
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanism and ligand effects of single atom rhodium supported on ZSM-5 for the selective oxidation of methane to methanol†
Rhys J.
Bunting
,
Jillian
Thompson
 and
P.
Hu
*
and
P.
Hu
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast BT9 5AG, UK. E-mail: p.hu@qub.ac.uk
First published on 7th May 2020
Abstract
The mechanism for the partial oxidation of methane to methanol on single atom rhodium supported on ZSM-5 is investigated by DFT. The most favoured mechanism for methane activation is shown to be via oxidative addition at an undercoordinated rhodium metal centre and not through a typical metal oxo intermediate. The formation of a C–OH bond, and not methane activation, is found to be the rate determining step. CO coordinated to the rhodium centre is observed to strongly promote this bond formation. Water is required in the system to help prevent catalyst poisoning by CO, which greatly hinders the methane activation step, and to protonate an intermediate RhOOH species. These results suggest that more focus is required on methyl–oxygen bond formation and that exclusive consideration of methane activation will not completely explain some methane partial oxidation systems.
1 Introduction
Single-site catalysts are gathering ever-increasing interest within the field of catalysis.1 Traditional heterogenous catalysts involve a bulk material with a catalytically active surface composed of multiple atoms with several possible binding sites. In stark contrast, single sites only allow one type of catalytic site for binding which in-turn offers unique chemistries compared to traditional catalyst surfaces. Methane functionalisation has been receiving increased attention2,3 and single site catalysts have been utilised for this purpose. Palladium single atoms supported on ZSM-54 were found to be active for the partial oxidation of methane to methanol. Additionally, two recent papers5,6 reported that rhodium single atoms supported on ZSM-5, Rh@ZSM-5, had notable activity for continuous acetic acid production from methane and CO. Specific to this work, some selectivity was observed towards the arguably more ideal product, methanol.The formally best systems for the partial oxidation of methane to methanol, those with the highest conversions and selectivities, typically use Cu7–10 or Fe11–13 mono-, di-, or tri-metal atom species supported on ZSM-5 or another viable zeolite. These processes require pre-reaction oxidation of the catalyst, where an oxidant (NOX, H2O2, O2) is used to oxidise the metal centre to form a catalytically active metal oxo species. Subsequently, methane is activated with the formed metal oxo species and the catalyst is washed with water to liberate the methanol product. For more methanol to be synthesised, the stepwise process must be repeated. It should be noted that efforts have been made towards using this class of system in a non-stepwise process.14 However, the low yield of product obtained and the cost of oxidants other than oxygen impedes the likelihood of industrial viability.
Whilst there has been considerable theoretical research reported for the stepwise Cu/Fe zeolite systems,15–20 there has been little theoretical research for the rhodium based single site catalyst system. It remains unknown whether the mechanism mirrors those found for Cu/Fe systems or if a new pathway is more favourable. In contrast to the work done on the Cu/Fe ZSM-5 catalysts, which were carried out in the gas phase and using O2 as an oxidant, the Rh@ZSM-5 system is required to be carried out in aqueous solution. In addition, in the research of Flytzani-Stephanopoulos and colleagues,6 they noticed a peculiar requirement for CO to be present for the conversion of methane to methanol over the Rh@ZSM-5 catalyst despite not having an active role within the catalytic cycle: if no CO is present in the system, no oxygenates are formed. A fundamental understanding of the mechanistic pathway and the role of CO in influencing activity and selectivity towards methanol, as well as the role of water, is required to better understand and develop this system further.
For traditional heterogeneous catalysts, adsorption of different surface species such as CO can affect the energetics of a reaction, having a profound effect on the kinetics and thermodynamics of the system, regardless of whether this surface species is required for that elementary step.21 Significant concentrations of a surface species must be present in order for there to be a notable effect. However, for a single site in a heterogeneous catalyst, the coverage effect could be more accurately described as a ligand effect.22,23 This is distinct from a traditional heterogeneous catalyst, as instead of having several possible sites across a uniform surface where the surface species can adsorb, only one site is present for each individual single atom. In addition, the increased ratio of bound atoms to metal atoms on a single site will have an increased effect compared to the same number of bound atoms at a traditional catalyst site. Their effect must be considered for a representative model of the true system to be made. Current models of single site catalysts do not consider these effects, modelling the reaction as occurring on a clean surface. Whilst the effects of water within the sphere of influence of a metal centre have been investigated for methane activation in zeolites, coordination of metal centres with spectator molecules, such as CO and H2O, has not yet been thoroughly studied.24,25 The effect of interacting molecules found in the reaction medium must be considered for a true mechanistic pathway to be developed.
Herein, with Rh@ZSM-5 used as a model system, various pathways to convert methane to methanol have been calculated and a mechanism for the partial oxidation of methane to methanol is proposed for this catalyst. To address the mysteries of the requirement for CO and H2O to be present in the catalytic system, the effect that CO and H2O have on the energetics of the catalytic cycle is also investigated.
