
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular environment and reactivity in gels and colloidal solutions under identical conditions†
Philip
Groves
a,
Jin
Huang
b,
Andreas
Heise
 c,
Jennifer
Marsh
d and
Victor
Chechik
*a
c,
Jennifer
Marsh
d and
Victor
Chechik
*a
aDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: victor.chechik@york.ac.uk
bSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland
cDepartment of Chemistry, Royal College of Surgeons in Ireland, 123 St. Stephens Green, Dublin 2, Ireland
dThe Procter & Gamble Company, Mason Business Center, 8700 Mason-Montgomery Road, Mason, 45040, USA
First published on 7th May 2020
Abstract
A PEG–Tyr block copolymer forms a kinetically stable colloidal solution in water at room temperature which undergoes an irreversible conversion to a gel phase upon heating. A micellar solution and a gel can therefore be studied under identical experimental conditions. This made it possible to compare physical properties and chemical reactivity of micelles and gels in identical chemical environments and under identical conditions. EPR spectra of the spin-labelled copolymer showed that tyrosine mobility in gels was slightly reduced compared to micelles. Chemical reactivity was studied using photochemical degradation of tyrosine and tyrosine dimerization, in the absence and in the presence of an Fe(III) salt. The reactivity trends were explained by reduced tyrosine mobility in the gel environment. The largest reactivity difference in gels and micelles was observed for bimolecular dityrosine formation which was also attributed to the reduction in molecular mobility.
Introduction
Supramolecular assembly has a profound effect on molecular environment. Solvation and dynamics are key factors determining many chemical and physical properties of supramolecular structures such as host–guest complexes, micelles, or gels.1 Chemical properties of functional groups can also be significantly affected by intermolecular interactions in supramolecular assemblies. The most remarkable examples of unusual chemical reactivity are in host–guest complexes,2 but the reactivity can also be altered in less well defined systems such as micelles or gels.3 Obtaining quantitative understanding of the factors determining reactivity in such supramolecular assemblies is challenging, not least because reference points do not exist under identical conditions.Importantly, supramolecular assembly of large (e.g., polymeric) building blocks is often almost irreversible at room temperature.4 These assemblies may become kinetically trapped and do not spontaneously convert into a more thermodynamically stable form. Such systems present a unique opportunity to examine the properties of chemically-identical supramolecular systems which are kinetically trapped in different morphologies.
PEG–Tyr block copolymer (Fig. 1) is an example of such a supramolecular system.5 The hydrophilicity of the PEG block and hydrophobicity of the Tyr block make the copolymer self-assemble into kinetically-trapped, stable colloidal structures upon dissolution in water. Upon heating, the solutions spontaneously convert into more thermodynamically-stable gels. Hence, the colloidal solutions and the gels of this copolymer can be examined under identical experimental conditions. Here, we report the results of our study on the comparative reactivity and molecular dynamics (assessed by EPR spectroscopy) of colloidal solutions and gels of PEG–Tyr block copolymers.
