
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomistic QM/MM simulations of the strength of covalent interfaces in carbon nanotube–polymer composites†
Jacek R.
Gołębiowski
 ab,
James R.
Kermode
c,
Peter D.
Haynes
abd and
Arash A.
Mostofi
*abd
ab,
James R.
Kermode
c,
Peter D.
Haynes
abd and
Arash A.
Mostofi
*abd
aDepartment of Materials, Imperial College London, London SW7 2AZ, UK. E-mail: a.mostofi@imperial.ac.uk
bThomas Young Centre for Theory and Simulation of Materials, Imperial College London, London SW7 2AZ, UK
cWarwick Centre for Predictive Modelling, School of Engineering, University of Warwick, Coventry, CV4 7AL, UK
dDepartment of Physics, Imperial College London, London SW7 2AZ, UK
First published on 5th May 2020
Abstract
We investigate the failure of carbon-nanotube/polymer composites by using a recently-developed hybrid quantum-mechanical/molecular-mechanical (QM/MM) approach to simulate nanotube pull-out from a cross-linked polyethene matrix. Our study focuses on the strength and failure modes of covalently-bonded nanotube–polymer interfaces based on amine, carbene and carboxyl functional groups and a [2+1] cycloaddition. We find that the choice of the functional group linking the polymer matrix to the nanotube determines the effective strength of the interface, which can be increased by up to 50% (up to the limit dictated by the strength of the polymer backbone itself) by choosing groups with higher interfacial binding energy. We rank the functional groups presented in this work based on the strength of the resulting interface and suggest broad guidelines for the rational design of nanotube functionalisation for nanotube–polymer composites.
1 Introduction
Functionalised carbon-nanotube/polymer composites (CNPC) are a promising structural material for demanding applications such as ballistic protection1,2 and in the aerospace industry. The current generation of CNPCs, however, fall short of their theoretical limit3,4 for mechanical properties, indicating that there is room for improving the efficiency of transferring external load to the interface between the carbon nanotubes (CNT) and the polymer matrix, and thereby more effectively realising the potential of CNT reinforcement. Key factors affecting the quality of load transfer in CNPCs include the dispersion3,5–8 and alignment3,6 of the fibres, as well as the strength of interfacial bonding.7,9–14 The latter is of critical importance3,15 and is the focus of this work.Experimental studies of CNPCs have shown that the quality of the interface can be significantly improved by the introduction of chemical cross-links between CNTs and the polymer matrix. CNT pull-out tests using atomic-force microscopy have shown that functionalisations that promote nanotube-matrix cross-links can improve the interfacial shear strength (ISS) from 40 MPa to 150 MPa16 and similar trends have been observed by analysing CNPC Raman spectra10,17,18 and in fracture tests.19 The conclusions of these experiments are supported by computational investigations: Frankland et al.20 have simulated CNT pull-out from crystalline polyethene (PE), finding that the ISS can be improved from 2.8 MPa to 110 MPa by introducing nanotube–polymer cross-links; Namilae et al.21 have reported an increase of ISS from 12.5 MPa to 1000 MPa; Chowdhury et al.22 report an ISS of 250–300 MPa for a crystalline CNT–PE interface and, while they do not provide a corresponding value for a non-bonded interface, their result can be contrasted with other CNT pull-out simulations which give ISS within the range of 1–150 MPa.20–33
Whilst it has been shown that CNT functionalisation can have a significant impact on CNPC properties3,15,34 and the effect of varying functionalisation has been studied extensively from the perspective of CNT dispersion in the polymer matrix,7,8,35 less effort has been devoted to discriminating between various functionalisation strategies based on the strength of the resulting covalently-bonded interface, with most studies focusing on the comparison between bonded and non-bonded interfaces.17,18,20–23 Experimental studies concerned with development of high-performance functionalised CNT–polymer composites often rely on CNTs functionalised with carboxyl,16–19,36–39 amine10,38 or carbene36,40,41 groups to create covalently bonded interfaces while better performing candidates are available.
