
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilization of arrestin-3 on different biosensor platforms for evaluating GPCR binding†
Saziye Yorulmaz
Avsar‡
a,
Larisa E.
Kapinos‡
b,
Cora-Ann
Schoenenberger
 a,
Gebhard F. X.
Schertler
cd,
Jonas
Mühle
c,
Benoit
Meger
c,
Roderick Y. H.
Lim
b,
Martin K.
Ostermaier
e,
Elena
Lesca
*cd and
Cornelia G.
Palivan
*a
a,
Gebhard F. X.
Schertler
cd,
Jonas
Mühle
c,
Benoit
Meger
c,
Roderick Y. H.
Lim
b,
Martin K.
Ostermaier
e,
Elena
Lesca
*cd and
Cornelia G.
Palivan
*a
aDepartment of Chemistry and the Swiss Nanoscience Institute, University of Basel, 4002 Basel, Switzerland. E-mail: Cornelia.Palivan@unibas.ch
bBiozentrum and the Swiss Nanoscience Institute, University of Basel, 4056 Basel, Switzerland
cDepartment of Biology and Chemistry, Laboratory of Biomolecular Research, Paul Scherrer Institute, 5303 Villigen-PSI, Switzerland. E-mail: Elena.Lesca@psi.ch
dDepartment of Biology, ETH Zürich, 8093 Zürich, Switzerland
ePaul Scherrer Institute, 5303 Villigen-PSI, Switzerland
First published on 29th September 2020
Abstract
G protein-coupled receptors (GPCRs) are a large and ubiquitous family of membrane receptors of great pharmacological interest. Cell-based assays are the primary tool for assessing GPCR interactions and activation but their design and intrinsic complexity limit their application. Biosensor-based assays that directly and specifically report GPCR-protein binding (e.g. arrestin or G protein) could provide a good alternative. We present an approach based on the stable immobilization of different arrestin-3 proteins (wild type, and two mutants, mutant X (arrestin-3 I386A) and mutant Y (arrestin-3 R393E)) via histidine tags on NTA(Ni2+)-coated sensors in a defined orientation. Using biolayer interferometry (BLI), surface plasmon resonance (SPR), and quartz crystal microbalance with dissipation (QCM-D), we were able to follow the interaction between the different arrestin-3 proteins and a representative GPCR, jumping spider rhodopsin-1 (JSR1), in a label-free manner in real-time. The interactions were quantified as binding affinity, association and dissociation rate constants. The combination of surface-based biosensing methods indicated that JSR1 showed the strongest binding to arrestin mutant Y. Taken together, this work introduces direct label-free, biosensor-based screening approaches that can be easily adapted for testing interactions of proteins and other compounds with different GPCRs.
1 Introduction
G protein-coupled receptors (GPCRs) are the largest family of membrane proteins that sense the extracellular environment in animals and transduce the information into the cell in order to coordinate a broad range of physiological processes including vision, olfaction, neural signal transmission, and pain.1,2 The stimuli that activate GPCRs are physically and chemically diverse, for example neurotransmitters, hormones, odorants, ions, and light.3 All GPCRs have a similar structure: an extracellular N-terminus, seven α-helices, and an intracellular C-terminus. The extracellular domain contains a pocket for the ligand, while the intracellular region interacts with binding proteins such as G-protein or arrestin. Upon activation, conformational changes occur from the ligand-binding pocket to the cytosolic region, starting G protein-mediated signaling cascades. In order to control the number of activated receptors, GPCR kinases (GRKs) phosphorylate the cytoplasmic region (e.g. the C-tail) of the receptor triggering the binding of arrestin, which prevents G protein binding to the receptor.Most vertebrates express four arrestin subtypes. Rods and cones visual arrestins (respectively arrestin-1 and arrestin-4) are selectively expressed in the retina, while non-visual β-arrestins 1 and 2 (also known as arrestin-2 and arrestin-3, respectively) are ubiquitously expressed and regulate many different GPCRs.4 Arrestins have no enzymatic activity but can assemble in multi-protein signaling complexes or deactivate GPCRs and other receptor types.5 Arrestin interacts with GPCR predominantly at two sites: the arrestin finger-loop penetrates the cytosolic side of the receptor, while elements of the N-terminal domain engage the phosphorylated C-tail of the receptor.6 These interactions prevent GPCR activation through G-protein and simultaneously promote receptor internalization, directing GPCRs to lysosomes for degradation or recycling them back to the plasma membrane where they are reused in signaling. Therefore, targeting specific arrestin-GPCR interactions has therapeutic potential. Different strategies are being considered in the context of developing arrestin-related therapeutics:5 (i) proteins that compete with GPCRs for binding arrestin, (ii) peptides or small molecules that disrupt GPCR-arrestin interactions, or (iii) the use of arrestin mutants to control excessive signaling by GPCRs. However, in order to develop therapeutics targeting GPCR–arrestin interactions, the precise identification of interaction sites as well as knowledge of the binding affinity between arrestin and the respective binding partners are necessary.
Cell-based assays are mainly used to investigate the GPCR-arrestin interactions because GPCRs need to be stabilized by the phospholipid bilayer and phosphorylated prior to binding arrestin.7,8 Approaches such as bioluminescence and fluorescence resonance energy transfer (BRET and FRET, respectively) are used to study different GPCR functions in intact cells either statically or in real-time.9–11 However, protein labeling might interfere with GPCR–arrestin interactions and thus, might influence experimental outcomes. In addition, the activity of the receptor on the cell surface has been monitored by a reporter assay, which indirectly measures changes in the concentration of downstream signaling molecules such as second messengers.12,13 Although using reporters or indirect measurements in cells provide qualitative evidence of GPCR–arrestin interactions, it is very difficult to quantify the kinetic parameters of such interactions due to the interference from other cellular components of GPCR signaling and exogenous protein overexpression.
Alternatively, label-free biosensor technologies lend themselves to quantifying GPCR interactions. Commonly used label-free biosensor technologies are biolayer interferometry (BLI), surface plasmon resonance (SPR) and quartz crystal microbalance with dissipation monitoring (QCM-D).14 In order to investigate GPCR–protein interactions, several methods for the immobilization of GPCR on biosensors have been proposed including antibody-mediated attachment, avidin–biotin based capture procedures, or the reconstitution of GPCR within physiologically relevant environments (e.g. liposomes, and nanodiscs).15–18 By these means, the interactions of G protein with immobilized receptors have been monitored. For example, immobilized bovine rhodopsin (BRho) enabled exploring G protein activation and receptor deactivation using SPR.19 In order to study the association of β-arrestin-1 with the C–C chemokine receptor 5 (CCR5), different immobilization strategies were pursued. Either an N-terminal cysteine residue of CCR5-derived peptides was coupled to the thiol groups of a CM5 biosensor or peptides corresponding to the CCR5 IL-3 cytoplasmic loop were synthesized with an N-terminal biotin moiety and immobilized to a streptavidin sensor surface.20 However, the immobilization of native GPCR receptors on the sensor surface might have certain disadvantages, such as random orientation, inaccessibility of the binding site or conformational changes interfering with receptor function. Since specific conformational changes are a prerequisite for arrestin binding and GPCRs require lipids or detergent molecules to be stabilized, it might be better to immobilize binding protein and allow the receptor to be free in order to trigger protein–protein interactions of interest. Besides, immobilization of the GPCRs on the biosensor requires extensive optimization procedures and thus, large amounts of GPCR which in itself is a big challenge due to difficulties in purifying GPCRs without altering their functional characteristics. Here we present a straightforward platform to study the interaction between GPCRs and arrestin-3 requiring minimal amounts of recombinantly expressed, functional proteins. This strategy is based on the surface immobilization of arrestin-3, a GPCR partner that appears to also interact with non-phosphorylated receptors.21,22 We combine label-free biosensor technologies, specifically BLI, SPR and QCM-D in order to provide an overview of the changes in GPCR/arrestin-3 interactions when arrestin-3 is immobilized on different surfaces.