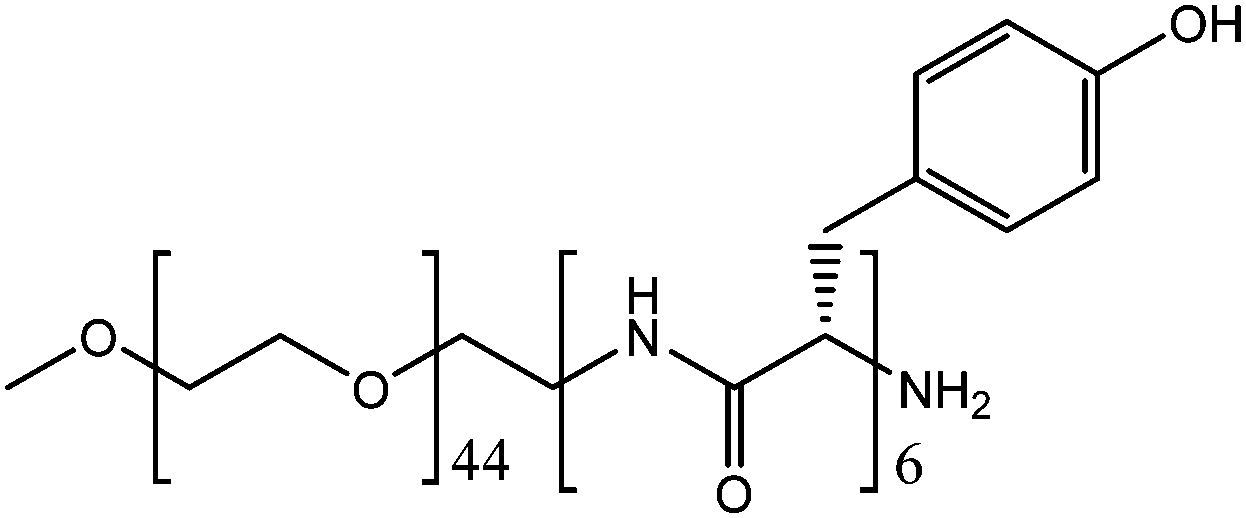 | ||
| Fig. 1 PEG–L-Tyr block copolymer system PEG2000–Tyr6. | ||
Results and discussion
Characterisation of PEG–Tyr self-assembled aggregates
Aqueous solutions of the PEG2000–Tyr6 copolymer was analysed by dynamic light scattering (Fig. 2). The polymer was found to form aggregates at room temperature with ca. 50–80 nm hydrodynamic diameter. As the volume of the long PEG chain relative to the overall surfactant volume is large, the colloidal structures are likely to be spherical micelles as suggested by TEM studies of PEG–Tyr copolymers.6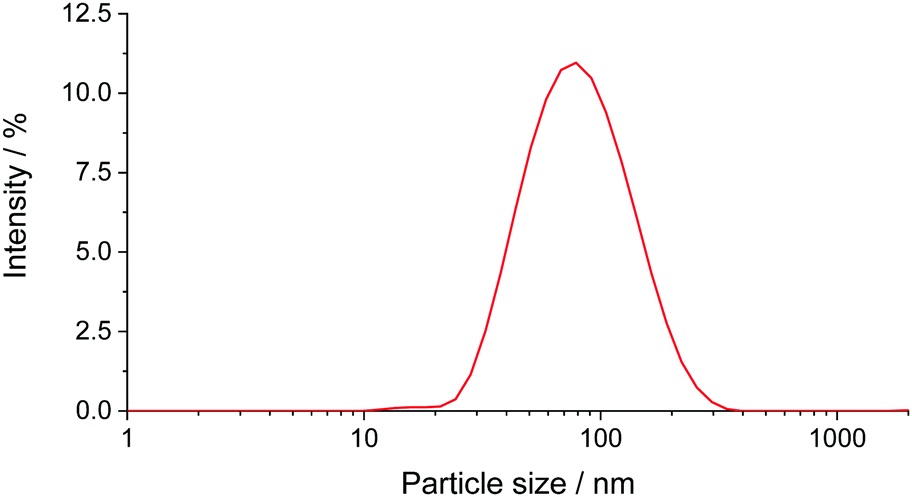 | ||
| Fig. 2 DLS size distribution plot (by intensity) for PEG2000–Tyr6 polymer sample in water. The plot is an average of 3 measurements. | ||
The critical micelle concentrations (cmc) of PEG2000–Tyr6 was estimated from DLS measurements by plotting particle count rate vs. polymer concentration as 0.6 × 10−5 mol dm−3, which is equivalent to 0.017 mg mL−1 (ESI,† Fig. S1). Although this measurement only provides a crude estimate of cmc, it is ca. 3 orders of magnitude lower than the concentrations used in all experiments reported here; hence it was safe to assume that polymer chains in our experiments are predominantly found in the micelles. Colloidal solutions of polymers were indefinitely stable at room temperature but formed gels upon heating to 60 °C. The gels remained stable after cooling to room temperature.
Gel structures formed upon heating of PEG2000–Tyr6 were previously characterised by Heise et al.5 The cryo-TEM images showed a continuous, interconnected network of fibres with Tyr block adopting a typical β-sheet conformation as evidenced by FT-IR and CD spectroscopy.