2 Computational method
Spin-polarized density functional theory calculations were performed with the Perdew–Burke–Ernzerhof (PBE)26 functional within the generalized gradient approximation (GGA) in the Vienna ab initio simulation package (VASP).27–29 The projector-augmented wave (PAW) method was used to represent the core–valence electron interaction.30,31 The D3 correction method with Becke–Jonson damping was employed to include the van der Waals interactions32 to model the porous nature of zeolites.33 Structures were optimised until the forces on all atoms were below 0.05 eV Å−1. Transition states were searched with the constrained minimization technique.34,35 Structures were verified with frequency calculations, where no imaginary frequencies are associated with an initial or final state and one imaginary frequency is associated with the transition state.For the DFT calculations, the cut-off energy of the plane wave basis set was 450 eV. Because of the large unit cell, the Brillouin zone was sampled with a 1 × 1 × 1 Monkhorst–Pack k-point grid, which is similar to work in the literature.36 Zero-point energy and entropic contributions for surface species were calculated from frequency calculations within VASP. Entropic energy contributions of gas phase molecules and H2O2 decomposition were calculated with Gaussian 09 with the Becke, 3-parameter, Lee–Yang–Parr (B3LYP) hybrid functional with the 6-311++G(d,p) basis set. All free energies were calculated at standard pressure and 423 K. The free energy of gas molecules in the zeolite were assumed to have their entropic contributions limited by a factor of a half due to the restricted free movement of gas molecules in zeolites.37
The structure for ZSM-5 was obtained from the IZA structure commission database. The T7 site of the main zeolite channel was selected as the substitution site of aluminium. The Rh+ atom catalyst species is bound across the aluminium at this site. The T7 site was selected due to its prevalence and its notably strong oxygen adsorption for other metal species.38
3 Results and discussion
To calculate pathway energies, the influences of the most commonly available binding molecules, CO and water, on the rhodium metal centre were determined by calculation of their adsorption energies (ESI1†).These calculations show CO as strongly binding to the metal centre with an average free energy of adsorption of −1.86 eV per CO molecule. The rhodium centre becomes saturated once two molecules bind in a square planar fashion, causing any further adsorption of molecules to become endergonic. Water also binds favourably, yet much weaker, to the supported rhodium atom with an average free energy of adsorption of −0.20 eV. For both CO and H2O, the binding energy of the second molecule is weaker than the first molecule by 0.10 eV and 0.18 eV, respectively. Interestingly, CO and H2O enhance the binding energies of their partner molecule when one of each molecule is bound to the metal centre in a mutualistic relationship: CO binding energy is enhanced by −0.17 eV in the presence of one molecule of water, whilst the binding energy strengthens by a more pronounced amount of −0.49 eV for H2O in the presence of one molecule of CO. Despite this enhancement, adsorption with two CO molecules remains favoured. These energies show that Rh@ZSM-5 coordinated with two CO molecules, as displayed in Fig. 1, is the most thermodynamically favoured rhodium species, suggesting that it is the dominant catalytic species in situ. This mirrors experimental findings, where Rh1+(CO)2 was found to be the prevalent form of the catalyst.6 This catalyst species was then used to investigate possible pathways.
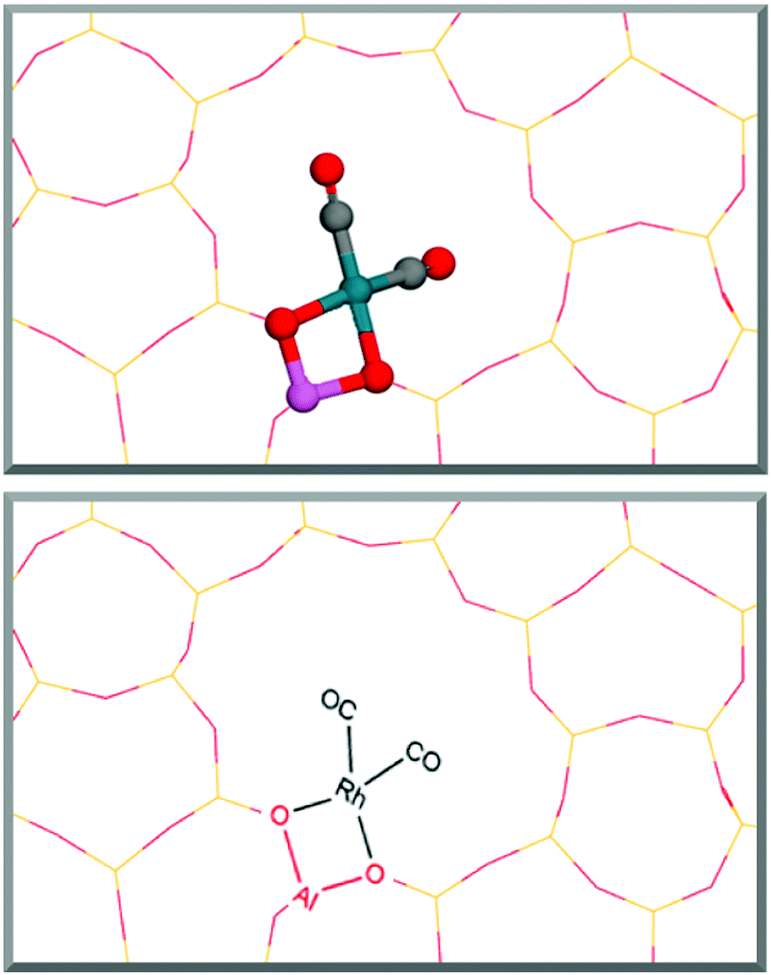 | ||
| Fig. 1 Structure of the most stable Rh@ZSM-5 single atom species when CO is readily available in the environment. Rhodium is bound at the T7 site where an aluminium is substituted into the zeolite structure. The 3D structure inside the main zeolite channel is shown above whilst the skeletal structure, drawn to clarify what consecutive skeletal structures represent, is displayed below. | ||
As a mechanism for Rh@ZSM-5 has not yet been fully investigated, a variety of reasonable mechanisms were initially investigated. As methane activation must inevitably occur, three plausible pathways for this step were considered. As a starting point, the metal oxo mechanism (Fig. 2d) that is observed for Fe/Cu zeolites was trialled. This involves the addition of methane across a Rh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species to form a Rh(CH3)(OH) species. The next two pathways take inspiration from pathways for palladium hydride oxidation. This oxidation step is important for the regeneration of the active palladium catalytic species in alcohol oxidation reactions.39–41 In the first pathway, methane undergoes oxidative addition across the rhodium metal centre to form a rhodium–methyl-hydride as shown in Fig. 2a–c. The hydride is subsequently oxidised via a hydrogen atom abstraction (HAA) with oxygen, forming a peroxo species that can readily be protonated with water to form a hydroxyl group. As CO is proven to poison Rh@ZSM-5,5 this pathway was considered with the rhodium centre coordinated to none, one, and two CO molecules. The final pathway is the hydrogen ‘X’ reductive elimination (HXRE) mechanism, where for this system ‘X’ is CH3; oxygen adsorbs to the supported rhodium as molecular oxygen and methane is activated through an oxygen promoted pathway, as shown in Fig. 2e.