For our assays we chose to immobilize non-visual arrestin-3 (Arr3) because it is the least selective member of the arrestin family and binds to many different GPCRs.23 In addition to wild-type arrestin-3 (Arr3-WT), two arrestin-3 mutants, I386A (Arr3-X) and R393E (Arr3-Y) were studied. According to cell-based assays, these two variants have been suggested to show a higher sensitivity towards activated GPCRs than Arr3-WT.24,25 Each of the mutations affects one of two stabilizing binding sites of arrestin-3 (Fig. 1): R393E results in a change of the arrestin polar core and I386A impinges on three-element interaction. Both are predicted to result in a “pre-activated” form of arrestin leading to an increased receptor coupling affinity compared to Arr3-WT.
 | ||
| Fig. 1 Schematic representation of possible interactions between DDM-stabilized JSR1 and immobilized Arr3. Arrestin-3 (PDB ID: 3p2d4) is immobilized via a histidine-tag to multivalent chelator headgroups on a supported lipid bilayer (SLB) or monolayer (SLM). Two distinct interaction interfaces with JSR1 (PDB ID: 6i9k26) are indicated by dotted circles: (1) JSR1 C-tail interactions with the N domain of Arr3 and (2) the hydrophobic inter-domain interactions. The replacement of the positively charged (blue) arginine to the negatively charged (red) glutamic acid (R393E mutation in the polar core, which is indicated by shaded area) interferes with charge interactions, which normally keep the binding inaccessible for the C-terminus of the receptor, thus likely inducing a pre-activated state of Arr3. The I386A mutation in Arr3-X is located in the region involved in three-element-interactions (TE-interaction, shaded area) and might help the N-terminus of Arr3 to disengage from an intramolecular binding, thus achieving a pre-activated mutant. | ||
As a model GPCR we selected Jumping Spider Rhodopsin-1 (JSR1), a light-sensitive bistable class A GPCR, which has recently been shown to be highly thermostable in its wild-type form in vitro26 and is an interesting target for the development of optogenetic tools.26,27 In fact, as other rhodopsins, JSR1 is easily activated by illumination without the addition of ligands. Moreover, invertebrate rhodopsins are known to be able to bind arrestin in a phosphorylation-independent way upon illumination.28 Finally, the high resolution crystal structure of JSR1 (PDB ID 6I9K)26 shows similarities between JSR1 and other non-light sensitive class A GPCRs26 suggesting JSR1 as a valid model for in vitro binding studies.26,27 At the same time, we establish here a platform, which allows an in-depth biophysical characterization of arrestin binding of any GPCR with pharmaceutical relevance.
We evaluated different surface-based methods, specifically BLI, SPR and QCM-D, which allow the quantitative evaluation of the interaction of WT and mutant arrestin-3 with JSR1. Specifically, Arr3 was immobilized on commercially available Ni2+–NTA-containing, portable BLI tips which have no lipid layer. Then, using vesicle fusion, we formed a Ni2+–NTA functional supported lipid bilayer (SLB) on silicon oxide (QCM-D sensor) and a lipid monolayer (SLM) on HPA chips (SPR sensor), respectively. The advantage of SLBs is that they enable lateral mobility and thus provide a biologically relevant environment. SLMs and functionalized BLI tips on the other hand, represent platforms with restricted translational fluidity and thus higher stability, which are more resistant to the detergents necessary to stabilize the GPCRs. We were able to evaluate the binding of JSR1 to Arr3 and its mutants and calculate the equilibrium dissociation constants (KDs), and association and dissociation rate constants (ka and kd, respectively) for these interactions. Such information will provide the basis for finding stronger binders among candidates in drug design approaches.
2 Experimental methods
2.1 Expression and purification of JSR1
Wild-type JSR1 with a C-terminal 1D4 epitope tag was expressed and purified as described in Ehrenberg et al.27 Briefly, a suspension culture of HEK293S GnTI-cell line stably expressing JSR1 was cultivated in FreeStyle 293 expression medium to 2 × 106 cells per mL and then harvested. Frozen cells were mechanically homogenized in 50 mM HEPES, 140 mM NaCl, and 3 mM MgCl2 with protease inhibitors (Roche complete EDTA-free Protease Inhibitor Cocktail; Sigma-Aldrich, Buchs, Switzerland) and incubated overnight with 30 μM 9-cis retinal (98% grade, Sigma-Aldrich) overnight at 4 °C in the dark for reconstitution with the chromophore. JSR1 membranes were solubilized in the same buffer supplemented with 1% n-dodecyl-b-D-malto-pyranoside (DDM) (Anagrade; Anatrace, Maumee, OH) for 2 h at 4 °C. Solubilized JSR1 was separated from the membrane fraction by ultracentrifugation at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 1 h at 4 °C. Subsequently, JSR1 was purified from the supernatant by 1D4-antibody affinity chromatography as described in Varma et al.26 JSR1 purity was assessed by SDS-PAGE and UV-vis spectroscopy. Purified JSR1 was stored in 50 mM HEPES, 150 mM NaCl, 1 mM MgCl2 with 0.01% DDM at pH 6.5.
000g for 1 h at 4 °C. Subsequently, JSR1 was purified from the supernatant by 1D4-antibody affinity chromatography as described in Varma et al.26 JSR1 purity was assessed by SDS-PAGE and UV-vis spectroscopy. Purified JSR1 was stored in 50 mM HEPES, 150 mM NaCl, 1 mM MgCl2 with 0.01% DDM at pH 6.5.
2.2 Cloning and expression of arrestin-3 proteins
Arr3-WT with an N-terminal hexahistidine followed by a TEV protease cleavage site (ENLYFQGGS) was cloned into the pET-15b vector (Novagen).29 Arr3-X (I386A) and Arr3-Y (R393E) (identified by InterAx Biotech) mutant constructs were produced by site-specific PCR mutagenesis using the pET-15b Arr3-WT plasmid as template.30,31 For arrestin-3 expression, chemically competent NiCo21(DE3) E. coli cells (New England Biolabs) were transformed with the corresponding plasmid. Expression was induced in high-density cultures strictly following the described procedure.2.3 Arrestin-3 protein purification
Histidine tagged Arr3-WT and Arr3 mutants were purified as described elsewhere29 with minor modifications. Briefly, cells were resuspended in solubilization buffer (50 mM Bis–Tris propane pH 7.0, 500 mM NaCl and 10% (v/v) glycerol) and lysed by 3 cycles of sonication. The clarified lysate was supplemented with 30 mM imidazole and purified over a 5 mL Ni–NTA FF crude column (GE Healthcare). The pooled protein fractions were dialyzed over night against dialysis buffer A (20 mM Bis–Tris propane pH 7.0, 120 mM NaCl, 10% (v/v) glycerol and 14.3 mM 2-mercaptoethanol) without adding TEV protease to preserve the N-terminal His-tag for immobilization. The dialyzed protein solution was diluted to a final salt concentration of 40 mM NaCl using dilution buffer (dialysis buffer without NaCl) and subsequently applied to a 15 mL Q sepharose column. After washing with Q sepharose buffer A (20 mM Bis–Tris propane pH 7.0, 40 mM NaCl, 10% (v/v) glycerol and 14.3 mM 2-mercaptoethanol) until a stable baseline was reached, the protein was eluted in Q sepharose buffer B (20 mM Bis–Tris propane pH 7.0, 200 mM NaCl, 10% (v/v) glycerol and 14.3 mM 2-mercaptoethanol). Finally, Arr3 proteins were purified over a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in SEC buffer (20 mM Bis–Tris propane pH 7.0, 300 mM NaCl, 10% (v/v) glycerol and 14.3 mM 2-mercaptoethanol). Pooled protein fractions were concentrated using a 30 kDa MWCO Millipore spin concentrator (Merck). Aliquots of Arr3 proteins were shock frozen in liquid nitrogen and stored at −80 °C.2.4 POPC:DGS-NTA(Ni2+) vesicle preparation