It is important to note that the chemical composition of the interface is not the only factor affecting the strength of carbon nanotube–polymer connection and tuning combination of facets can lead to greatly improved composite performance. For CNPCs based on functionalized nanotubes, the ISS has been shown to increase with the increase in matrix density,22 the crystallinity of the polymer20 and functionalization density.21,32 However, only weak dependence have been found between interfacial strength and CNT embedded length.23 For composites with non-bonded interface, it has been found that the ISS is positively correlated with nanotube diameter27,33 and only weakly coupled with CNT length.23,27
Mechanical failure of the CNPC originates from local changes in chemical bonding that occur at the nanoscale but propagate via stress transfer through the polymer matrix at micrometre lengths. Accurately modelling the former requires a quantum-mechanical (QM) treatment of bond-breaking processes, but fully quantum-mechanical methods are too computationally expensive to apply on a routine basis to systems and processes at these length- and time-scales. This issue is often addressed through the use of classical molecular mechanical (MM) models based on parametrised empirical force-fields such as ReaxFF,42 REBO43 or AIREBO.44 Such models can be calibrated to reproduce QM results,45 but laborious refitting is required for each different chemistry and transferability to configurations not featured in the training set is not guaranteed. The limitations of such potentials for this particular problem were demonstrated in earlier work46 where detachment of various functional groups from a CNT surface was simulated using ReaxFF and a fully QM approach. The results showed that bond-breaking processes were not described accurately with the reactive force-field, with significant qualitative differences as compared to the full QM simulations. Here, we use a concurrent hybrid QM/MM method46 to tackle the multiscale problem of critical failure. In this approach, the system evolves according to a relatively simple MM model, but with regions where bond-breaking occurs identified on the fly and the atomic forces in these regions are computed using a QM approach. This method gave excellent agreement with the fully quantum calculations of the functional group detachment46 at a fraction of computational cost. Similar QM/MM techniques have been used successfully to simulate crack propagation in silicon,47,48 dislocation motion in metals49 and molecular adsorption on a CNT surface.50,51
In this work, we study how the chemical structure at the CNT–polymer interface determines its failure characteristics by simulating CNT pull-out from a cross-linked PE matrix. We focus on CNT–polymer interfaces composed of commonly used amine, carbene and carboxyl functional groups as well as a [2+1] cycloaddition. The systems studied differ only in the choice of the functional group linking the polymer matrix to the nanotube, allowing a direct comparison of the interfacial strength and failure characteristics of each functionalisation. Our findings allow us to make practical recommendations that will guide further experimental work. Additionally, the molecular resolution offered by our computational study enables us to analyse and explain the underlying interfacial failure mechanisms.
The remainder of this paper is structured as follows: our hybrid QM/MM approach is briefly summarised in Section 2 alongside a detailed description of the simulation setup; Section 3 is devoted to the analysis of the results of our nanotube fibre pull-out simulation; and we summarise our main conclusions in Section 4.
2 Methods
The single CNT pull-out simulations conducted in this study were carried out using the QM/MM hybrid method introduced and demonstrated by Gołębiowski et al.46 In this approach, which builds on the ‘Learn on the Fly’ (LOTF) scheme originally introduced by De Vita and co-workers,47,52 the focus is placed on ensuring forces for all atoms are sufficiently accurate to ensure accurate atomic trajectories and hence accurate measurement of derived observables. Here, this is achieved by combining forces computed with classical and QM simulations using the adaptive buffered force mixing variant of QM/MM,53 rather than derived from a combined Hamiltonian as in the original LOTF method.Classical force-fields are typically designed to accurately describe atomic systems in the vicinity of equilibrium configurations making them capable of describing the general morphology of the system. However, when the structure is significantly disturbed such as in the case of interfacial failure, the description can be incorrect. In the classical picture, local departure from the stable configuration is marked by elevated potential energies, therefore in our hybrid scheme particles with energies exceeding a pre-defined threshold are flagged for quantum-mechanical treatment; additional details about the flagging mechanism can be found in the ESI.† Marked atoms are used to form the inner QM clusters by selecting all particles within a set number of bond hops of each flagged particle and expanded by including a buffer region with a specific width (also defined in terms of bond hops) to form full clusters. The resulting full clusters are carved out of the system, isolated and have all dangling bonds passivated by hydrogenation to prepare structures for quantum mechanical calculations. After the QM computation, classical forces on the inner cluster atoms are replaced by the quantum forces computed for those particles to form a complete QM/MM description and the total sum of forces is adjusted to zero to conserve momentum.
We used the generalised Amber force-field (GAFF),54 as implemented in LAMMPS,55 as our MM model. The standard GAFF model was refitted for bond and angle terms in the CNT and the polyethene chain, in order to match the lattice constant and elastic response around the equilibrium obtained with the force-field and the QM method for a (10,0) CNT and a 25 monomer PE chain as described by Gołębiowski et al.46 The QM method used is density-functional tight binding (DFTB),56 as implemented in the PLATO57 simulation package. DFTB calculations were performed with self-consistent charge equilibration with an electronic temperature of 0.2 eV and Pulay mixing, using the parameters by Krishnapriyan et al.58 optimised for atomization energy and interatomic forces in C, H, N, and O system. In this work we do not use the electrostatic embedding approach for our quantum calculations but rather use a mechanical embedding technique and rely on a wide buffer to ensure an accurate description of forces in the core of the QM regions, following the discussion in Peguiron et al.;59 see Gołębiowski et al.46 for more details. Throughout the pull-out simulations, an NVT ensemble was enforced using the Langevin thermostat with the friction coefficient of 0.01 as implemented in the atomic simulation environment.60 The simulations were carried out at a temperature of 100 K. The effect of changing the temperature from 100 K to 300 K was found to be negligible (see ESI† for details). DFTB calculations do not include a dispersion term and the forces on the atoms of the inner clusters were adjusted post hoc by including dispersion interactions with respect to all of the particles in the system as discussed by Zhechkov et al.61 The correction was obtained from the Lennard-Jones term of the GAFF, as described in the ESI.†
2.1 Simulation setup
A CNT embedded in a cross-linked PE matrix was gradually pulled out of the matrix and the response of the system was analysed. The preparation of the simulation cell is described briefly below, with further details provided in the ESI.†The initial polymeric structure was a slab with a thickness of 50 Å and density equal to the target density of 0.3 g cm−3. The slab was composed of fifty 48-monomer polyethene-like chains, each made out of six alternating units of propylene, acetylene and propylene organised in the following fashion: H–(–CH2–CH2–CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2–CH2–CH2–)6–H. The initial chain configuration was obtained using a Monte Carlo sampling procedure as implemented in the MedeA software package.62
CH–CH2–CH2–CH2–)6–H. The initial chain configuration was obtained using a Monte Carlo sampling procedure as implemented in the MedeA software package.62
The next step involved creating a void inside the polymer cell by introducing seven Lennard-Jones (LJ) repulsive beads spaced equally between the centre of the slab's free surface and a point inside the polymeric structure located at the midpoints of the periodic axis and 35 Å away from the surface in the non-periodic axis. The strength of LJ repulsion was increased quasi-statically by changing the depth of the potential well from 4 meV to 200 meV and the equilibrium distance from 1 Å to 8 Å over the course of ten iterations, each including parameters change and geometry optimisation with the FIRE algorithm.63 Finally the values were kept constant for 0.1 ns in an NVT simulation at 300 K to allow for local equilibration, after which the beads were removed from the matrix.