O species to form a Rh(CH3)(OH) species. The next two pathways take inspiration from pathways for palladium hydride oxidation. This oxidation step is important for the regeneration of the active palladium catalytic species in alcohol oxidation reactions.39–41 In the first pathway, methane undergoes oxidative addition across the rhodium metal centre to form a rhodium–methyl-hydride as shown in Fig. 2a–c. The hydride is subsequently oxidised via a hydrogen atom abstraction (HAA) with oxygen, forming a peroxo species that can readily be protonated with water to form a hydroxyl group. As CO is proven to poison Rh@ZSM-5,5 this pathway was considered with the rhodium centre coordinated to none, one, and two CO molecules. The final pathway is the hydrogen ‘X’ reductive elimination (HXRE) mechanism, where for this system ‘X’ is CH3; oxygen adsorbs to the supported rhodium as molecular oxygen and methane is activated through an oxygen promoted pathway, as shown in Fig. 2e.
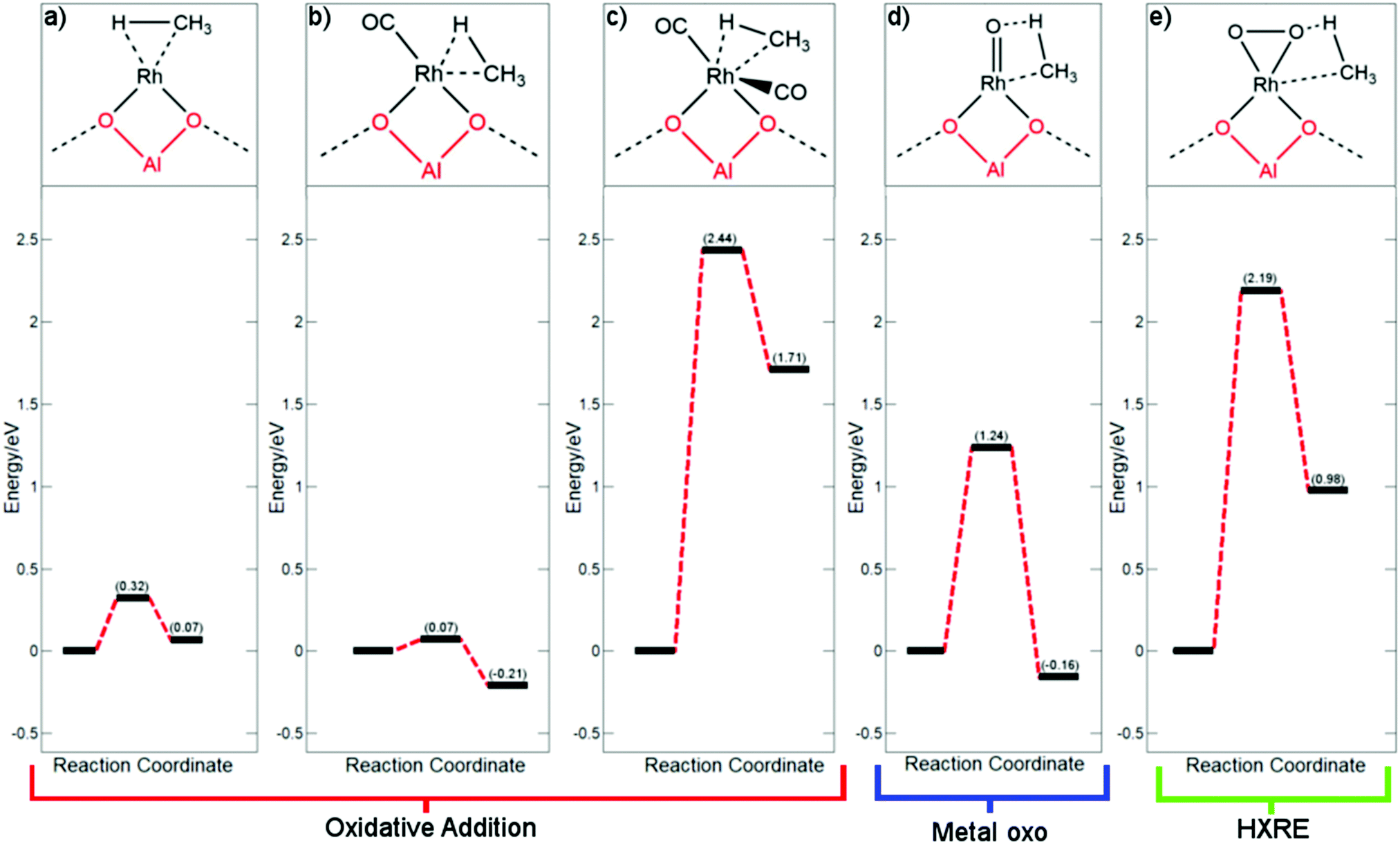 | ||
| Fig. 2 Free energy profiles for each of the possible mechanisms for C–H activation with the respective intermediate species drawn above: (a) methane oxidative addition across Rh; (b) methane oxidative addition across Rh(CO); (c) methane oxidative addition across Rh(CO)2; (d) o-promoted methane activation with Rh(O); and (e) o-promoted methane activation with Rh(O2). All pathways are calculated with respect to methane in the gas phase. | ||
As clearly shown in Fig. 2 for the species a–c, the oxidative addition pathway for the activation of methane occurs very readily and reversibly across supported Rh+ atoms that are not saturately coordinated. A very low barrier of 0.07 eV, with respect to methane in the gas phase, is observed for Rh(CO). Additionally, this process is favoured and exergonic for this species. However, if the metal centre is saturated with CO and there is no available site for activation, methane activation becomes extremely disfavoured with a very large barrier of 2.44 eV – a barrier that is very unlikely to be overcome at standard operating conditions. The implication is that for the reaction to take place through an oxidative addition mechanism, a CO molecule would have to first desorb from the rhodium centre. The effective C–H activation barrier of methane, whilst considering the desorption of a CO molecule, will still be 0.56 eV lower than that of a saturated rhodium centre with two CO molecules, under atmospheric pressure of CO. Under the conditions of the trace amounts of CO required for this system, the effective barrier of the Rh(CO) pathway would be even lower.