1-Palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) and 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) (DGS-NTA(Ni2+)) were obtained in chloroform from Avanti Polar Lipids (Alabaster, AL). Lipids were mixed at the desired molar ratio in a round-bottom glass flask. Dried lipid films were formed by evaporating the chloroform upon exposure to a gentle stream of nitrogen gas. The dried lipid films were placed in a vacuum desiccator overnight in order to remove residual chloroform. Subsequently, they were hydrated in 20 mM HEPES, pH 7.4 containing 150 mM NaCl, and vortexed for one minute. The resulting vesicle suspension was sequentially extruded through a track-etched polycarbonate membrane with 100 nm and then 50 nm diameter pores for a minimum of 21 times using a Mini Extruder apparatus (Avanti Polar Lipids).2.5 Dynamic light scattering (DLS)
The size distribution of lipid vesicles was characterized using a Zetasizer nano ZSP (Malvern Instruments, Malvern, UK). The scattering was recorded at a fixed and backscattering angle of 173°. The average effective hydrodynamic diameter of the vesicles and the sample polydispersity are recorded. All measurements were conducted at room temperature.2.6 Quartz crystal microbalance with dissipation (QCM-D)
A Q-Sense E1 instrument (Biolin Scientific, Gothenburg, Sweden) was used in order to monitor lipid and protein adsorption processes through changes in resonance frequency (ΔF) and dissipation (ΔD) of a silicon oxide-coated quartz crystal (model no. QSX303) as a function of time. The QCM-D measurement data were collected at the 3rd, 5th, 7th, 9th, and 11th odd overtones. The QCM-D data reported were all obtained at the 5th overtone and normalized accordingly (ΔFn = 5/5). Immediately before the experiment, the QCM-D sensor substrates were sequentially rinsed with 1% SDS, water, and ethanol followed by drying with nitrogen gas. The substrates were then subjected to oxygen plasma treatment (Harrick Plasma, Ithaca, NY) at the maximum radiofrequency (RF) power. All QCM-D measurements were conducted under continuous flow conditions controlled by a Reglo Digital peristaltic pump (Ismatec, Glattbrugg, Switzerland), with the flow rate set to 100 μL min−1 for bilayer formation and washing processes and 50 μL min−1 for protein addition and adsorption. The temperature of the flow cell was fixed at 24.0 ± 0.5 °C. The molecular ratio of adsorbed proteins was estimated by applying the Sauerbrey model to convert frequency shifts into mass density values as follows:| Δm = −C·ΔFn |
2.7 Surface plasmon resonance (SPR)
SPR binding assays were performed using a BiacoreT200 (GE Healthcare) and Biacore T200 Control software (version 2.0.1) at 25 °C with four parallel flow cells. A supported lipid monolayer (SLM) was established on the HPA chip (GE Healthcare), a flat hydrophobic surface consisting of long-chain alkanethiol molecules directly attached to the gold film, by fusion of POPC:DGS-NTA(Ni2+) (95:5) vesicles to the hydrophobic surface of this sensor chip in buffer (20 mM HEPES, pH 7.4 containing 150 mM NaCl). On the resulting SLM layer, His-tagged wild-type arrestin3 (Arr3) and its mutants X (Arr3-X) and Y (Arr3-Y) were immobilized. JSR1 was then injected into all for flow channels to measure its binding to arrestins (immobilized in the flow channels 2, 3 and 4, respectively). The first flow channel was used as a reference to compensate for the SPR signal change caused by unbound JSR1 during injection. The binding data were analyzed using Biacore T200 Evaluation software (version 3.0). The stoichiometric ratio of DDM stabilized JSR1 binding to Arr3 was calculated as follows;| (Rmax/MW of JSR1 in DDM)/(Rimm/MW of Arr3) |
2.8 Bio-layer interferometry (BLI)
Label-free bio-layer interferometry experiments were performed on the BLItz instrument (ForteBio, Inc., USA) with BLItz Pro™ analysis software. The his-tagged arrestin3 samples (4 μL of 1 μM each) were immobilized on the commercially available ForteBio Ni–NTA biosensors coated with a biocompatible matrix made of QIAGEN's agarose resin that is uniform and non-denaturing with minimal nonspecific binding. The provided ‘Dip and Read’ assay was used to monitor specific protein binding. Binding of the specific protein to the surface leads to the increase in the optical thickness of the bio-layer, resulting in the shift of the wavelength (in nm). All experiments were performed in 20 mM HEPES, pH 7.4 with 150 mM NaCl.3 Results and discussion
3.1 Supported lipid monolayer and bilayer formation on different sensors
In order to generate platforms with immobilized arrestin, we first created SLM and SLB platforms on SPR and QCM-D sensors, respectively, exposing Ni–NTA for histidine binding. SLM and SLB surfaces were prepared by vesicle adsorption, fusion, rupture, spreading, and lipid reassembly on the solid support.34,35 For SPR measurements, we used a hydrophobic HPA sensor surface for SLM formation whereas a hydrophilic silicon oxide QCM-D sensor served as an SLB support. Due to the different intrinsic properties of SLM and SLB, i.e. their fluidity and thickness, the interaction of arrestin with JSR1 stabilized in detergent micelles might vary depending on the platform. Similar to biological membranes, the SLB platform enables lateral mobility, which can be a crucial parameter for proper protein conformation. SLM on the other hand, is a more stable membrane that more likely resists GPCR stabilizing detergents. For SLM and SLB formation, the lipid vesicles were composed of a mixture of 95 mol% 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-choline (POPC) and 5 mol% Ni–NTA functionalized lipids (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt), DGS-NTA(Ni2+)), where the latter enables the immobilization of histidine-tagged proteins.36 The obtained vesicles were 77.23 ± 3.6 nm in diameter and had a polydispersity index (PDI) of 0.06 ± 0.016 as determined by dynamic light scattering.The SLM formation process was monitored by SPR (Fig. 2A and B). Real-time changes in the SPR angle were recorded as resonance units (RU), being proportional to the amount of the material deposited on the sensor.35 An optimal SLM coverage of the sensor was obtained when the lipid vesicles were injected at a low flow rate (2 μL min−1) immediately after a cleansing pulse of 40 mM octyl glucoside (n-octyl β-D-glucopyranoside) (2 μL min−1). This resulted in an SPR response within the range of 1500–2500 RU as is expected for a uniform lipid monolayer on the sensor.37 SLB formation on silicon oxide sensors was followed by QCM-D (Fig. 2C and D). Changes in mass (Δm) were derived from frequency (ΔF) shifts, which were simultaneously recorded in real-time (Fig. S1A, ESI†).38 In addition, viscoelastic properties of layers formed on sensors were represented as changes in dissipation (ΔD) (Fig. S1B, ESI†). Because the frequency shift is negatively proportional to the adsorbed mass, a Δm of approximately 450 ng cm−2 was calculated, indicating the formation of a uniform lipid bilayer. For all SLB platforms, the final QCM-D measurements corresponded to ΔF and ΔD values of −24.04 ± 2.17 Hz and <1 × 10−6, respectively, which agree well with literature values for SLB systems.34,39,40 In contrast to QCM-D and SPR sensors, BLI sensors are provided with polymer coatings that include Ni-NTA for histidine binding and thus, were not further modified.