Once the process was completed, a 30 Å long, (10,0) carbon nanotube fragment with hydrogen passivated ends was placed inside the newly created void; its position was chosen so that it aligned with the repulsive beads resulting in one end of the CNT inside the slab and the other end aligned with the slab free surface. CNT functionalisation was then achieved by introducing three additional PE chains, identical to those used to construct the polymer bulk, which were attached to the CNT surface using amine, carbene or carboxyl functional groups or a [2+1] cycloaddition resulting in a functionalisation density of one group per 100 CNT carbon atoms. Schematic representations of the four covalently-bonded CNT–polymer interfaces is shown in Fig. 1. After functionalisation, the system was equilibrated using the procedure of Kucukpinar and Doruker,64 which involves compression to 0.5 GPa over 0.3 ns and relaxation over an additional 0.3 ns, then heating the sample to 800 K over 0.5 ns and subsequent cooling to 300 K over the same period and a final compression–relaxation cycle identical to the first one. Finally, cross-linking of the polymer network was carried out by introducing atomic bonds between all carbon atoms bonded with three ions closer than 3.5 Å during a 0.5 ns simulation in an NVT ensemble at 800 K to generate a fully connected polymer network. All simulations in the above-described process were performed with the GAFF,54 as implemented in LAMMPS55 with the timestep of 1 fs. We believe that a fully classical model is sufficiently accurate to identify generally correct morphology.
 | ||
| Fig. 1 Schematic representation of interfacial structure between the CNT and polymer matrix. The four panels show different functional groups used to connect polyethene chains (represented as R) to the CNT.‡ | ||
The procedure described above yields a cross-linked PE matrix reinforced with a 30 Å functionalised CNT that accounts for 12% of the total weight (see Fig. 2). The average density of the resulting cross-linked PE was found to be approximately 0.945 g cm−3, which is consistent with experimental findings,65 and the cross-link density of the polymer network, defined as a ratio of cross-linked carbon atoms to total carbon atoms was found to be approximately 10%.
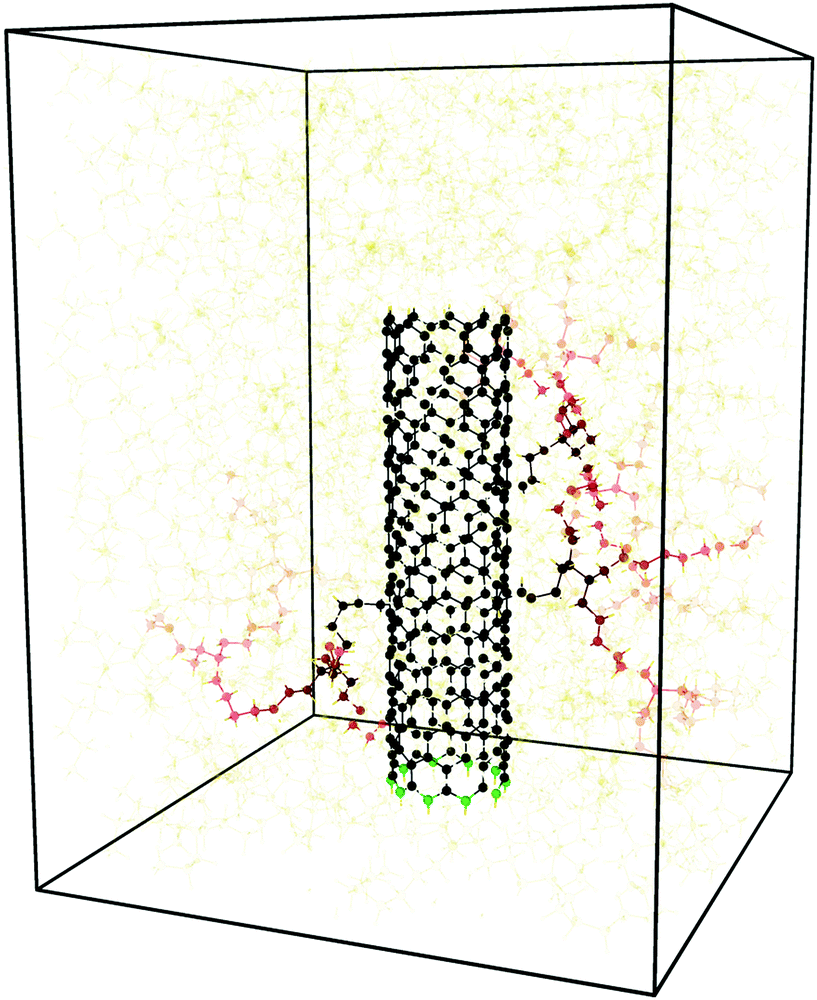 | ||
| Fig. 2 A representative atomic configuration for the CNT–PE system, composed of 53 48-monomer PE chains cross-linked by C–C bonds, reinforced with a 30 Å, (10,0) functionalised CNT; three polymer chains are covalently bonded to the CNT surface resulting in a functionalisation density of one group per 100 CNT carbon atoms. Amine, carbene or carboxyl functional groups or a [2+1] cycloaddition were used to link the CNT and polymer matrix throughout the study; this figure shows the carbene attachment. CNT atoms and functional groups are marked in black while the polymer atoms are coloured with a gradient changing from black to yellow depending on the distance from the attachment point measured in bond hops. The ring of carbon atoms at the bottom of the CNT marked in green was constrained to move at a constant velocity inducing nanotube pull-out. | ||
CNT pull-out was simulated with the QM/MM method by extracting the CNT from the polymer slab at a constant velocity while analysing the response of the system. After equilibration for 10 ps, the terminating ring of carbon atoms aligned with the composite slab free surface was constrained to move outwards of the slab with a velocity which was gradually increased over the course of 1 ps from 0 Å fs−1 to 10−3 Å fs−1 (equivalent to the strain rates that could be observed in ballistic tests1,2). Such pull-out velocity is high when compared to experimental pull-out tests but is comparable to velocities used in computational studies with classical potentials21,23,24 that do not the include the accurate and computationally expensive quantum mechanical description of bond-breaking. The constrained CNT atoms are marked in green in Fig. 2. Following the velocity ramp-up, the pull-out simulation was carried out by keeping the pull-out velocity constant and recording the forces acting on the constrained ring of atoms.