Upon consideration of the oxygen promoted pathways with a metal oxo species and molecular oxygen, reasonably large barriers are observed. The more reactive metal oxo species activates methane more readily than the adsorbed molecular oxygen rhodium species, with a free energy barrier of 1.24 eV compared to a barrier of 2.19 eV, respectively. Our calculations suggest that the oxygen promoted methane activation for both metal–oxygen species is less favoured compared to that of an oxidative addition mechanism. This result is akin to the findings for the respective oxygen covered metal surface systems, where it was found that methane activates preferentially through a surface stabilised mechanism on Rh(111) and an oxygen promoted mechanism on Cu(111).42,43 Despite this, the barrier observed for a metal oxo species remains moderately reasonable, therefore we must consider the facility of the formation of a rhodium metal oxo species. For the Cu–zeolite system, oxidation of the copper centre is understood as being the rate determining step,44 and therefore, we computed the thermodynamic favourability of forming a RhO species with respect to CuO formation. The formation of the rhodium metal oxo species was found to be 1.65 eV more endergonic than copper metal oxo formation, suggesting that this active species is unlikely to form.
With a rhodium metal oxo species being very thermodynamically disfavoured, the most energetically favourable mechanism of methane activation is calculated to be through the desorption of CO from the most stable Rh(CO)2@ZSM-5 species with subsequent oxidative addition of methane. Having recognised the most favourable pathway of methane activation being through an oxidative addition mechanism, a complete pathway is proposed. The pathway is considered for both a saturated (Rh(CO)2@ZSM-5) and an unsaturated (Rh(CO)@ZSM-5) rhodium complex. The first step is the activation of methane via oxidative addition. Subsequently, the formed rhodium hydride is oxidised with molecular oxygen to form rhodium peroxide. This species then degrades through protonation with water to form a rhodium hydroxide that is able to couple with the methyl group. The final step is the formation of methanol via methyl–hydroxyl coupling. Additionally, methanol desorption is considered for a closed catalytic cycle, as shown in Fig. 3.
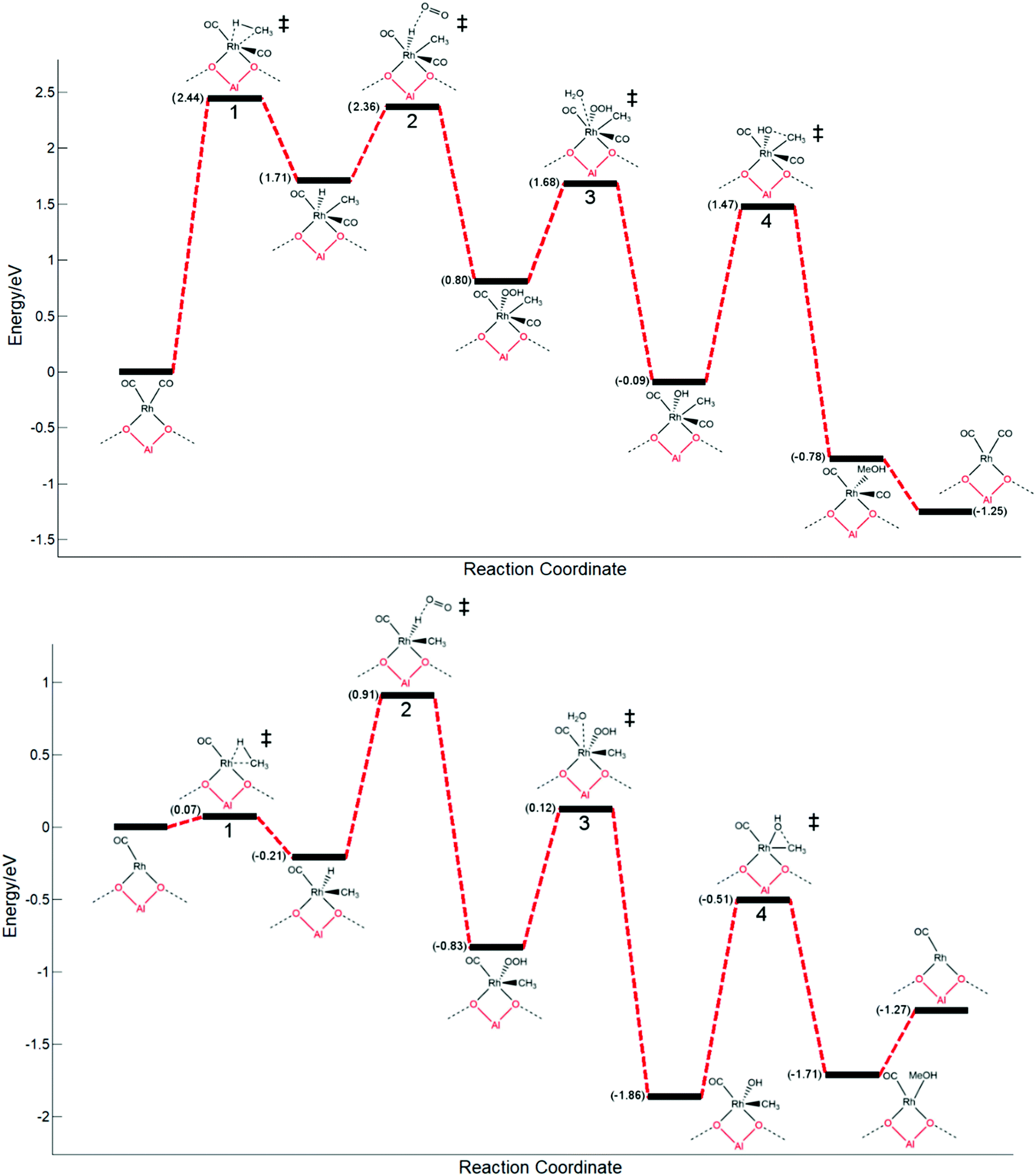 | ||
| Fig. 3 Free energy pathways for methane partial oxidation to methanol over Rh(CO)2@ZSM-5 (top) and Rh(CO)@ZSM-5 (bottom), respectively. The individual elementary steps are: 1. methane C–H activation; 2. hydrogen atom abstraction (HAA); 3. peroxide protonation with water; and 4. methyl-hydroxyl coupling. Desorption of methanol into the gas phase is also considered as the final step. Transition state species are designated with a double dagger. | ||
With methane activation already considered, generation of a hydroxyl group to form methanol at the rhodium centre is required. Activation of methane through the metal oxo pathway would form a Rh(CH3)(OH) species immediately. The HXRE pathway would also form a Rh(CH3)(OOH) species after methane activation, which can readily form into a Rh(CH3)(OH) species. However, as aforementioned, formation of a metal oxo species is disfavoured and activation of methane through an HXRE pathway is too kinetically hindered. A pathway that transforms the product of oxidative addition, Rh(CH3)(H), into Rh(CH3)(OH) is required. A mechanism for the oxidation of the metal hydride is needed for a complete catalytic cycle.