Spin labelling of PEG–Tyr copolymers
In order to characterise the molecular dynamics of supramolecular assemblies of PEG–Tyr copolymers, they were spin-labelled with TEMPO units following a modified Mannich-type reaction protocol reported by Belle et al. (Scheme 1).7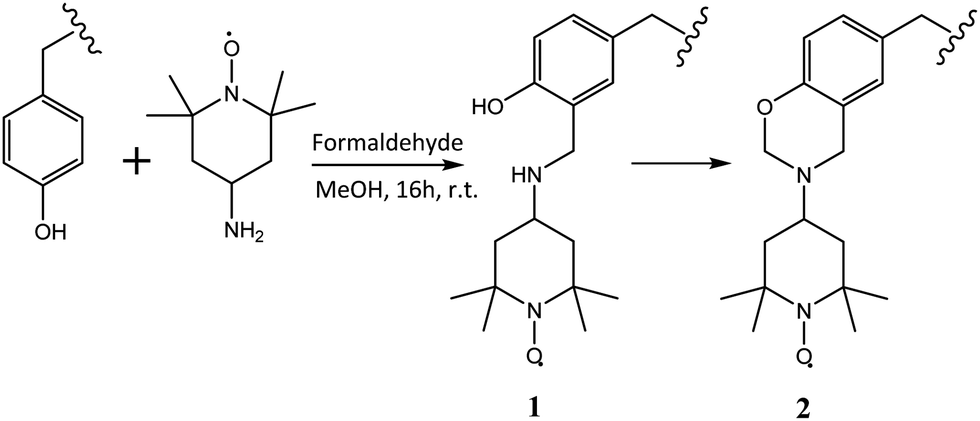 | ||
| Scheme 1 Spin labelling of PEG–Tyr block copolymer tyrosine side chains with 4-amino-TEMPO. | ||
Spin labelling of tyrosine residues within the micelle structure was not successful when the reaction was attempted in aqueous phosphate buffer solution. It was postulated that the micelle structures hinder access to the aromatic ring in tyrosine. The reaction was therefore repeated in methanol where copolymer aggregation was at least partially suppressed (ESI,† Fig. S7). Aqueous solutions of spin-labelled polymer were purified from excess spin label by dialysis against water, as the colloidal assemblies were retained by the membrane. Purification by dialysis was monitored by EPR. No loss in polymer was observed during dialysis as confirmed by tyrosine fluorescence at 310 nm.
The yield of the labelling reaction was estimated from the EPR signal intensity of the colloidal solution. The results indicate that 0.3% of available tyrosine residues were successfully labelled. Hence, a 5 mg mL−1 solution of the polymer has the average nitroxide concentration of ca. 25 μM. This low yield is advantageous for an EPR study as interactions between adjacent labels can be ignored, and the concentration of labelled residues is still sufficiently high to give strong EPR signals.
Characterisation of tyrosine microenvironment in micelles and gels
Spin labelled PEG2000–Tyr6 micelle and gel samples were analysed by X-band EPR spectroscopy. Spectra (Fig. 3) show overlapped contributions from several nitroxide environments. Rapidly tumbling radicals give rise to a characteristic three-line nitroxide spectrum, however both micelles and gels of the spin labelled polymer also exhibit nitroxides in significantly restricted environments, giving rise to broad features in the spectrum.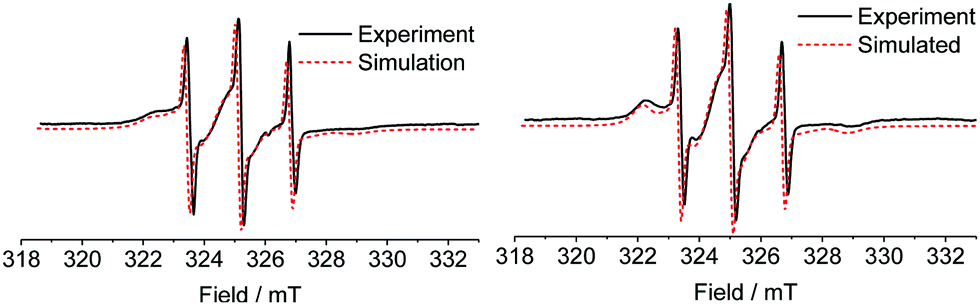 | ||
| Fig. 3 X-band EPR spectra of spin labelled PEG2000–Tyr6 micelles (left) and gel (right). Experimental data is shown in black solid line, fitted simulations in red dashed line. The simulations are offset for clarity. | ||
We postulate that the more restricted nitroxide environments observed in the spectra correspond to labelled tyrosine buried within a hydrophobic micelle core, or within a rigid gel fibre. There are notable qualitative differences between micelle and gel spectra, with the latter exhibiting clearer defined immobilised component peaks in low (≈322.5 mT) and high (≈329 mT) field regions. This indicates a slightly different tumbling environment in gel fibres compared to micelles.
