
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of holmium(III)-acetylacetonate complexes derived from therapeutic microspheres by infrared ion spectroscopy†
Kas J.
Houthuijs
 a,
Jonathan
Martens
*a,
Alexandra G.
Arranja
bcd,
Giel
Berden
a,
J. Frank W.
Nijsen
cd and
Jos
Oomens
*ae
a,
Jonathan
Martens
*a,
Alexandra G.
Arranja
bcd,
Giel
Berden
a,
J. Frank W.
Nijsen
cd and
Jos
Oomens
*ae
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7, 6525ED Nijmegen, The Netherlands. E-mail: jonathan.martens@ru.nl; j.oomens@science.ru.nl
bDepartment of Pharmaceutics, Utrecht Institute for Pharmaceutical Sciences (UIPS), Utrecht University, PO Box 80082, 3508 TB Utrecht, The Netherlands
cRadboudumc, Department of Radiology and Nuclear Medicine, Geert Grooteplein Zuid 10, 6525 GA Nijmegen, The Netherlands
dQuirem Medical B.V., Zutphenseweg 55, 7418 AH Deventer, The Netherlands
evan’t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 908, 1098 XH Amsterdam, The Netherlands
First published on 29th June 2020
Abstract
Microspheres containing radioactive 166holmium-acetylacetonate are employed in emerging radionuclide therapies for the treatment of malignancies. At the molecular level, details on the coordination geometries of the Ho complexes are however elusive. Infrared ion spectroscopy (IRIS) was used to characterize several 165Ho-acetylacetonate complexes derived from non-radioactive microspheres. The coordination geometry of four distinct ionic complexes were fully assigned by comparison of their measured IR spectra with spectra calculated at the density functional theory (DFT) level. The coordination of each acetylacetonate ligand is dependent on the presence of other ligands, revealing an asymmetric chelation motif in some of the complexes. A fifth, previously unknown constituent of the microspheres was identified as a coordination complex containing an acetic acid ligand. These results pave the way for IRIS-based identification of microsphere constituents upon neutron activation of the metal center.
Introduction
Over the past two decades, the lanthanide holmium has seen a steady increase in the number of radionuclide therapy applications. This interest is driven by the possibility to convert holmium-165 into radioactive holmium-166 upon neutron activation (165Ho + n → 166Ho).1,2 With a half-life of 26.8 hours, holmium-166 decays into erbium-166 through the emission of beta-minus particles (β−) and low-energy gamma photons.3,4 The β− particles can be used to kill tumor cells, while the emitted gamma photons enable visualization and quantification of the isotope distribution in vivo by single-photon emission computed tomography (SPECT).5–8 In addition, holmium has a high X-ray attenuation coefficient and paramagnetic properties, which allow its visualization by computed tomography (CT) and high-resolution magnetic resonance imaging (MRI), respectively.5,8Holmium microspheres with a defined size distribution (20–50 μm) are an ideal medical device for selective internal radiation therapy (SIRT) of liver malignancies.5–7 Microspheres based on holmium acetylacetonate complexes are particularly attractive from a clinical perspective because of their high holmium content of ∼47 wt%, giving them the possibility to achieve a high specific activity (MBq mg−1) and hence improved imaging and quantification performance in MRI and CT.8–10 Moreover, they pave the way towards other clinical applications such as radiation segmentectomy and interstitial micro brachytherapy.9,11–13 The holmium-acetylacetonate microspheres are formed using as precursor holmium-acetylacetonate crystals, Ho(acac–H)3·H2O. The crystalline structure of this precursor has been previously elucidated by X-ray crystallography.14 The chemical composition of the microspheres (Ho2(acac–H)3·2H2O) and their amorphous structure has also been recently described.9 It has been hypothesized that in these microspheres, deprotonated acac and holmium form a network, with additional water molecules coordinating to the holmium.10 A detailed investigation of the coordination structures in the microspheres has not been reported thus far.
Here, coordination complexes of Ho and [acac–H]− isolated from microspheres are structurally characterized in the gas phase of a mass spectrometer. Although different from the condensed phase of the microspheres, the gas phase can provide fundamental insight in structural properties of the complexes. This is in contrast to X-ray crystallography, which cannot be readily applied to the amorphous microspheres. Previous research on gas-phase holmium-ligand complexes includes a mass-spectrometric (MS) study on the chelation with fluorinated acac15 and glucose.16 The former was combined with solid-state infrared transmission measurements that qualitatively described the binding of the ligands. Recent work has employed MS in combination with density functional theory (DFT) to investigate the mechanisms of using gas-phase Ho(I) as a catalyst for carbon bond activation and the formation of dibenzothiophene.17–19 However, as these studies are solely based on MS, precise geometries were not experimentally verified. Here we employ infrared ion spectroscopy (IRIS), a combination of MS and IR spectroscopy, to determine the coordination motifs of the complexes by mass-selective probing of their vibrational fingerprints. This approach has previously been used to probe metal–ligand interactions to investigate, for instance, the factors influencing ligand denticity.20,21 Systems studied involve a wide variety metal centers including transition metals,22 lanthanides23 and other f-block metals, such as uranyl complexes, where the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO asymmetric stretch frequency is a diagnostic probe of the coordination interactions.21,24–27
