
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen adsorption on In2O3(111) and In2O3(110)†
Alvaro
Posada-Borbón
* and
Henrik
Grönbeck
 *
*
Department of Physics and Competence Centre for Catalysis, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: palvaro@chalmers.se; ghj@chalmers.se
First published on 26th June 2020
Abstract
In2O3-Based catalysts have been measured to have a high activity for CO2 hydrogenation to H3COH. Here, we use density functional theory calculations with and without Hubbard-U corrections in combination with ab initio thermodynamics to investigate the dissociative adsorption of H2 over In2O3(111) and In2O3(110). H2 is found to dissociate heterolytically with a moderate barrier on both facets. Diffusion of hydrogen leads to the preferred homolytic adsorption configuration. Vacancy formation by water formation is thermodynamically preferred at high hydrogen coverages. Both surfaces are found to be hydroxylated at typical reaction conditions with the highest coverage predicted for In2O3(110). O 1s core level shifts are calculated for different coverages. The hydroxylated surfaces show two distinct shifts corresponding to different types of OH-groups. The presence of surface oxygen vacancies is not visible in the O 1s signatures. The results show that hydroxylation of the surfaces results in changes of the oxidation state of In-ions, which suggests that the redox properties on In2O3 are important for catalytic reduction of CO2 to added value chemicals.
1 Introduction
The search for alternative energy solutions that reduce carbon emissions and facilitate the depart from fossil-fuels has accelerated during the last decades.1–7 Catalytic conversion of CO2 to methanol (H3COH) is one proposed path for synthesis of a fuel with high energy density and simultaneous reduction of greenhouse emissions.8–12 Conversion of CO2 to methanol could ideally, if renewable energy sources are used during conversion, allow for a closed carbon-loop with a net-zero carbon emission production of a valuable fuel or stock chemical.9–12In2O3 has recently attracted significant attention as a viable catalyst for conversion of CO2 to H3COH.13–21 In2O3 supported on ZrO2 has been reported to be highly active and selective under industrially relevant synthesis conditions (T = 573 K, P = 50 Bar, H2/CO2= 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).13 Very recently, improved catalytic activity for this reaction was reported for Pd dispersed on In2O3.16 The increase in activity in ref. 16 was attributed to facile H2 dissociation as compared to the case without Pd.
:1).13 Very recently, improved catalytic activity for this reaction was reported for Pd dispersed on In2O3.16 The increase in activity in ref. 16 was attributed to facile H2 dissociation as compared to the case without Pd.
The degree of surface hydroxylation on In2O3(111) upon H2 adsorption was investigated in ref. 14 Based on density functional theory (DFT) calculations, hydroxylation of the surface was suggested to be thermodynamically unfavorable at T = 573 K, PH2= 0.96 Bar and ΔμH2O = −2.0 eV, whereas creation of vacancies through water formation was calculated to be preferred.14 The relative thermodynamic stability of hydrogen adsorption was in ref. 14 investigated at a high hydrogen coverage with all oxygen atoms protonated together and H-atoms also on the In-sites.
Dissociation of H2 on In2O3(111) has been investigated both experimentally and theoretically.14–16 The apparent activation energy for H2 dissociation was in ref. 14 measured to be 0.9 eV. The barrier for dissociation was calculated to be ∼0.85 eV on the pristine surface.14–16 The presence of oxygen vacancies has been calculated to slightly reduce the barrier for dissociation.14,16
In a recent study using DFT-calculations,21 it was suggested that activation of H2 on pristine In2O3(110) is facile, with a barrier of only 0.3 eV. Moreover, it was proposed that the surface is heavily hydroxylated under typical reaction conditions (T = 573 K, PH2 = 10–50 Bar). The calculations also indicated that oxygen vacancies are relevant only at high hydrogen coverages and that hydroxylation induces a change in oxidation-state of undercoordinated In-ions. Facile H2 uptake on In2O3 has been measured at 300 K together with substantial surface reduction and reordering at higher temperatures.22 A change in oxidation state of the In-ions during CO2 reduction to H3COH has been measured using X-ray spectroscopy.20
The different results reported for In2O3(110) and In2O3(111) may suggest that H2 activation and surface hydroxylation depend on the surface orientation. However, the different coverages and thermodynamic conditions considered in the two studies14,21 might influence the results. Thus, an investigation that accounts for different coverages is needed. Additionally, the DFT results on In2O3 surface properties reported so far in the literature14–16,21 have been performed with the general-gradient approximation (GGA). The incomplete cancellation of the electron self-interaction in GGA is known to underestimate oxide band-gaps and give a spurious over-delocalization. Hubbard corrected (DFT+U) calculations are generally believed to describe better the electronic structure of metal-oxides, including In2O3.23
Herein, density functional theory (DFT) with and without Hubbard-U corrections and ab initio thermodynamics are used to investigate the dissociative adsorption of H2 on In2O3(111) and In2O3(110). The results suggest that surface hydroxylation is thermodynamically favorable under typical methanol synthesis reaction conditions, regardless of surface orientation. Hydroxylation is found to induces changes in the oxidation states of the In-cations. The O 1s core level shifts are evaluated to obtain spectroscopic signatures of different adsorbates. Comparison with experimental data suggests the presence of different OH-groups. The qualitative results are found not to depend on the inclusion of the Hubbard-U correction.
2 Computational details
Density Functional Theory (DFT) calculations are performed using the Vienna ab initio simulation package.24–27 The Kohn–Sham orbitals are expanded with a plane wave basis, truncated at a kinetic energy of 500 eV. Projector augmented wave potentials (PAW) are used to describe the interaction between the valence electrons and the core.28,29 Hydrogen, oxygen and indium are described with one, six and thirteen valence electrons, respectively. Thus, the valence of indium includes the 4d10 states. Integration over the Brillouin zone is approximated with finite sampling using the Monkhorst–Pack scheme.30 A 3 × 3 × 3 and 3 × 3 × 1 k-point grid is used for the bulk and surface calculations, respectively. The exchange–correlation energy is calculated with the Perdew, Burke, Ernzerhof (PBE) formulation.31 Given the semi-local nature of the approximation, electronic self-interaction might cause artificial delocalization of the electronic states. To localize the In 4d electrons, the effect of a Hubbard correction is investigated.32 The value of the Hubbard correction Ueff (7 eV) is adopted from ref.23 where it was determined by calculations of the lattice constant and the position of the In 4d states. The performance of the chosen U-value is studied by comparative calculations using a functional including exact exchange.33,34 The energies for oxygen vacancy formation and hydrogen adsorption are found to be in good agreement using the chosen U-value (see ESI†).Convergence in energy is assumed when a criteria of 10−6 eV is reached for the electronic optimization. Structural optimization is performed with the use of the conjugate gradient method and convergence is assumed when forces are smaller than 0.01 eV Å−1. Activation energies for H2 dissociative adsorption and Ov formation are calculated with use of the climbing-image nudge elastic band (CI-NEB)35 technique. Seven to ten images are used along the reaction coordinate to describe the minimum energy path. Vibrational frequency analysis is used to confirm the transition states. A force criteria of 0.05 eV Å−1 is used for the transition states.