 | ||
| Fig. 2 Supported lipid monolayer and bilayer formation by vesicle fusion. (A) Schematic illustration of SLM platform on an SPR sensor. (B) A representative SPR response of SLM formation on HPA sensor (number of replicates, n = 3). Arrows indicate first, an injection of vesicles made of POPC:DGS-NTA(Ni2+) (95:5), and then an injection of buffer (20 mM HEPES, 150 mM NaCl at pH 7.4). 2280 RU correspond to 175 ng cm−2 of adsorbed lipids. (C) Schematic representation of SLB platform on a QCM-D sensor. (D) Changes in mass were recorded as function of time upon formation of SLBs. The numbers at the top denote the injection of (1) buffer (20 mM HEPES, 150 mM NaCl at pH 7.4) containing 5 mM MgCl2, (2) vesicles made of POPC:DGS-NTA(Ni2+) (95:5), (3) buffer, and (4) buffer without MgCl2. | ||
3.2 Immobilization of Arr3 on different biosensors
The capture of Arr3, Arr3-X, and Arr3-Y on the sensor surfaces was achieved through interaction between the 6-histidine tag (His6) of the arrestin and nickel charged tris-nitrilotriacetic (NTA(Ni2+)) exposed on the respective biosensors. Because surface immobilization occurs via the N-terminal His6 tag, the finger loop structure in arrestin presumably is not perturbed and thus, is available for the interaction with JSR1 (Fig. 1).For BLI measurements, 1 μM of Arr3 was added to the NTA(Ni2+) functionalized sensors (Fig. 3A) and immobilization of Arr3 changed the interference pattern of the reflected light, causing a wavelength shift that was recorded as a function of time.
 | ||
| Fig. 3 Immobilization of WT and mutant Arr3 on BLI and SPR biosensors. (A) Capture of different arrestins (1 μM) to BLI Ni2+–NTA tips. (B) Change of the SPR response upon Arr3-WT (1 μM) addition to SLMs with different molar percentage of DGS-NTA(Ni2+). (C) Change of the SPR response upon Arr3-WT, mutants Arr3-X and Arr3-Y binding to SLM with 5% of DGS-NTA(Ni2+). Curves representing one of three independent measurements are shown. | ||
For the immobilization of Arr3 to SLM-coated SPR biosensors, 1 μM of Arr3 was injected to the platforms. Immobilization experiments for Arr3 were repeated at least 3 times. In order to control the amount of immobilized Arr3 on the SPR sensor, we changed the amount of DGS-NTA(Ni2+) content (1, 5 and 20%) in the vesicles used for SLM production. Representative sensorgrams recorded upon addition of Arr3-WT are presented in Fig. 3B. The binding response was related to the content of DGS-NTA(Ni2+) in SLM (Table 1).
| DGS-NTA(Ni2+) content (%) | SPR response (RU) | Adsorbed mass (ng cm−2) |
|---|---|---|
| 1 | 306 ± 52 | 23.5 ± 5.2 |
| 5 | 522 ± 95 | 40.2 ± 4 |
| 20 | 3692 ± 1800 | 284 ± 138 |
At the highest DGS-NTA(Ni2+) content, the change in SPR response corresponded to 3692 ± 1800 RU, from which, using the relation 1300 RU = 1 ng mm−2, a surface adsorption of 284 ± 138 ng cm−2 Arr3-WT was calculated.41 Increasing DGS-NTA(Ni2+) lipids within the SLM layer up to 20% results in an estimated distance between NTA-lipids of less than 3 nm. In this case, the arrestin immobilization capacity would be maximal and limited only by the size of arrestin itself. In order to compare binding profiles of wild type and mutant Arr3, we fixed the concentration of DGS-NTA(Ni2+) in the vesicles to 5 mol% and added 1 μM of the different arrestins (Fig. 3C). The results indicated that Arr3-WT led to a final response of 306 ± 52 RU whereas the final responses from Arr3-X and Arr3-Y (about 1000 RU) were similar but higher than the response obtained from Arr3-WT.
For QCM-D measurements, we limited the fraction of functional DGS-NTA(Ni2+) to 10% maximum since higher amounts of functionalized lipids might hinder the formation of SLBs on silicon oxide. After SLB formation on QCM-D biosensors with 1–10% of DGS-NTA(Ni2+), the QCM-D readouts for mass signals were re-equilibrated and set to zero to specifically focus on the immobilization of Arr3-WT. 120 nM Arr3-WT were injected to DGS-NTA(Ni2+) containing SLBs and the Δm as a function of time (Fig. 4A) were calculated from ΔF. Upon injection of Arr3-WT, an increase in mass was observed.
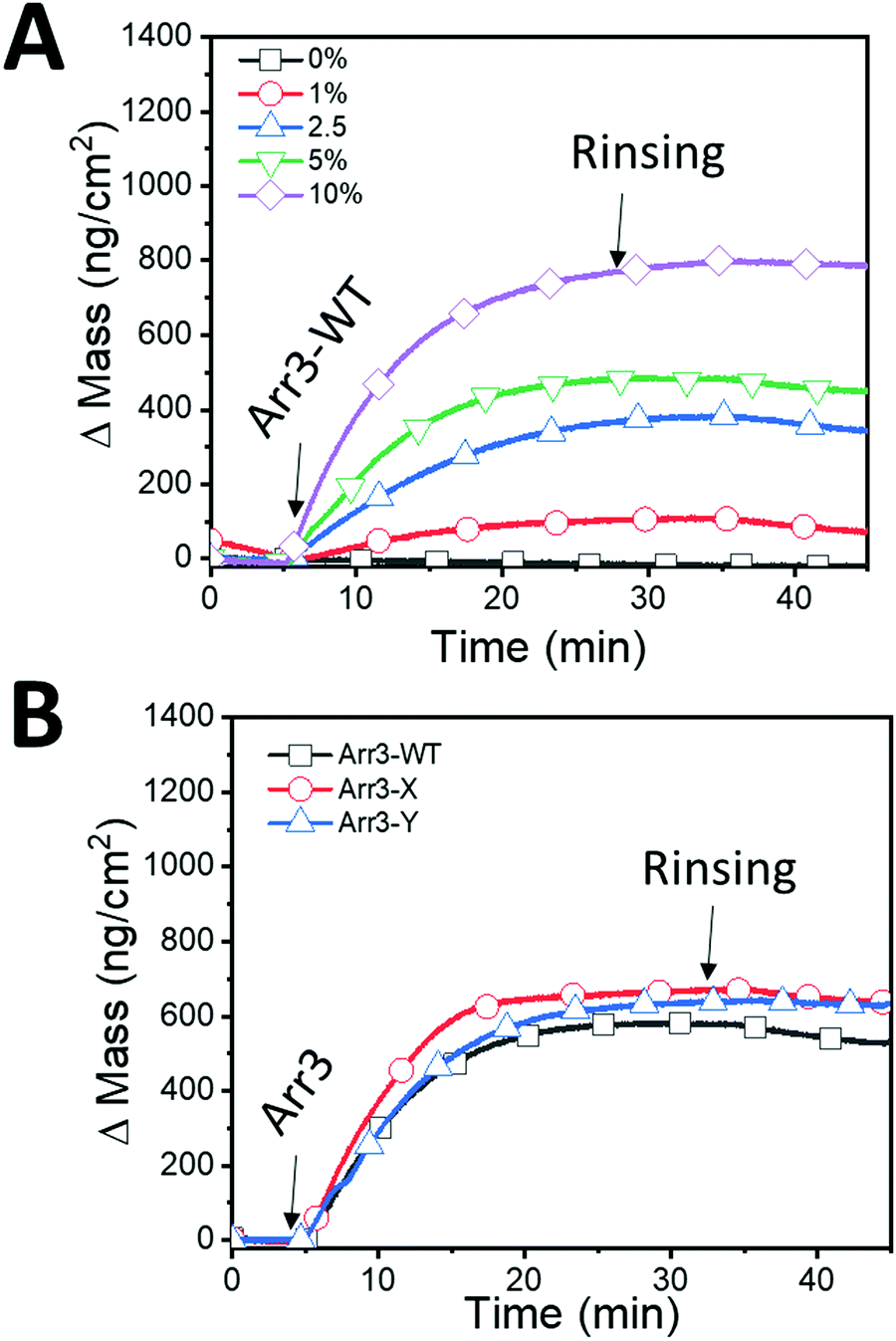 | ||
| Fig. 4 Immobilization of Arr3 proteins on SLB. (A) Changes in mass were recorded upon sequential addition of Arr3-WT on SLBs made up from 0 to 10% DGS-NTA(Ni2+) lipids. Arrows denote the injection of 120 nM Arr3 and the rinsing with buffer (20 mM HEPES, 150 mM NaCl at pH 7.4). (B) Changes in mass were recorded upon addition of Arrestin-3 protein. After the SLB formation, Arr3-WT (black squares), Arr3-X (red circles), and Arr3-Y (blue triangles), were immobilized on SLB with 5% DGS-NTA(Ni2+). | ||
The ΔF ranged from −6.03 ± 0.17 Hz at 1 mol% DGS-NTA(Ni2+) to −43.10 ± 2.5 at 10 mol% DGS-NTA(Ni2+) (Table 2). Increasing the amount of DGS-NTA(Ni2+) in the SLB did not result in a significant change of dissipation energy over time, suggesting that the conformation of bound Arr3-WT, which is related to the viscoelastic properties, is unchanged for the concentrations used. Additionally, since ΔD was around 1 × 10−6 for DGS-NTA(Ni2+) from 1% to 10%, Sauerbrey equation has been applied to quantify the surface density of attached Arr3-WT.32 The mass of Arr3-WT varied from 106.73 ± 3.003 ng cm−2 at 1 mol% DGS-NTA(Ni2+) up to 762.78 ± 44.43 ng cm−2 at 10 mol% DGS-NTA(Ni2+) (Table 2) indicating that higher amounts of chelator lipids in SLBs led to higher amounts of immobilized of Arr3-WT. In the absence of DGS-NTA(Ni2+), i.e. at 100% POPC, both ΔF and ΔD did not change upon addition of Arr3-WT. This confirms that the immobilization of Arr3-WT on the SLB platforms occurs specifically through the interaction between histidine tag and NTA(Ni2+). Initially, Arr3-WT in solution monovalently binds to the chelator lipid. Due to the fluidity of bilayers, Arr3-WT will encounter additional chelator lipids, resulting in polyvalently bound, stable Arr3-WT/DGS-NTA(Ni2+) lipid complexes. Thus, in addition to varying the DGS-NTA(Ni2+) fractions in SLBs, incubation conditions of Arr3-WT (e.g. time or concentration) can also be modified to tune the amount of bound proteins on SLBs.36