3 Results and discussion
For each attachment chemistry shown in Fig. 1, the initial structure was prepared as discussed in Section 2. The simulation procedure including generating the initial atomic structure and simulating the CNT pull-out has been performed for an ensemble of ten independent configurations for each functionalisation chemistry.For each simulation, the component parallel to the pull-out direction of the force acting on the constrained ring of atoms was recorded at each timestep as the pull-out force and plotted as a function of CNT displacement. The resulting curve was smoothed using a 100 fs time window with a top-hat smoothing filter to average out high-frequency vibrations associated with internal modes of the CNT. The smoothed curve displays local peaks that are associated with bond-breaking events; we use the value of the force at each of these peaks (which we refer to as the “critical force”) as an indicator of the strength of the interface. The method for determining the exact location of each bond-breaking event is discussed in more detail in the ESI.† The top panel of Fig. 3 shows a typical example of the smoothed force–displacement curve, with peaks corresponding to failure marked with stars; there are three such events per simulation as each system is prepared with three functionalised attachments between the CNT and the polymer matrix. The bottom panel of the same figure shows the number of atoms flagged for QM calculation at each point of the simulation. It can be seen that the more computationally-demanding QM calculations are only performed when necessary, i.e., just before, during and just after each series of bond rearrangements and that atoms are promptly de-flagged following a failure event. It can also be seen that at any point during the simulation, the number of atoms for which QM forces are calculated is a small fraction of the total number of atoms in the system (typically less than 1% of the total number of atoms at any given time).
 | ||
| Fig. 3 Top panel: Pull-out force vs. displacement plot for a single representative simulation of the CH2-functionalised system. The force peaks associated with bond-breaking events are highlighted (stars). Shaded areas show times when at least one atom was treated quantum mechanically. Bottom panel: The total number of atoms marked as quantum mechanical at a given time. | ||
The critical forces recorded from each pull-out simulation have been aggregated and the distributions describing the strength of the interface in each case forms the key result of this work: the histograms and aggregate statistics are shown in Fig. 4. We find that interfaces based on amine (NH), carbene (CH2) and carboxyl (COOH) functional groups are characterised by similar strength; there is, however, an improvement in ISS of approximately 50% when the [2+1] cycloaddition (CH) is used to attach the polymer to the CNT. The first three functionalisations rely on a single covalent bond to graft PE chains to the CNT, whereas the cycloaddition is grafted with a pair of single bonds. The systems that we have investigated are identical except for the chemistry of the attachments, indicating that the functionalisation can indeed be chosen to enhance the ISS of the composite. In their study, Milowska et al.66 have shown that the first group of attachments (NH, CH2 and CH) have similar binding energies of approximately 1.3 eV while the [2+1] cycloaddition are characterised by a higher value of approximately 2.3 eV. This indicates that the binding energy of a functional group can be seen as a proxy for the critical force of the attachment and therefore used to gauge the strength of the resulting interface.