UO asymmetric stretch frequency is a diagnostic probe of the coordination interactions.21,24–27
Methods
Holmium-acetylacetonate microspheres preparation
Microspheres were prepared by an emulsification and solvent evaporation method as previously reported.9 Briefly, 10 g of holmium acetylacetonate crystals2 were dissolved in 186 g of chloroform and the solution was added to 1 liter of an aqueous solution of poly-vinyl alcohol (2% w/w). Overhead four blades propeller stirrers (IKA Eurostar power digi-visc) were used to vigorously stir the mixture in two liters baffled beakers to obtain an oil-in-water (o/w) emulsion which was stirred at a constant speed of 300 rpm and temperature of 25 °C. A constant flow of nitrogen (12 L min−1) was applied for 72 h. After evaporation of the organic solvent, the holmium acetylacetonate microspheres were formed and sieved to the desired size (20–50 μm) using an electronic sieve vibrator (TOPAS EMS 755). The sieved microspheres were dried at room temperature for 5 hours under ambient pressure followed by vacuum drying at room temperature for 72 hours. For infrared ion spectroscopy analysis, dried powders of the holmium acetylacetonate microspheres were dissolved in methanol.Infrared ion spectroscopy
Infrared spectra of the positively charged holmium complexes were measured in a quadrupole ion trap (QIT) mass spectrometer (Bruker, AmaZon Speed ETD, Bremen, Germany),28 which was coupled to the beamline of the infrared free-electron laser FELIX.29 An approximately 10 μM solution of the dried powder of the holmium acetylacetonate microspheres in methanol was injected and ionized using electrospray ionization (ESI) to generate positively charged holmium complexes (see Fig. S1 and S2, ESI†). After isolation of the precursor ion of interest, the ions were irradiated with two 6 μs long macropulses from FELIX at a repetition rate of 10 Hz, each pulse having an energy of 20–100 mJ and a bandwidth of about 0.5% of the center frequency. Wavelength dependent absorption of infrared radiation increases the internal energy of the precursor ion, until the dissociation threshold is reached and the precursor ion dissociates (see Fig. S3, ESI†). The wavelength dependent fragmentation is converted into an IR spectrum by calculating the fragmentation yield: −ln(1 − ∑I(fragment ions)/∑I(all ions)).30 This yield is linearly corrected for wavelength dependent variations in laser power and the wavelength is calibrated using a grating spectrometer.Computational methods
Density functional theory (DFT) calculations were carried out in Gaussian16,31 using the B3LYP density functional. The Stuttgart–Dresden small core relativistic effective core-potential (ECP28MWB) and its associated basis set was used for the holmium atom32,33 and the 6-31++G(d,p) basis set for the first and second row atoms. After geometry optimization of the complexes, without symmetry constraints, a harmonic frequency calculation was performed. To correct for anharmonic effects, frequencies were scaled by a uniform factor of 0.975.34 To aid comparison to experimental spectra, a Gaussian line broadening of 20 cm−1 full width at half maximum was applied to the calculated stick spectrum. Computed IR fundamental frequencies are given in Tables S1 and S2 (ESI†). Relative energies were based on the sum of the electronic and thermal free energies of the B3LYP frequency calculations.Results and discussion
Infrared spectral assignment of the holmium-ligand complexes
Holmium-ligand complexes are generated by ESI from a solution of holmium-acetylacetonate containing microspheres dissolved in methanol. This mainly produced singly charged ions corresponding to a Ho(III) center chelated by two [acac–H]− ligands, resulting in an overall 1+ ion at m/z 363. Three other ions in the mass spectrum (shown in Fig. S1, ESI†) at m/z values of 381, 395, and 463 were tentatively assigned as [Ho(acac–H)2L]+ species, where L is either water, methanol, or acac, respectively. Multinuclear holmium complexes were not observed, which likely relates to the acetylacetonate geometry that favours bidentate binding to a single metal centre (see Scheme 1).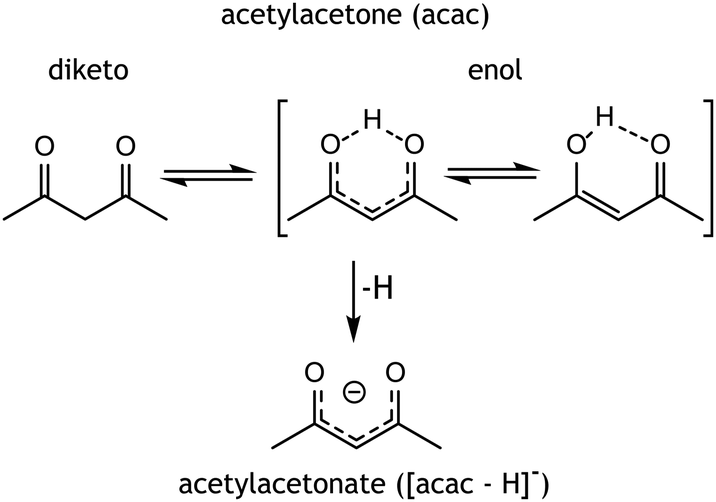 | ||
| Scheme 1 Possible tautomers and their nomenclature of acetylacetone and its conjugated base acetylacetonate. | ||
Mass-to-charge ratios provide the stoichiometry of the observed MS features, but do not reveal the precise chelation geometry, e.g. which ligands are deprotonated and which of their atoms act as donors to the central Ho ion. For instance, in the [Ho(acac)2L − 2H]+ systems, both acac ligands may be deprotonated, but depending on the acidity of L relative to that of acac, alternative deprotonation isomers are conceivable. Apart from acidity, the nucleophilicity of the ligands towards Ho3+ and steric effects likely influence the overall coordination structure. The acac ligand presents a variety of possible chelating motifs, both mono- and bidentate, and both as neutral (acac) or as anion ([acac–H]−); diketo and cis-enol binding are most common (see Scheme 1).35 Although coordination in the diketo tautomeric form is energetically more favourable, the enol tautomer is more abundant in both methanol solution and in the gas phase,36,37 having a free-energy advantage of approximately 10 kJ mol−1 at 298 K. It may therefore be a reasonable candidate to be observed upon gradual solvent evaporation in the ESI process. To derive the chelation structure of the complexes, IRIS spectra were compared with quantum-chemically computed IR spectra of candidate structures.