Both In2O3(111) and In2O3(110) are modelled with (1 × 1) surface cells. The surface slabs are described with five [(111)] and four [(110)] layers. The bottom two layers of the slab models are fixed to their bulk positions during the geometry optimizations. Barriers are calculated for a three layer system for (111) to reduce computational cost. The slabs are separated by at least 15 Å of vacuum. The bond length of H2 in gas phase are calculated to be 0.75 Å, which is in good agreement with the experimental value of 0.74 Å.
To investigate the relative stability of surface hydroxylation and presence of oxygen vacancies at methanol synthesis conditions (573 K, 10–50 Bar, H2/H2O:10–50/1), we calculate the surface free energy (Δγ(T,p)) as a function of H2 chemical potential (μH2(T,p)) using ab initio thermodynamics.36,37 The surface free energy is computed according to:
 | (1) |
 | (2) |
 | (3) |
Thus, a negative value indicates exothermic adsorption.
The O 1s core-level shifts (CLS) are calculated in reference to an O atom in the bulk of the slab:
| ECLS = E* − Ebulk* | (4) |
Charge analysis of the structures of interest is performed via Bader charges as implemented with the code developed by Henkelman and co-workers.41
3 Results and discussion
3.1 In2O3 bulk
The (110) and (111) surfaces have been constructed from the bixbyite (space group 206) bulk structure, which has 80 atoms in the unit cell. The bixbyite structure is composed of two types of six-fold coordinated In-ions, with oxygen being four-fold coordinated. Density functional theory calculation with the general-gradient approximation are know to spuriously delocalize electronic states on metal–oxides. Bulk structure was optimized both with PBE and PBE+U. We find the equilibrium lattice constant calculated with PBE [PBE+U] functional to be 10.299 [10.030] Å. The experimental value is 10.121 Å. The corresponding values for the band gap are 0.94 (PBE) and 1.77 eV (PBE+U), which are in agreement with previous reports using a similar approach.23 The experimental band gap is 3.2 eV.42The effect of the Hubbard correction on the bulk electronic structure is visible in a Density of States (DOS) analysis, Fig. 1. With the PBE functional (top panel), the DOS is divided into three regions. The valence band spreads over approximately 5 eV and is mainly composed of O 2p states, with hybridization with some In 5s and In 5p. The second region, composed of mainly In 4d states with some hybridization with O 2s and O 2p states, is situated at an energy of approximately 11 eV below the Fermi energy and spans ∼2 eV. The third region, which is dominated by O 2s states is located at an energy of circa 17 eV below the Fermi energy. Applying the Hubbard-U correction (bottom panel) shifts the In 4d states in the second region to lower energies by approximately 2 eV and increases the spread of this region by ∼1 eV. In this way, the Hubbard-U correction places the 4d-states close to the experimental position.43 Moreover, a higher degree of hybridization between the In 4d and O 2s states is observed in both the second and third region. The valence band, which is dominated by O 2p and In states, is only weakly affected by the Hubbard correction. The effect of the Hubbard-U correction on the electronic structure is consistent with previous reports.23
 | ||
| Fig. 1 Density of states for bulk In2O3 using PBE and PBE+U. | ||
3.2 In2O3(111) and In2O3(110)
Stoichiometric In2O3(111) and In2O3(110) surfaces were cleaved from the bulk structure at the theoretical equilibrium lattice constant. We calculate the surface free energy with PBE [PBE+U] to be 0.78 [1.04] and 1.05 [1.36] J m−2 for (111) and (110), respectively. These results are in quantitative agreement with previous reports using a similar methodology.44The surface structures for In2O3(111) and In2O3(110) are shown in Fig. 2 and 3, respectively. The In2O3(111) surface is composed of stoichiometric layers, where each layer constitutes an O–In–O trilayer. In2O3(111) exhibits three slightly different geometric terminations, however, they are all energetically equivalent. The trilayer consist of an In-layer with 16 atoms sandwiched between two oxygen layers with 12 atoms in each. The In-atoms in the topmost layer can be grouped into six semi-equivalent types, labeled Ina–Inf in Fig. 2, while the oxygen can be grouped into four types, labeled Oa–Od. Inb and Ind are six-fold coordinated to oxygen, whereas the rest have a five-fold coordination. The six-fold coordinated In-ions are present in one region of the (1 × 1) surface cell, giving rise to a heterogeneous surface. The oxygen atoms in the topmost layer are three-fold coordinated to indium, whereas the rest are fourfold coordinated. The bonds at the surface are expanded with respect to the bulk values. The tri-layer at the surface is on average expanded by about 0.06 Å.