The faster tumbling component likely represents spin labels at the surface of micelles/gel fibres; due to low cmc (vide infra), the concentration of dissolved unaggregated polymer is low and cannot give rise to this signal. The more flexible, acyclic Mannich adduct 1 (Scheme 1) which has been reported to incompletely convert to 2 in some cases8 could also contribute to this component.
The experimental X-band EPR data were simulated using EasySpin9 to determine rotational diffusion parameters for the spin labelled micelles and gels. As the polarity of the spin label in the micelles/gels is unknown (and different from water), the principal values of A and g tensors of the spin-labelled systems could not be taken from the literature. They were instead determined from the simulations of EPR spectra of frozen solution at Q-band (Table 1 and ESI,† Fig. S2a).
| x | y | z | |
|---|---|---|---|
| g | 2.00917 | 2.00579 | 2.00214 |
| A/MHz | 21.7 | 17.5 | 102.9 |
Attempts to simulate X-band EPR spectra of micelles and gels with a two-site model (with two different diffusion parameters, either isotropic or anisotropic) failed to give a satisfactory fit. This is not unexpected as there is clearly a complex distribution of labelling sites in the micelles and gels. A satisfactory fit was obtained with a simple three-site model which considers one fast tumbling component and two slow tumbling components. The tumbling of all components was assumed to be axially-symmetric. The results of simulation are shown in Fig. 3. The accuracy of our approach was tested by multifrequency analysis: a room-temperature Q band spectrum of a micellar sample was compared with a simulated spectrum with the same parameters, giving a reasonable fit (ESI,† Fig. S3).
Although the three-component simulation offers a reasonable fit of experimental data, it is an approximation of the distribution of different environments observed. The fitting algorithm compensates for this by broadening spectral linewidth. Therefore, accurate analysis of line shape is not possible. However, effective rotational diffusion rates and the relative contributions of different components could be qualitatively compared, as summarised in Table 2.
| “Fast” | “Slow A” | “Slow B” | ||||
|---|---|---|---|---|---|---|
| D av/s−1 | % | D av/s−1 | % | D av/s−1 | % | |
| Micelle | 3.9 × 109 | 7 | 1.9 × 107 | 48 | 1.5 × 108 | 45 |
| Gel | 2.5 × 109 | 8 | 2.0 × 107 | 52 | 1.1 × 108 | 40 |
The tumbling rates in Table 2 are the averages of the perpendicular and parallel axial terms. Full data and linewidth for simulations is given in ESI,† Table S1a and c. Overall, tumbling is more restricted in gel fibres, as both the “fast” and “slow B” components possess lower tumbling rates D (Table 2) than those of the micelles. The relative contributions of the two “slow” components in the gel system highlights the slightly narrower distribution of immobilised nitroxide species compared to micelles, indicative of a more homogenous environment in gels. Overall, PEG–Tyr gels provide a more consistent tyrosine environment, which is slightly more restrictive than the micellar system.
As EPR studies of site-directed spin-labelled proteins reported in literature usually use different nitroxide spin labels with different flexibility,10 the scope for direct comparison of Tyr environment in model systems and protein structures is limited. However, labelled tyrosine near a flexible C-terminus in a small chloroplastic protein CP12 gives X-band EPR spectra similar to those in Fig. 38 and faster than the “slow” component in gels/micelles. This suggests that the environment of tyrosine within micellar and gel model systems is comparable to that in protein structures.
