
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV-visible absorption spectrum of FAD and its reduced forms embedded in a cryptochrome protein†
Karno
Schwinn
,
Nicolas
Ferré
 and
Miquel
Huix-Rotllant
*
and
Miquel
Huix-Rotllant
*
Aix-Marseille Univ, CNRS, ICR, Marseille, France. E-mail: miquel.huixrotllant@univ-amu.fr
First published on 7th May 2020
Abstract
Cryptochromes are a class of flavoproteins proposed as candidates to explain magnetoreception of animals, plants and bacteria. The main hypothesis is that a biradical is formed upon blue-light absorption by flavin adenine dinucleotide (FAD). In a protein milieu, the oxidized form of FAD can be reduced, leading to four redox derivative forms: anionic and neutral semi-reduced radicals, and anionic and neutral fully reduced forms. All these forms have a characteristic electronic absorption spectrum, with a strong vibrational resolution. Here, we carried out a normal mode analysis at the electrostatic embedding QM/MM level of theory to compute the vibrationally resolved absorption spectra of the five redox forms of FAD embedded in a plant cryptochrome. We show that explicitly accounting for vibrational broadening contributions to electronic transitions is essential to reproduce the experimental spectra. In the case of the neutral radical form of FAD, the absorption spectrum is reproduced only if the presence of a tryptophan radical is considered.
1 Introduction
Cryptochromes are a class of flavoproteins sensitive to blue light.1 A blue sensor is found in their active center, and they usually consist of a flavin adenine dinucleotide (FAD) group. Cryptochromes are found in most living systems, from plants, to animals and bacteria.2,3 However, their exact biological role is unknown. They are well known for regulating the circadian clock.4 Recently, they have attracted much attention due to their potential role in magnetoreception of certain animals.5,6FAD in protein can exist in five forms: fully oxidized (hereafter simply called FAD), an anionic radical form (FAD˙−), a neutral radical form (FADH˙), an anionic reduced form (FADH−) and a neutral reduced form (FADH2). The anionic and neutral forms are connected via acid–base reactions (FAD˙ + H+ ↔ FADH˙ and FADH− + H+ ↔ FADH2), while the radical and non-radical forms are connected via a redox reaction (FAD + e− ↔ FAD˙− and FADH˙ + e− ↔ FADH−). The redox reactions are favored in the excited state (photocatalyst).7 The source of protons and electrons to reduce FAD in protein is still uncertain. The most widely accepted hypothesis for plant cryptochromes is that aromatic amino acids in the protein (probably tryptophan) near the cavity of the cryptochromes transfer electrons and protons to FAD in the excited state.8 However, the mechanism might be different in other types of cryptochromes.9
The UV-visible absorption spectrum is an essential tool for detecting the different redox states of FAD in protein. Indeed, the five forms have a characteristic absorption spectrum and lifetime.10 Simulations of the absorption spectra of FAD and its redox and acid–base derivatives require explicit account of vibrations in order to reproduce the color of absorption and emission. This was first shown by Saalfrank and coworkers for the fully oxidized form of riboflavin.11 Recently, several groups have computed the vibrationally resolved absorption spectra for five redox forms of lumichrome-10H in the gas phase,12 lumiflavin in several solvents,13 and the absorption and emission spectra for isoalloxazine, lumichrome, lumiflavin and riboflavin in solution using implicit continuum solvent models.7
Even though they are detectable in solution, most of the redox derivatives of FAD (especially for the radical forms) are stabilized in protein.14 Currently, a systematic simulation of the absorption spectra of FAD and its redox derivatives in a protein milieu is missing, which would be essential to interpret the origin of the peaks. Cannuccia and coworkers,15 and more recently Solov'yov and coworkers16 have performed a theoretical study of the vertical transitions at the QM/MM level for the oxidized form of the flavin mononucleotide and FAD, respectively. However, none of these studies included explicitly the effect of nuclear vibrations in their simulations.
Here, we perform, for the first time, the simulations of the vibrationally resolved electronic absorption spectrum of FAD in its five redox forms in a plant cryptochrome. For this purpose, we have implemented our recently developed method for computing the analytic second derivatives of electrostatic embedding QM/MM energy to unrestricted single-determinant wavefunctions,17 necessary to simulate the vibrationally resolved spectra of radical species. Our theoretical absorption spectra are in good agreement with the experimental spectra. For FADH˙, the spectra of tyrosil and tryptophanyl radicals have to be included to reproduce the experimental spectra.