The energy pathways for both an unsaturated and saturated rhodium centre are distinguishably different from each other. The unsaturated rhodium species (Rh(CO)) has a profile with energies that are reasonable for a low TON system, whilst the energies involved for a saturated rhodium centre (Rh(CO)2) would be indicative of an extremely unreactive system where no product would form under standard operating conditions.
On the unsaturated rhodium species (Rh(CO)), the C–H activation (Fig. 3, step 1) is readily accessible and favoured with a barrier of 0.07 eV, whilst for Rh(CO)2 the C–H activation process would be extremely disfavoured and rate determining, with a very large barrier of 2.44 eV. There is an inherent requirement for there to be an active site available on the rhodium atom for C–H activation, where the metal centre is not saturately coordinated. Similar scenarios can be observed for activation of C–H bonds on metal surfaces with undercoordinated metal atoms, such as FCC (211) B5 sites being more active with lower methane activation barriers compared to more coordinated FCC (111) top sites.45
On the saturated rhodium species (Rh(CO)2), the metal hydride oxidation step (Fig. 3, step 2) is a highly studied process with great importance throughout the field of catalysis. Whilst it is mainly studied in palladium hydrides,39,40 some research has been directed towards rhodium hydrides.43 Our calculated energy barriers for this step can readily be overcome under the typical reaction conditions of 110 °C to 170 °C; a barrier of 0.65 eV is observed. For Rh(CO), a higher, yet still reasonable, barrier of 1.22 eV is calculated. Unlike C–H activation, this process is not easily reversible and is a strongly endergonic step.
The subsequent reaction, the degradation of the formed peroxide by protonation with water (Fig. 3, step 2), would readily occur in the zeolite framework. Although other mechanisms, such as one electron pathways without water, may be more favoured, the relative height of the energy barrier compared to other steps means that the kinetics of this step would not affect the overall rate of the reaction significantly.46
The methyl functionalisation step, where the methyl group couples with a hydroxyl group (Fig. 3, step 4), is likely the rate determining step of the reaction for the active Rh(CO) species. The largest barrier along the energy pathway is observed at this step with a calculated value of 1.35 eV. Notably, an energy well is observed at this step; a relatively stable intermediate is formed. The calculated pathway suggests that a significant ratio of this intermediate will be formed in situ. It is possible that if only methane and oxygen were present, and CO is not present to minimise this barrier, this species will form but no methanol will be produced; the reaction will halt at this step. This coupling step readily occurs for Cu/Fe centres supported in zeolites and methanol can be readily liberated from its adsorption site at room temperature upon washing.25 However, for the described rhodium species, the energetics of the pathway suggest that this process cannot readily occur at low temperatures.
As both the methane C–H activation and methyl–hydroxyl coupling steps are rate determining elementary steps, dependent on the coordination of the rhodium centre, a further investigation of these two steps was performed. Additionally, as water is typically used as a solvent, the effects of coordination with H2O were also considered.
In Fig. 4, the energy profile is compared for Rh@ZSM-5 whilst coordinated to none, one, or two molecules of CO or H2O. Using this scenario as an example provides a better understanding of the dynamics of key elementary processes. Consideration is first given to the methane activation step. From the previous discussion of CO coordinated to the rhodium single atom, it is expected that methane activation will readily occur if an active site is vacant on the metal centre; methane would be readily activated for all undercoordinated systems. A bare Rh@ZSM-5 site had a calculated barrier of 0.32 eV, whilst the barriers for rhodium coordinated to one molecule are similar, regardless of whether the molecule is CO or H2O, with barriers found to be 0.07 eV and 0.08 eV, respectively. If the rhodium becomes saturated with CO, C–H activation cannot readily occur and a very large barrier of 2.44 eV is observed.
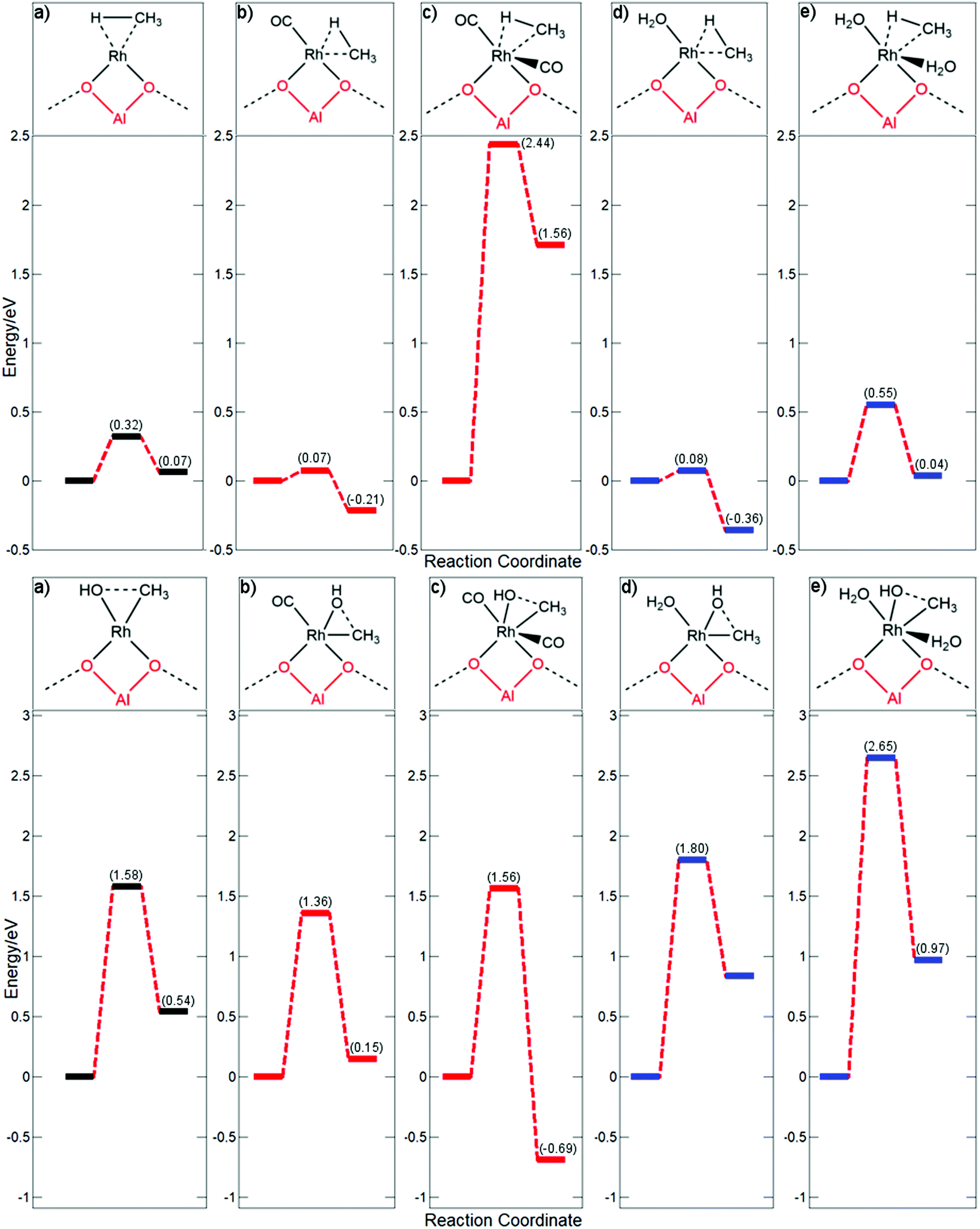 | ||
| Fig. 4 Free energy profiles for methane activation (top) and methyl hydroxyl coupling (bottom) on Rh@ZSM-5 whilst coordinated to varying numbers of CO and H2O molecules with the respective intermediate species drawn above: (a) Rh@ZSM-5; (b) Rh(CO)@ZSM-5; (c) Rh(CO)2@ZSM-5; (d) Rh(H2O)@ZSM-5; and (e) Rh(H2O)2@ZSM-5. The initial state for methane activation is with respect to methane in the gas phase. Initial states are considered from the respective Rh(CH3)(OH) species for the methyl hydroxyl coupling step. All energies are reported in eV. | ||