Fig. 1 presents the experimental IRIS spectrum of the isolated m/z 381 ion (black trace in all panels). To establish the deprotonation state and the tautomeric form of the ligands in [Ho(acac)2(H2O)–2H]+, the IR spectrum is contrasted against computed spectra of possible chelation geometries represented by the coloured traces. Fig. 1a presents the best match, which suggests that the complex involves an intact water ligand and two [acac–H]− ligands. The absence of the symmetric CO/CO stretch of diketo acac at 1720 cm−1 (Fig. 1b) and the absence of the hydrogen-bonded OH bend of enol acac at 1580 cm−1 (Fig. 1c) indicate that the coordination complex does not contain a neutral acac ligand. This is in line with the computed relative free energies of the structures, given in each panel, and with what is expected on the basis of the higher acidity of acac relative to water.
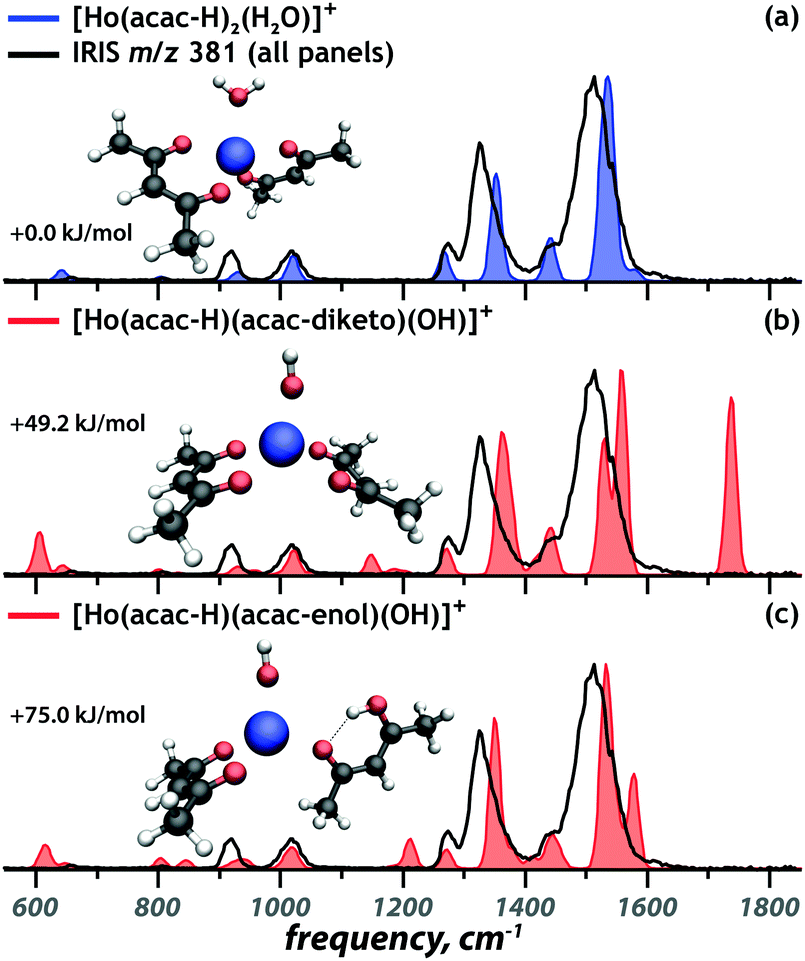 | ||
| Fig. 1 Comparison of the calculated spectra (coloured) of three candidate structures to the experimental spectrum of the m/z 381 ion (black). Relative free energies are given. | ||
Although previous studies have shown that IR spectra of transition metal coordination complexes can be sensitive to the spin state of metal center,38,39 this is not the case for lanthanides. f-Orbitals barely engage in ligand binding, so that their occupation barely affects chelation and hence IR spectra; f-orbitals in lanthanides show negligible crystal field splitting (approximately 1 kJ mol−1)40 and here we expect that the Ho3+ center is in its atomic ground electronic state, which has quintet spin multiplicity ([Xe]4f10, 5I8). This is indeed confirmed by computed relative free energies for different spin states (see Fig. S4, ESI†) and in line with previous DFT investigations on Ho(III).41 Computed IR spectra in Fig. 1 correspond to complexes with quintet spin multiplicity.
The remaining coordination complexes were structurally characterized using an analogous approach. Diagnostic vibrational bands were used to assign the coordination motif of the ions at m/z 363 [Ho(acac)2–2H]+, m/z 395 [Ho(acac)2(MeOH)–2H]+ and m/z 463 [Ho(acac)3–2H]+, as presented in Fig. 2a, c and d, respectively. The experimental IR spectra (black) are overlaid onto the best matching calculated spectrum (blue) for each of the complexes. In all cases, the lowest-energy structure provides the best match with experiment. Computed spectra of alternative candidate structures, including alternative spin multiplicities, are shown in Fig. S5–S7 (ESI†). These computations follow the same trend as that for [Ho(acac)2(H2O)–2H]+.
