
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bond-forming and electron-transfer reactivity between Ar2+ and O2†
Sam
Armenta Butt
and
Stephen D.
Price
 *
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: s.d.price@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 7679 4606
First published on 31st March 2020
Abstract
The reactivity, energetics and dynamics of the bimolecular reactions between Ar2+ and O2 have been studied using a position sensitive coincidence methodology at a collision energy of 4.4 eV. Four bimolecular reaction channels generating pairs of product ions are observed, forming: Ar+ + O2+, Ar+ + O+, ArO+ + O+ and O+ + O+. The formation of Ar+ + O2+ is a minor channel, involving forward scattering, and generates O2+ in its ground electronic state. This single electron transfer process is expected to be facile by Landau–Zener arguments, but the intensity of this channel is low because the electron transfer pathways involve multi-electron processes. The formation of Ar+ + O+ + O, is the most intense channel following interactions of Ar2+ with O2, in agreement with previous experiments. Many different combinations of Ar2+ and product electronic states contribute to the product flux in this channel. Major dissociation pathways of the nascent O2+* ion involve the ion's first and second dissociation limits. Unusually, the experimental results clearly show the involvement of a short-lived collision complex [ArO2]2+ in this channel. The formation of O+ and ArO+ involves direct abstraction of O− from O2 by Ar2+. There is scant evidence of the involvement of a collision complex in this bond forming pathway. The ArO+ product appears to be formed in the first excited electronic state (2Π). The formation of O+ + O+ results from dissociative double electron transfer via an O22+ intermediate. The exoergicity of the dissociation of the nascent O22+ intermediate is in good agreement with previous work investigating the unimolecular dissociation of this dication.
1 Introduction
Planetary ionospheres are composed of a variety of atomic and molecular species which can be ionised by absorption of energetic photons and by collisional processes. Recent studies have indicated that that di-positive atomic and molecular ions (dications) should be included in models of these environments.1 Indeed, dications have been detected in the ionospheres of the Earth, Venus and Io.2–5 Dications are also predicted to be present, in chemically significant concentrations, in the atmospheres of Titan and Mars.6–8Recent work has shown that both atomic and molecular dications exhibit significant reactivity following collisions with neutral species.9–11 Such collisional processes are expected to be the major route which limits the lifetimes of atomic dications in planetary ionospheres.12 In addition, despite their inherent thermodynamic instability, the metastable electronic states of molecular dications have been shown to possess lifetimes sufficient to allow collisions with other species in planetary environments.9 The reactive nature of dications, coupled with their significant abundance in ionospheres, suggests that the bimolecular chemistry of these species could play a role in ionospheric chemistry.13 For example, dication reactions could be involved in the chemistry of complex molecule assembly.12 Indeed, carbon–carbon coupling reactions have been observed following interactions of aromatic dications with methane,14,15 ethyne16 and benzene.17
Despite their potential significance in ionospheric chemistry, the potential influence of dications on a variety of energized media is often neglected, due in part to the difficulty of unambiguous detection of dications by simple mass spectrometry.9 The development of a catalogue of known dication reactions, emphasizing those reactions where bond-forming steps are present, coupled with additional identification of dications in planetary atmospheres, should help reveal potential ionospheric processes.18 Indeed, the influence of the unimolecular chemistry of molecular dications in atmospheric erosion has been recently recognized.19–22
This paper presents a detailed investigation of the interactions between Ar2+ and O2, giving information on dicationic energetics, reactivity and the associated reaction mechanisms. This detailed information allows a better understanding of the relevance and influence of Ar2+/O2 collisions in planetary environments. Argon constitutes ∼1% of the Earth's atmosphere and is one of the most abundant elements in the universe.23,24 Argon is also abundant in the atmospheres of the Moon, Mercury and Mars.25–27 In these atmospheres, the formation of the Ar2+ dication is likely, as recognised by Thissen et al.12 The bimolecular reactivity of Ar2+ was one of the first dicationic collision systems to be investigated, as beams of Ar2+ are relatively easy to generate using electron ionisation.28–31 In most of these early investigations of Ar2+-neutral collisions, only the dominant single-electron transfer (SET) and double-electron transfer (DET) channels were observed. These early experiments were usually carried out at high laboratory translational energies (0.1–20 keV) and involved rare gases or simple molecules as the neutral collision partner (e.g. He, H2, N2, CO2, C2H6, C6H6).29,30,32–34 More recent experiments, at lower collision energies (<100 eV), led to the observation of bond-forming chemistry following the interactions of Ar2+ with various neutral species, revealing, for example, the formation of Ar–O, Ar–N and Ar–C bonds.35–40 Indeed, the bimolecular reactivity of rare gas dications is now recognized as an effective route to forming these unusual chemical bonds.