Using spin labelled system to monitor transformation between micelle and gel
The unique properties of the PEG–Tyr polymer system, coupled with the observable changes in spin label environment upon gelation, provide an opportunity to monitor the process of gel formation in situ. We followed gelation on three different scales: on a bulk scale using tube inversion test, on a supramolecular scale with DLS, and on a molecular scale with EPR spectroscopy. When a 10 mg mL−1 colloidal solution of PEG2000–Tyr6 was rapidly heated to 60 °C and then kept at this temperature, a stable gel was formed after 8–10 minutes (tube inversion test). DLS measurements of samples rapidly heated and then kept at 60 °C, on the other hand, indicate a rapid transformation of micelles into larger aggregate structures after only 30 seconds, with longer incubation at 60 °C leading to larger average particle size as gel fibres form (see ESI,† Fig. S4a and b). This suggests the rapid formation of loosely aggregated structures such as β-sheets (predicted by circular dichroism studies), significantly before bulk gelation occurs.5 DLS data become unreliable in the presence of very large particles, therefore only the initial phase of micelle breakdown could be accurately monitored.Molecular changes during micelle–gel transformation of spin-labelled polymer solution were monitored by EPR. Following gelation in situ at 60 °C proved challenging as the EPR spectra at this temperature show rapid tumbling and gelation is not accompanied by an appreciable change in the spectral lineshape. Therefore, samples were rapidly heated to 60 °C and then kept at this temperature for 30–120 second time intervals for a period of 20 minutes, with spectra recorded at 20 °C after rapid cooling. The relative contribution of immobilised spectral components was found to change significantly with heating time as micelle structures collapsed and gel fibres formed, as outlined in Fig. 4.
 | ||
| Fig. 4 Change in combined slow components contribution to EPR spectrum relative to fast component upon heating over time (left). Selected normalised EPR spectra (right). | ||
We believe that changes reported in Fig. 4 reflect evolution of molecular environment during micelle–gel transition. A potential artefact which would have been caused by thermal conversion of 1 to 2 under these conditions can be ruled out as (i) this conversion requires formaldehyde which is not present in the reaction mixture, and (ii) the changes to EPR spectra upon gelation are completely reversible (e.g., dissolution of gel in methanol followed by evaporation and resuspension in water gives EPR spectra identical to those of original micelles, see ESI,† Fig. S5).
Interestingly, the initial heating stage leads to a rapid loss in immobilised nitroxide environment (22% drop) over 120 seconds (Fig. 4) as micelle structures break down. This loose aggregate intermediate is probably more mobile than micelle or gel components, and could therefore correspond to a sheet-type structure, consistent with DLS data. There is still significant immobilised component, likely due to the formation of larger aggregate structures observed at longer timescales by DLS. Continued heating leads to a gradual increase in this immobilised component as loose aggregates form gel fibres up to a plateau after 10 minutes. This is in good agreement with macroscopic observations of gel formation. EPR results thus reveal a two-stage process of micelle–gel transformation: (i) breaking down of micelles, and (ii) re-assembly of loose aggregates into gel fibres.
Photochemical degradation of gels and micelles
In order to compare the reactivity of polymers in gels and micelles under identical reaction conditions, the reactions need to be triggered in both systems at a defined time. We argue that a photochemical reaction is best suited for such studies. As tyrosine undergoes a well-understood photodegradation which can be conveniently monitored by fluorescence, the gel and micelle samples were exposed to UV light (using high pressure UV lamp) for 24 h. The characteristic tyrosine fluorescence was then analysed by fluorometry relative to a control sample kept in the dark. This provided information on relative reactivity of photoexcited tyrosine in micelles and gels. Dityrosine formation (a common tyrosine photooxidation product)11 was also monitored by fluorometry. This reported on bimolecular reactivity between two components of the supramolecular assemblies.Gels and micelles have similar UV spectra. Although increased scattering in gels leads to somewhat higher absorbance, the absorbance at 276 nm (corresponding to tyrosine λmax) is very high and nearly identical in both systems (transmittance is 0.133% for micelles and 0.116% for gels, see ESI,† Fig. S6). Therefore, most of the light is absorbed by tyrosine rather than scattered, and the differences between micelles and gel absorbance are minimal.