2 Methodology
Simulations of the vibrationally resolved spectra require the computation of the Hessian for the whole QM/MM system. Currently, there are few methods capable of computing efficiently the QM/MM Hessian for a large macromolecule like a protein, mostly relying on approximations.18–22 We have recently developed one of the first analytic electrostatic embedding QM/MM Hessians, which is both efficient and without approximations.23 Here, we summarize the main equations following the electrostatic potential fitting (ESPF) method and its derivatives, focusing on the new implementation for unrestricted wavefunctions. The reader is referred to previous literature for further details on the closed shell ESPF equations and analytic QM/MM interaction energy derivatives.17,23–252.1 Electrostatic potential fitting method
The ESPF method is an electrostatic embedding QM/MM method,24 which adds a one-electron potential in the usual (gas-phase) Fock operator (F0),![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) = 0 + ĥ. = 0 + ĥ. | (1) |
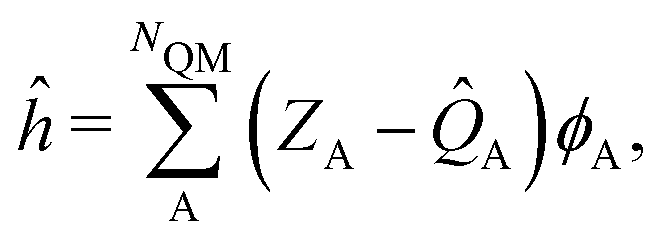 | (2) |
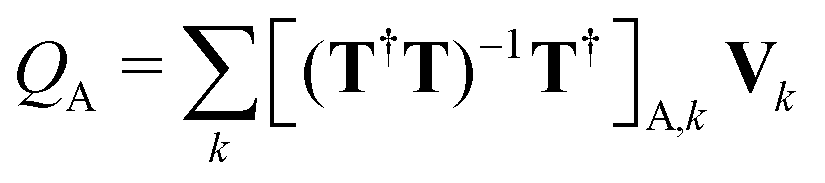 . Here, Vk are the electrostatic integrals calculated on a grid surrounding the QM subsystem and T the electrostatic kernel.24 Usually, the lowest order approximation is a good approximation for the QM/MM interaction.25 Hereafter, we focus on the lowest order, despite being straightforward to generalize it to higher orders in the multipolar expansion.23
. Here, Vk are the electrostatic integrals calculated on a grid surrounding the QM subsystem and T the electrostatic kernel.24 Usually, the lowest order approximation is a good approximation for the QM/MM interaction.25 Hereafter, we focus on the lowest order, despite being straightforward to generalize it to higher orders in the multipolar expansion.23
The ESPF charge operator is spin independent, so the only spin dependency comes from the density matrix. For a single determinant wavefunction expanded in molecular orbitals, the ESPF QM/MM energy is then obtained as
| E = Tr[PF0] + EMM + Tr[Ph] = E0 + EMM + EESPF, | (3) |
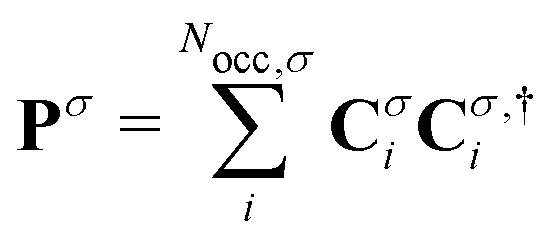 (σ = α, β). The C terms are the molecular orbital coefficients obtained from the solution of the secular equation with the Fock operator defined in eqn (1). The ESPF energy interaction term can be rewritten as
(σ = α, β). The C terms are the molecular orbital coefficients obtained from the solution of the secular equation with the Fock operator defined in eqn (1). The ESPF energy interaction term can be rewritten as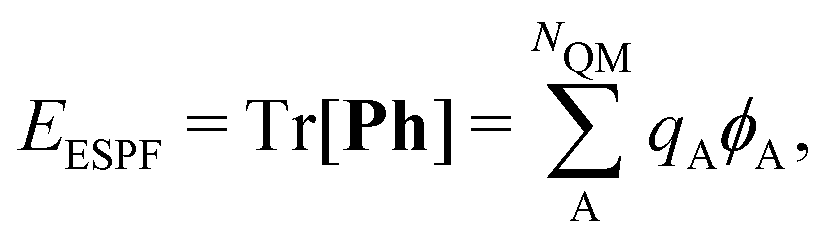 | (4) |
| qA = ZA − Tr[PQA]. | (5) |
In the case of unrestricted wavefunctions, the α and β contributions to the ESPF charges can be defined as QA,σ = Tr[PσQA]. Since classical force fields are spin-independent, the ESPF energy expression eqn (4) remains unchanged, and the total atomic charge approximating the QM atom A charge distribution is simply the sum of the α and β components,
| qA = ZA − QA,α − QA,β. | (6) |
2.2 Analytic second derivatives of the ESPF energy
The analytic first and second derivatives of the ESPF energy have been described in ref. 24 and 17, respectively. The ESPF energy depends on the coordinates of the QM and MM atoms. Therefore, its second derivative with respect to these coordinates contains four blocks,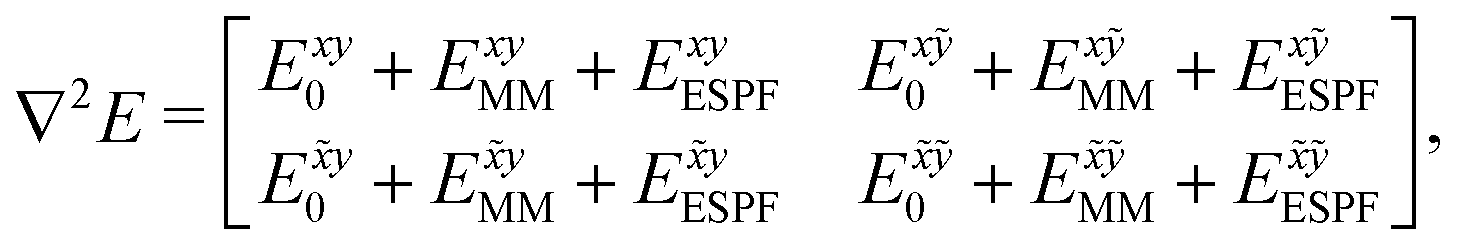 | (7) |
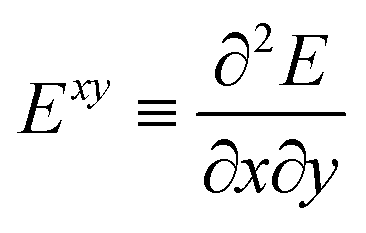 . x corresponds to the x coordinate of a QM atom, while the tilde symbolizes an MM atom. The analytic second derivatives of E0 have been discussed in depth in the literature,26 while the EMM derivatives have a trivial expression. Here we focus on the derivative of the ESPF energy term, which can be written in orders of external potential derivatives as
. x corresponds to the x coordinate of a QM atom, while the tilde symbolizes an MM atom. The analytic second derivatives of E0 have been discussed in depth in the literature,26 while the EMM derivatives have a trivial expression. Here we focus on the derivative of the ESPF energy term, which can be written in orders of external potential derivatives as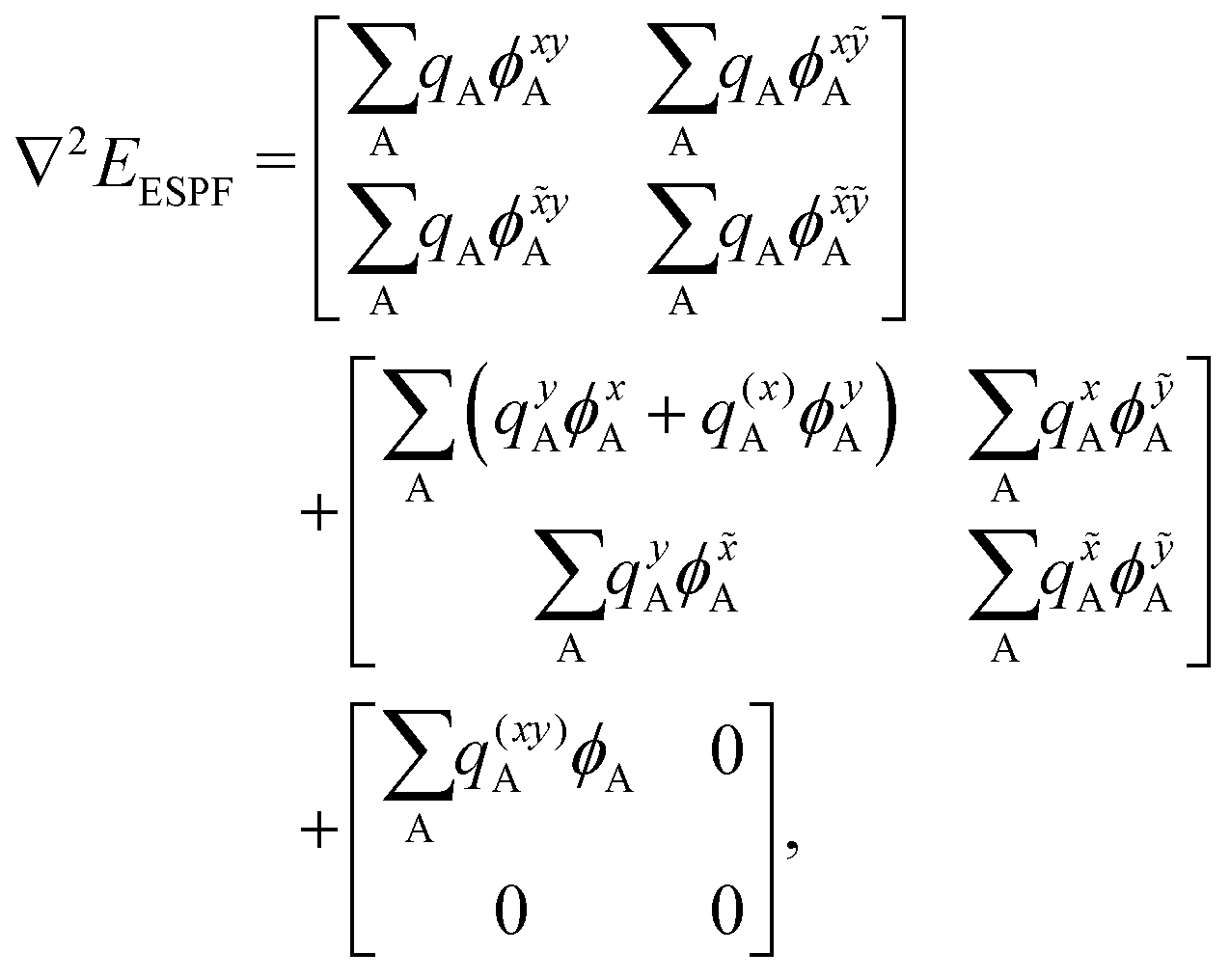 | (8) |
The atomic charge derivatives qxA and q![[x with combining tilde]](https://www.rsc.org/images/entities/i_char_0078_0303.gif) A include the derivatives of the density matrix and of the ESPF charge matrix,
A include the derivatives of the density matrix and of the ESPF charge matrix,
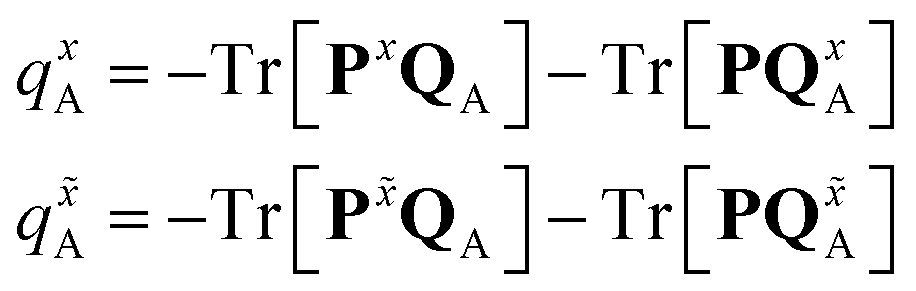 | (9) |
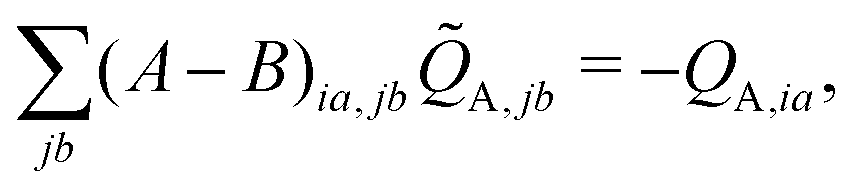 | (10) |
![[Q with combining tilde]](https://www.rsc.org/images/entities/i_char_0051_0303.gif) A,jb, is then used to construct the ESPF charge derivative with respect to the MM atoms,