| DGS-NTA(Ni2+) content (%) | ΔF (Hz) | Mass (ng cm−2) |
|---|---|---|
| 0 | 1.18 ± 0.62 | −20.79 ± 10.88 |
| 1 | −6.03 ± 0.17 | 106.73 ± 3.003 |
| 2.5 | −21.01 ± 0.66 | 371.877 ± 11.76 |
| 5 | −33.71 ± 3.7 | 596.69 ± 65.56 |
| 10 | −43.10 ± 2.5 | 762.78 ± 44.43 |
In order to further compare the immobilization profiles of WT and mutant Arr3, we fixed the concentration of DGS-NTA(Ni2+) in the vesicles to 5 mol % and added 120 nM of the different arrestins (Fig. 4B). Under these conditions, WT and mutant Arr3 showed similar mass adsorption.
3.3 Probing the interaction between JSR1 and immobilized Arr3
To explore the interaction of immobilized arrestins with JSR1 by QCM-D, we first tested the binding activity of the platform using the negatively charged polysaccharide heparin as a model system since it has been shown that heparin competes with light-activated phosphorylated rhodopsin for binding to arrestin.42When heparin was injected to SLBs with immobilized Arr3-WT, a ΔF was recorded and converted to the number of heparin molecules bound by taking into account molecular weights of Arr3-WT (48 kDa) and heparin (5 kDa) (Fig. 5A) The numbers of Arr3-WT bound to SLBs and heparin bound to Arr3-WT increased as a function of DGS-NTA(Ni2+) content. At 1 mol% DGS-NTA(Ni2+), 13.34 ± 0.37 × 1011 molecules of Arr3-WT per cm2 were bound whereas the number of bound heparin molecules was 13.53 ± 2.56 × 1011 cm−2, indicating that each heparin was bound to one Arr3-WT molecule. Increasing the mol% of DGS-NTA(Ni2+) changed the stoichiometry between heparin and Arr3 WT. At 10% of DGS-NTA(Ni2+), each Arr3-WT bound approximately 2.3 heparin molecules. However, this calculation might overestimate the stoichiometry between Arr3-WT and heparin since in QCM-D bound water in addition to mass of the protein cannot be subtracted.
 | ||
| Fig. 5 Interaction of heparin with Arr3. (A) Summary of the number of immobilized Arr3-WT molecules and bound heparin as a function of DGS-NTA(Ni2+) content (B) summary of the bound heparin mass in the presence/absence of immobilized Arr3 proteins. | ||
To compare the immobilization of Arr3-WT with mutant Arr3-X and Arr3-Y, SLB membranes with 5 mol % of DGS-NTA(Ni2+) were prepared. Absolute changes upon binding of heparin with Arr3 are presented in Fig. 5B. A similar Δm of heparin was obtained for Arr3-X and Arr3-Y, and it was higher compared to Arr3-WT. Specifically, the bound mass of heparin was 208.74 ± 17.72 ng cm−2 on Arr3-X containing SLBs while it was 148.33 ± 09.3 ng cm−2 on Arr3-WT containing SLBs. In the absence of Arr3 there was no binding of heparin to SLB platforms.
Interaction of heparin with Arr3 was verified using BLI, and an equilibrium dissociation constant of 400 ± 103 nM (ka = 1.6 ± 0.41 × 105 M−1 s−1 and kd = 0.064 ± 0.002 s−1) was determined, indicative of a very stable binding (Fig. S2 and Table S1, ESI†). The presented data on the interaction arrestin/heparin are a functional indication of the activity of our recombinant arrestin.42
We next characterized JSR1–arrestin-3 interactions in different environments, either by mimicking a cell membrane (SLB platform) or by providing more stability (SLM platform) which is important for the further development of screening assays. To test the interaction between immobilized Arr3 and JSR1, we first activated JSR1 by illuminating it with a blue laser (495 nm) for 1.5 minutes in all experiments.
In the case of BLI, tips derivatized with WT and mutant Arr3 were dipped into five different concentrations of JSR1 (0.125, 0.250, 0.500, 1, and 4.3 μM) stabilized in DDM micelles. The binding of JSR1 to Arr3 on the BLI sensor (association) followed by a rinsing with buffer (dissociation) was monitored in real-time. Binding association and dissociation rate constants (ka and kd, respectively) for the JSR1 binding to the different Arr3 on BLI biosensors were obtained by fitting the binding and dissociation curves according to 1:1 binding model (Fig. 6).43 This model is based on an equimolar interaction between ligand and analyte,44 assuming that JSR1 is homogenous, monomeric, does not aggregate and only one molecule is bound per molecule of Arr3 on the surface. This assumption was supported by stoichiometric ratios between maximum binding response for JSR1 and Arr3 ≤1 obtained with all platforms and thus, 1:1 binding model was applied to all JSR1–Arr3 interactions. The equilibrium dissociation constants (KD) for the JSR1 complexes with different Arr3-BLI sensors were calculated from the fitted kinetic constants as summarized in Table 3. The highest KD was observed for the Arr3-X (389 ± 59 nM) and the lowest KD for the Arr3-Y (112 ± 12 nM). Association and dissociation kinetic constants show that JSR1 binds relatively fast to Arr3-WT and its mutants. However, in the BLI setup, the half-life of the JSR1·Arr3 complexes is only around 3–4 s as was estimated from the kinetic dissociation constants shown in Table 3. The kinetic association constant for the JSR1-Arr3-Y complex was the fastest. The exchange of positively charged arginine to negatively charged glutamic acid in Arr3-Y might work as a switch in the polar core that regulates the affinity of the adjacent finger loop of Arr3 towards JSR1. In contrast, the I386A mutation in Arr3-X may make the interaction of the finger loop with JSR1 helices V and VI less probable. Accordingly, the kinetic association constant was the smallest.