Tyrosine degradation was slower in gel systems than micelles (Fig. 5, left). However, the difference is small. As the chemical environment and polymer concentration are identical in gel and micelle measurements, and the degradation reactions are kinetically limited by the absorption of light, the similar photodegradation rates are not unexpected. The small difference must arise from the slightly more restricted physical environment of tyrosine in gel systems, which could affect processes after photoexcitation, e.g., reactions with oxygen and reactive oxygen species such as hydroxyl radicals, singlet oxygen and superoxide.12 These reactive species do not diffuse by more than a few nanometers in biological systems.13
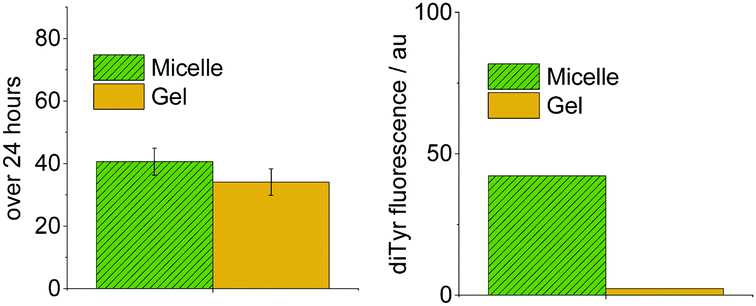 | ||
| Fig. 5 Comparison of photochemical reactivity in 5 mg mL−1 micelle and gel systems for PEG2000–Tyr6. Measured by tyrosine degradation (left) and dityrosine formation (right). | ||
The difference in reactivity is considerably more pronounced when dityrosine formation is monitored (Fig. 5, right). Gel systems show considerably lower formation of dimerised product. This is attributed to the slightly more restrictive physical environment in gels. As two tyrosyl radicals need to combine to form dityrosine, the dependence upon tyrosine mobility is much greater than for overall tyrosine degradation.
To test if gel systems limited diffusion of reactive oxygen species through the fibres, comparative photodegradation experiments were carried out on micelle and gel systems containing 0.1 ppm tris(acetylacetonato)iron(III). This significantly increases the reactive oxygen species (ROS) flux through photo-Fenton processes. In addition, metal complexes could act as sensitisers for ROS production.14 The relatively hydrophobic metal complex likely incorporates into micelle cores and hydrophobic gel pockets. Direct interaction between metal centres and tyrosine is possible, however as the concentration of metal (1.79 μmol dm−3) is very low compared to the polymer (1.72 mmol dm−3), this is likely to be diffusion limited.
The presence of the redox active metal significantly increases tyrosine degradation in micelle systems, as shown in Fig. 6, however gel systems show only a modest increase in activity. The lower tumbling rates of gels compared to micelles in our EPR studies (vide supra) suggested that gel fibres have lower mobility compared to micelles. Hence they restrict diffusion of reactive oxygen species generated at metal centres, limiting their effectiveness over larger distances. Diffusion of the metal complex is likely also restricted leading to further reduction in gel reactivity.
 | ||
| Fig. 6 Comparison of photochemical reactivity in PEG2000–Tyr6 gel and micelle polymer systems in the presence and absence of 0.1 ppm Fe(III), measured by decrease in tyrosine fluorescence after 24 hours UV exposure. | ||
Conclusions
An amphiphilic PEG–Tyr copolymer forms two distinct physical environments (a micellar solution and a gel) which are kinetically stable under ambient conditions. This provided a unique opportunity to study the effect of physical and molecular environment upon chemical reactivity, in chemically identical systems. Using EPR spectroscopy of spin-labelled copolymers, we observed co-existence of a relatively mobile and a relatively immobile environments in both morphologies. Two stages were observed during the transformation of micelles to gels: breaking up of micelles into loose aggregates and their assembly into fibres. Overall, tyrosine units were slightly less mobile in a gel than in a micelle morphology, although the microenvironments are quite similar.Photochemical degradation of tyrosine and formation of ditorysine was then studied in gels and micelles in the absence and in the presence of a relatively hydrophobic Fe(III) salt. Reactivity was fairly similar in both morphologies, with reactions in gels consistently slower than in micelles. This is consistent with the slightly lower tumbling rates (and hence lower mobility) of spin-labelled tyrosine units observed by EPR. The largest reactivity difference was observed for formation of dityrosine which requires two tyrosine units on polymer chains to come together: ditorysine formation in gels was much less pronounced than in micelles. This is also consistent with the lower tyrosine mobility in the gel environment.
