ab initio potential energy surface of methane with a spectroscopic accuracy, J. Chem. Phys., 2016, 145, 114309 CrossRef.
- B. Nelander, An infrared spectroscopic study of the rotation of methane in solid nitrogen, J. Chem. Phys., 1985, 82, 5340 CrossRef CAS.
- M. G. Govender and T. A. Ford, The infrared spectrum of matrix-isolated methane—rotation or dimerization?, J. Mol. Struct., 2000, 550–551, 445–454 CrossRef CAS.
- K. Nauta and R. E. Miller, Rotational and vibrational dynamics of methane in helium nanodroplets, Chem. Phys. Lett., 2001, 350, 225–232 CrossRef CAS.
- O. Gálvez, A. Zoermer, A. Loewenschuss and H. Grothe, A combined matrix isolation and ab initio study of bromine oxides, J. Phys. Chem. A, 2006, 110, 6472–6481 CrossRef PubMed.
- J. Bernard, M. Seidl, I. Kohl, K. R. Liedl, E. Mayer, Ó. Gálvez, H. Grothe and T. Loerting, Spectroscopic observation of matrix-isolated carbonic acid trapped from the gas phase, Angew. Chem., Int. Ed., 2011, 50, 1939–1943 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: a general purpose quantum chemistry program package, WIREs Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, III, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro quantum chemistry package, J. Chem. Phys., 2020, 152(14), 144107 CrossRef PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B–Ne, and Al–Ar, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- K. E. Yousaf and K. A. Peterson, Optimized auxiliary basis sets for explicitly correlated methods, J. Chem. Phys., 2008, 129, 184108 CrossRef PubMed.
- B. Ziegler and G. Rauhut, Efficient generation of sum-of-products representations of high-dimensional potential energy surfaces based on multimode expansions, J. Chem. Phys., 2016, 144(11), 114114 CrossRef PubMed.
- T. Petrenko and G. Rauhut, A new efficient method for the calculation of interior eigenpairs and its application to vibrational structure problems, J. Chem. Phys., 2017, 146, 124101 CrossRef PubMed.
- F. Weigend, A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- F. Weigend, A. Köhn and C. Hättig, Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations, J. Chem. Phys., 2002, 116, 3175–3183 CrossRef CAS.
- J. G. Hill, S. Mazumder and K. A. Peterson, Correlation consistent basis sets for molecular core-valence effects with explicitly correlated wave functions: The atoms B–Ne and Al–Ar, J. Chem. Phys., 2010, 132, 054108 CrossRef PubMed.
- C. Hättig, Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core–valence and quintuple-ζ basis sets for H to Ar and QZVPP basis sets for Li to Kr, Phys. Chem. Chem. Phys., 2005, 7, 59–66 RSC.
- B. Stoicheff, High Resolution Raman Spectroscopy of Gases: XI. Spectra of CS2 and CO2, Can. J. Phys., 1958, 36, 218–230 CrossRef CAS.
- L. Henry, N. Husson, R. Andia and A. Valentin, Infrared absorption spectrum of methane from 2884 to 3141 cm−1, J. Mol. Spectrosc., 1970, 36, 511–520 CrossRef CAS.
- T. Shimanouchi, NSRDS-NBS 39. Tables of molecular vibrational frequencies. Consolidated volume I, National Bureau of Standards, 1972 Search PubMed.
- A. Chedin, N. Husson, N. A. Scott and D. Gautier, ν 4 Band of methane (12CH4 and 13CH4). Line parameters and evaluation of Jovian atmospheric transmission at 7.7 μm, J. Mol. Spectrosc., 1978, 71, 343–368 CrossRef CAS.
- D. F. Dinu, B. Ziegler, M. Podewitz, K. R. Liedl, T. Loerting, H. Grothe and G. Rauhut, The interplay of VSCF/VCI calculations and matrix-isolation IR spectroscopy – Mid infrared spectrum of CH3CH2F and CD3CD2F, J. Mol. Spectrosc., 2020, 367, 111224 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Erich Knözinger. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02121k |
| This journal is © the Owner Societies 2020 |