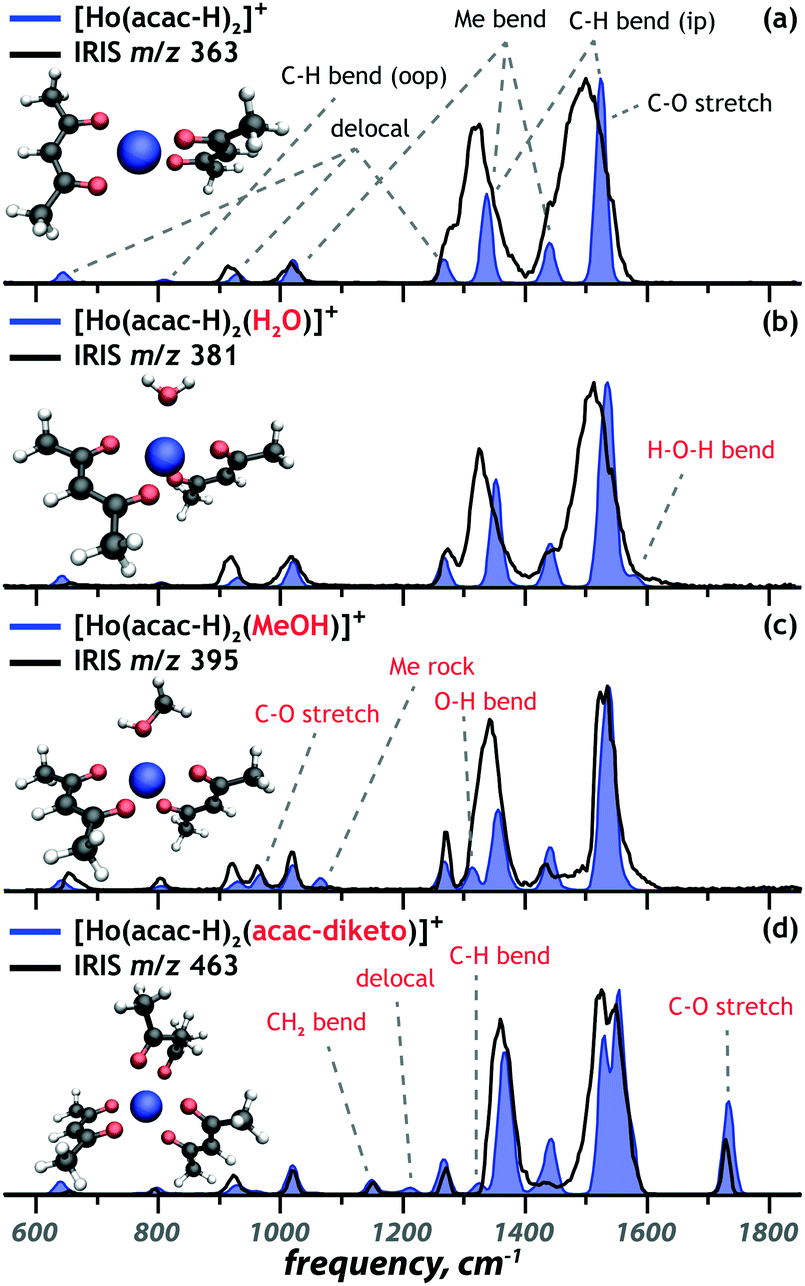 | ||
| Fig. 2 Comparison of calculated spectra (blue) of the lowest-energy structures to the experimental spectra of the [Ho(acac)2L–2H]+ ions (black). Selected vibrational mode assignment for the two acetylacetonate ligands (black) and the additional neutral ligands (red) are given. | ||
Effect of third ligand on geometry and IR spectra
Closer inspection shows that the geometries are determined by maximized interaction of the ligand donor atoms with the metal centre and ligand–ligand repulsion. This leads to the following coordination geometries: tetrahedral for [Ho(acac–H)2]+, trigonal bipyramidal for [Ho(acac–H)2(H2O)]+ and [Ho(acac–H)2(MeOH)]+ and octahedral for the [Ho(acac–H)2(acac)]+ ion. These coordination geometries are slightly distorted due to the limited angle that bidentate [acac–H]− can accommodate and the repulsion between the third ligand and the [acac–H]− ligands (Fig. 2b–d). The [acac–H]− ligands in all complexes adopt a planar OCC(H)CO structure as a consequence of electron delocalization in the conjugated π-orbitals that is typical for deprotonated 1,3-diketones (see Scheme 1). The neutral acac ligand, on the other hand, has an sp3-hybridized central carbon atom, which results in the CH2 sticking out-of-plane (Fig. 2d).The DFT computations not only allow for the structural identification of the coordination complexes, but also give insight into the binding properties of the ligands and how these are affected by the other ligands. The [Ho(acac–H)2]+ coordination complex is taken as the unperturbed system, with its experimental and computed IR spectrum shown in Fig. 2a. The vibrational bands are classified as one of five types of modes: delocalized modes (645 cm−1, 930 cm−1, 1270 cm−1), CH out-of-plane and in-plane bending modes (810 cm−1, 1340 cm−1, 1525 cm−1), methyl rocking (1020 cm−1), methyl wagging (1340 cm−1, 1440 cm−1) and CO stretching modes (1525 cm−1). These modes are clearly observed in the experimental IR spectrum, despite some broadening due to a relatively high dissociation threshold for this complex. Binding of a third (neutral) ligand to the [Ho(acac–H)2]+ ion reduces this threshold and introduces additional bands as indicated in red in Fig. 2b–d. Binding of water introduces an H–O–H bending mode at 1580 cm−1. Binding of methanol introduces three significant bands: C–O stretching at 965 cm−1, methyl rocking at 1065 cm−1 and COH bending at 1315 cm−1. Binding of a neutral acac ligand introduces four new bands: CH2 bending at 1150 cm−1, a delocalized mode at 1210 cm−1, CH in plane bending at 1320 cm−1 and a symmetric CO stretch at 1735 cm−1.