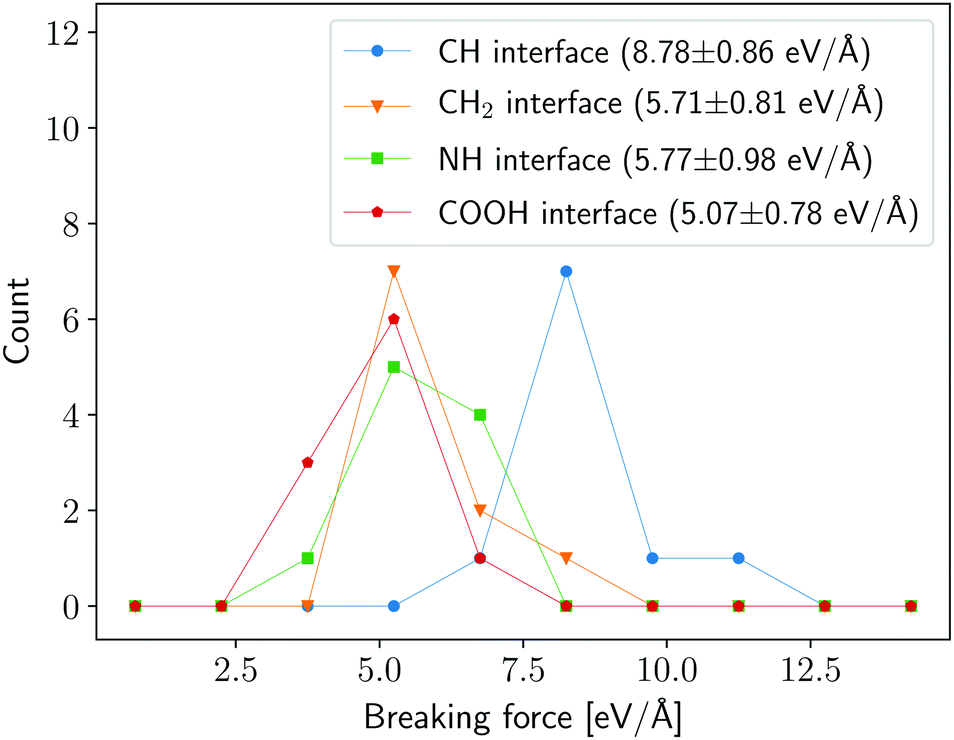 | ||
| Fig. 4 Histograms of maximum forces necessary to pull out a CNT embedded in a polymeric matrix. Different colours represent results for composite systems with different interfaces as discussed in Section 2. The mean and standard deviation of each functionalisation is shown in parenthesis in the legend. The bins used in the histogram have a width of 1.5 eV Å−1 with the left-most edge of the first bin located at 0; the data points are shown at the midpoint of each bin. | ||
For each pull-out simulation, the area under the pull-out force–CNT displacement curve provides a measure of the interfacial fracture energy; the distribution of fracture energy across the ensemble of ten simulations for each functional group are portrayed in Fig. 5. The results show that interfaces based on amine, carbene and carboxyl functional groups have similar fracture energy; however, an improvement of approximately 70% is found when a [2+1] cycloaddition is used to anchor the polymer chains to the CNT. The difference between fracture energy values of the carboxyl-based interface and amine- or carbene-based ones was analysed with a two-sided t-test for the null hypothesis that two independent samples (with the COOH-based interface as the first sample and combined results for NH and CH2 interfaces as the second sample) have identical expected values. The test concluded with a p-value of 0.037. In comparison, a similar test performed with amine, carbene and carboxyl interfaces in one sample and the [2+1] cycloaddition-based connection as the second resulted in a p-value of 5 × 10−7, reinforcing the conclusion that our findings can be used to clearly divide the interfaces into two major classes with significantly different fracture energy performance.
 | ||
| Fig. 5 Histogram of interfacial fracture energy for composite systems with various interfaces. Numbers in parenthesis represent mean and standard deviation of the results in eV, respectively. The bins used in the histogram have the width of 45 eV with the leftmost edge of the first bin located at 0; the markers are placed at the midpoint of each bin. | ||
The mechanism of interfacial failure has been further studied by inspecting the yield strength and atomistic structure of individual attachments in each of the composite systems. The analysis reveals that in systems where CH2, NH and COOH functional groups were used to form the interface, critical failure occurred by the failure of the functional group and detachment of polymer chains from the CNT surface demonstrating that the grafting point itself is the weakest element of the interface. On the other hand, in the case of [2+1] cycloadditions (CH), two equally common failure modes were observed: detachment of the functional group from the CNT and bond-breaking within the polymer matrix. The distribution of the strength of individual attachments, discriminated by their failure mode is shown in Fig. 6. The [2+1] cycloaddition-based interface and the polymer backbone fail at a similar load, demonstrating that their strengths are comparable and a further increase of the strength of the interface would not yield any effective improvement without fortifying the polymer matrix itself. Our results show that the functionalisation chemistry can be tuned to improve the ISS up to a limit defined by the strength of the polymer backbone.