 | ||
| Fig. 6 Detection of DDM-stabilized JSR1 binding to Arr3-BLI sensors. Representative JSR1 binding curves measured by BLI for (A) binding to Arr3-WT (JSR1 concentrations: 0.2, 0.4, 0.8, 1.6, 3.2, 4.3 μM), (B) binding to Arr3-X (JSR1 concentrations: 0.125, 0.25, 0.5, 1, and 4.3 μM), and (C) for binding to Arr3-Y (JSR1 concentrations: 0.125, 0.250, 0.500, 1, and 4.3 μM) immobilized on the Ni2+–NTA tips. Black curves are the experimental data whereas red curves are the best fits. | ||
| Proteins | K D, nM | k a × 105, M−1 s−1 | k d, s−1 | Model for kinetic fit |
|---|---|---|---|---|
| Arr3-WT | 213 ± 41 | 9.9 ± 1.5 | 0.21 ± 0.025 | 1:1 binding |
| Arr3-X | 389 ± 59 | 7.2 ± 0.8 | 0.28 ± 0.028 | 1:1 binding |
| Arr3-Y | 112 ± 12 | 11.3 ± 0.9 | 0.13 ± 0.009 | 1:1 binding |
For SPR measurements, all flow channels were first functionalized with NTA(Ni2+)-SLM followed by Arr3 except for one channel that was kept without Arr3 as a reference. The specificity of the Arr3 binding to NTA(Ni2+)-SLM was validated by a return to the SLM base line after the injection of 0.5 M EDTA or 0.5 M imidazole that disrupts the histidine–Ni2+ interactions (not shown). For more quantitative equilibrium and kinetic binding analysis, a number of JSR1 concentrations (16.5, 31.25, 62.5, 125, 250, 500, 1000 nM) were injected into all flow channels followed by 0.2 M NaOH to remove JSR1. After the reference subtraction, the sensograms were fitted using 1:1 binding model to obtain the kinetic constants (Fig. 7A). Additionally, the equilibrium responses at different JSR1 concentrations were plotted and fitted to the Langmuir binding isotherm (Fig. 7B). The obtained values for the kinetic and equilibrium dissociation constants from SPR measuremens show binding of JSR1 to all Arr3 (Tables 4 and 5, respectively). However, the complexes dissociate slower than was observed in the BLI measurements. It is possible that an interaction of Arr3 with the charged lipid head groups of the SLM on the SPR sensor chip might induce slight conformational changes that influence the dissociation and association kinetic rate of the JSR1–Arr3 complex formation. Therefore, we have also fitted the SPR data with the ‘two-state’ model that assumes a structural re-arrangement after the fist binding step (see Fig. S4 and Table S2, ESI†). However, the equilibrium binding constants calculated from the kinetic constants using either model are comparable with those obtained by BLI which could indicate similar interactions but under different conformational conditions. Since JSR1 was stabilized in DDM micelles, the measurements at high JSR1 concentrations show some disruption of the lipid monolayer due to the DDM exchange with SLM. Thus, the equilibrium dissociation constants obtained from the equilibrium fit (Fig. 7B) are less accurate than those calculated based on the kinetic constants. Nevertheless, they are comparable to those obtained using BLI within error limits.
 | ||
| Fig. 7 Analysis of JSR1 binding to Arr3-SLM. (A) Representative kinetic fit to 1:1 model for JSR1 binding to Arr3 immobilized on the Ni2+–NTA SLM of HPA sensor chip. (B) Equilibrium binding analysis for the JSR1 binding to Arr3-WT, Arr3-X and Arr3-Y. The solid lines represent a best fit to the Langmuir binding isotherms. Black curves are the experimental data whereas red curves are the fitted data. | ||
| Proteins | K D, nM | k a × 105, M−1 s−1 | k d × 10−3, s−1 | Model for kinetic fit |
|---|---|---|---|---|
| Arr3-WT | 170.3 ± 55.7 | 0.33 ± 0.27 | 4.55 ± 0.02 | 1:1 binding |
| Arr3-X | 152.6 ± 27.5 | 2.97 ± 0.06 | 10.97 ± 0.50 | 1:1 binding |
| Arr3-Y | 96.7 ± 6.6 | 0.76 ± 0.06 | 5.08 ± 0.14 | 1:1 binding |
| Proteins | K D,a nM | Type of analysis |
|---|---|---|
| Arr3-WT | 347.7 ± 120.7 | Equilibrium fit |
| Arr3-X | 528 ± 468 | Equilibrium fit |
| Arr3-Y | 616.3 ± 319.3 | Equilibrium fit |
The interaction of light-activated, DDM-stabilized JSR1 with different Arr3 immobilized on SLBs was monitored by QCM-D. Note that flowthrough experiments by QCM-D consume considerable amounts of protein because the measurement chamber is bigger than in BLI and SPR. To conserve JSR1, we injected 1 μM DDM-stabilized JSR1 to SLBs with/without Arr3 under continous flow for only 2 min, stopped the flow, and then continued the incubation for 30 min, after which stable frequency and dissipation signals were observed. Unbound proteins were removed by rinsing with buffer under continous flow. To first evaluate the effect of the DDM micelles on SLBs with and without Arr3, we performed QCM-D measurements with 0.01% DDM micelles alone. Δm upon addition of DDM micelles was 55.57 ± 29.22 ng cm−2 (Fig. 8A), independent of the presence or absence of Arr3 on SLBs. In contrast, injection of DDM-stabilized JSR1 to Arr3-WT SLBs caused a increase in mass to 959.51 ± 384.97 ng cm−2 (Fig. 8B). The Δm of 655.43 ± 168.15 ng cm−2 for Arr3-X and Δm of 462.85 ± 20.53 ng cm−2 for Arr3-Y were in a similar range. In the absence of Arr3 on the SLB platforms, the Δm shift was considerably smaller (286.38 ± 25.84 ng cm−2). The significant increase in mass indicates the specific interactions between JSR1 and all three Arr3. The binding of JSR1 to Arr3 was analyzed by fitting the data to a 1:1 binding model (Fig. S3, ESI† and Table 6). The equilibrium dissociation constants were derived from the kinetic constants obtained from the fits.