A peculiarity occurs when the rhodium atom becomes saturated with H2O; a relatively low barrier of 0.55 eV is observed instead of an expected much higher barrier. However, it is unclear whether both H2O molecules remain coordinated throughout the entirety of the step, which is analysed below. For the initial Rh(H2O)2@ZSM-5 complex, a bond length of 2.09 Å is obtained for both H2O–Rh coordinations. For the final state, bond lengths of 2.08 Å and 2.37 Å are calculated for the planar and axial water molecules coordinated to the rhodium, respectively. However, at the transition state, a large extension of the axial water is observed, with a coordination distance of 3.39 Å being calculated whilst the planar water maintains a bond length of 2.09 Å. At a distance of 3.39 Å, this water molecule is only very weakly coordinated to the rhodium atom. For comparison, for Rh(CO)2@ZSM-5, the axial CO bond extends by a meagre 0.15 Å through the transition state. It is more energetically feasible to dissociate a water molecule and then activate methane.
In order to address this, the axial water is fixed in place at its final state distance of 2.37 Å for the transition state search and not allowed to dissociate. An energy barrier of 1.40 eV is observed instead of the lessened barrier of 0.55 eV. This barrier is much higher and is an expected barrier for a coordinately saturated rhodium centre. Whilst this is not a true transition state, as the structure is not completely relaxed, it would indicate what the energy barrier would be if the axial water remained coordinated to the metal centre instead of dissociating. Whilst CO poisons the C–H activation step by blocking active sites, this effect is not observed for H2O due to its weak coordination. It is important to emphasise this point for future catalyst design: a good ligand for this system would have a certain degree of lability to allow an active site for the C–H activation step.
For methane activation, an available active site is the most important factor affecting the efficacity of this step. For the actual formation of methanol through a methyl-hydroxyl coupling step, the effect of spectator ligands is much more pronounced. From the calculated energy barriers, this step is expected to be the rate determining step for most of the coordinated species with the exception of Rh(CO)2@ZSM-5 where methane activation is the rate determining step. For the free Rh@ZSM-5 species, a barrier of 1.58 eV is observed. However, if a molecule is coordinated to the centre, the energetics of the step change drastically. If the rhodium centre is coordinated to water, a large elevation in the kinetic barrier is observed, going from 1.58 eV to 1.80 eV which is much too large to be overcome at the regular reaction conditions of approximately 423 K. The barrier increases even further if the complex is saturately coordinated with water, with a huge barrier of 2.65 eV being calculated. Water extremely inhibits this part of the reaction, with the electron donation enrichening the Rh-methyl species making it more stable and less inclined to couple. For CO, however, the trend is somewhat reversed. If rhodium is coordinated to a single CO molecule, the barrier is decreased from 1.58 eV to 1.36 eV. If the centre becomes saturated with CO, only a small decrease in the barrier to 1.56 eV is observed compared to the clean supported rhodium atom. This is due to the coordination of the Rh centre also affecting the energetics of this step. The coupling step with only one CO ligand goes from the less stable 16 electron pentavalent structure to the more stable tetravalent 16 electron square planar species. Whilst for the two ligand species, the coordination is going from an already stable hexavalent octahedral 18 electron complex to a similarly stable pentavalent distorted trigonal bi-pyramid 18 electron structure.
With these steps in mind, a total catalytic cycle can be proposed (Fig. 5). Methane will first be activated at the metal centre if there is an active site. If the rhodium atom is saturated, the reaction will not progress. The formed rhodium-hydride will then undergo oxygen insertion to form a rhodium–peroxide species which can degrade to rhodium–hydroxide. Avoiding the oxygen insertion step by taking a HXRE pathway or a traditional metal oxo pathway is unlikely due to the very high barrier for oxygen promoted methane activation and difficulty to form a RhO reactive species. Finally, the rate determining step, the formation of methanol through a methyl–hydroxyl coupling step, requires coordination with a CO ligand. This barrier is minimised by CO and maximised with the presence of water or another electron donating compound. This step is probably the underlying reason why CO is required for this system.