Our study of micelle and gel reactivity in identical chemical environments showed relatively small differences, despite very different bulk appearance (“solid” gels vs. “liquid” colloidal solutions). The reactivity can be readily explained by the molecular mobility in the microenvironment around the reactive functionalities.
Experimental part
Unless otherwise stated, all chemicals and solvents were of analytical grade and used without further purification.DLS measurements
DLS measurements were performed at 22 °C using a 173° non-invasive backscatter method on a Malvern Instruments Zetasizer Nano ZS particle size analyser using 1 mg mL−1 solutions in Milli-Q water filtered through 450 nm PTFE filters unless stated otherwise. The data are averages of 3 measurements with each experiment repeated at least twice.Synthesis of PEG–Tyr block copolymer
PEG2000–Tyr6 (Mn = 2959 g mol−1) was prepared according to published literature procedures.5Spin labelling of PEG–Tyr
PEG–Tyr copolymer was spin labelled using a procedure adapted from literature.7 PEG2000–Tyr6 (1.33 mg) was dissolved in methanol (100 μL) to give a 4.58 mM solution. A 58.6 mM amino-TEMPO solution in methanol (50 μL) was added to the sample, followed by 3.5 M formaldehyde solution in methanol (8.4 μL). The solution was stirred at room temperature for 16 h, and the solvent was removed under vacuum. The residue was redissolved in Milli-Q water (333 μL) to give an end polymer concentration of 1.38 mM. The crude product was then dialysed in deionised water (3 L) to remove any unreacted nitroxide using a dialysis membrane (Mw cut-off 12–14 kDa), with 2 water changes over 24 hours.EPR analysis of spin labelled samples
5 mg mL−1 (1.72 mM) aqueous spin labelled PEG2000–Tyr6 polymer solutions in Milli-Q water were placed into a quartz flat cell and X-band EPR spectra were recorded at room temperature on JEOL JES-X320 spectrometer. Typical EPR parameters: microwave power = 2 mW, frequency = 9.128 GHz, sweep width = 15 mT, modulation frequency = 100 kHz, modulation width = 0.1 mT, sweep time = 240 s, number of scans = 5, time constant = 0.1 s.Gel samples were prepared by heating the micellar solutions within the flat cell in a water bath at 60 °C. Gelation was confirmed by introducing a small metal (lead solder) ball to the top of the sample within the flat cell. Following gelation, the increased macroscopic viscosity of the sample prevented the ball sinking through the sample.
EPR spectra were simulated using EasySpin (chili function)9 to determine rotational correlation times of different spectral components.
Low temperature Q-band EPR measurements
5 mg mL−1 (1.72 mM) Aqueous spin labelled PEG2000–Tyr6 polymer solutions in Milli-Q water were placed into a quartz EPR tube (inner diameter 1.1 mm, Wilmad WG-221T-RB). The samples were then frozen in the EPR cavity. Spectra were recorded at a temperature of 88 K. Typical EPR parameters: microwave power = 124 μW, frequency = 34.69 GHz, sweep width = 30 mT, modulation frequency = 100 kHz, modulation width = 0.1 mT, sweep time = 480 s, number of scans = 10, time constant = 0.3 s.EPR spectra were simulated using EasySpin (pepper function)9 to determine principal values of g- and A-tensors.