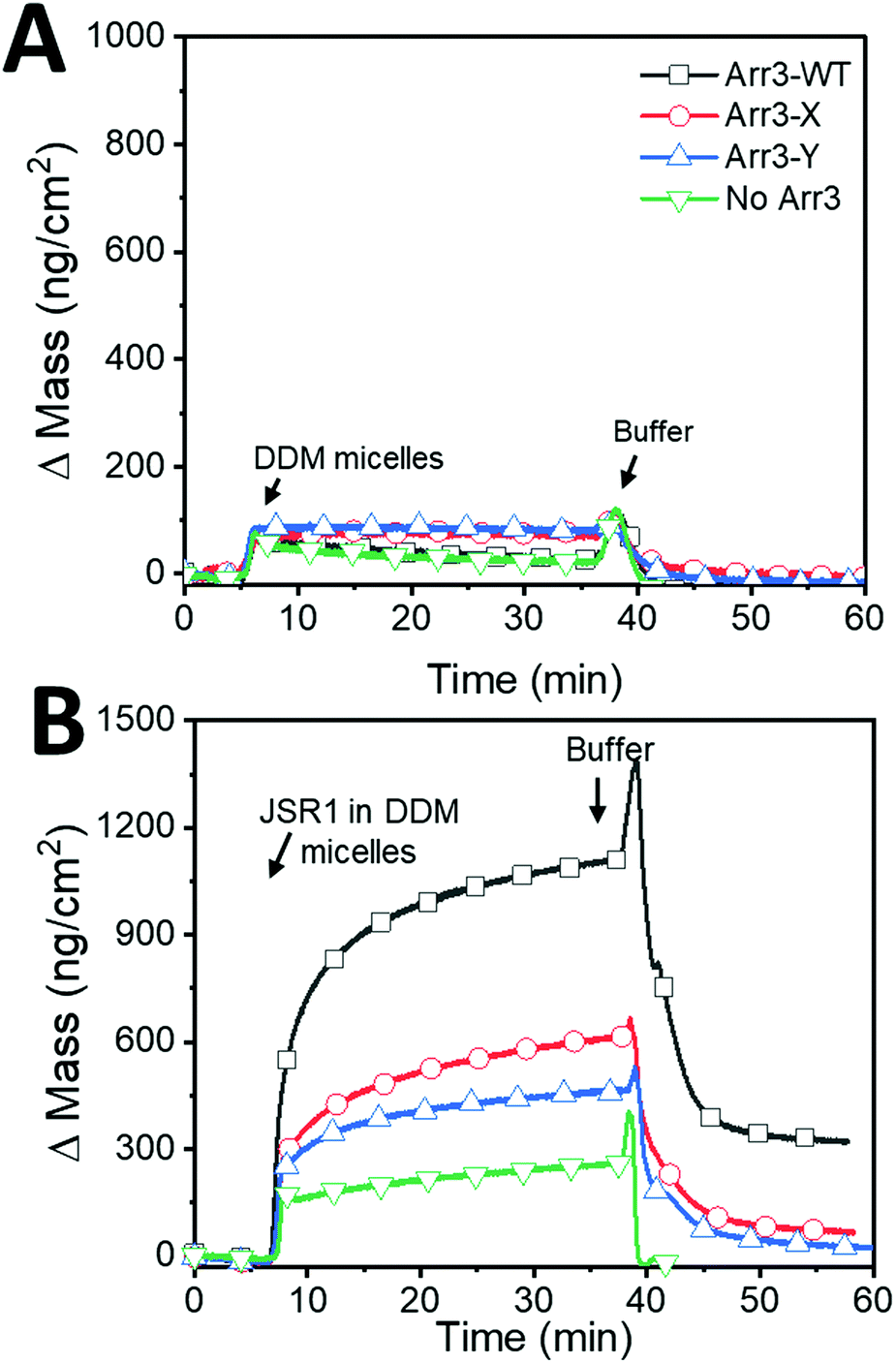 | ||
| Fig. 8 Interaction of DDM-stabilized JSR1 s with immobilized Arr3. After SLB formation and Arr3 immobilization, the system was re-equilibrated and the mass baseline set to 0. Changes in mass were recorded over time, after addition of (A) DDM micelles and (B) DDM stabilized-JSR1 to Arr3 immobilized SLBs. | ||
| Proteins | K D, nM | k a × 105, M−1 s−1 | k d × 10−3, s−1 | Model for kinetic fit |
|---|---|---|---|---|
| Arr3-WT | 3845 ± 727 | 0.019 ± 0.003 | 7.6 ± 0.12 | 1:1 binding model |
| Arr3-X | 3518 ± 296 | 0.035 ± 0.001 | 7.4 ± 0.73 | 1:1 binding model |
| Arr3-Y | 914 ± 74 | 0.028 ± 0.002 | 2.4 ± 0.04 | 1:1 binding model |
As expected, the obtained equilibrium dissociation constants and the kinetic association and dissociation rates for Arr3–JSR1 complexes differ depending on the characterization method, specifically on the different type of platforms. SPR and BLI experiments led to small differences in the kinetic association and dissosication rates indicating that on the SLM platform the JSR1 association with Arr3 is faster and dissociation is slower compared to complex formation on the SLB platform. This finding results from the different properties of the membranes on the sensor: SLM on the HPA chip is less fluidic than SLB on the surface of the QCM-D sensor.45 It is well known that a water layer is present between a lipid bilayer and silicon dioxide substrate.46 On the contrary, HPA surfaces are composed of long chain octadekanethiol molecules that are covalently bound to gold, forming a flat, quasi-crystalline hydrophobic layer. Thus, the lateral mobility of the SLM and bound Arr3 is restricted on HPA surfaces.47 This may impact on the conformation of Arr3 and its interactions with JSR1. In particular, the kinetic association constants are much lower which might be due to a delay in the recognition of the binding site that is linked to a certain mobility of arrestin. The equilibrium dissociation constant values obtained from QCM-D are higher compared to those of the other two methods. Hence, the observed differences in binding kinetics likely arise from intrinsic differences between the mono- and bilayer membrane. In addition, the increase in the equilibrium dissociation constant values obtained from QCM-D might be due to differences in the extent of coupled water molecules that contribute to the mass of proteins adsorbed on the sensors.
The kinetic association and dissociation constants derived from BLI measurements suggest a faster complex formation than observed with SPR and QCM-D, but equilibrium dissociation constants are comparable with SPR values. It is also possible that the fast binding kinetics arise from a lower protein density on the tip surface, which improves the accessibility of the binding sites compared to Arr3 on SLM or SLB platforms. Also, the tip surface in this case might influence the binding less because DDM micelles do not interact with BLI polymer coating. The binding kinetics could also be influenced by the size of the flow cell and the flow rate of the sample, both of which are different in all three methods. Overall, the differences observed in the JSR1–Arr3 interaction indicate that the complex formation is influenced by local conditions, such as the intrinsic property of the membrane that hosts the Arr3. While all three methods are suitable for measuring changes in the binding affinities between GPCRs and arrestins, and can be used to quantitatively assess changes in the complex stability caused by specific drugs, it is important to take into account such local parameters when comparisons between biosensing platforms are made. Therefore, a combination of methods is necessary to ascertain how changes to the environment impact on the interactions of GPCRs and their associated proteins.
4 Conclusions
To overcome the challenges associated with the isolation, reconstitution and oriented immobilization of GPCRs on biosensors, we immobilized distinct variants of arrestin-3 by means of histidine–NTA(Ni2+) interactions on different types of biosensors. Using a combination of surface-based platforms, we demonstrate for the first time that biophysical characterization of the selective recognition and binding of JSR1 and wild type arrestin-3 and two mutants by BLI, SPR and QCM-D is feasible. The characterization of the kinetic properties of JSR1–arrestin-3 complex formation in terms of binding constant, association and dissociation rates revealed specific differences in JSR1 interaction with the arrestin-3 variants. Interestingly, while disturbing access to the polar core did result in an increased affinity in our system, in line with what has been observed in cellular systems, the mutation affecting the three-element interaction did not. It would be interesting to assess whether this is also true for other GPCRs. If yes, it could mean that additional proteins interacting with arrestin may play a role in regulating arrestin–receptor interactions in cells. JSR1 has also been suggested to be an appropriate basis for engineering of novel optogenetic tools allowing for an optical control of diverse GPCR signaling pathways.22,26,48 Establishing a reliable assay allowing for a robust biophysical characterization of JSR1 with its cellular binding partners is a critical step towards realizing the optogenetic potential of this receptor. Our system can be readily used, not only to assess other GPCR–arrestin interactions, but it also provides the basis for probing binding properties of GPCRs with other protein partners.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the Swiss Nanoscience Institute (NanoArgovia project), the Swiss National Science Foundation and the University of Basel. We acknowledge InterAx Biotech for providing the arrestin-3 WT and mutants and for the coordination of the NanoArgovia project, a part of which is presented here. All authors thank Dr. Martin K. Ostermaier acting as the NanoArgovia project leader in the joint collaboration. CGP, SYA and CAS thank Prof. Wolfgang Meier (University of Basel) for useful discussions, Dr Csaba Fodor (University of Basel) for input and support for the synthesis reactions and Gaby Persy (University of Basel) for initial characterization of the membranes. We acknowledge the SNF grant #310030B_173335 to EL and GFXS. EL and GFXS thank Dr Gregor Cicchetti for project support, and Dr Valerie Panneels for support to in the receptor sample preparation.References
- M. C. Lagerström and H. B. Schiöth, Nat. Rev. Drug Discovery, 2008, 7, 339 CrossRef.