Oxygen is the third most abundant element in the universe, it is a major component of the atmosphere of the Earth and is also present in the atmosphere of other planets and satellites.24,25 The reactions between Ar2+ and O2 have been studied in experiments over a range of supra-thermal energies.29,30,32–34 In the earliest work, no chemical bond formation was observed, and the ion yields were explained using models involving varying contributions from SET and DET. In later work, Ascenzi et al.38 studied the reaction of Ar2+ with O2 using a combination of experiments and ab initio calculations. This work revealed a reactivity involving SET as well as the formation of two different products involving Ar–O bonds: ArO2+ and ArO+.
Whilst the ionic products of the reaction between Ar2+ and O2 are now reasonably well established,38 there has been little investigation of the dynamics and kinematics of these chemical processes. To address this issue, this paper reports an investigation of the reactivity of Ar2+ and O2, at collision energies of 2.7 and 4.4 eV in the centre-of-mass frame, using position-sensitive coincidence mass spectrometry (PSCO-MS). The PSCO-MS technique couples a crossed-beam collisional methodology with the coincident detection of the cationic products of dication-neutral collisions.41 Capable of investigating reactivity at collision energies well below 10 eV, the PSCO-MS experiment can provide a comprehensive insight into the reactivity and dynamics involved in dication-neutral interactions. For example, this technique has been used to show that ArC+ is formed following collisions of Ar2+ with C2H2, and that the ArC+ ion forms via dissociation of a nascent ArCH+ product.28
In the study of Ar2+/O2 collisions detailed in this paper we observe that chemical bond formation occurs via a direct mechanism rather than complexation. Conversely, we find strong evidence of complex formation ([ArO2]2+) in the dynamics of the dissociative SET channel; a reaction that typically occurs via long-range electron transfer.
2 Experimental
Coincidence techniques involve the simultaneous detection of two or more products from a single reactive event. Bimolecular reactions of dications with neutral species often generate pairs of monocations and these pairs of ions are detected in coincidence in the PSCO-MS experiment. The PSCO-MS apparatus used in this study has been described in detail in the literature.41–43 Briefly, a pulsed beam of dications is produced and directed into the field-free source region of a time-of-flight mass spectrometer (TOF-MS). In the source region, the dications interact with a jet of the neutral reactant. Subsequent application of an extraction voltage to the source region allows the TOF-MS to detect the cation pairs generated in the dication-neutral interactions. The detection of these ions involves recording their time of flight and arrival position at a large microchannel-plate detector. From this raw data, a list of flight times and arrival positions of the ions detected in pairs, a two-dimensional mass spectrum, can be generated revealing the different reactive channels. The positional data accompanying the ionic detections can be processed to reveal the relative motion of the products of each reactive event, providing a detailed insight into the mechanisms of each reactive channel.43In this work the Ar2+ ions are generated, along with Ar+, by electron ionisation of Ar (BOC, 99.998%) by 100 eV electrons in a custom-built ion source. The positively charged argon ions are extracted from the ion source and pass through a hemispherical energy analyser to restrict the translational energy spread of the final Ar2+ beam to ∼0.3 eV. The continuous beam of ions exiting the hemispherical analyser is then pulsed, using a set of electrostatic deflectors, before being accelerated and focussed into a commercial velocity filter. The velocity filter is set to transmit just the 40Ar2+ (m/z = 20) ions. The resulting pulsed beam of energy-constrained Ar2+ ions is then decelerated to less than 10 eV in the laboratory frame, using a commercial electrostatic decelerator, before entering the source region of the