 | ||
| Fig. 6 Distribution of critical strength of individual attachments for simulations with interfaces exhibiting various modes of rupture. Mean and standard deviation of the results are shown in the legend. The bins used in the histogram have a width of 1.5 eV Å−1 with the left-most edge of the first bin located at 0; the data points are placed at the midpoint of each bin. | ||
Additional effort has been devoted to the analysis of the [2+1] cycloadditions (CH) based interface by exploring the effect of the attachment point geometry on the yield strength. Due to the structure of carbon nanotubes, [2+1] cycloaddition can be found in two configurations, attached to two CNT atoms located parallel to the CNT axis or at an angle of 45°. Fig. 7 shows the normalised distribution of the strength of individual attachments divided into these two configurations. It can be seen that the orientation of the attachments is not a major factor for the designing of high-strength CNT–polymer interfaces based on functional groups attached with two bonds.
 | ||
| Fig. 7 Distribution of critical strength of individual attachments for simulations with interface composed of CH groups in various orientations. The total number of sideways attachments is 24 while the number of parallel ones is 6, therefore the counts are normalised for ease of comparison. The mean and standard deviation of the results are shown in the legend. The bins used in the histogram have a width of 1.5 eV Å−1 with the left-most edge of the first bin located at 0; the data points are placed at the midpoint of each bin. | ||
Quantitative comparison of our calculated ISS with experimental measurements is very challenging for systems as complex as real CNT–polymer composites. The pull-out velocity commonly used in computational pull-out studies21,23,24 is significantly higher than is experimentally achievable and limits the ability for the polymer to respond to the CNT motion. This is exacerbated by the differences in CNT diameters and lengths which vary by orders of magnitude between simulated and experimental systems. However, we have compared our results to the experimental work of Barber et al.16 who performed pull-out experiments of carboxyl-functionalised CNTs. The experiments of Barber et al.16 demonstrate that the pull-out force is proportional to the embedded surface area of the CNT and recorded the ISS (maximum pullout force over CNT area) of around 50 MPa. For comparison, our simulations give a value of around 1000 MPa, i.e., a factor of 20 larger. Putting aside the difference in chemistry, pull-out velocity and the size§ between the experimental system and our model, a crucial piece of information is missing to enable us to relate these two values, namely the density of covalent attachments between the CNT and the polymer matrix in the experiment. In our model we have three attachments over the surface area of a 300 carbon atoms CNT, but the equivalent grafting ratio is not known for the experimental system in Barber et al.,16 and this makes it very difficult to make a reasonable attempt at a comparison. Other experimental studies concerned with processing of carbon nanotubes–polymer composites have measured grafting ratios between a factor of 1.5 to a factor of 30 smaller than in our study.67 Such differences in grafting ratios are likely to be a significant determinant of the discrepancy between our simulated values of Fmax/A and the experimental measurements of Barber et al.16
4 Conclusions
We investigated the critical failure of covalently bonded CNT–polymer interfaces by simulating pull-out of a CNT fibre from a cross-linked polyethene matrix. We focused on the strength and failure modes of composite interfaces composed of polymer chains grafted to the CNT surface by amine, carbene and carboxyl functional groups and via [2+1] cycloaddition. We used a hybrid QM/MM simulation method whereby the system evolves according to a relatively simple classical force-field, but regions that stray outside of the scope of accuracy of the force-field (e.g., where bond-breaking events are about to occur) are identified on the fly as the dynamical simulation progresses and the atomic forces in these regions are computed using a more accurate QM approach.We found that interfaces based on CH2, NH and COOH functional groups exhibit similar strength, suggesting that other factors, such as CNT dispersion, should take precedence when discriminating between them. With the [2+1] cycloaddition, however, a significant improvement in interfacial shear strength and fracture energy was observed, with the resulting interface found to be 50% stronger and 70% tougher than the other cases studied. The [2+1] cycloaddition is characterised by a higher binding energy than the other functional groups,66 indicating a correlation between the functional group binding energy and the resulting ISS. Furthermore, in the case of the [2+1] cycloaddition, we observed two distinct failure modes in our simulations: detachment of the functional group from the CNT, and rupture of the polymer backbone inside the matrix. Both were found to occur at similar critical strengths, demonstrating that the grafting strength is on a par with the strength of the polymer backbone for the [2+1] cycloaddition-based interfaces studied here, effectively maximising the ISS for the composite.
This is the first time this on-the-fly hybrid QM/MM approach has been used to simulate failure in CNT pull-out at time- and length-scales usually only accessible to purely classical methods, and expands the simulation toolkit available for studying these inherently multiscale systems.
Conflicts of interest
The authors declare no competing financial or non-financial interests.Acknowledgements
We thank Andrew Horsfield for his help with the DFTB simulations. This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant Agreement no. 642890 through TheLink research and training project. This work was supported through a studentship in the Centre for Doctoral Training on Theory and Simulation of Materials at Imperial College London, funded by the EPSRC (EP/L015579/1). Calculations were performed on the Imperial College London High-Performance Computing Service. We acknowledge Materials Design for providing a licence for the MedeA simulation toolkit, and the Thomas Young Centre under grant number TYC-101.Notes and references
- S. Laurenzi, R. Pastore, G. Giannini and M. Marchetti, Compos. Struct., 2013, 99, 62–68 CrossRef.