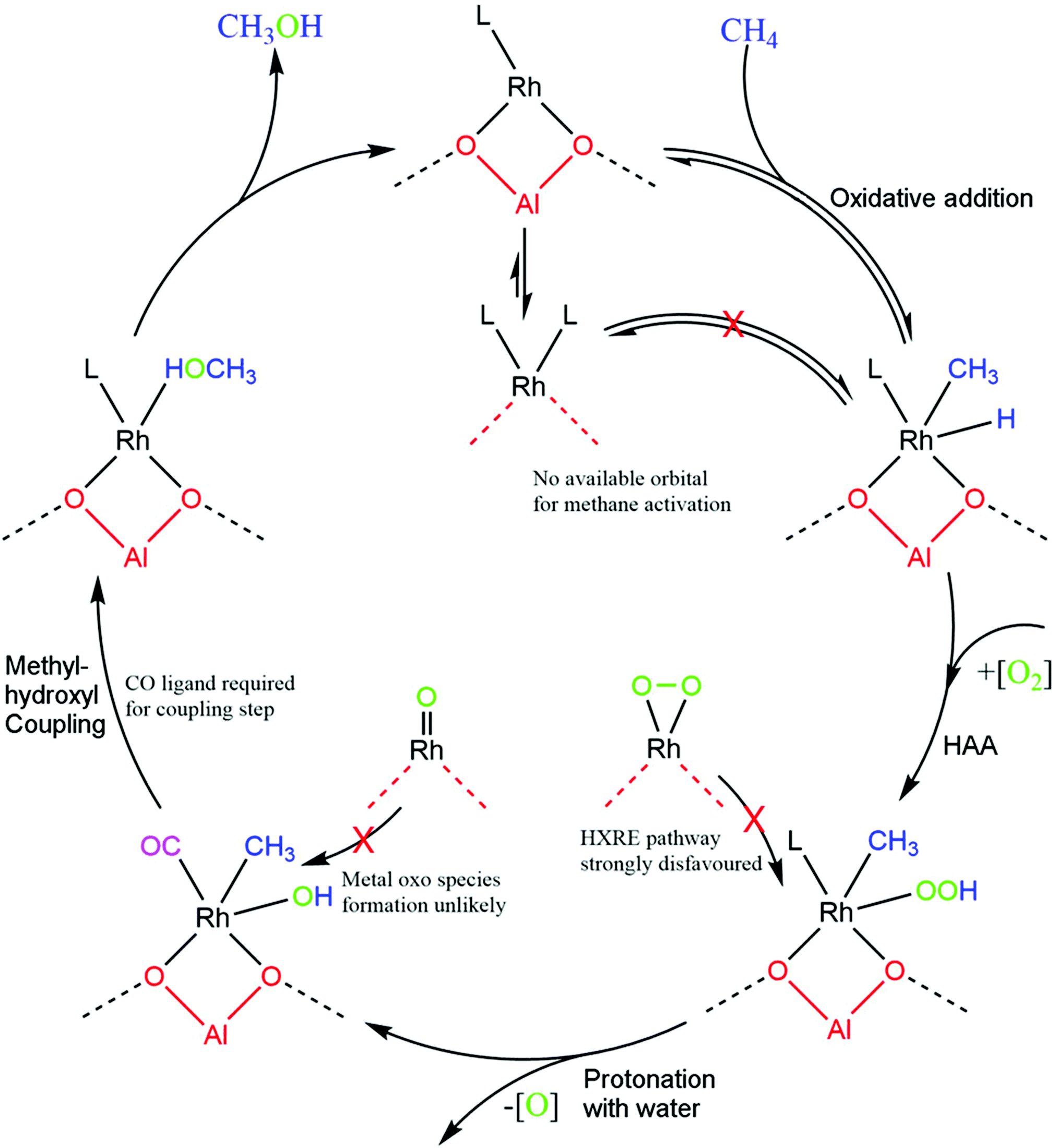 | ||
| Fig. 5 Proposed catalytic cycle for methane partial oxidation to methanol over Rh@ZSM-5. The steps, going clockwise, are methane activation, oxygen insertion, peroxide decomposition, methyl–hydroxyl coupling, and methanol desorption. CO is required for a ligand effect for the methanol formation step. Coordinated molecules are unimportant for all other steps except for the rhodium atom being unsaturated for methane activation. | ||
As a final note, we would like to bring emphasis to the similarity of this system to homogeneous systems for C–H activation. C–H activation is of great interest not just for industrial chemistry but also in synthetic organic chemistry47,48 where single atom chemistry, in the form of metal complexes, is also used. Our work attempts to bring emphasis to aspects of C–H activation homogeneous catalyst modelling to heterogeneous systems. The low yields obtained in the homogeneous systems requires a high molar ratio of catalyst to be used, resulting in the formation of in situ metal nanoparticles.47 Unfortunately, metal nano-particles are adept catalysts for methane total combustion.49 This is one of the issues with using homogeneous catalysts for methane to methanol partial oxidation. With a single atom supported system, where the single atom is immobilised and thus it cannot conglomerate with other metal atoms, these challenges may be overcome. Additionally, current methods of modelling heterogeneous catalysts may have to be adapted to better describe supported single atom systems.50,51 Our calculations considered the rhodium centre with a variety of coordination numbers with different coordinating molecules to mimic the active species under different experimental conditions. For instance, the Rh centre with two CO adsorbed responds the experimental condition with high CO pressures and the Rh centre with one CO adsorbed models the experimental condition with low CO pressures or trace amount of CO in the system. Ideally, full kinetic simulations are needed to quantitatively solve these problems in the future. It would be exciting to see the same kind of development that homogeneous C–H activation catalysis has had be realised for supported catalysts as well, specifically with ligand design.
4 Conclusions
The catalytic cycle for partial methane oxidation to methanol has been investigated and an energy profile calculated. Methane activation is readily accessible and can reverse, whilst formation of a carbon oxygen bond is more difficult. The requirement for CO has been found to be for the methanol formation step. Yet, it is possible that conventional ligands can replace CO. This could remove the issue where CO poisons the catalyst by saturating the metal centre. Additionally, as the HAA pathway is favoured, water is required specifically to accelerate the degradation of the formed metal peroxide species, and possibly to help displace CO from the metal centre.A greater focus on the formal oxidation, where methane is oxygenated, is required to accurately describe these systems. Just as for total combustion, where dehydrogenation and oxidation steps must both be considered, selective oxidation requires the same treatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for computational support from the UK national high performance computing service, ARCHER, for which access was obtained via the UKCP consortium and funded by EPSRC grant ref EP/P022561/1. We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1). We are grateful for access to the Queen's University Belfast Kelvin HPC service, which is partially funded by ESPRC.References
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
.