TOF-MS. As noted above, in the TOF-MS source region the beam of dications is crossed with an effusive jet of O2 (BOC, 99.5%). Single-collision conditions44 are achieved by employing a low pressure of O2; hence, most dications do not undergo a collision, whilst only a small percentage experience one collision. Such a pressure regime ensures no reactivity due to successive collisions with two O2 molecules influences the Ar2+ reactivity we observe. An electric field is applied across the TOF-MS source when the dication pulse reaches the centre of this region. This electric field accelerates positively charged species into the second electric field (acceleration region) of the TOF-MS and then on into the flight tube. At the end of the flight tube, the cations are accelerated onto a position-sensitive detector comprising a chevron-pair of microchannel plates located in front of a dual delay-line anode.41 The voltage pulse applied to the source region also starts the ion timing circuitry, to which the signals from the detector provide stop pulses. The experiments in this work employed both high (183 V cm−1) and low (28.5 V cm−1) TOF-MS source fields. As discussed in more detail below, the lower source field results in better energy resolution in the resulting PSCO-MS data. However, in these low field spectra ions with high transverse (off-axis) velocities do not reach the detector.
Signals from the detector are amplified and discriminated before being passed to a PC-based time-to-digital converter. If two ions are observed in the same TOF cycle, a coincidence event is recorded and each ion's arrival time and impact position on the detector is stored for off-line analysis. The use of single-collision conditions ensures ‘false’ coincidences are kept to a minimum. The ion pairs data can be plotted as a 2D histogram, a ‘pairs spectrum’, where the time of flights (t1,t2) of each ion in the pair are used as the (x,y) co-ordinates. Peaks in the pairs spectrum readily identify bimolecular reaction channels that result in a pair of positively charged product ions. Each such peak, the group of events corresponding to an individual reaction channel, can then be selected for further off-line analysis.
As shown in previous work, the positional and time of flight information for each ion of a pair can be used to generate their x, y and z velocity vectors in the laboratory frame; here the z-axis is defined by the principal axis of the TOF-MS.41 The x and y vectors are determined from the positional information and flight time; the z vector is determined from the deviation of the observed TOF from the expected TOF of the same ion with zero initial kinetic energy. The laboratory frame velocities are then converted into the centre-of-mass (CM) frame using the initial dication velocity. Often the pair of monocations resulting from the reaction between a dication and a neutral are accompanied by a neutral species: a three-body reaction. A powerful feature of the PSCO-MS experiment is that the CM velocity of such a neutral product can be determined from the CM velocities of the detected ionic products via conservation of momentum.41
To reveal the dynamics of a given dication-neutral reaction channel, a CM scattering diagram (Fig. 1) can be generated from the velocities of the product ions. Such CM scattering diagrams are radial histograms that, for each event collected for a given reaction channel, plot the magnitude of the CM velocity |wi| as the radial co-ordinate and the scattering angle θ between wi and the CM velocity of the incident dication as the angular coordinate. In our CM scattering diagrams, since 0° ≤ θ ≤ 180°, the data for one product is shown in the upper semi-circle of the figure and the data for another product in the lower semi-circle, as the scattering of each ion is azimuthally symmetric. In addition, for three-body reactions, internal-frame scattering diagrams can be a powerful aid in interpreting the reaction dynamics. In this class of scattering diagram |wi| is again the radial coordinate, but the angular coordinate is now the CM scattering angle with respect to CM velocity of one of the other product species.