The modes associated with the [acac–H]− ligands below 1300 cm−1 remain largely unaffected by coordination of additional ligands. Some of the modes above 1300 cm−1, however, shift significantly in frequency upon additional ligand binding. The carbonyl stretch of [acac–H]− blueshifts by about 30 cm−1 upon binding of an additional neutral acac, as is seen in Fig. 3 and 2a, d. The additional ligand reduces the electron density donation of the other, anionic ligands to the metal center, increasing the electron density in their CO bonds, blue-shifting the CO stretch frequencies. This is also reflected in the increased HoO and reduced CO bond lengths upon coordination of an additional ligand (see Fig. 3). The value of the shifts for the three neutral ligands qualitatively follows the order of their proton affinity, with PA(H2O) < PA(MeOH) < PA(acac), reflecting the ability of these ligands to donate electron density to the metal ion.
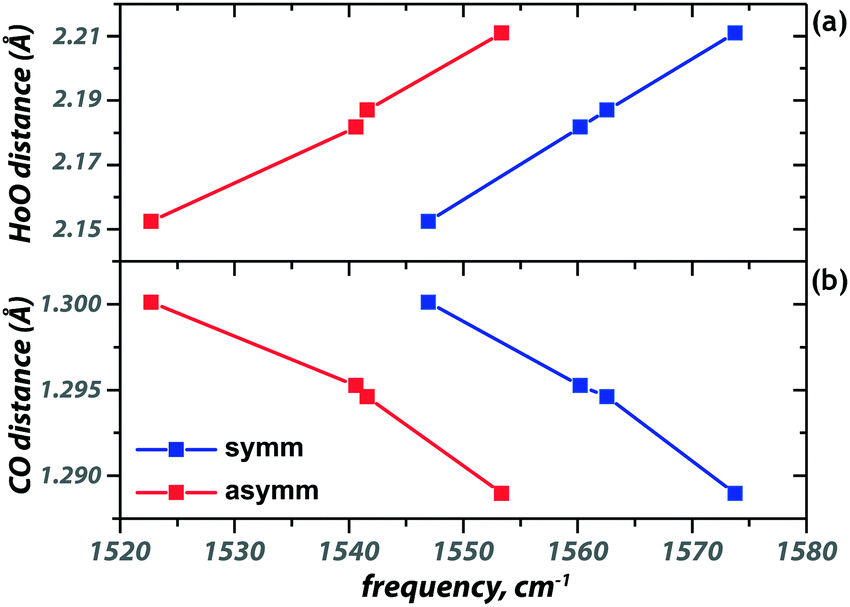 | ||
| Fig. 3 Correlation of the symmetric (blue) and antisymmetric (red) CO stretch frequencies with the Ho–O (a) and C–O (b) bond lengths. The squares represent the following ions, from left to right: [Ho(acac–H)2]+, [Ho(acac–H)2(H2O)]+, [Ho(acac–H)2(MeOH)]+ and [Ho(acac–H)2(acac)]+. | ||
The geometry of the [acac–H]− ligands is also affected by the binding of the other ligands. Where the ligands in [Ho(acac − H)2]+ coordinate symmetrically, with all four C–O and all four Ho–O bond lengths being equal, this is no longer the case upon coordination of an additional ligand, see Table 1. In both [Ho(acac–H)2(H2O)]+ and [Ho(acac–H)2(MeOH)]+, one of the CO bonds in [acac–H]− is 0.007 Å shorter than the other. Together with a small difference in the backbone CC bond lengths (0.006 Å), this suggests that the [acac–H]− ligands surrender their symmetric bidentate binding and adopt a slightly “enol-fashion” binding. Although these bond length differences are small, they are reproduced in all [acac–H]− ligands in the two coordination complexes. This shift towards enol character parallels the ongoing discussion concerning the symmetry of the hydrogen sharing in the cis enol tautomer of acac (see Scheme 1). Experimental studies have been unable to resolve this issue, as evidence for both equal and unequal OH bond lengths, resulting in C2v and Cs symmetry respectively, has been reported.42,43
| Bond | Third ligand | ||||
|---|---|---|---|---|---|
| — | acac | acac | H2O | MeOH | |
| Ho–O | 2.153 | 2.268 | 2.209 | 2.214 | 2.221 |
| O–C | 1.300 | 1.289 | 1.288 | 1.292 | 1.292 |
| C–C | 1.407 | 1.407 | 1.408 | 1.410 | 1.409 |
| C–C | 1.407 | 1.407 | 1.406 | 1.403 | 1.404 |
| C–O | 1.300 | 1.288 | 1.290 | 1.299 | 1.297 |
| O–Ho | 2.152 | 2.183 | 2.184 | 2.149 | 2.153 |
| Symmetric | Asymmetric | ||||
Coordination complex at m/z 423: [Ho(acac)2(CH3COOH)–2H]+