A,jb, is then used to construct the ESPF charge derivative with respect to the MM atoms,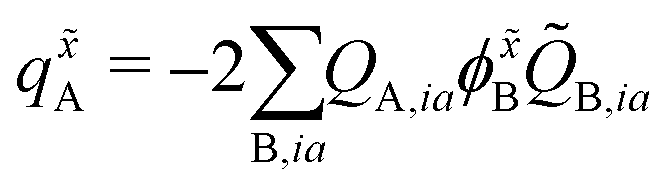 | (11) |
In the case of the QM/MM energy second derivatives based on an unrestricted determinant, we can use similar arguments as we did in eqn (6). Since the force field derivatives do not depend on the spin, all charge and charge derivatives of eqn (8) involve the sum of the α and β contributions. The Q-vector equation in eqn (10) splits into two expressions
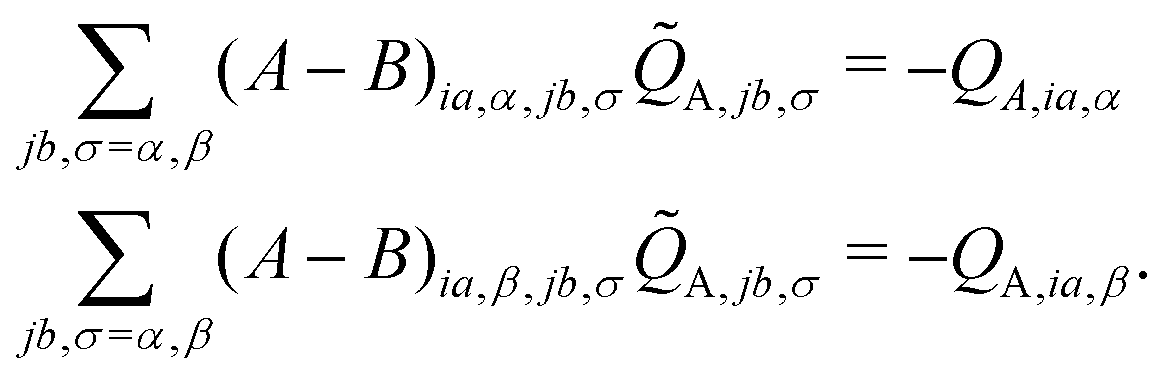 | (12) |
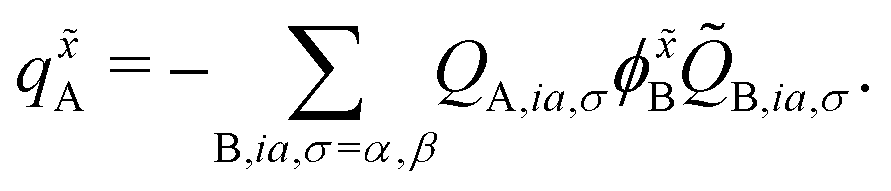 | (13) |
3 Computational details
The ESPF QM/MM hessian for unrestricted wavefunctions described above has been implemented in a modified version of Gaussian16 interfaced with a modified version of Tinker 6.3.3.17,23,24,27,28 The X-ray structure of Arabidopsis Thaliana plant cryptochrome (RCSB PDBID: 2J4D) has been used as a molecular model. Hydrogen atoms have been added to the standard protonation state at physiological pH. For the QM computations, the B3LYP/TZVP/TZVPFit level of theory has been employed for DFT and TD-DFT calculations.29–35 For the MM computations, the Amber99 force field has been used,36 with van der Waals parameters for FAD taken from ref. 37. The vibrationally resolved spectrum for FAD has been obtained by Fourier transforming the time-dependent auto-correlation function for the multi-dimensional harmonic oscillator, as implemented in Gaussian16.27,38 The vertical gradient approximation has been used in all cases.394 Results
The QM/MM model is defined as FAD (QM subsystem) and the protein scaffold (MM subsystem), from which crystallographic water molecules have been excluded (see Fig. 1). The overall system has been optimized with a quadratically convergent algorithm for the QM part and microiterations for the MM part. From the minimum energy structure of FAD, the subsequent structures have been constructed for the reduced forms and each of them reoptimized. All structures have been confirmed via a frequency calculation, leading to a positive semi-definite internal Hessian matrix in all cases.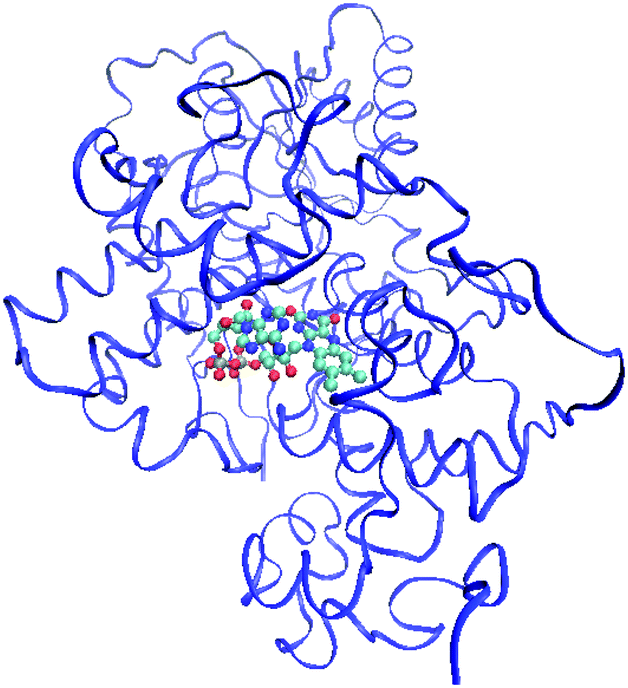 | ||
| Fig. 1 QM/MM subsystems for Arabidopsis Thaliana cryptochrome (PDBID: 2J4D). The QM fragment representing FAD is shown as balls and sticks, while the MM fragment representing the protein scaffold is shown as ribbons. | ||
4.1 Geometries
In the series , the lowest occupied molecular orbital (LUMO) becomes a singly occupied molecular orbital (SOMO) after the first electron attachment and the highest occupied molecular orbital (HOMO) after the second electron attachment. The orbital involved in this transformation is essentially anti-bonding between N5–C4a and bonding between C4a–C10a. This transforms the α-diimine bond (N5
, the lowest occupied molecular orbital (LUMO) becomes a singly occupied molecular orbital (SOMO) after the first electron attachment and the highest occupied molecular orbital (HOMO) after the second electron attachment. The orbital involved in this transformation is essentially anti-bonding between N5–C4a and bonding between C4a–C10a. This transforms the α-diimine bond (N5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C4a–C10aN1) of the oxidized form in ethene-1,2-diamine (N5–C4aC10a–N1) into the fully reduced form.7
C4a–C10aN1) of the oxidized form in ethene-1,2-diamine (N5–C4aC10a–N1) into the fully reduced form.7
In Fig. 2, the effect of the protein scaffold on the chromophore optimized (local) minimum energy structure is compared with respect to the gas phase minimum energy structure. For the protein, this corresponds to a local minimum of the internal energy, which is taken as reference to do the harmonic expansion. Overall, one observes only minor structural differences when the chromophore is embedded in the protein compared to the gas phase. This is evident for bond distances, in which changes of less than 0.1 Å are observed for all five forms. Conversely, angles and dihedral angles show important variations for all cases. This is especially important in the case of the fully reduced forms, FADH− and FADH2. These forms contain secondary amines at centre N5 and/or N1 atoms. Secondary amines tend to break the planarity of the chromophore both in the gas phase or in solution (see Fig. 3).7,13 Indeed, in the gas phase these two forms find their minimum energy structure in the so-called “butterfly” bending conformation. This structural feature is hindered when the chromophore is embedded in the protein, which favours a rather planar structure. Despite the weak electrostatic effects on the π system and thus the chromophore excitations,13 the conformational constraints have a strong impact on the peak position (see further in the next sections).