Monitoring gelation in situ by EPR
10 mg mL−1 (3.44 mM) Aqueous spin labelled PEG2000–Tyr6 polymer solutions were heated to 60 °C by immersing the sealed quartz flat cell in a water bath at the required temperature. After heating for 30 seconds, the sample was removed, allowed to cool to room temperature for 5 minutes before reanalysing by EPR as described above. This process was repeated multiple times to monitor the change by EPR over 30 minutes heating.Photochemical degradation measurements
5 mg mL−1 (1.72 mM) aqueous samples of PEG2000–Tyr6 (280 μL) were prepared in 0.1 mM acetate buffer at pH 5. Samples were then split into two quartz cuvettes: one sample (140 μL) was left in the dark as a control, the other sample was irradiated for 24 hours with a 100 W high pressure Hg lamp. Following irradiation, the samples were diluted to a volume of 1 mL with ethanol (140 μL, to break up micelle/gel structures), 0.1 mol dm−3 acetate buffer at pH 5 (400 μL) and 0.4 mol dm−3 borate buffer at pH 8.5 (400 μL), to bring the end pH to 8.5 (for improved observation of singly deprotonated dityrosine fluorescence).15 Samples were analysed by fluorometry for characteristic tyrosine fluorescence: excitation scan 240–300 nm with λem = 303 nm, emission scan 270–500 nm with λex = 276 nm. Samples were also analysed for dityrosine fluorescence: excitation scan 240–400 nm with λem = 405 nm, emission scan 325–500 nm with λex = 320 nm. The degradation of tyrosine fluorescence and increase in dityrosine fluorescence was determined relative to the dark control for every experiment.Gel samples were prepared as described above, with heating in a water bath at 60 °C for one hour after splitting the PEG–Tyr sample into two cuvettes. Gelation was confirmed after this time by inverting cuvettes, and samples were analysed as described above.
Hydrophobic iron(III) acetylacetonate was added to some samples. Fe(acac)3 stock solution at a metal concentration of 50 ppm was prepared in water. This stock solution (0.56 μL) was added to 5 mg mL−1 (1.72 mM) samples of PEG–Tyr copolymer (280 μL) in 0.1 M pH 5 acetate buffer to give a metal concentration of 0.1 ppm. Photochemical experiments were then performed as described above.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge The Procter & Gamble Company for support of this work.Notes and references
- K. Bhattacharyya, Acc. Chem. Res., 2003, 36, 95–101 CrossRef CAS PubMed; A. V. Davis, R. M. Yeh and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4793–4796 CrossRef PubMed; N. J. Buurma, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2012, 108, 316–333 RSC.
- A. Galana and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC; P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- D. G. Whitten, Acc. Chem. Res., 1993, 26, 502–509 CrossRef CAS; A. Dey and K. Biradha, Isr. J. Chem., 2019, 59, 220–232 CrossRef.
- J. L. Santos and M. Herrera-Alonso, Macromolecules, 2014, 47, 137–145 CrossRef CAS.
- J. Huang, C. L. Hastings, G. P. Duffy, H. M. Kelly, J. Raeburn, D. J. Adams and A. Heise, Biomacromolecules, 2013, 14, 200–206 CrossRef CAS PubMed.
- G. H. Van Domeselaar, G. S. Kwon, L. C. Andrew and D. S. Wishart, Colloids Surf., B, 2003, 30, 323–334 CrossRef CAS.
- M. Lorenzi, C. Puppo, R. Lebrun, S. Lignon, V. Roubaud, M. Martinho, E. Mileo, P. Tordo, S. R. A. Marque, B. Gontero, B. Guigliarelli and V. Belle, Angew. Chem., Int. Ed., 2011, 50, 9108–9111 CrossRef CAS PubMed.
- E. Mileo, E. Etienne, M. Martinho, R. Lebrun, V. Roubaud, P. Tordo, B. Gontero, B. Guigliarelli, S. R. A. Marque and V. Belle, Bioconjugate Chem., 2013, 24, 1110–1117 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- J. P. Klare, Biol. Chem., 2013, 394, 1281–1300 CAS.
- C. Giulivi, N. J. Traaseth and K. J. A. Davies, Amino Acids, 2003, 25, 227–232 CrossRef CAS PubMed.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 RSC.
- W. A. Pryor, Annu. Rev. Physiol., 1986, 48, 657–667 CrossRef CAS PubMed; R. B. Mikkelsen and P. Wardman, Oncogene, 2003, 22, 5734–5754 CrossRef PubMed.
- G. Ruppert, R. Bauer and G. Heisler, J. Photochem. Photobiol., A, 1993, 73, 75–78 CrossRef CAS; P. Chábera, Y. Liu, O. Prakash, E. Thyrhaug, A. E. Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695–699 CrossRef PubMed.
- M. Correia, M. T. Neves-Petersen, P. B. Jeppesen, S. Gregersen and S. B. Petersen, PLoS One, 2012, 7, e50733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, EPR spectra and simulation details. See DOI: 10.1039/d0cp01956a |
| This journal is © the Owner Societies 2020 |