In addition to the complexes discussed above, a Ho containing ion at m/z 423 was observed, for which the experimental IR spectrum is presented in black in Fig. 4. We hypothesize that this mass peak corresponds to [Ho(acac–H)2]+ with an additional acetic acid ligand. Acetic acid was also identified as one of the degradation products of acac after neutron activation,9 but its presence here indicates that it is also a constituent of the non-irradiated microspheres. In contrast to the complexes discussed above, the acetylacetone and acetic acid ligands have comparable acidities and it is not a priori clear which of them deprotonate in the formation of the 1+ coordination complex. Computed IR spectra for possible proto-isomers – in different conformational configurations – are presented as the coloured spectra in Fig. 4. The energetically most favourable complex is predicted to possess two deprotonated acac ligands. The neutral acetic acid coordinates to the holmium center in a monodentate fashion and forms a hydrogen bond between its OH group and the ketone oxygen atom of one of the [acac–H]− ligands (Fig. 4a). Two other conformations possess a free OH group tilted away from the Ho center, adopting either a cis (Fig. 4b, +17.8 kJ mol−1) or trans (Fig. 4d, +26.3 kJ mol−1) configuration. For the proto-isomer featuring an acetate and a neutral acac ligand, both the diketo (Fig. 4c) and enol (Fig. 4e) form of the acac ligand were considered.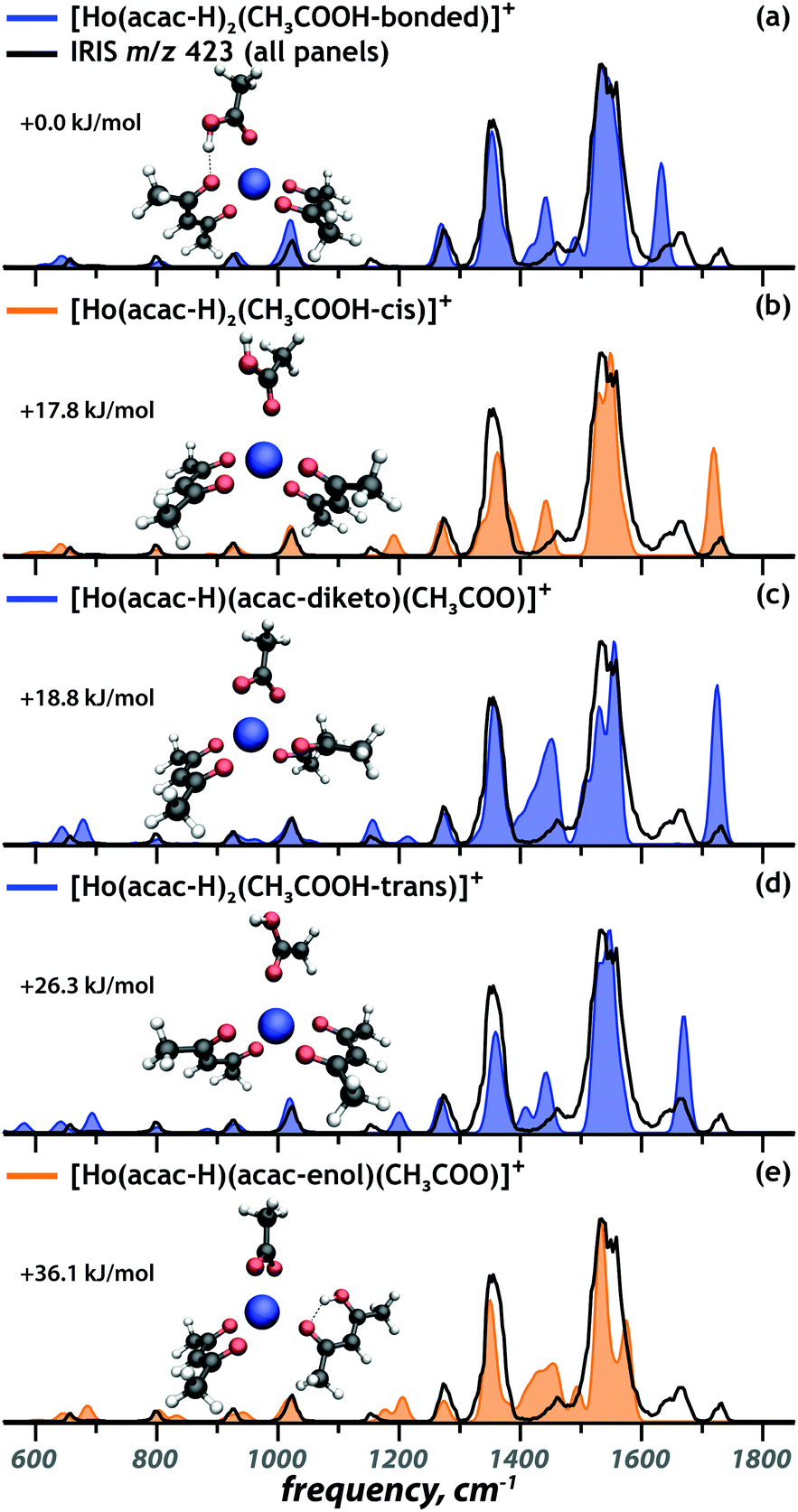 | ||
| Fig. 4 Comparison of the calculated spectra (coloured) of selected tautomers and conformers of the [Ho(acac)2(CH3COOH)–2H]+ ion to the experimental spectrum of the m/z 423 ion (black), with computed relative free energies. The complexes with spectra in blue and orange correspond to presence confirmed and ambiguous, respectively. | ||
In the IR spectrum of the [Ho(acac)2(CH3COOH) − 2H]+ ion, three absorptions are observed above 1600 cm−1, two distinct bands at 1720 cm−1 and 1660 cm−1 and a small shoulder at 1630 cm−1. This suggests the presence of at least three different isomers, since all computed spectra show only one band in this spectral range. The band at 1630 cm−1 can be assigned as the acetic acid CO stretch of the lowest-energy structure (Fig. 4a). The band at 1660 cm−1 is identified as the CO stretch of trans acetic acid (Fig. 4d). The 30 cm−1 redshift of this mode in the lowest-energy structure is caused by a stronger coordination of the acetic acid ligand, weakening the CO bond, as is reflected in its 0.014 Å elongation. The hydrogen-bond gives partial carboxylate character to the acetic acid ligand, making it a stronger nucleophile, which results in a stronger coordination. Concurrently, the hydrogen bond weakens the interaction between [acac–H]− and the metal center.