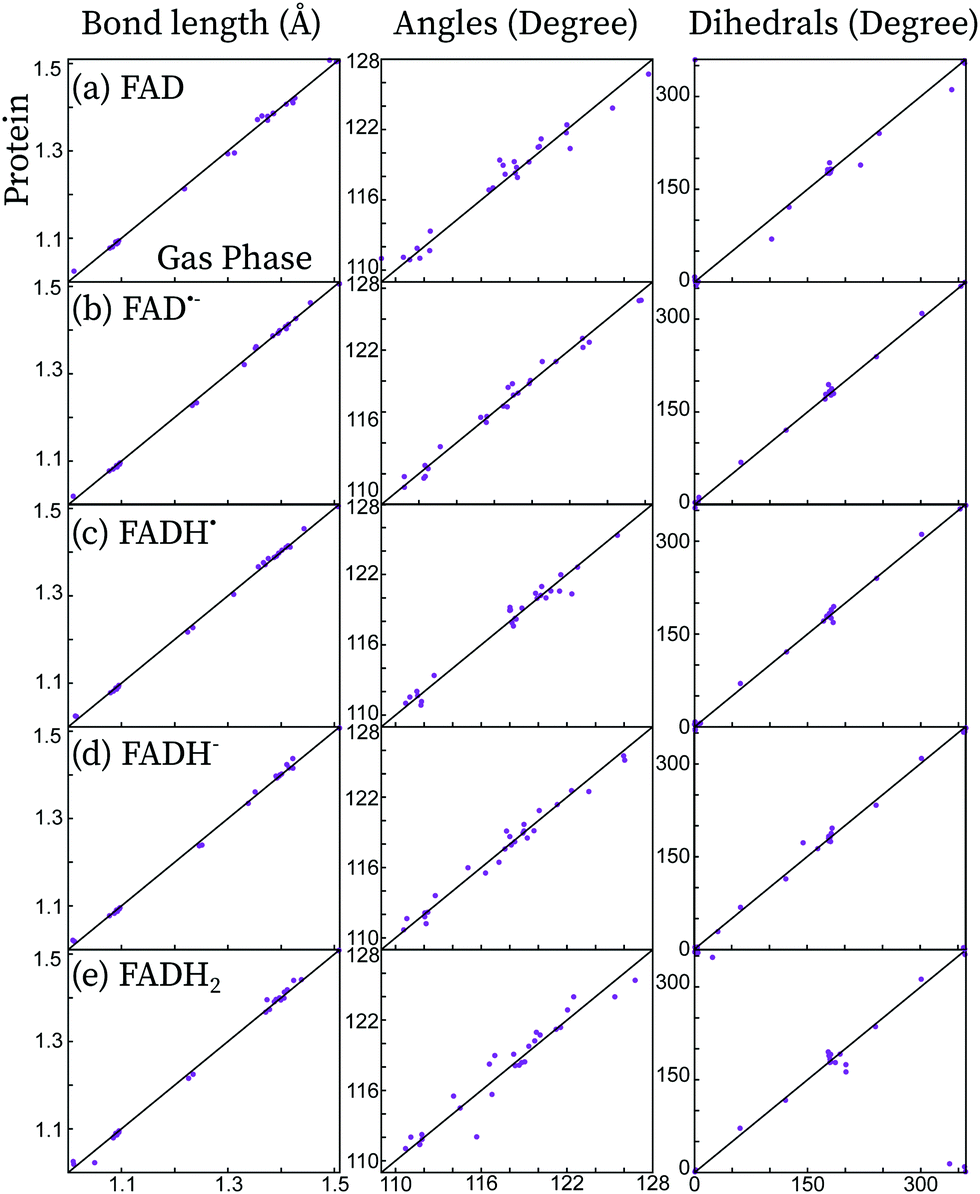 | ||
| Fig. 2 Comparison of the distances, angles and dihedral angles of the isoalloxazine chromophore optimized in protein and in the gas phase for the five redox forms considered here. The diagonal line (y = x) is shown as guideline. | ||
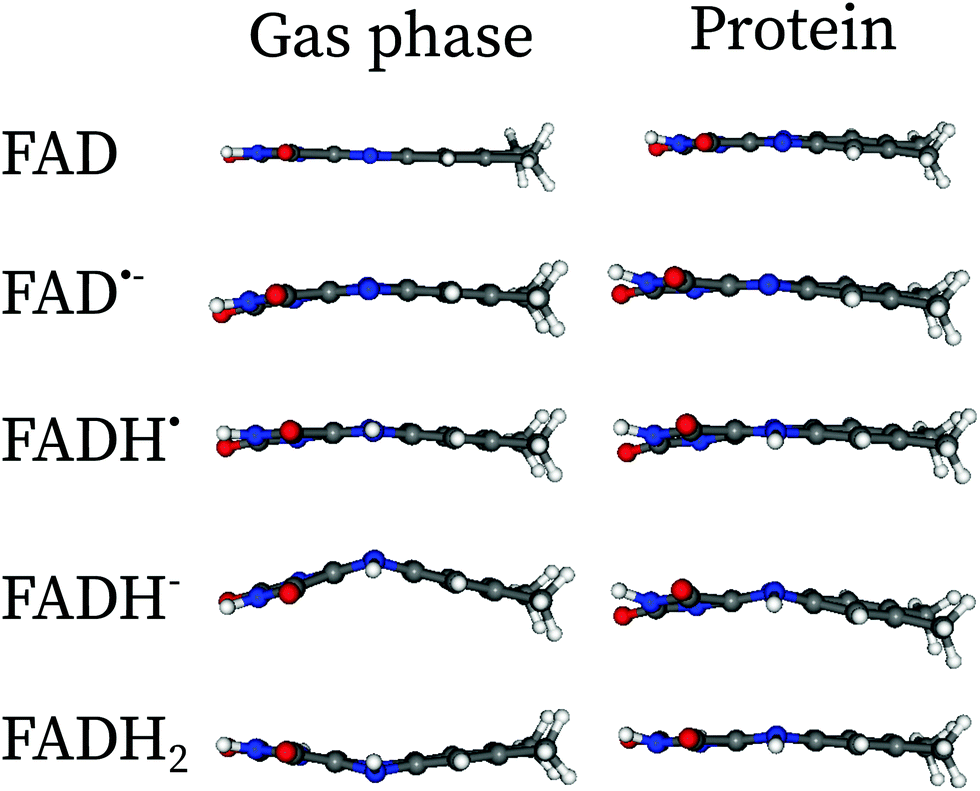 | ||
| Fig. 3 Comparaison of the minimum energy structures in the gas phase and in protein embedding the isoalloxazine fragment of FAD along the xz-plane. The butterfly structures in the gas phase are hindered by the presence of the protein. The Cartesian coordinates of the optimized structures in protein can be found in the ESI.† | ||
4.2 Absorption spectra of FAD and its redox derivatives embedded in the protein
In Fig. 4, the total theoretical vibrationally resolved absorption spectrum is shown in the range between 300 nm and 800 nm. Besides, the contributions of the most important electronic configurations are also explicitly shown. For all redox forms, the lowest-energy peak is represented by a single electronic transition of the type π → π* centred on the isoalloxazine ring. The second peak of FAD, FAD˙− and FADH˙ shows a complex pattern, involving three to four electronic transitions. For FADH− and FADH2 the second transition finds its maximum above 300 nm, and it corresponds to a π → π* transition in both cases.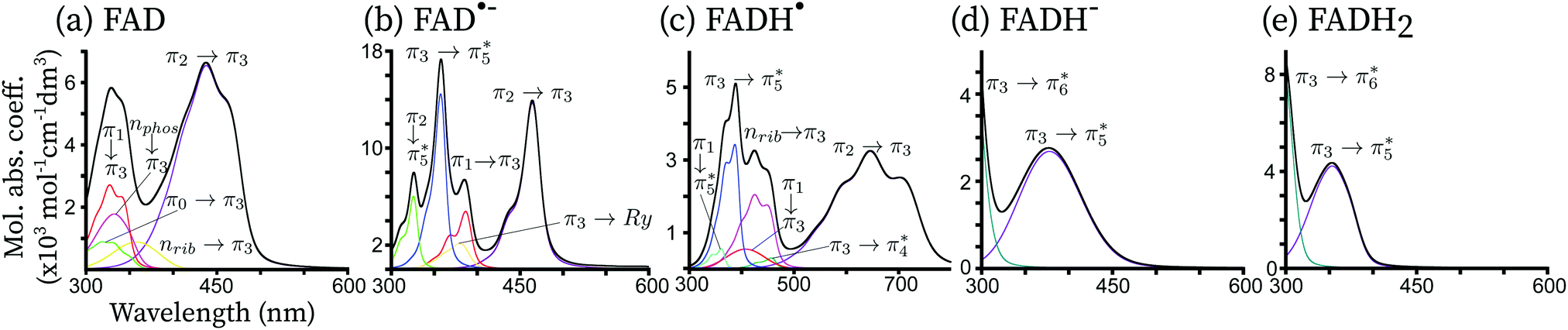 | ||
| Fig. 4 Theoretical vibrationally resolved spectrum of FAD and its redox derivatives. All excitations with a non-negligible oscillator strength up to 300 nm are shown. The x-axis is in nm while the y-axis is in units of molar absorption coefficient (dm3 cm−1 mol−1). The individual electronic excitation contributions are shown in several colors with an interpretation of the electronic excitations in terms of single orbital excitations indicated on top of each contribution. The total spectrum (black) corresponds to the sum of all individual contributions. | ||
For all redox forms of FAD, the frontier molecular π3 orbital plays a central role. This orbital exhibits bonding character between C4a–C10a and anti-bonding character between N10C10a. For FAD, π3 is the lowest unoccupied molecular orbital (LUMO). For the radical forms, π3 is a singly occupied molecular orbital (SOMO). For the reduced forms, π3 is the highest occupied molecular orbital (HOMO). For the oxidized and anionic radical forms, a π2 → π3 transition is observed in the region of 450 nm. This transition is red-shifted in the series FAD → FAD˙− → FADH˙ since the energetic gap between π2 and π3 becomes smaller when FAD is reduced. Indeed, the absorption maxima are predicted at around 460 nm, 440 nm and 650 nm for FAD, FAD˙− and FADH˙, respectively, corresponding to an absorption color ranging from dark blue, to blue and finally red. For the fully reduced forms, π3 → π5* becomes the bright transition, which is blue-shifted with respect to the first transition of the oxidized and semi-reduced forms. Indeed, the absorption maxima are found in the near-UV, around 325 nm for FADH− and 345 nm for FADH2. All these transitions have a similar intensity, corresponding to a broad bright peak of 100 nm.