- P. Jindal, S. Pande, P. Sharma, V. Mangla, A. Chaudhury, D. Patel, B. P. Singh, R. B. Mathur and M. Goyal, Composites, Part B, 2013, 45, 417–422 CrossRef CAS.
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- Y. Liu and S. Kumar, ACS Appl. Mater. Interfaces, 2014, 6, 6069–6087 CrossRef CAS PubMed.
- M. L. Manchado, L. Valentini, J. Biagiotti and J. Kenny, Carbon, 2005, 43, 1499–1505 CrossRef CAS.
- X. L. Xie, Y. W. Mai and X. P. Zhou, Mater. Sci. Eng., R, 2005, 49, 89–112 CrossRef.
- P. C. Ma, N. A. Siddiqui, G. Marom and J. K. Kim, Composites, Part A, 2010, 41, 1345–1367 CrossRef.
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS.
- B. Arash, Q. Wang and V. K. Varadan, Sci. Rep., 2014, 4, 6479 CrossRef CAS PubMed.
- P. C. Ma, Q. B. Zheng, E. Mader and J. K. Kim, Polymer, 2012, 53, 6081–6088 CrossRef CAS.
- M. R. Loos, S. H. Pezzin, S. C. Amico, C. P. Bergmann and L. A. F. Coelho, J. Mater. Sci., 2008, 43, 6064–6069 CrossRef CAS.
- F. Buffa, G. A. Abraham, B. P. Grady and D. Resasco, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 490–501 CrossRef CAS.
- T. Chang, L. Jensen, A. Kisliuk, R. Pipes, R. Pyrz and A. Sokolov, Polymer, 2005, 46, 439–444 CrossRef CAS.
- D. Qian, E. C. Dickey, R. Andrews and T. Rantell, Appl. Phys. Lett., 2000, 76, 2868 CrossRef CAS.
- M. Rahmat and P. Hubert, Compos. Sci. Technol., 2011, 72, 72–84 CrossRef CAS.
- A. H. Barber, S. R. Cohen, A. Eitan, L. S. Schadler and H. D. Wagner, Adv. Mater., 2006, 18, 83–87 CrossRef CAS.
- N. Lachman, C. Bartholome, P. Miaudet, M. Maugey, P. Poulin and H. D. Wagner, J. Phys. Chem. C, 2009, 113, 4751–4754 CrossRef CAS.
- D. Roy, S. Bhattacharyya, A. Rachamim, A. Plati and M.-L. L. Saboungi, J. Appl. Phys., 2010, 107, 043501 CrossRef.
- N. Lachman and H. Daniel Wagner, Composites, Part A, 2010, 41, 1093–1098 CrossRef.
- S. J. V. Frankland, A. Caglar, D. W. Brenner and M. Griebel, J. Phys. Chem. B, 2002, 106, 3046–3048 CrossRef CAS.
- S. Namilae, U. Chandra, A. Srinivasan and N. Chandra, Comput. Model. Eng. Sci., 2007, 22, 189–202 Search PubMed.
- S. Chowdhury and T. Okabe, Composites, Part A, 2007, 38, 747–754 CrossRef.
- S. Bagchi, A. Harpale and H. B. Chew, Proc. R. Soc. A, 2018, 474(2216), 20170705, DOI:10.1098/rspa.2017.0705.
- S. Frankland and V. Harik, Surf. Sci., 2003, 525, L103–L108 CrossRef CAS.
- J. Gou, B. Minaie, B. Wang, Z. Liang and C. Zhang, Comput. Mater. Sci., 2004, 31, 225–236 CrossRef CAS.
- S. Haghighatpanah and K. Bolton, Comput. Mater. Sci., 2013, 69, 443–454 CrossRef CAS.
- Y. Li, Y. Liu, X. Peng, C. Yan, S. Liu and N. Hu, Comput. Mater. Sci., 2011, 50, 1854–1860 CrossRef CAS.
- K. Liao and S. Li, Appl. Phys. Lett., 2001, 79, 4225–4227 CrossRef CAS.
- K. Sharma, K. Sen Kaushalyayan and M. Shukla, Comput. Mater. Sci., 2015, 99, 232–241 CrossRef CAS.
- M. Wong, M. Paramsothy, X. J. Xu, Y. Ren, S. Li and K. Liao, Polymer, 2003, 44, 7757–7764 CrossRef CAS.
- Q. Xiong and S. Meguid, Eur. Polym. J., 2015, 69, 1–15 CrossRef CAS.
- Q. Zheng, Q. Xue, K. Yan, X. Gao, Q. Li and L. Hao, Polymer, 2008, 49, 800–808 CrossRef CAS.
- Q. Zheng, Q. Xue, K. Yan, X. Gao, Q. Li and L. Hao, J. Appl. Phys., 2008, 103, 44302 CrossRef.
- J. Chen, L. Yan, W. Song and D. Xu, Composites, Part A, 2018, 114, 149–169 CrossRef CAS.