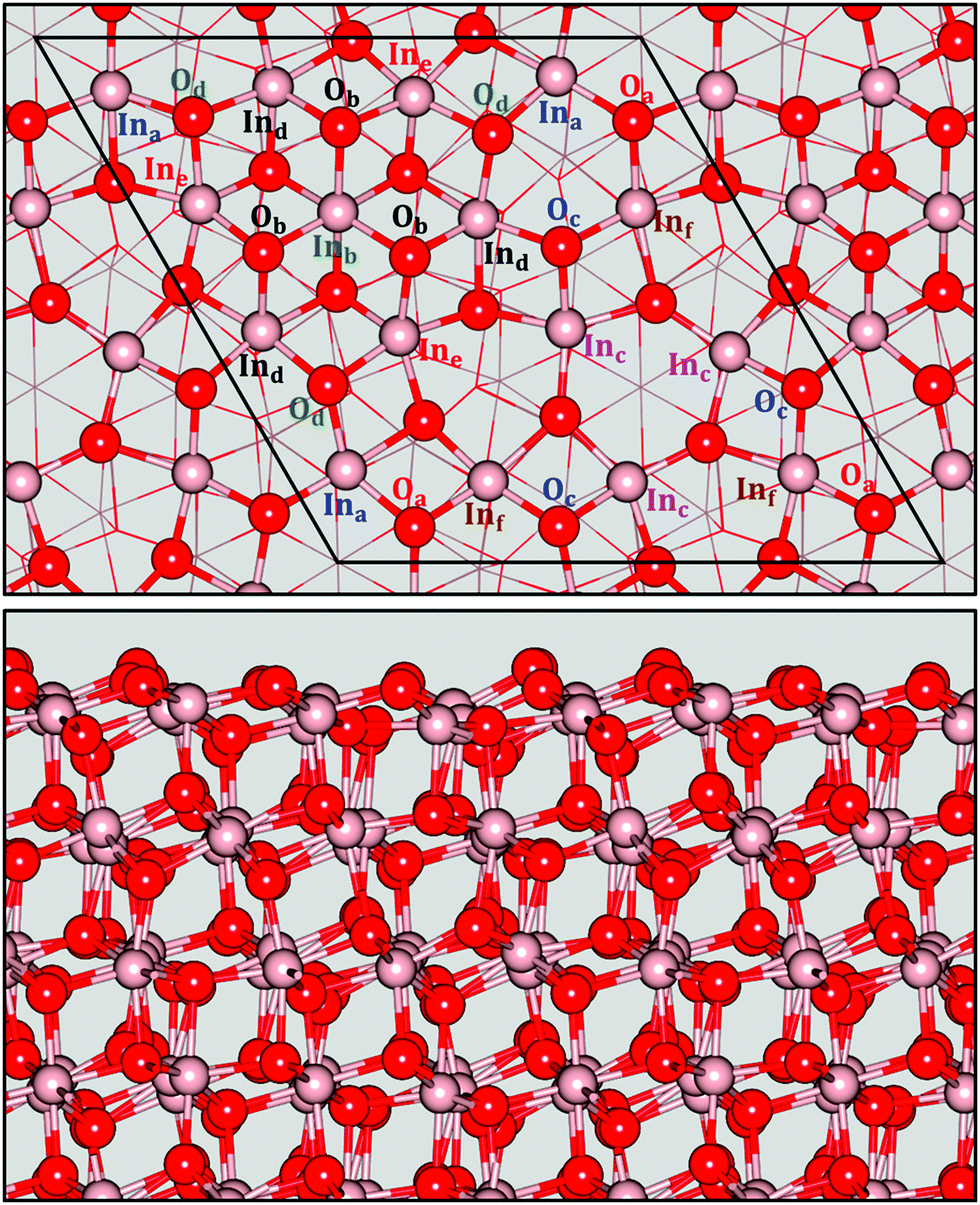 | ||
| Fig. 2 Structural model of In2O3(111). Top panel shows the oxygen and indium sites, labeled Oa–Od and Ina–Inf. A ball and stick model is used to show the topmost layer, whereas the rest are presented by lines. The surface cell is shown by black lines. The bottom panel shows a side view of the slab, using the ball and sticks representation. Atomic color code: Indium (brown), oxygen (red). | ||
 | ||
| Fig. 3 Structural model of In2O3(110). Top panel shows the oxygen and indium sites, labeled O1–O6 and In1–In4. A ball and stick model is used to show the topmost layer, whereas the rest are presented as lines. The (1 × 1) surface cell is indicated with black lines. The bottom figure shows a side view using the ball and stick representation. Atomic color code as in Fig. 2. | ||
In similarity with In2O3(111), In2O3(110) consists of stoichiometric layers. The surface is composed of a repeating chain of four In-ions, In1–In4 in Fig. 3, and six oxygen, O1–O6. All atoms are non-equivalent in the repeating chain. There are 12 oxygen and 8 In-ions in each layer of the surface cell. In1 and In2 are four-fold coordinated to oxygen, whereas the rest are five-fold coordinated. All oxygen atoms in the topmost layer are three-fold coordinated to indium atoms. The relaxation for the In2O3(110) surface is more complex than for the In2O3(111) case. The topmost layer contracts on average by 0.14 Å, with respect to the bulk-constrained layer, whereas the second layer expands by ∼0.06 Å. The (110) orientation has two slightly different geometric terminations, which are energetically equivalent. In one case the O3 and O4 are on the same side of the In–O chain (as in Fig. 3), whereas they are on opposite sides in the other case.
The oxygen vacancy formation energy for the non-equivalent oxygen types is calculated for the two surfaces with PBE and PBE+U (see Table 1). We find that the energy to create an oxygen vacancy is systematically higher with PBE+U by about 1 eV. This shows that the In–O bond is stronger when the Hubbard correction is applied, which is consistent with the higher surface energy calculated with PBE+U. The higher stability of the (111) facet is manifested by higher vacancy formation energies as compared to (110).
| (110) (U = 0 eV) | (110) (U = 7 eV) | (111) (U = 0 eV) | (111) (U = 7 eV) | ||
|---|---|---|---|---|---|
| O1 | 1.97 | 2.84 | Oa | 2.81 | 3.81 |
| O2 | 2.20 | 3.06 | Ob | 1.88 | 2.86 |
| O3 | 2.47 | 3.20 | Oc | 2.65 | 3.61 |
| O4 | 2.29 | 3.04 | Od | 2.15 | 3.15 |
| O5 | 1.83 | 2.80 | — | — | — |
| O6 | 1.74 | 2.69 | — | — | — |
3.3 Surface hydroxylation
The relative stability of In2O3(111) and In2O3(110) as a function of hydrogen chemical potential (μH2) is investigated with the use of ab initio thermodynamics. For In2O3(111), nine different situations are considered (See Fig. 4), namely the pristine surface, five hydrogen coverages, one oxygen vacancy in the pristine surface, and two hydrogen coverages in the presence of a vacancy. The coverage is defined as the percentage of oxygen atoms in the topmost layer being occupied with hydrogen. Hence, 16% coverage corresponds to dissociating one H2 molecule homolytically. | ||
| Fig. 4 Structural models of In2O3(111) with and without oxygen vacancies for different hydrogen (white) coverages. Atomic color code as in Fig. 2. Position of oxygen vacancy is highlighted as a purple circle in the relevant structures. | ||
The number of possible hydrogen configurations are high at low coverages. To reduce the computational cost, we explored different situations at 16% coverage using a three layer system and only the PBE functional. For In2O3(111), we find that adsorption on the Ob–Ob pair to be preferred, with an adsorption energy of −2.70 eV. This is followed by the Ob–Od and Ob–Oc, with adsorption energies of −2.30 and −1.98 eV, respectively. The adsorption energy at Od–Oa, Od–Oc, Oa–Oa and Oa–Oc are calculated to be −1.78, −1.68, −1.46 and −1.38 eV, respectively. A similar trend is found for heterolytic adsorption, where average adsorption energy on Ine–Ob site is −0.63 eV, followed by Ine–Od (−0.32 eV), Inc–Oc (−0.23 eV) and Inf–Oa (−0.26 eV). The trends in adsorption energies show that the Ob and Od are more reactive to hydrogen adsorption than are Oc and Oa. The exothermicity in the adsorption is found to be inversely correlated to the oxygen vacancy formation energy (See Table 1). It should noted that the most reactive adsorption sites, Ob and Od, are bonded to the sixfold coordinated In-ions, see Fig. 2.