Finally, the CO stretch band at 1720 cm−1 is reproduced well by the computed spectra of the complex that includes a cis acetic acid ligand (Fig. 4b) and of the complex involving an acetate anion and neutral acac in its diketo form (Fig. 4c). Of those possibilities, only the acetate complex correctly reproduces the weak band at 1155 cm−1. For the two remaining complexes, the cis acetic acid (Fig. 4b) and the enol tautomer of acac (Fig. 4e), there is no direct spectroscopic evidence of their presence. However, their computed spectra do not disagree severely with the experimental IR spectrum, so that their contribution to the ion population cannot be ruled out definitively.
Conclusions
In conclusion, five different Ho3+–acetylacetonate coordination complexes, prepared from clinically relevant holmium-acetylacetonate microspheres, have been characterized using IRIS. The coordination geometries of these complexes have been established on the basis of the good match between experimentally observed and theoretically predicted vibrational bands. This assignment resulted in a qualitative description of how the binding properties of [acac–H]− depend on the presence of a third ligand, inducing a slightly asymmetric binding of the [acac–H]− ligands, reminiscent of the enol tautomer of neutral acac. Moreover, infrared ion spectroscopy was able to identify an unknown ligand binding to the holmium complex ion as acetic acid. The DFT calculations confirmed that the IR spectra are minimally affected by the spin multiplicity of the metal center, which reflects the marginal crystal field splitting in f-block metals. Future studies will exploit IRIS to identify the byproducts formed upon neutron activation of the microspheres.Conflicts of interest
A. G. Arranja is working for Quirem Medical since 1st September 2019. J. F. W. Nijsen is inventor on the patents related to the holmium-acetylacetonate microspheres which are assigned to University Medical Center Utrecht Holding BV and/or Quirem Medical (patent families: USA Patent No. 6373068 B1, PCT/NL03/00485, EP07112807.8, 10190254.2, P114198PC00, P112614NL00). He is co-founder and chief scientific officer of Quirem Medical, and has a minority share in the company Quirem Medical. The activities of J. F. W. Nijsen within Quirem Medical are approved and supported by Dirkjan Masman (Director Technology Transfer Office Radboudumc) and Mathias Prokop (Head of Radiology and Nuclear Medicine at Radboudumc).Acknowledgements
We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. Financial support for this project was provided by NWO Roadmap grant nr. 184.034.022 and by NWO Innovation Fund for Chemistry (IFC) under the Launchpad for Innovative Future Technology (LIFT) project nr. 731.015.411. Computations were carried out at the CARTESIUS cluster of SURFsara in Amsterdam; access was provided through NWO Rekentijd grant 2019.062.References
- M. A. D. Vente, J. F. W. Nijsen, R. de Roos, M. J. van Steenbergen, C. N. J. Kaaijk, M. J. J. Koster-Ammerlaan, P. F. A. de Leege, W. E. Hennink, A. D. van het Schip and G. C. Krijger, Biomed. Microdevices, 2009, 11, 763–772 CrossRef CAS PubMed.
- J. F. W. Nijsen, B. A. Zonnenberg, J. R. W. Woittiez, D. W. Rook, I. A. Swildens-van Woudenberg, P. P. Van Rijk and A. D. Van het Schip, Eur. J. Nucl. Med., 1999, 26, 699–704 CrossRef CAS PubMed.
- J. F. W. Nijsen, G. C. Krijger and A. D. Van het Schip, Anticancer Agents Med. Chem., 2007, 7, 271–290 CrossRef CAS PubMed.
- A. F. van den Hoven, D. Y. Sze and M. G. E. H. Lam, Handbook of Radioembolization, CRC Press, 2016, pp. 21–48 Search PubMed.
- M. L. J. Smits, M. Elschot, M. A. A. J. van den Bosch, G. H. van de Maat, A. D. Van het Schip, B. A. Zonnenberg, P. R. Seevinck, H. M. Verkooijen, C. J. Bakker, H. W. A. M. de Jong, M. G. E. H. Lam and J. F. W. Nijsen, J. Nucl. Med., 2013, 54, 2093–2100 CrossRef CAS PubMed.
- M. L. J. Smits, J. F. W. Nijsen, M. A. A. J. van den Bosch, M. G. E. H. Lam, M. A. D. Vente, W. P. T. M. Mali, A. D. Van het Schip and B. A. Zonnenberg, Lancet Oncol., 2012, 13, 1025–1034 CrossRef CAS PubMed.
- J. F. Prince, M. A. A. J. van den Bosch, J. F. W. Nijsen, M. L. J. Smits, A. F. van den Hoven, S. Nikolakopoulos, F. J. Wessels, R. C. G. Bruijnen, M. N. G. J. A. Braat, B. A. Zonnenberg and M. G. E. H. Lam, J. Nucl. Med., 2018, 59, 582–588 CrossRef CAS PubMed.
- P. R. Seevinck, J.-H. Seppenwoolde, T. C. de Wit, J. F. W. Nijsen, F. J. Beekman, A. D. Van het Schip and C. J. G. Bakker, Anticancer Agents Med. Chem., 2007, 7, 317–334 CrossRef CAS PubMed.
- A. G. Arranja, W. E. Hennink, A. G. Denkova, R. W. A. Hendrikx and J. F. W. Nijsen, Int. J. Pharm., 2018, 548, 73–81 CrossRef CAS PubMed.
- W. Bult, P. R. Seevinck, G. C. Krijger, T. Visser, L. M. J. Kroon-Batenburg, C. J. G. Bakker, W. E. Hennink, A. D. Van het Schip and J. F. W. Nijsen, Pharm. Res., 2009, 26, 1371–1378 CrossRef CAS PubMed.
- R. C. Bakker, M. G. E. H. Lam, S. A. van Nimwegen, A. J. W. P. Rosenberg, R. J. J. van Es and J. F. W. Nijsen, J. Radiat. Oncol., 2017, 6, 323–341 CrossRef CAS PubMed.