A second intense peak appears between 300 nm and 800 nm for the FAD, FAD˙− and FADH˙ species. For the reduced forms, this peak is located outside the considered range, between 200 nm and 300 nm, corresponding to a π3 → π6* transition. For the oxidized and radical forms, this peak is found around 300 nm and 500 nm and it includes 3 to 4 different electronic transitions. For the radical forms, the most intense transition corresponds to the electron transfer from the SOMO π3 to the vacant orbital π5*. This transition is the same as the first bright transition for the reduced forms, although it is around 50 nm blue-shifted for the radical species. Indeed, the π3 → π5* absorption maximum is found at around 340 nm for FAD˙−. The same transition is found at 360 nm for FADH˙. This type of transition is not present in FAD, as in this form, π3 is a vacant orbital. For FAD, π3 accepts electrons from the lower lying π orbitals and from the oxygen lone-pairs located on the phosphate (nphos) and on the ribitol (nrib). These transitions, which would normally be dark, appear as the peaks of a similar intensity of the π → π* transitions due to two main reasons: (i) these transitions are strongly mixed with the π → π* transitions, thus becoming slightly bright thanks to an intensity-borrowing mechanism, and (ii) the wavefunction overlap between the ground and excited states is large. A similar trend is observed for the radical forms.
5 Discussion
The UV-visible absorption spectra of flavins and derivatives (riboflavin, lumichrome, lumiflavin, isoalloxazine, etc.) have been explored in depth in the literature, both experimentally10,40–46 and theoretically.11–13,15,16 Theoretically, these have been studied mainly in the gas phase and within an implicit description of the solvent. Some of these forms, especially for the radical forms, are usually stabilized when embedded in the protein. To our knowledge, only two theoretical studies have attempted to compute the absorption spectrum in protein, the work of Cannuccia et al.15 and the work of Solov'yov et al.,16 albeit only for the oxidized form and without explicitly accounting for the vibrational structure. Here, we attempt to compare the theoretical absorption spectra in protein and in the gas phase with the experimental spectra of FAD and its redox derivatives in protein. This is shown in Fig. 5. Since many flavoproteins share similar spectral properties,47 some of the experimental data have been taken from different forms of cryptochromes or photolyases: data for FAD and FAD˙− have been taken from Mosquito Cryptochrome 1 (Anopheles gambiae), and FADH˙ and FADH− from a photolyase of E. Coli. The four forms are similar to the absorption spectrum of Arabidopsis Thaliana cryptochrome 1 and have been taken from ref. 45. The spectrum of FADH2 has been taken from solution measurements, and it is reproduced from the data taken from ref. 10. In the following, we discuss each form separately.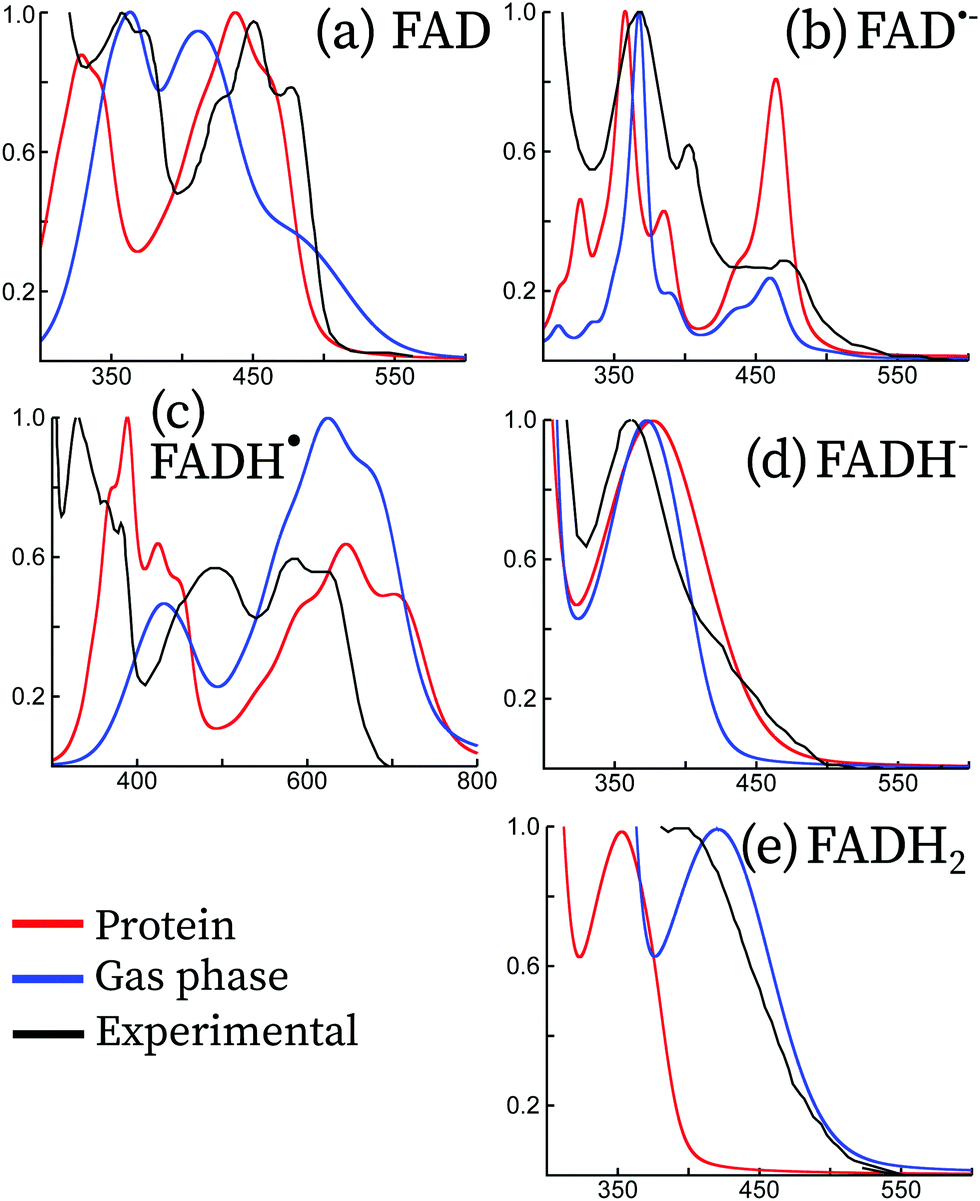 | ||
| Fig. 5 Comparison between the experimental (black) and theoretical absorption spectra of the five redox forms of the flavin adenine dinucleotide. Vibrationally resolved theoretical spectra have been computed in the gas phase (blue) and in protein (red). For the sake of comparison, all spectra have been normalized to the most intense peak between 300 nm and 800 nm. Experimental spectral data for FAD and FAD˙− have been taken from mosquito cryptochrome 1 (Anopheles gambiae), and FADH˙ and FADH− from a photolyase of E. Coli. The four forms are similar to the absorption spectrum of Arabidopsis Thaliana cryptochrome 1 and have been taken from ref. 45. The spectrum of FADH2 has been taken from solution measurements, and it is reproduced from the data taken from ref. 10. | ||
FAD
The oxidized form of FAD is the most stable one, both in solution and in protein. For this reason, it has been extensively studied in the literature both experimentally41,44,46,48–55 and theoretically.7,11,13,16,56–64 FAD has two main absorption bands, the first band in the blue visible light region and the second band in the UV-A region. In protein, the first is centered around 450 nm with a well-resolved three peak vibrational structure, while the second peak is centred at 368 nm and has two resolved vibrational peaks. This is true in solution as well, however the peak features are less apparent.10Our simulations taking into account the vibrational effects in protein are in good agreement with the experimental spectra. Indeed, the vibrational structure and the peak broadening are well reproduced for both the first and second peaks. The QM/MM spectrum leads to a slight blue-shift of the first transition of around 10 nm, and an important blue-shift of the second transition of 30–40 nm. The spectra calculated in the protein are distinct from the spectra calculated in the gas phase, indicating that the electrostatic interaction and the structure confinement due to the protein scaffold are essential to capture the vibrational structure of the absorption spectrum.