Based on the experience for the 16%-case, we explored four-five different configurations for the higher hydrogen coverages. It was found that hydrogen preferably adsorbs close to the six-fold coordinated In-atoms. The results demonstrates the heterogeneity of the surface with two regions having different chemical properties. This has been noted previously in connection to water adsorption for In2O3(100)44 and In2O3(111).45 The difference in reactivity of the oxygen atoms has been correlated with the proximity of their O 2p states with respect to the valence band maximum.45
The OH-groups that are formed upon hydrogen adsorption are at higher coverages (≥50%) displaced from the original oxygen position occupying bridge sites between two In-ions. This is an effect of the increased oxygen ionic radii upon the change in formal charge by hydroxylation. The displacement by hydrogenation has been reported previously for In2O3(110).21
The average hydrogen adsorption energy is −2.59, −1.69, −1.10, −1.22, −0.75, −0.87 and −0.25 eV for 16%, 33%, 33% + Ov, 50%, 50% + Ov, 66% and 100% covered, respectively. It should be noted that the adsorption of hydrogen takes place on different oxygen sites as the coverage is increased. For 16% coverage, the stable configuration is hydrogen adsorbed on Ob sites. In case of 33% coverage, all Ob sites are occupied together with one Od site. Finally, all Ob and all Od sites are occupied at 50% coverage. Once the Ob and Od are occupied, increased stability of adsorption can only be achieved at high hydrogen pressures (ΔμH2 > 0). This is the case of the 66% covered surface, where the preferred site for adsorption is a Oa–Oc pair (see in Fig. 4).
On In2O3(110), seven cases of hydrogen coverage are considered of which one case is investigated with an oxygen vacancy. The corresponding surface structures are shown in Fig. 5. The average hydrogen adsorption energy is −2.24, −1.65, −1.34, −1.44, −1.41, −1.24 and −1.14 eV for 16%, 50%, 66%, 83%, 83% + Ov, 92% and 100% coverage, respectively. The average binding energies on (110) surface are less affected by increasing hydrogen coverage than on the (111) surface. This is rationalized by the higher reactivity of the (110) surface.
 | ||
| Fig. 5 Structural models of In2O3(110) with and without oxygen vacancies for different hydrogen (white) coverages. Atomic color code as in Fig. 2. Position of oxygen vacancy is highlighted as a purple circle in the relevant structures. | ||
The stability of the hydrogen covered surfaces as a function of hydrogen chemical potential at methanol synthesis conditions (573 K, H2/H2O:10–50/1) is shown in Fig. 6. The surface free energy is calculated with respect to the pristine surface, indicated as a dashed line at zero energy. The chemical potential corresponding to H2 partial pressure of 10–50 bar is highlighted in the figures as a purple fringe. Partial pressure of water, relevant for the Ov reduced surfaces, is set to 1 bar. The phase diagram in Fig. 6 (a) shows that the (111) surface is predicted within PBE to be hydroxylated (33% covered) at relevant conditions. The hydroxylation of the surface as a function of hydrogen chemical potential proceeds from a pristine surface, to a 16% at a chemical potential of −2.32 eV, to 33% at −0.52 eV until reducing to Ov + 33% at −0.18 eV. The relevant hydrogen chemical potential at methanol synthesis conditions covers the span from −0.33 to −0.25 eV (10–50 bar), suggesting a hydrogen coverage close to 33% at reaction conditions. Note that the mean coverage does not take the heterogeneity of the surface into account. In fact, at 50% coverage, all oxygen sites close to the six-fold coordinate In atoms are hydrogen covered.
 | ||
| Fig. 6 Surface stability of In2O3(111) and In2O3(110) as a function of hydrogen chemical potential (μH2) and Hubbard parameter at a temperature of 573 K. The results are calculated with respect to the pristine surface at zero energy. Experimental conditions, PH2 = 10–50 bar, are indicated by a purple fringe. Water chemical potential is set at a pressure of PH2O = 1 bar. Labels are color-coded in accordance to Fig. 4. | ||
The Ov + 33% covered surface is found to be close in stability to the hydroxylated surface. If a lower partial pressure of water (0.1 Bar) is considered, this surface becomes isoenergetic with the only hydroxylated surface at a chemical potential of −0.30 eV. This results suggest that oxygen vacancies may exist when the surface is hydrogen covered. We find that vacancies on the pristine surface are thermodynamically unfavorable.
The phase diagram for In2O3(110) (PBE) is shown in Fig. 6(b). The (110) surface is predicted to be slightly reduced and heavily hydroxylated (83% + Ov) at all relevant reaction conditions. Surface hydroxylation as a function of hydrogen chemical potential proceeds from the pristine surface, to a 16% at −1.98 eV, to 50% at −1.08 eV, to the mildly reduced 83% + Ov at −0.87 eV, well before methanol synthesis conditions are reached. The stability of oxygen vacancies is clearly promoted by a high hydrogen coverage unlike the In2O3(111) surface.