- A. Eitan, K. Jiang, D. Dukes, R. Andrews and L. S. Schadler, Chem. Mater., 2003, 15, 3198–3201 CrossRef CAS.
- C. J. An, Y. C. Lee, Y. H. Kang and S. Y. Cho, Carbon, 2017, 124, 662–668 CrossRef CAS.
- Y. Li, C. Chen, J. Xu, Z. Zhang, B. Yuan and X. Huang, J. Mater. Sci., 2015, 50, 1117–1128 CrossRef CAS.
- A. Hameed, M. Islam, I. Ahmad, N. Mahmood, S. Saeed and H. Javed, Polym. Compos., 2015, 36, 1891–1898 CrossRef CAS.
- N. G. Sahoo, H. K. F. Cheng, J. Cai, L. Li, S. H. Chan, J. Zhao and S. Yu, Mater. Chem. Phys., 2009, 117, 313–320 CrossRef CAS.
- A. A. Koval'chuk, V. G. Shevchenko, A. N. Shchegolikhin, P. M. Nedorezova, A. N. Klyamkina and A. M. Aladyshev, Macromolecules, 2008, 41, 7536–7542 CrossRef.
- R. E. Gorga, K. K. S. Lau, K. K. Gleason and R. E. Cohen, J. Appl. Polym. Sci., 2006, 102, 1413–1418 CrossRef CAS.
- A. C. T. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- D. W. Brenner, O. A. Shenderova, J. A. Harrison, S. J. Stuart, B. Ni and S. B. Sinnott, J. Phys.: Condens. Matter, 2002, 14, 783–802 CrossRef CAS.
- S. J. Stuart, A. B. Tutein and J. A. Harrison, J. Chem. Phys., 2000, 112, 6472 CrossRef CAS.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, npj Comput. Mater., 2016, 2, 15011 CrossRef CAS.
- J. R. Gołębiowski, J. R. Kermode, A. A. Mostofi and P. D. Haynes, J. Chem. Phys., 2018, 149, 224102 CrossRef PubMed.
- G. Csányi, T. Albaret, M. C. Payne and A. De Vita, Phys. Rev. Lett., 2004, 93, 1–4 CrossRef PubMed.
- J. R. Kermode, T. Albaret, D. Sherman, N. Bernstein, P. Gumbsch, M. C. Payne, G. Csányi and A. De Vita, Nature, 2008, 455, 1224–1227 CrossRef CAS.
- F. Bianchini, A. Glielmo, J. R. Kermode and A. De Vita, Phys. Rev. Mater., 2019, 3, 043605 CrossRef CAS.
- P. N. Samanta and K. K. Das, Int. J. Quantum Chem., 2016, 116, 411–420 CrossRef CAS.
- K. Hu, Q. Zhuang, X. Liu and Z. Han, Polym. Compos., 2015, 36, 1454–1461 CrossRef CAS.
- A. De Vita and R. Car, Mater. Res. Soc. Symp. Proc., 1998, 491, 473–480 CrossRef CAS.
- N. Bernstein, J. R. Kermode and G. Csányi, Rep. Prog. Phys., 2009, 72, 26501 CrossRef.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- S. D. Kenny and A. P. Horsfield, Comput. Phys. Commun., 2009, 180, 2616–2621 CrossRef CAS.
- A. Krishnapriyan, P. Yang, A. M. N. Niklasson and M. J. Cawkwell, J. Chem. Theory Comput., 2017, 13(12), 6191–6200, DOI:10.1021/acs.jctc.7b00762.
- A. Peguiron, L. Colombi Ciacchi, A. De Vita, J. R. Kermode and G. Moras, J. Chem. Phys., 2015, 142, 064116 CrossRef PubMed.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- L. Zhechkov, T. Heine, S. Patchkovskii, G. Seifert and H. A. Duarte, J. Chem. Theory Comput., 2005, 1, 841–847 CrossRef CAS PubMed.
- Materials Design, Inc., Angel Fire, NM, USA, MedeA.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 1–4 CrossRef PubMed.
- E. Kucukpinar and P. Doruker, Polymer, 2006, 47, 7835–7845 CrossRef CAS.
- L. van Krevelen, Properties of Polymers, Elsevier, Amsterdam, 4th edn, 2009 Search PubMed.
- K. Z. Milowska and J. A. Majewski, J. Chem. Phys., 2013, 138, 194704 CrossRef PubMed.
- A. J. Clancy, H. S. Leese, N. Rubio, D. J. Buckley, J. L. Greenfield and M. S. Shaffer, Langmuir, 2018, 34, 15396–15402 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01841d |
| ‡ Figure partially reprinted from J. R. Gołębiowski, J. R. Kermode, A. A. Mostofi and P. D. Haynes, J. Chem. Phys., 2018, 149, 224102, DOI: 10.1063/1.5035508, with the permission of AIP Publishing. |
| § The authors of Barber et al.16 estimate their CNT diameters to be 80 ± 30 nm and their lengths to be 5–10 μm, whereas in our model we have a diameter that is two orders of magnitude smaller (0.8 nm) and a length that is over three orders of magnitude smaller (3 nm). |
| This journal is © the Owner Societies 2020 |