FAD˙−
From Arabidopsis Thaliana cryptochrome 1 (AtCry1), an anion radical is obtained after 450 nm photoexcitation of FAD via an electron transfer from a tryptophan residue found nearby the protein cavity.65 For some animal cryptochromes it can even be the primary photoproduct after blue light irradiation.43,45 Its spectroscopic signatures are three clearly distinguished peaks at 372, 402 and 490 nm.40From our results, we observe overall good agreement between the experimental and theoretical spectra. The transition energies in all the spectra range up to 300 nm. In contrast, the intensities are only approximately reproduced, despite the vibrational structure being in good accordance with experimental spectra. On the one hand, the simulated absorption spectrum in protein largely overestimates the intensity of the lowest-energy peak, although the vibrational progression and broadening match well the experimental peak. The relative intensity obtained using the simulations in the gas phase better matches with that obtained using the experiment. On the other hand, the vibrational structure of the peaks above 400 nm is well reproduced by the simulations in protein, while the gas phase simulations do not correctly describe this region.
In the literature, it has been argued that the peak around 370 nm corresponding to π1 → π3 could arise or have a contribution from a tyrosil radical (TyrO˙),46,66 which would be formed by a deprotonation of tyrosil and an electron transfer to the tryptophane radical previously formed.42,66 In Fig. 6, we show the vibrationally resolved theoretical spectrum of tyrosine429 in its radical form. The spectrum is shifted by about 30 nm with respect to the TyrO• in solution. Indeed, the peak position matches the region of the π1 → π3 and it has a similar shape. However, our simulations of the FAD˙− spectrum show a peak in the same region originating from an isoalloxazine transition. Therefore, our simulations are not able to confirm or refute the hypothesis that TyrO• is present in the absorption spectrum of FAD˙.
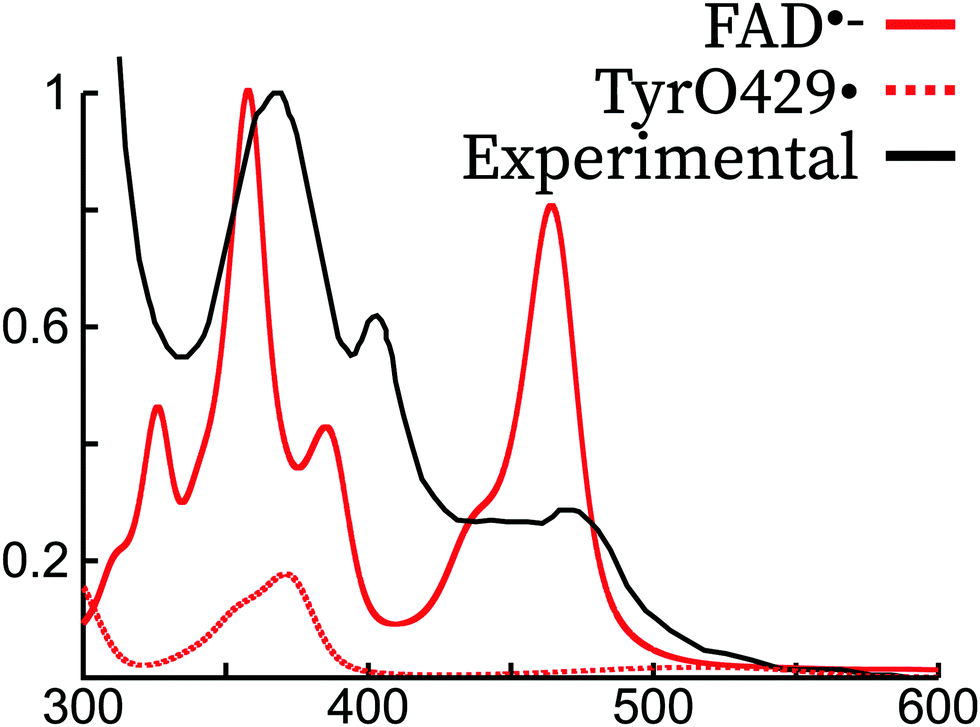 | ||
Fig. 6 Comparison of the experimental spectrum of FAD˙− (ref. 45) with the theoretical spectrum of FAD˙− and Tyr429˙ in the Arabidopsis Thaliana cryptochrome. The experimental and theoretical FAD˙− spectra have been normalized to 1 for the largest intensity peak. The TyrO429˙ intensity was scaled with the same scaling factor as FAD˙−, supposing thus a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two species. :1 mixture of the two species. | ||
FADH˙
Absorption difference spectra clearly show the formation of the neutral radical in AtCry1.41,42,44 It can be obtained after proton transfer coming from TrpH˙+ to the anionic radical. Alternatively, the nearby aspartate Asp396 may also function as a proton donor.8 Müller et al. reported an absorption spectrum for AtCry1 extracted from a mixture of the oxidized FAD and neutral radical.46 Contrary to the oxidized and anionic radical forms, the neutral radical has a nonzero absorbance in the green/red visible spectral region, between 500 and 650 nm.Our simulations only qualitatively reproduce the absorption spectrum of FADH˙, but the overall spectrum shows a large red shift of about 100 nm both for the gas phase and the protein embedded simulations with respect to experiments. We attribute this to the chosen level of theory, attributed to the lack of derivative discontinuity of the exchange–correlation potential, which underestimates the fundamental gaps especially of radical systems.67 As far as the peak intensities are concerned, the protein simulations are in better accordance than the gas phase with respect to experiments. The simulations in protein seem to reproduce the relative intensities and the peak structure of the lowest energy and the highest energy peaks.
Both the gas phase and protein simulations completely miss the peak around 500 nm. A peak was obtained in this region in previous theoretical simulations of FADH˙ in polar solvents using continuum models,7,13 but the origin is probably different from the peak appearing in protein. Rather, we attribute this peak as a radical signature of a tryptophan radical. In the experimental literature, it has been argued that a contribution from the Trp˙ spectrum is present in the absorption spectrum of FADH˙.46,66 In Fig. 7, we show the shifted absorption spectrum of FADH˙ together with the spectrum of the deprotonated radical tryptophan409. As it can be observed, the position of the tryptophan spectrum falls in the region of 500 nm, which would explain the extra peak missing in our simulated spectrum. The two peaks that were appearing in the FADH˙ spectrum would therefore correspond to the first and third peak in the experimental spectrum. Owing to the relative intensities of the FADH˙ and Trp˙, the concentration of both species would be 1:1, therefore excluding the formation of TyrO˙, which if formed from Trp˙ and would otherwise decrease its concentration in protein.