The phase diagrams for (111) and (110) calculated with the Hubbard correction are shown in Fig. 6(c) and (d), respectively. (The adsorption energies are reported in the ESI.†) The relative stabilities of all hydrogen coverages are shifted by ∼0.9 eV with respect to the PBE results. The trends are, however, not affected by the choice of functional. On the (111) surface, we find that hydroxylation at 16% is favorable at reaction conditions. The hydroxylated surface becomes thermodynamically favorable over the pristine surface at a hydrogen chemical potential of −1.37 eV and remains the preferred phase up to zero chemical potential. The presence of oxygen vacancies are destabilized when the Hubbard-U correction is applied. From the phase diagram of the (110) surface, Fig. 6(d), we observe a similar behaviour as on the (111) surface. The favorable surface coverage is the 50% covered case. The hydroxylation proceeds similarly as with PBE, however, with transitions occurring at higher chemical potentials and reaching a lower overall hydrogen coverage. Hydroxylation proceeds from the pristine surface, to a 16% at −1.25 eV, to 50% at −0.33 eV, to 83% at −0.11 eV. As for the (111) surface, the creation of an oxygen vacancy at the surface becomes unfavorable with the Hubbard-U correction. The destabilization of the surface upon oxygen vacancy formation is consistent with the increased Ov formation energy (Table 1).
A Bader charge analysis is performed on In2O3(111) within PBE. The Bader charge for an In-ion in the In2O3 bulk is 11.17 electrons, which should be compared to the number of valence electrons being 13. The formal oxidation state is +3, which gives that the Bader analysis yields 0.61 electrons per formal charge. The charge on the pristine surface is 11.23, which is close to the bulk value. The oxidation state of the In-ions changes marginally in the presence of an oxygen vacancy, Fig. 7(a). Changes are instead calculated when the surface is hydroxylated. The charge on two In-ions change to 12.12 and 12.06 electrons in the case of 33% + Ov. Fig. 7(b). This change corresponds to a formal change in oxidation state to +1. The results suggest that the vacancies alone do not reduce the In-ions, but allow for undercoordination that facilitates a change of the charge upon hydroxylation.
 | ||
| Fig. 7 Bader charge analysis of relevant In2O3(111) structures. Charge localization is shown in dark blue. The charge on the pale blue atoms is about ± 0.1 e. Hydrogen atoms are shown in white. A vacancy is indicated by a red circle. | ||
On the 50% covered structure, Fig. 7(c), charge of a single Ine changes from 11.24 to 12.16 electrons, whereas the rest of the atoms change within 0.1 electrons. Similarly, on the 66% structure, Fig. 7(d), we find a greater degree of surface reduction. At this coverage the charge of two Ine and a Inf changes upon hydroxylation in the same way as the 50% covered structure. The change in oxidation state is structurally observable in the change of In–O and In–In distance and leads to the creation of adsorbed OH groups in bridge position between neighboring In-ions.
A bond valence sum analysis46,47 of the charged Ine at 50% shows a change from 2.79 in the pristine surface to 0.96 upon hydroxylation. The charge and BVS analysis suggest a change in oxidation state from +3 to close to +1 for the reduced In-ions, in agreement with previous reports on change of oxidation state upon hydroxylation on In2O3(110).21 The results are in agreement with the change of oxidation state of In-ions during reaction.20 A charge analysis was also performed with Hubbard correction, with similar results.
3.4 H2O formation by H2 adsorption