- N. Agarwal, S. Freakley, R. McVicker, S. Althahban, N. Dimitratos, Q. He, D. Morgan, R. Jenkins, D. Willock, S. Taylor, C. Kiely and G. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS
- W. Huang, S. Zhang, Y. Tang, Y. Li, L. Nguyen, Y. Li, J. Shan, D. Xiao, R. Gagne, A. Frenkel and F. Tao, Angew. Chem., Int. Ed., 2016, 55, 13441–13445 CrossRef CAS PubMed
- Y. Tang, Y. Li, V. Fung, D. Jiang, W. Huang, S. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Zhang, A. Frenkel and F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed
- J. Shan, M. Li, L. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16–22 CrossRef CAS
- D. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed
- T. Sheppard, C. Hamill, A. Goguet, D. Rooney and J. Thompson, Chem. Commun., 2014, 50, 11053–11055 RSC
- V. Sobolev, K. Dubkov, O. Panna and G. Panov, Catal. Today, 1995, 24, 251–252 CrossRef CAS
- E. Starokon, M. Parfenov, S. Arzumanov, L. Pirutko, A. Stepanov and G. Panov, J. Catal., 2013, 300, 47–54 CrossRef CAS
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed
- J. Xu, R. Armstrong, G. Shaw, N. Dummer, S. Freakley, S. Taylor and G. Hutchings, Catal. Today, 2016, 270, 93–100 CrossRef CAS
- A. Latimer, A. Kakekhani, A. Kulkarni and J. Nørskov, ACS Catal., 2018, 8, 6894–6907 CrossRef CAS
- A. Arvidsson, V. Zhdanov, P. Carlsson, H. Grönbeck and A. Hellman, Catal. Sci. Technol., 2017, 7, 1470–1477 RSC
- Z. Zhao, A. Kulkarni, L. Vilella, J. Nørskov and F. Studt, ACS Catal., 2016, 6, 3760–3766 CrossRef CAS
- A. Kulkarni, Z. Zhao, S. Siahrostami, J. Nørskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 RSC
- M. Mahyuddin, A. Staykov, Y. Shiota and K. Yoshizawa, ACS Catal., 2016, 6, 8321–8331 CrossRef CAS
- S. Li, Y. Wang, T. Wu and W. Schneider, ACS Catal., 2018, 8, 10119–10130 CrossRef CAS
- K. Hahn, M. Iannuzzi, A. Seitsonen and J. Hutter, J. Phys. Chem. C, 2013, 117, 1701–1711 CrossRef CAS
- D. Kaphan, R. Klet, F. Perras, M. Pruski, C. Yang, A. Kropf and M. Delferro, ACS Catal., 2018, 8, 5363–5373 CrossRef CAS
- S. Vummaleti, A. Genest, N. Kuriakose and N. Rösch, ACS Catal., 2018, 8, 9836–9846 CrossRef CAS
- T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, ACS Catal., 2016, 6, 2487–2495 CrossRef CAS
- Z. Zhao, A. Kulkarni, L. Vilella, J. Nørskov and F. Studt, ACS Catal., 2016, 6, 3760–3766 CrossRef CAS
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- P. Blöchl, O. Jepsen and O. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed
- A. Alavi, P. Hu, T. Deutsch, P. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650–3653 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- Y. Mao, Z. Wang, H. Wang and P. Hu, ACS Catal., 2016, 6, 7882–7891 CrossRef CAS
- A. Michaelides and P. Hu, J. Am. Chem. Soc., 2001, 123, 4235–4242 CrossRef CAS PubMed
- Z. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS PubMed
- H. Monkhorst and J. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef
- M. Jørgensen, L. Chen and H. Grönbeck, J. Phys. Chem. C, 2018, 122, 20351–20357 CrossRef
- S. Siahrostami, H. Falsig, P. Beato, P. Moses, J. Nørskov and F. Studt, ChemCatChem, 2016, 8, 767–772 CrossRef CAS
- J. Keith, R. Nielsen, J. Oxgaard and W. Goddard, J. Am. Chem. Soc., 2005, 127, 13172–13179 CrossRef CAS PubMed
- M. Denney, N. Smythe, K. Cetto, R. Kemp and K. Goldberg, J. Am. Chem. Soc., 2006, 128, 2508–2509 CrossRef CAS PubMed
- B. Xing, X. Pang and G. Wang, J. Catal., 2011, 282, 74–82 CrossRef CAS
- A. Latimer, H. Aljama, A. Kakekhani, J. Yoo, A. Kulkarni, C. Tsai, M. Garcia-Melchor, F. Abild-Pedersen and J. Nørskov, Phys. Chem. Chem. Phys., 2017, 19, 3575–3581 RSC
- R. Halbach, T. Teets and D. Nocera, Inorg. Chem., 2015, 54, 7335–7344 CrossRef CAS PubMed
- A. Arvidsson, V. Zhdanov, P. Carlsson, H. Grönbeck and A. Hellman, Catal. Sci. Technol., 2017, 7, 1470–1477 RSC
- R. Bunting, X. Cheng, J. Thompson and P. Hu, ACS Catal., 2019, 9, 10317–10323 CrossRef CAS
- C. Yoon, K. Hirsekorn, M. Neidig, X. Yang and T. Tilley, ACS Catal., 2011, 1, 1665–1678 CrossRef CAS
- X. Chen, K. Engle, D. Wang and J. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed
- J. He, M. Wasa, K. Chan, Q. Shao and J. Yu, Chem. Rev., 2016, 117, 8754–8786 CrossRef PubMed
- D. Ciuparu, M. Lyubovsky, E. Altman, L. Pfefferle and A. Datye, Catal. Rev., 2002, 44, 593–649 CrossRef CAS
- J. Wang, H. Wang and P. Hu, Sci. China: Chem., 2017, 61, 336–343 CrossRef
- M. Yang, H. Yuan, H. Wang and P. Hu, Sci. China: Chem., 2017, 61, 457–467 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01284j |
| This journal is © the Owner Societies 2020 |