- S. A. van Nimwegen, R. C. Bakker, J. Kirpensteijn, R. J. J. van Es, R. Koole, M. G. E. H. Lam, J. W. Hesselink and J. F. W. Nijsen, Vet. Comp. Oncol., 2018, 16, 114–124 CrossRef CAS PubMed.
- R. C. Bakker, R. J. J. van Es, A. J. W. P. Rosenberg, S. A. van Nimwegen, R. Bastiaannet, H. W. A. M. de Jong, J. F. W. Nijsen and M. G. E. H. Lam, Nucl. Med. Commun., 2018, 39, 213 CrossRef CAS PubMed.
- H. Kooijman, F. Nijsen, A. L. Spek and F. van het Schip, Acta Crystallogr. Sect. C: Cryst. Struct. Commun., 2000, 56, 156–158 CrossRef PubMed.
- R. Belcher, J. Majer, R. Perry and W. I. Stephen, J. Inorg. Nucl. Chem., 1969, 31, 471–478 CrossRef CAS.
- I. Dalmazio and T. P. Campos, Appl. Radiat. Isot., 2019, 145, 193–197 CrossRef CAS PubMed.
- S. Zhou, M. Schlangen, J. Li, X. N. Wu and H. Schwarz, Chem. – Eur. J., 2015, 21, 14305–14308 CrossRef CAS PubMed.
- S. Zhou, J. Li, M. Schlangen and H. Schwarz, Chem. – Eur. J., 2016, 22, 4336–4339 CrossRef CAS PubMed.
- S. Zhou, M. Schlangen and H. Schwarz, Phys. Chem. Chem. Phys., 2015, 17, 9564–9568 RSC.
- S. Le Caër, M. Heninger, J. Lemaire, P. Boissel, P. Maıtre and H. Mestdagh, Chem. Phys. Lett., 2004, 385, 273–279 CrossRef.
- G. S. Groenewold, W. A. De Jong, J. Oomens and M. J. Van Stipdonk, J. Am. Soc. Mass Spectrom, 2010, 21, 719–727 CrossRef CAS PubMed.
- J. Oomens, N. C. Polfer, G. Berden and J. R. Eyler, Eur. J. Mass Spectrom., 2019, 25, 86–96 CrossRef CAS PubMed.
- G. L. Tripodi, T. C. Correra, C. F. F. Angolini, B. R. V. Ferreira, P. Maître, M. N. Eberlin and J. Roithová, Eur. J. Org. Chem., 2019, 3560–3566 CrossRef CAS PubMed.
- J. K. Gibson, H.-S. Hu, M. J. Van Stipdonk, G. Berden, J. Oomens and J. Li, J. Phys. Chem. A, 2015, 119, 3366–3374 CrossRef CAS PubMed.
- G. S. Groenewold, A. K. Gianotto, K. C. Cossel, M. J. Van Stipdonk, D. T. Moore, N. Polfer, J. Oomens, W. A. De Jong and L. Visscher, J. Am. Chem. Soc., 2006, 128, 4802–4813 CrossRef CAS PubMed.
- S.-X. Hu, J. K. Gibson, W.-L. Li, M. J. Van Stipdonk, J. Martens, G. Berden, B. Redlich, J. Oomens and J. Li, Chem. Commun., 2016, 52, 12761–12764 RSC.
- J. Jian, S.-X. Hu, W.-L. Li, M. J. van Stipdonk, J. Martens, G. Berden, J. Oomens, J. Li and J. K. Gibson, Inorg. Chem., 2018, 57, 4125–4134 CrossRef CAS PubMed.
- J. Martens, G. Berden, C. R. Gebhardt and J. Oomens, Rev. Sci. Instrum., 2016, 87, 103108 CrossRef PubMed.
- D. Oepts, A. F. G. Van der Meer and P. W. Van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- G. Berden, M. Derksen, K. J. Houthuijs, J. Martens and J. Oomens, Int. J. Mass Spectrom., 2019, 443, 1–8 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.01, 2016 Search PubMed.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS.
- A. A. El-Azhary and H. U. Suter, J. Phys. Chem., 1996, 100, 15056–15063 CrossRef CAS.
- S. Kawaguchi, Coord. Chem. Rev., 1986, 70, 51–84 CrossRef CAS.
- J. N. Spencer, E. S. Holmboe, M. R. Kirshenbaum, D. W. Firth and P. B. Pinto, Can. J. Chem., 1982, 60, 1178–1182 CrossRef CAS.
- M. M. Folkendt, B. E. Weiss-Lopez, J. P. Chauvel Jr and N. S. True, J. Phys. Chem., 1985, 89, 3347–3352 CrossRef CAS.
- D. T. Moore, J. Oomens, J. R. Eyler, G. von Helden, G. Meijer and R. C. Dunbar, J. Am. Chem. Soc., 2005, 127, 7243–7254 CrossRef CAS PubMed.
- L. MacAleese and P. Maitre, Mass Spectrom. Rev., 2007, 26, 583–605 CrossRef CAS PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, The f-block metals: lanthanoids and actinoids, Pearson, Barcelona, Spain, 4th edn, 2012, pp. 1002–1032 Search PubMed.
- S. Schinzel, M. Bindl, M. Visseaux and H. Chermette, J. Phys. Chem. A, 2006, 110, 11324–11331 CrossRef CAS PubMed.
- W. Caminati and J.-U. Grabow, J. Am. Chem. Soc., 2006, 128, 854–857 CrossRef CAS PubMed.
- S. A. Broadbent, L. A. Burns, C. Chatterjee and P. H. Vaccaro, Chem. Phys. Lett., 2007, 434, 31–37 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01890b |
| This journal is © the Owner Societies 2020 |