A reaction mechanism for dissociative adsorption of H2 is examined on the pristine (111), Fig. 8 (a), and (110), Fig. 8(b). Two possible routes are considered after H2 heterolytic dissociation, (i) Hydroxylation of the surface to make OH* + OH* and (ii) water formation by oxygen vacancy creation (Ov + H2O*), where H2O is adsorbed on the surface. On (111), we find that the first step of the reaction, heterolytic dissociative adsorption, has a barrier of 0.48 eV. This step is exothermic by 0.58 eV with respect to H2(g). From here, InH + OH, the reaction path splits into homolytic adsorption or H2O formation. For path (i) we find that forming two OH-groups is exothermic by 1.62 eV with respect to the InH + OH state. The process has a barrier of 0.78 eV. For path (ii) we find that the creation of an oxygen vacancy is exothermic by 0.50 eV with respect to the heterolytic adsorption and has a barrier of 0.73 eV. The barriers of both path (i) and (ii) on In2O3(111) is associated with the diffusion of hydrogen over the In-ion. The two transition states are geometrically similar. The effect of an oxygen vacancy was also investigated for the H2 dissociative adsorption. We find that the vacancy has an almost negligible effect on the activation energy, as it reduces the barrier by 0.1 eV.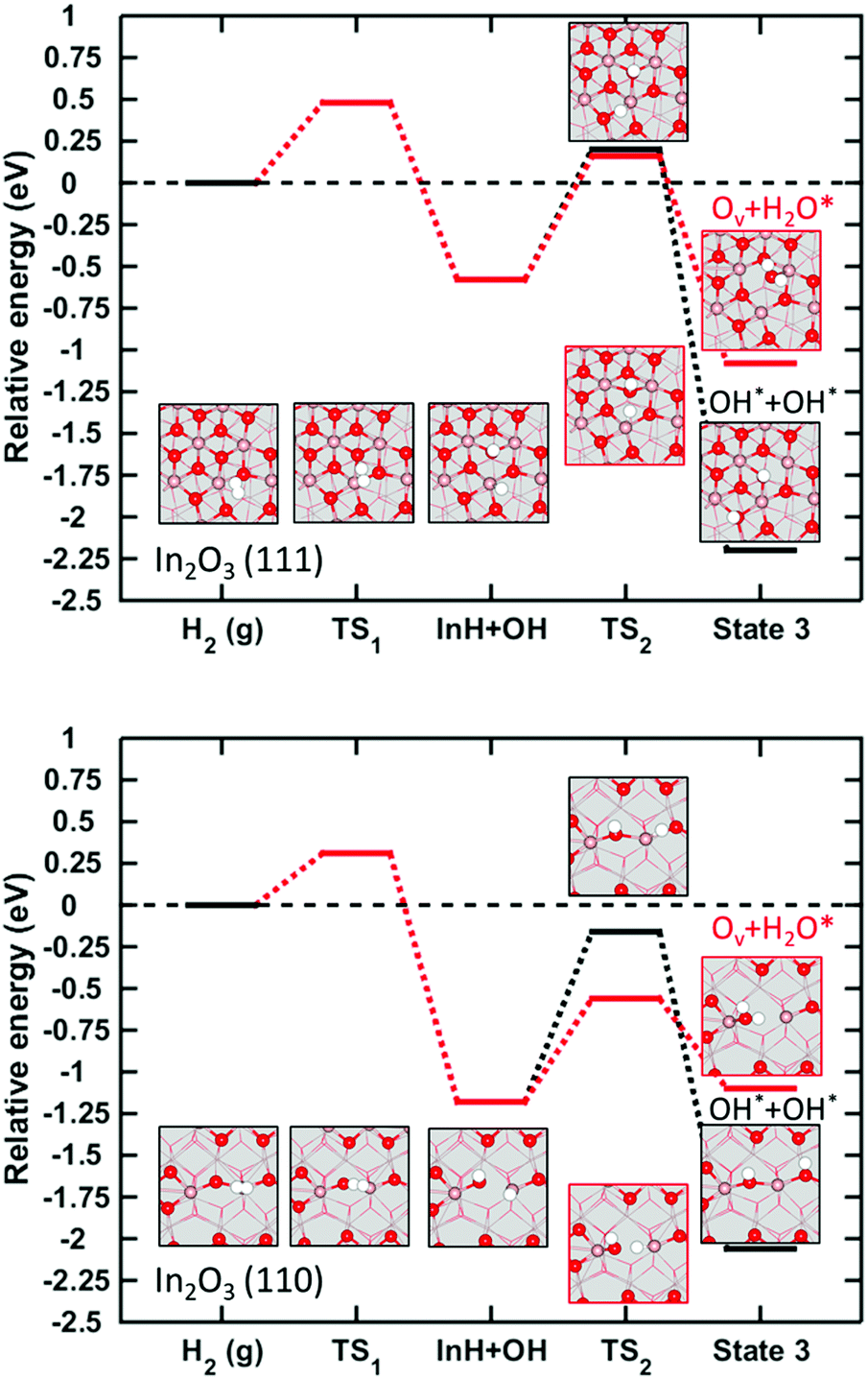 | ||
| Fig. 8 Relative energy landscapes for H2 dissociative adsorption and H2O formation on In2O3(111) and In2O3(110). | ||
The barrier for H2 dissociation in the pristine surface is slightly lower than reported previously.16 This can be attributed to different paths for dissociation, which in ref. 16 was chosen to be homolytic. Our result for dissociation over the vacancy is similar to the one reported in ref. 16. The barrier for H2 dissociation on the pristine surface is in quantitative agreement with a previous investigation following heterolytic dissociative adsorption.48 This is also the case for the formation of adsorbed water on the surface. However, we find a slightly lower energy barrier for the formation of two OH groups at the surface.48
Heterolytic dissociation into InH + OH is on the (110) surface exothermic by 1.18 eV with respect to the initial state, having a barrier of only 0.31 eV. For path (i), we find that the formation of OH* + OH* is exothermic by 0.88 eV with respect to the initial InH + OH configuration. The formation of two OH-groups is associated with a barrier of 1.02 eV. The higher barrier as compared to (111) is due to a stronger hydride bond on (110). For Path (ii), we find that the formation of water and an Ov at the surface is endothermic by 0.08 eV with respect to the heterolytically dissociated hydrogen, with an energy barrier of 0.62 eV. The lower energy barrier for water formation can be explained by structural arguments as a shorter elongation of the In–H bond at the transition state is required in the formation of the H2O than for the two OH-groups.
Similarly to In2O3(111), the heights of the barriers on the (110) appear to be related to the energy penalty for hydrogen diffusion over the In-ion. Our results are in quantitative agreement with a previous report.48 We note that the calculated activation energies are moderate for the experimental conditions (573 K, 10–50 Bar) and that there is no thermodynamic driving force to form oxygen vacancies on any of the investigated surfaces.
It is experimentally known that H2 may reduce In2O3 surfaces.13,20,22 Our results show that a reduction of the surface requires a certain coverage of hydrogen. The phase diagram in Fig. 6, shows that structures with vacancies are competitive at a coverage of 0.33 and 0.83% for In2O3(111) and In2O3(110), respectively.
3.5 Hydrogen induced O 1s core-level shifts
The chemical properties and degree of surface hydroxylation of In2O3 have been investigated previously using O 1s X-ray photoelectron spectroscopy.13,49,50 The measured O 1s spectrum has been deconvoluted into three main features,13 namely lattice oxygen (530.3 eV), signatures of oxygen vacancies (531.7 eV) and surface OH-groups (532.5 eV). Here we evaluate the O 1s core level shift for different oxygen species with respect to oxygen atoms in the bulk of In2O3, see Fig. 9.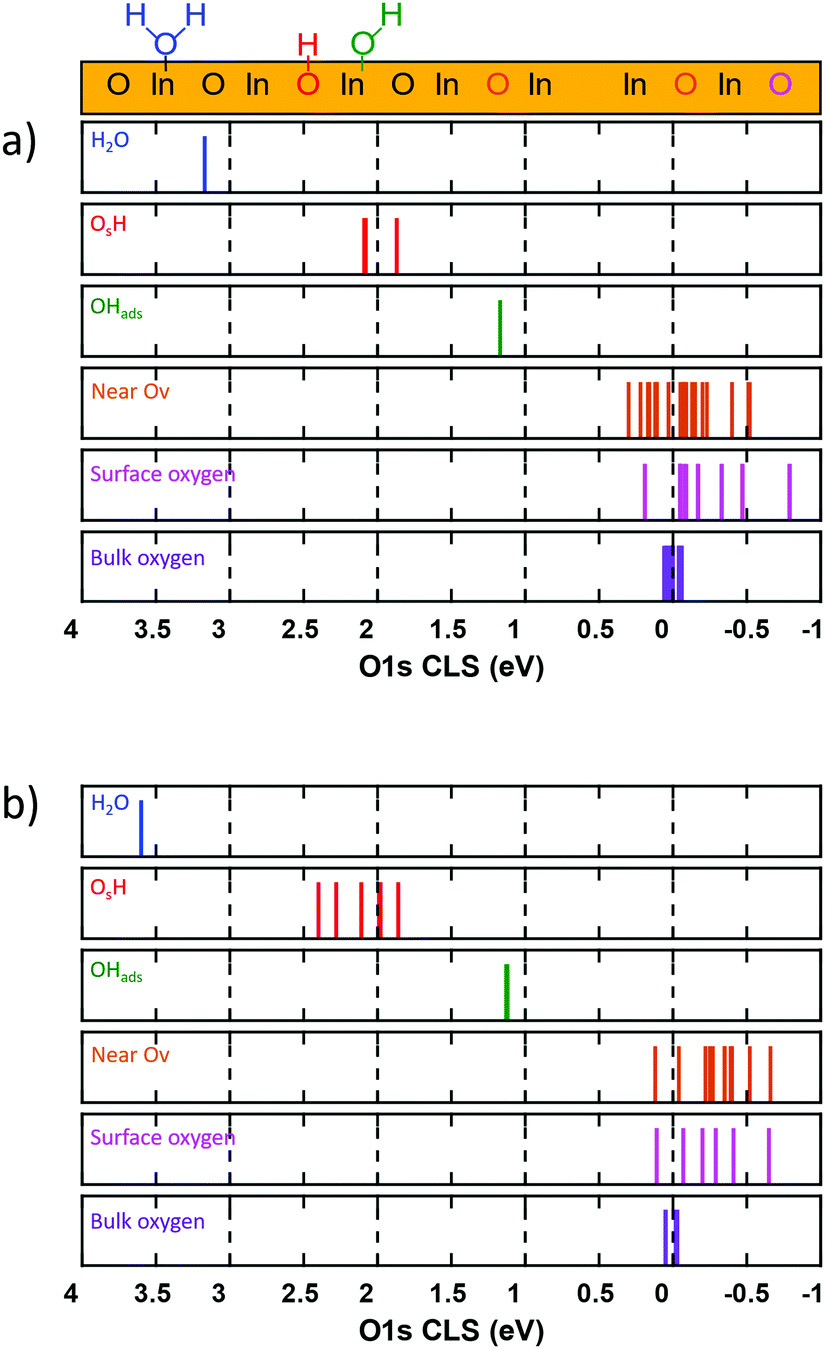 | ||
| Fig. 9 Calculated O 1s core-level shifts for In2O3(111) and In2O3(110). Top panel: O 1s CLS of In2O3(111), OH species are investigated at 16% hydrogen coverage. Bottom panel: O 1s CLS of In2O3(110), OH species are investigated at 16% hydrogen coverage. | ||