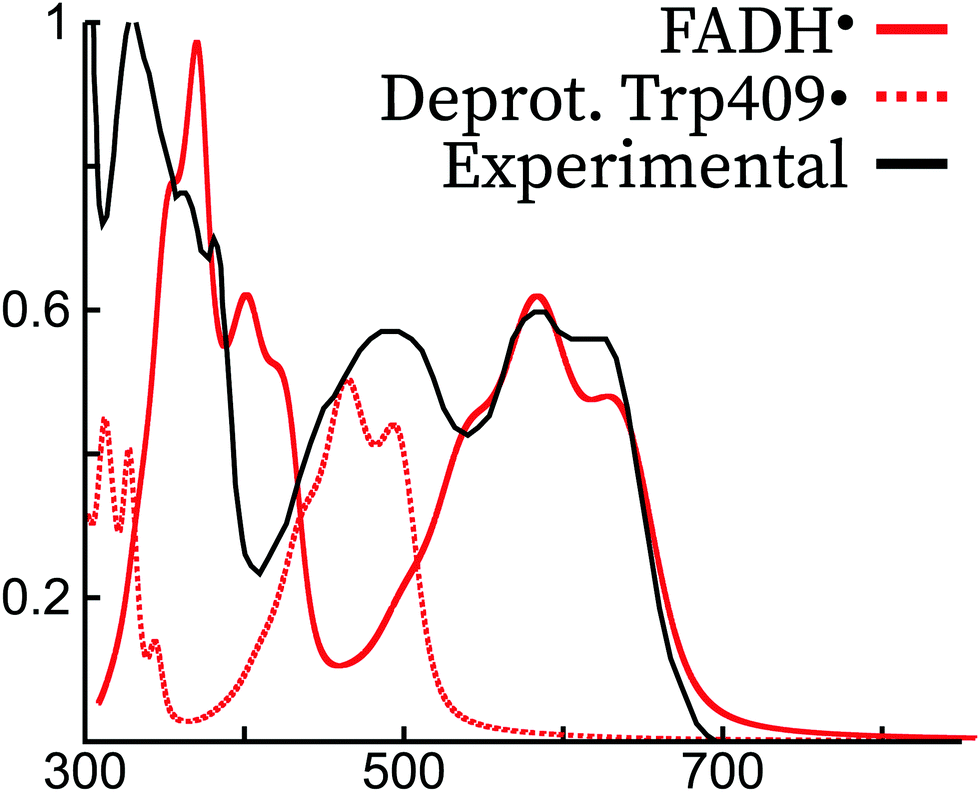 | ||
| Fig. 7 Comparison of the experimental spectrum of FADH˙ (E. Coli photolyase, ref. 45) with the theoretical spectrum of FADH˙ and Trp409˙ in the Arabidopsis Thaliana cryptochrome. The experimental and theoretical FADH˙ spectra have been normalized to 1 for the largest intensity peak, and the theoretical spectrum of FADH˙ was blue-shifted by ca. 70 nm to match the most intense peak of the lowest energy experimental transition. Trp409• was not shifted, and its intensity was scaled with the same scaling factor as FADH˙, supposing thus a 1:1 mixture of the two species. | ||
FADH−
The fully reduced state is not favored in vivo and in vitro requires a strong reducing agent.68,69 In protein the reduced anion is formed after a second electron transfer from tryptophan, usually after a second photon absorption from FADH˙.70 Reoxidation occurs in the presence of molecular oxygen. In solution, the anionic reduced form has a maximum absorption peak at 256 nm and shoulders at 288 and 350 nm.71 A study in the E. Coli photolyase showed an absorption band around 360 nm, with very weak absorbance in the visible region.10,45Our simulations both in protein and in the gas phase match the experimental spectrum, both in terms of shape and vibrational progression. In the experimental spectrum, a shoulder appears at around 450 nm, which is probably due to some residual species (like a small concentration of Trp•) or an almost dark π → π* transition as found in the experimental Stark spectra and theoretical simulations of FADH− in solution.13,72 This feature is not captured by our QM/MM simulations, probably due to an underestimation of the intensity of this transition.
FADH2
The neutral reduced form is formed after a second proton transfer from tryptophan from the FADH− form. In protein, its spectrum hardly differs from FADH−43 and its characterization is mostly limited to solution studies, where it has a distinguished shoulder at 400 nm.10,71
The experimental measurements for this form shown in Fig. 5 were done in solution. These match well with the gas-phase calculations, but the protein calculations show a blue-shift of this peak of around 100 nm, leading to a similar excitation energy to that of FADH−. This large blue-shift does not have an electrostatic origin, but it can be rather attributed to the enforced planar structure due to the steric hindrance of the protein, since the electronic transition of FADH2 in the gas phase at the protein optimized conformation leads to the same absorption maximum. Unfortunately, there is no clear experimental evidence of this form in the cryptochrome protein to determine whether this shift of the QM/MM simulations is realistic.
6 Conclusions
The simulations of the UV-visible absorption spectrum in complex media are a difficult task in theoretical chemistry. Here, we have developed and implemented the analytic second-derivatives of the electrostatic embedding QM/MM ESPF method24 for unrestricted wavefunctions, extending our previous developments17,23 to open-shell systems. This method allows, among other molecular properties, the vibrational analysis of minimum energy structures and the simulations of the vibrationally resolved spectra in protein. We have applied this methodology to simulate the absorption spectrum of FAD and its four redox derivatives (two radicals and two reduced forms, one of each anionic and one neutral) embedded in the Arabidopsis Thaliana cryptochrome protein.The theoretical absorption spectrum in protein shows a perfect agreement with the experimental spectrum for the non-radical forms, reproducing well both the intensity, peak position and vibrational peak structure of the electronic transitions found between 300 nm and 600 nm. In contrast, the agreement in the case of the radical forms is only moderate. As discussed recently in the literature,46,66 the signature of tryptophan and tyrosine radicals could be found in the absorption spectra of FAD˙− and FADH˙. On the one hand, for FAD˙−, a tyrosil peak appears at the same position corresponding to the π → π* transition. The position and shape of both peaks are similar, and therefore we cannot conclude that a tyrosil signature is present. On the other hand, the theoretical FADH˙ absorption spectrum in protein can only be matched to the experimental spectrum considering the lowest intense peak of the Trp˙ radical at around 500 nm. We estimate the proportion between the two species as 1:1.
In conclusion, we demonstrate that vibrational analysis at the electrostatic embedding QM/MM level is a valuable tool to interpret the complex absorption spectra arising in proteins. In the future, we plan to extend our method to the simulations of vibrational spectroscopy (infra-red, non-resonant Raman, etc.) as well as extending the vibrational analysis to other kinds of wavefunctions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the “Agence Nationale pour la Recherche” through the project BIOMAGNET (ANR-16CE29-0008-01). “Centre de Calcul Intensif d'Aix-Marseille” is acknowledged for granting access to its high performance computing resources.Notes and references
- X. Yu, H. Liu, J. Klejnot and C. Lin, The Arabidopsis Book, 2010, vol. 2010 Search PubMed.
- S. Braatsch and G. Klug, Photosynth. Res., 2004, 79, 45–57 CrossRef CAS PubMed.
- I. Chaves, R. Pokorny, M. Byrdin, N. Hoang, T. Ritz, K. Brettel, L.-O. Essen, G. T. J. van der Horst, A. Batschauer and M. Ahmad, Annu. Rev. Plant Biol., 2011, 62, 335–364 CrossRef CAS PubMed.
- C. Lin, D. Top, C. C. Manahan, M. W. Young and B. R. Crane, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3822–3827 CrossRef CAS PubMed.
- M. Liedvogel and H. Mouritsen, J. R. Soc., Interface, 2010, 7, S147–S162 CrossRef CAS PubMed.
- A. Pinzon-Rodriguez, S. Bensch and R. Muheim, J. R. Soc., Interface, 2018, 15, 20180058 CrossRef PubMed.
- P. Mondal, K. Schwinn and M. Huix-Rotllant, J. Photochem. Photobiol., A, 2020, 387, 112164 CrossRef CAS.
- I. A. Solov'yov, T. Domratcheva, A. R. Moughal Shahi and K. Schulten, J. Am. Chem. Soc., 2012, 134, 18046–18052 CrossRef PubMed.
- P. Mondal and M. Huix-Rotllant, Phys. Chem. Chem. Phys., 2019, 21, 8874–8882 RSC.
- Y.-T. Kao, C. Saxena, T.-F. He, L. Guo, L. Wang, A. Sancar and D. Zhong, J. Am. Chem. Soc., 2008, 130, 13132–13139 CrossRef CAS PubMed.
- B. Klaumünzer, D. Kröner and P. Saalfrank, J. Phys. Chem. B, 2010, 114, 10826–10834 CrossRef PubMed.
- Y. Ai, C. Zhao, J. Xing, Y. Liu, Z. Wang, J. Jin, S. Xia, G. Cui and X. Wang, J. Phys. Chem. A, 2018, 122, 7954–7961 CrossRef CAS PubMed.