- A. J. Venkatakrishnan, X. Deupi, G. Lebon, F. M. Heydenreich, T. Flock, T. Miljus, S. Balaji, M. Bouvier, D. B. Veprintsev and C. G. Tate, Nature, 2016, 536, 484 CrossRef CAS.
- D. Wacker, R. C. Stevens and B. L. Roth, Cell, 2017, 170, 414–427 CrossRef CAS.
- D. P. Staus, H. Hu, M. J. Robertson, A. L. Kleinhenz, L. M. Wingler, W. D. Capel, N. R. Latorraca, R. J. Lefkowitz and G. Skiniotis, Nature, 2020, 579, 297–302 CrossRef CAS.
- V. V. Gurevich and E. V. Gurevich, Advances in Protein Chemistry and Structural Biology, Elsevier, 2020, vol. 121, pp. 169–197 Search PubMed.
- V. V. Gurevich and E. V. Gurevich, Front. Pharmacol., 2019, 10, 125 CrossRef CAS.
- H. S. Chan, Y. Li, T. Dahoun, H. Vogel and S. Yuan, Trends Biochem. Sci., 2019, 44, 312–330 CrossRef CAS.
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schiöth and D. E. Gloriam, Nat. Rev. Drug Discovery, 2017, 16, 829 CrossRef CAS.
- R. S. Haider, A. Godbole and C. Hoffmann, Curr. Opin. Cell Biol., 2019, 57, 16–24 CrossRef CAS.
- L.-P. Picard, A. M. Schönegge, M. J. Lohse and M. Bouvier, Commun. Biol., 2018, 1, 106 CrossRef.
- H. Schihada, S. Vandenabeele, U. Zabel, M. Frank, M. J. Lohse and I. Maiellaro, Commun. Biol., 2018, 10 Search PubMed.
- R. Zhang and X. Xie, Acta Pharmacol. Sin., 2012, 33, 372 CrossRef CAS.
- W. Thomsen, J. Frazer and D. Unett, Curr. Opin. Biotechnol., 2005, 16, 655–665 CAS.
- S. G. Patching, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 43–55 CrossRef CAS.
- I. Navratilova, M. Dioszegi and D. G. Myszka, Anal. Biochem., 2006, 355, 132–139 CrossRef CAS.
- C. A. Shepherd, A. L. Hopkins and I. Navratilova, Prog. Biophys. Mol. Biol., 2014, 116, 113–123 CrossRef CAS.
- I. Navratilova, J. Besnard and A. L. Hopkins, ACS Med. Chem. Lett., 2011, 2, 549–554 CrossRef CAS.
- P. Stenlund, G. J. Babcock, J. Sodroski and D. G. Myszka, Anal. Biochem., 2003, 316, 243–250 CrossRef CAS.
- C. Bieri, O. P. Ernst, S. Heyse, K. P. Hofmann and H. Vogel, Nat. Biotechnol., 1999, 17, 1105 CrossRef CAS.
- F. Hüttenrauch, A. Nitzki, F.-T. Lin, S. Höning and M. Oppermann, J. Biol. Chem., 2002, 277, 30769–30777 CrossRef.
- A. K. Shukla, J. D. Violin, E. J. Whalen, D. Gesty-Palmer, S. K. Shenoy and R. J. Lefkowitz, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9988–9993 CrossRef CAS.
- R. H. Oakley, S. A. Laporte, J. A. Holt, L. S. Barak and M. G. Caron, J. Biol. Chem., 2001, 276, 19452–19460 CrossRef CAS.
- X. Zhan, L. E. Gimenez, V. V. Gurevich and B. W. Spiller, J. Mol. Biol., 2011, 406, 467–478 CrossRef CAS.
- M. Vrecl, R. Jorgensen, A. Pogačnik and A. Heding, J. Biomol. Screening, 2004, 9, 322–333 CrossRef CAS.
- T. A. Key, C. M. Vines, B. M. Wagener, V. V. Gurevich, L. A. Sklar and E. R. Prossnitz, Traffic, 2005, 6, 87–99 CrossRef CAS.
- N. Varma, E. Mutt, J. Mühle, V. Panneels, A. Terakita, X. Deupi, P. Nogly, G. F. Schertler and E. Lesca, Proc. Natl. Acad. Sci. U. S. A., 2019, 201902192 Search PubMed.
- D. Ehrenberg, N. Varma, X. Deupi, M. Koyanagi, A. Terakita, G. F. Schertler, J. Heberle and E. Lesca, Biophys. J., 2019, 116, 1248–1258 CrossRef CAS.
- A. Bandyopadhyay, N. Van Eps, B. T. Eger, S. Rauscher, R. S. Yedidi, T. Moroni, G. M. West, K. A. Robinson, P. R. Griffin and J. Mitchell, J. Mol. Biol., 2018, 430, 4102–4118 CrossRef CAS.
- D. Mayer, F. F. Damberger, M. Samarasimhareddy, M. Feldmueller, Z. Vuckovic, T. Flock, B. Bauer, E. Mutt, F. Zosel and F. H. Allain, Nat. Commun., 2019, 10, 1261 CrossRef.
- D. Sun, M. K. Ostermaier, F. M. Heydenreich, D. Mayer, R. Jaussi, J. Standfuss and D. B. Veprintsev, PLoS One, 2013, 8 Search PubMed.
- R. S. Haider, F. Wilhelm, A. Rizk, E. Mutt, X. Deupi, C. Peterhans, J. Mühle, P. Berger, G. F. Schertler and J. Standfuss, Sci. Rep., 2019, 9, 439 CrossRef.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- B. A. Handbook, Biacore AB, GE Healthcare Life Sciences, Uppsala, 1997.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096 CrossRef CAS.
- M. A. Cooper, A. C. Try, J. Carroll, D. J. Ellar and D. H. Williams, Biochim. Biophys. Acta, Biomembr., 1998, 1373, 101–111 CrossRef CAS.
- J. A. Nye and J. T. Groves, Langmuir, 2008, 24, 4145–4149 CrossRef CAS.
- R. B. Schasfoort, Handbook of surface plasmon resonance, Royal Society of Chemistry, 2017 Search PubMed.
- I. Reviakine, F. F. Rossetti, A. N. Morozov and M. Textor, J. Chem. Phys., 2005, 122, 204711 CrossRef.
- S. Yorulmaz Avsar, J. A. Jackman, M. C. Kim, B. K. Yoon, W. Hunziker and N.-J. Cho, Langmuir, 2017, 33, 7332–7342 CrossRef CAS.
- S. Yorulmaz, J. A. Jackman, W. Hunziker and N.-J. Cho, Biomacromolecules, 2015, 16, 3594–3602 CrossRef CAS.
- R. L. Schoch and R. Y. Lim, Langmuir, 2013, 29, 4068–4076 CrossRef CAS.
- K. Palczewski, A. Pulvermüller, J. Buczyłko and K. Hofmann, J. Biol. Chem., 1991, 266, 18649–18654 CAS.
- V. Kairys, L. Baranauskiene, M. Kazlauskiene, D. Matulis and E. Kazlauskas, Expert Opin. Drug Discovery, 2019, 14, 755–768 CrossRef CAS.
- S. Kumaraswamy and R. Tobias, Protein-Protein Interactions, Springer, 2015, pp. 165–182 Search PubMed.
- C. Draghici, V. Mikhalevich, G. Gunkel-Grabole, J. Kowal, W. Meier and C. G. Palivan, Langmuir, 2018, 34, 9015–9024 CrossRef CAS.
- T. M. Bayerl and M. Bloom, Biophys. J., 1990, 58, 357–362 CrossRef CAS.
- K. Glasmästar, C. Larsson, F. Höök and B. Kasemo, J. Colloid Interface Sci., 2002, 246, 40–47 CrossRef.
- E. Lesca, J. Membr. Biol., 2020, 253, 81–86 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01464h |
| ‡ Equal contribution. |
| This journal is © the Owner Societies 2020 |