For In2O3(111), Fig. 9(a), we find a slight energetic spread (0.1 eV) of the calculated O 1s CLS on the bulk oxygen. As bulk In2O3 in the bixbyite 206 phase has only one unique oxygen atom, the spread is caused by the finite thickness of the slab and can be viewed as the uncertainty of the computations arising from the structure. We find a significant spread in the O 1s CLS for the surface atoms, ranging from 0.18 to −0.73 eV, with respect to the bulk average. The semi-equivalent oxygen atoms have the same O 1s CLS, showing that these oxygen sites are chemically equivalent. For the topmost oxygen layer, we calculate the O 1s CLS for Oa, Ob, Oc and Od to be −0.08, −0.73, −0.05 and −0.43 eV, respectively. It should be noted that the O-atoms bonded to the six-fold coordinated In-ions (Ob) have a clear shift of the O 1s binding energy to lower energies. The oxygen atoms in the bottom of the In2O3(111) topmost trilayer, can be grouped into four groups of semi-equivalent oxygen (see supporting information). The calculated O 1s CLS for these groups are −0.28, −0.14, −0.04 and 0.18 eV, respectively. The results for the bulk and surface oxygen suggest that measurements of the O 1s spectrum of the pristine surface should have a broad feature close to the value for oxygen in the bulk.
To investigate the effect of an oxygen vacancy on the O 1s CLS on neighbouring surface oxygen, we created and oxygen vacancy in the Ob site and calculated the CLS on the remaining oxygen atoms in the topmost layer. We find that the presence of oxygen vacancies does not change significantly the O 1s binding energies for the remaining oxygen atoms. The calculated O 1s CLS for the oxygen atoms close to the Ob oxygen vacancy spans energies from 0.29 to −0.49 eV. We do not find any general trend of the small shifts induced by the vacancy and the chemical equivalency for the different types of O-sites is virtually maintained (see ESI†). The largest effect of the Ov on the surface oxygen is calculated for the Ob and Od sites. The average calculated O 1s CLS for these sites are −0.48 and −0.25 eV, respectively. The energy span for surface oxygen near an Ov is roughly within the same energy spread as surface oxygen on the pristine, making these sites indistinguishable oxygen sites originating from a pristine surface.
We also investigated the effect of hydrogen adsorption on the O 1s CLS of the formed species. We find that adsorbed hydrogen gives rise to two distinct OH features. OHads, which refers to an OH group adsorbed on an In-ion, obtained from H2O dissociation, has a CLS of 1.27 eV. OsH, which refers to OH groups with oxygen in a lattice position, has an O 1s CLS of 1.96–2.11 eV. The difference in O–In bonds for the two OH species rationalizes the difference in binding energy. For reference, we calculated the O 1s CLS for adsorbed molecular H2O, which was found to be 3.30 eV.
For In2O3(110), Fig. 9(b), the results are similar to the (111) surface. A seven layered surface was used to calculate accurate O 1s CLS. We find that the bulk oxygen O 1s CLS spans also in this case has a span of 0.1 eV. The surface oxygen, calculated on the pristine surface, show a spread in the calculated CLS, ranging from 0.06 to −0.59 eV. The calculated O 1s CLS for O1, O2, O3, O4, O5 and O6 is −0.26, −0.18, 0.06, −0.11, −0.35 and −0.59 eV, respectively. The results suggest, similarly to the (111) surface, that the measured O 1s spectra should have a broadening around the binding energy for lattice oxygen. The effect of oxygen vacancies on neighbouring O-sites is investigated by removing an O3 site from the topmost layer of the surface. We find that the Ov induce a minor shift to lower binding energies for neighboring oxygen. The calculated O 1s CLS near an Ov for O-sites in the same chain [and contiguous chain] for O1, O2, O3, O4, O5 and O6 are calculated to be, −0.39 [−0.25], −0.47 [−0.22], Ov [0.05], −0.40 [−0.10], −0.38 [−0.22] and −0.64 [−0.48] eV, respectively.
Similarly to the (111), the O 1s shift for oxygen near an Ov is within the same energy spread as surface oxygen on the pristine, suggesting that the effect of Ov on neighbouring O-sites would be difficult to detect. Also for this surface, we investigated the effect of hydrogen adsorption on the surface oxygen. The results are qualitatively the same as for In2O3(111). Two OH features are clearly distinguished. For OHads, we calculated the O 1s CLS to be 1.21–1.27 eV, with respect to the bulk reference, whereas the O 1s CLS for OsH is calculated to be 1.82–2.39 eV. The shift for molecular water on the (110) is calculated to be 3.64 eV.