- M. P. Kabir, Y. Orozco-Gonzalez and S. Gozem, Phys. Chem. Chem. Phys., 2019, 21, 16526–16537 RSC.
- F. Müller, Free Radical Biol. Med., 1987, 3, 215–230 CrossRef.
- E. Cannuccia, O. Pulci, R. D. Sole and M. Cascella, Chem. Phys., 2011, 389, 35–38 CrossRef CAS.
- C. Nielsen, M. S. Norby, J. Kongsted and I. A. Solov'yov, J. Phys. Chem. Lett., 2018, 9, 3618–3623 CrossRef CAS PubMed.
- K. Schwinn, N. Ferré and M. Huix-Rotllant, J. Chem. Theory Comput., 2020 DOI:10.1021/acs.jctc.9b01145.
- S. Dapprich, I. Komáromi, K. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct. THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS.
- Q. Cui and M. Karplus, J. Chem. Phys., 2000, 112, 1133–1149 CrossRef CAS.
- Q. Cui and M. Karplus, J. Phys. Chem. B, 2000, 104, 3721–3743 CrossRef CAS.
- A. Ghysels, H. L. Woodcock, J. D. Larkin, B. T. Miller, Y. Shao, J. Kong, D. V. Neck, V. V. Speybroeck, M. Waroquier and B. R. Brooks, J. Chem. Theory Comput., 2011, 7, 496–514 CrossRef CAS PubMed.
- T. Giovannini, L. Grazioli, M. Ambrosetti and C. Cappelli, J. Chem. Theory Comput., 2019, 15, 5495–5507 CrossRef CAS PubMed.
- K. Schwinn, N. Ferré and M. Huix-Rotllant, J. Chem. Phys., 2019, 151, 041102 CrossRef PubMed.
- N. Ferré and J. G. Ángyán, Chem. Phys. Lett., 2002, 356, 331–339 CrossRef.
- F. Melaccio, M. Olivucci, R. Lindh and N. Ferré, Int. J. Quantum Chem., 2011, 111, 3339–3346 CrossRef CAS.
- J. A. Pople, R. Krishnan, H. B. Schlegel and J. S. Binkley, Int. J. Quantum Chem., 1979, 16, 225–241 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- J. W. Ponder, TINKER: Software tools for molecular design, version 6.3, 2010, http://dasher.wustl.edu/tinker/.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- J. Antony, D. Medvedev and A. Stuchebrukhov, J. Am. Chem. Soc., 2000, 122, 1057 CrossRef CAS.
- A. Baiardi, J. Bloino and V. Barone, J. Chem. Theory Comput., 2013, 9, 4097–4115 CrossRef CAS.
- P. Macak, Y. Luo and H. Ågren, Chem. Phys. Lett., 2000, 330, 447–456 CrossRef CAS.
- Y. S. Choong and V. Massey, J. Biol. Chem., 1980, 255, 8672–8677 CAS.
- C. Lin, D. Robertson, M. Ahmad, A. Raibekas, M. Jorns, P. Dutton and A. Cashmore, Science, 1995, 269, 968–970 CrossRef CAS PubMed.
- B. Giovani, M. Byrdin, M. Ahmad and K. Brettel, Nat. Struct. Mol. Biol., 2003, 10, 489–490 CrossRef CAS.
- S.-H. Song, B. Dick, A. Penzkofer, R. Pokorny, A. Batschauer and L.-O. Essen, J. Photochem. Photobiol., B, 2006, 85, 1–16 CrossRef CAS PubMed.
- T. Kottke, A. Batschauer, M. Ahmad and J. Heberle, Biochemistry, 2006, 45, 2472–2479 CrossRef CAS PubMed.
- B. Liu, H. Liu, D. Zhong and C. Lin, Curr. Opin. Plant Biol., 2010, 13, 578–586 CrossRef CAS PubMed.
- P. Müller, J.-P. Bouly, K. Hitomi, V. Balland, E. D. Getzoff, T. Ritz and K. Brettel, Sci. Rep., 2014, 4, 5175 CrossRef PubMed.
- P. Macheroux, UV-Visible Spectroscopy as a Tool to Study Flavoproteins, Humana Press, 1999, pp. 1–7 Search PubMed.
- J. Koziol, Experientia, 1965, 21, 189–190 CrossRef CAS PubMed.
- J. Koziol, Photochem. Photobiol., 1966, 5, 41–54 CrossRef CAS.
- J. Koziol, Photochem. Photobiol., 1969, 9, 45–53 CrossRef CAS PubMed.
- A. Weigel, A. L. Dobryakov, M. Veiga and J. L. P. Lustres, J. Phys. Chem. A, 2008, 112, 12054–12065 CrossRef CAS PubMed.
- P. Zirak, A. Penzkofer, T. Mathes and P. Hegemann, Chem. Phys., 2009, 358, 111–122 CrossRef CAS.
- T. Kottke, J. Heberle, D. Hehn, B. Dick and P. Hegemann, Biophys. J., 2003, 84, 1192–1201 CrossRef CAS PubMed.
- E. Sikorska, I. V. Khmelinskii, J. Koput, J. L. Bourdelande and M. Sikorski, J. Mol. Struct., 2004, 697, 137–141 CrossRef CAS.
- A. Penzkofer, J. Photochem. Photobiol., A, 2016, 314, 114–124 CrossRef CAS.
- C. B. Martin, X. Shi, M.-L. Tsao, D. Karweik, J. Brooke, C. M. Hadad and M. S. Platz, J. Phys. Chem. B, 2002, 106, 10263–10271 CrossRef CAS.
- M. Takahashi, Y. Ishikawa, J.-I. Nishizawa and H. Ito, Chem. Phys. Lett., 2005, 401, 475–482 CrossRef CAS.
- S. Salzmann, J. Tatchen and C. M. Marian, J. Photochem. Photobiol., A, 2008, 198, 221–231 CrossRef CAS.
- S. Salzmann and C. M. Marian, Chem. Phys. Lett., 2008, 463, 400–404 CrossRef CAS.
- J.-Y. Hasegawa, S. Bureekaew and H. Nakatsuji, J. Photochem. Photobiol., A, 2007, 189, 205–210 CrossRef CAS.
- M. M. N. Wolf, C. Schumann, R. Gross, T. Domratcheva and R. Diller, J. Phys. Chem. B, 2008, 112, 13424–13432 CrossRef CAS PubMed.
- C. Neiss, P. Saalfrank, M. Parac and S. Grimme, J. Phys. Chem. A, 2003, 107, 140–147 CrossRef CAS.
- M. Dittric, P. L. Freddolino and K. Schulten, J. Phys. Chem. B, 2005, 109, 13006–13013 CrossRef PubMed.
- Y.-K. Choe, S. Nagase and K. Nishimoto, J. Comput. Chem., 2007, 28, 727–739 CrossRef CAS PubMed.
- K. Maeda, A. J. Robinson, K. B. Henbest, H. J. Hogben, T. Biskup, M. Ahmad, E. Schleicher, S. Weber, C. R. Timmel and P. J. Hore, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4774–4779 CrossRef CAS PubMed.
- F. Lacombat, A. Espagne, N. Dozova, P. Plaza, P. Müller, K. Brettel, S. Franz-Badur and L.-O. Essen, J. Am. Chem. Soc., 2019, 141, 13394–13409 CrossRef CAS PubMed.
- E. J. Baerends, O. V. Gritsenko and R. van Meer, Phys. Chem. Chem. Phys., 2013, 15, 16408–16425 RSC.
- P. Müller and M. Ahmad, J. Biol. Chem., 2011, 286, 21033–21040 CrossRef PubMed.
- L. J. van Wilderen, G. Silkstone, M. Mason, J. J. van Thor and M. T. Wilson, FEBS Open Bio., 2015, 5, 885–892 CrossRef CAS PubMed.
- J. Wang, X. Du, W. Pan, X. Wang and W. Wu, J. Photochem. Photobiol., C, 2015, 22, 84–102 CrossRef CAS.
- P. Hemmerich, C. Veeger and H. C. S. Wood, Angew. Chem., 1965, 4, 671–687 CrossRef CAS PubMed.
- R. F. Pauszek, G. Kodali, M. S. U. Siddiqui and R. J. Stanley, J. Am. Chem. Soc., 2016, 138, 14880–14889 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the optimized QM/MM minimum energy structures for oxidized, anionic and neutral semi-reduced and reduced forms. See DOI: 10.1039/d0cp01714k |
| This journal is © the Owner Societies 2020 |