The results suggest that the experimentally assigned13 peak at 531.7 eV could be assigned to adsorbed OH groups on In-ions. Moreover, the peak assigned at 532.5 eV is probably caused to the presence of OH groups in the oxygen lattice position. The CLS were also calculated with Hubbard U-correction, and are in qualitative agreement to the ones without the U-correction (see ESI†).
4 Conclusions
We have used density functional theory calculations to investigate hydrogen adsorption on In2O3(111) and In2O3(110). The degree of surface hydroxylation has been investigated and O 1s core level shifts calculated for representative structures. The calculations have been performed with PBE as well as PBE augmented with a Hubbard-U correction to the In 4d states.Dissociative hydrogen adsorption is found to be facile, occurring via a heterolytic dissociation mechanism. Thermodynamic analysis suggests that surface hydroxylation is favorable under experimentally relevant conditions. Moreover, presence of oxygen vacancies is predicted to become thermodynamic relevant upon hydroxylation. Surface hydroxylation is, moreover, found to induce a change of oxidation state, from +3 to close to +1, on surface In-ions on both surfaces.
Calculations of O 1s core level shifts suggest the existence of two distinct types of OH groups on the In2O3 catalyst. Furthermore, we do not find any clear spectroscopic signature originating from oxygen vacancies. The calculated CLS are in good agreement with previous XPS measurements.
The Hubbard correction is found to accurately describe bulk characteristics with respect to experimental observations of the In 4d states. We find that the oxygen vacancy formation energies to be higher when the Hubbard-U term is used. Similarly, the adsorption strength of hydrogen is lowered with the Hubbard-U correction. However, the trends in surface stability upon hydroxylation as well as the calculated O 1s CLS, and change of oxidation state upon hydroxylation, are in qualitative agreement with the PBE results. Comparisons with reference calculations including exact exchange, suggest that calculations with a Hubbard-U correction provide a better description of the surface electronic properties than standard PBE calculations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Knut and Alice Wallenberg Foundation through the project “Atomistic design of catalysts” (No: KAW 2015.0058) is gratefully acknowledged. Additional support has been obtained from Swedish Research Council and Swedish Energy Agency. The calculations have been performed at C3SE (Göteborg), Uppmax (Uppsala) and PDC (Stockholm) through a SNIC grant.Notes and references
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- T. Reichenbach, K. Mondal, M. Jaeger, T. Vent-Schmidt, D. Himmel, V. Dybbert, A. Bruix, I. Krossing, M. Walter and M. Moseler, J. Catal., 2018, 360, 168–174 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- M. Hus, D. Kopac and B. Likozar, ACS Catal., 2019, 9, 105–116 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- J. Artz, T. E. Mueller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- G. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- O. Martin, A. J. Martin, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferre and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- D. Albani, M. Capdevila-Cortada, G. Vile, S. Mitchell, O. Martin, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2017, 56, 10755–10760 CrossRef CAS PubMed.
- M. S. Frei, M. Capdevila-Cortada, R. Garcia-Muelas, C. Mondelli, N. Lopez, J. A. Stewart, D. C. Ferre and J. Perez-Ramirez, J. Catal., 2018, 361, 313–321 CrossRef CAS.
- M. S. Frei, C. Mondelli, R. Garcia-Muelas, K. S. Kley, B. Puertolas, N. Lopez, O. V. Safonova, J. A. Stewart, D. C. Ferre and J. Perez-Ramirez, Nat. Commun., 2019, 10, 3377 CrossRef PubMed.
- J. Ye, C. Liu and Q. Ge, J. Phys. Chem. C, 2012, 116, 7817–7825 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- C.-Y. Chou and R. F. Lobo, Appl. Catal., A, 2019, 583, 117144 CrossRef CAS.
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M.-G. Willinger, A. Fedorov and C. R. Mueller, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS PubMed.
- A. Posada-Borbón and H. Grönbeck, Phys. Chem. Chem. Phys., 2019, 21, 21698–21708 RSC.
- T. Bielz, H. Lorenz, W. Jochum, R. Kaindl, F. Klauser, B. Kloetzer and S. Penner, J. Phys. Chem. C, 2010, 114, 9022–9029 CrossRef CAS.
- P. Erhart, A. Klein, R. G. Egdell and K. Albe, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153205 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Dudarev, G. Botton, S. Savrasov, C. Humphreys and A. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- G. Henkelman, B. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 035406 CrossRef.
- Q. Sun, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205424 CrossRef.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69; National Institute of Standards and Technology: Gaithersburg, MD, http://webbook.nist.gov, 2005, Online; Accessed 3-December-2018.
- E. Pehlke and M. Scheffler, Phys. Rev. Lett., 1993, 71, 2338 CrossRef CAS PubMed.
- M. V. den Bossche, N. M. Martin, J. Gustafson, C. Hakanoglu, J. Weaver, E. Lundgren and H. Grönbeck, J. Chem. Phys., 2014, 141, 034706 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- A. Walsh, J. L. F. Da Silva, S.-H. Wei, C. Koerber, A. Klein, L. F. J. Piper, A. DeMasi, K. E. Smith, G. Panaccione, P. Torelli, D. J. Payne, A. Bourlange and R. G. Egdell, Phys. Rev. Lett., 2008, 100, 1674021 Search PubMed.
- A. Klein, Appl. Phys. Lett., 2000, 77, 2009–2011 CrossRef CAS.
- P. Agoston and K. Albe, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045311 CrossRef.
- M. Wagner, P. Lackner, S. Seiler, A. Brunsch, R. Bliem, S. Gerhold, Z. Wang, J. Osieck, K. Schulte, L. A. Boatner, M. Schmid, B. Meyer and U. Diebold, ACS Nano, 2017, 11(11), 11531–11541 CrossRef CAS PubMed.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- J. A. Enterkin, A. E. Becerra-Toledo, K. R. Poeppelmeier and L. D. Marks, Surf. Sci., 2012, 606, 344–355 CrossRef CAS.
- B. Qin and S. Li, Phys. Chem. Chem. Phys., 2020, 22, 3390–3399 RSC.
- L. B. Hoch, T. E. Wood, P. G. O'Brien, K. Liao, L. M. Reyes, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400013 CrossRef PubMed.
- T. Yan, L. Wang, Y. Liang, M. Makaremi, T. E. Wood, Y. Dai, B. Huang, A. A. Jelle, Y. Dong and G. A. Ozin, Nat. Commun., 2019, 10, 2521 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01749c |
| This journal is © the Owner Societies 2020 |