 | ||
| Fig. 1 CM scattering diagram for the reaction Ar2+ + O2 → Ar+ + O2+ at a CM collision energy of 4.4 eV. The black dot indicates the position of the CM. See text for details. | ||
From the CM velocities of the product species the total kinetic energy release (KER) T for a given reactive event can also be determined using the individual CM velocities of the products.41 The exoergicity of the reaction ΔE can then be determined from T and the CM collision energy, Ecom:
| ΔE = T − Ecom = Eproducts − Ereactants | (1) |
 | ||
| Fig. 2 Experimental exoergicity spectrum for the reaction Ar2+ + O2 → Ar+ + O2+. The exoergicities for potential SET pathways calculated from literature values are also shown: (x) Ar2+(1S) + O2(3Σ−g) → Ar+(4D) + O2+(X2Πg), (y) Ar2+(1D) + O2(3Σ−g) → Ar+(2S) + O2+(X2Πg), (z) Ar2+(1S) + O2(3Σ−g) → Ar+(2S) + O2+(X2Πg). The error bars represent two standard deviations of the associated counts. | ||
3 Results and discussion
PSCO-MS spectra were recorded following the collisions of Ar2+ with O2 at Ecom = 4.4 eV. Four significant product ions were detected in the coincidence spectrum: Ar+, O2+, O+ and ArO+. Of course, O22+ could also be present, although no sharp peak indicative of a dication is visible in the m/z = 16 region of the mass spectrum. The experiments were repeated at Ecom = 2.7 eV and, as we discuss below, no significant differences in the ratios of products or the derived reaction dynamics were apparent.Table 1 lists the product channels observed in the coincidence spectrum following the collisions of Ar2+ with O2. The most intense channel (Rxn. B) is dissociative single-electron transfer (DSET), producing Ar+ + O+ + O. Non-dissociative SET is also observed (Rxn. A), albeit with only 1% of the intensity of the DSET channel. The second most intense channel involves the production of O+ + O+ (Rxn. D) and results from double electron transfer (DET): the dissociation of a nascent O22+ ion into O+ and O+. Finally, a bond forming reaction forming ArO+ + O+ is also observed at low intensity (Rxn. C). Reaction B makes up 94.5% of the total counts, indicating a significantly larger reaction cross section for this (DSET) reaction than the other channels, in agreement with previous experiments.38
| Reaction | Products | Relative intensity/% | Modal experimental ΔE/eV |
|---|---|---|---|
| A | Ar+ + O2+ | 0.9 | 4.0 |
| B | Ar+ + O+ + O | 94.5 | 9.5 |
| C | ArO + + O+ | 0.2 | 10.5 |
| D | Ar + O+ + O+ | 4.4 | 8.5 |
As noted above, PSCO-MS data were also recorded at low TOF-MS source field to yield a higher energy resolution in the exoergicity spectrum (Ecom = 4.0 eV). As discussed below, these experiments reveal that Reaction B initially involves the population of a number of electronic states of the product Ar+ and O2+* ions, the O2+* states then dissociating to O+ + O.
3.1 Formation of Ar+ and O2+
Fig. 1 shows a CM scattering diagram for the Ar+ and O2+ products of the non-dissociative SET reaction (Rxn. A). Fig. 1 reveals strong forward scattering, where the velocity of the Ar+ ion is strongly oriented with the incident dication velocity, w(Ar2+). This strong forward scattering results in the O2+ product ion's velocity being directed anti-parallel to w(Ar2+), and strongly oriented with w(O2). This form of scattering has been commonly observed for other non-dissociative SET processes and arises from a direct electron transfer mechanism, where the electron is transferred between the reactants at a significant interspecies separation (3–6 Å).42,43,45,46 Such electron transfer processes are well-represented by a Landau–Zener formalism.47From analysis of the product ion velocities, as previously discussed, the exoergicity distribution of Reaction A can be determined (Fig. 2). The exoergicity for the formation of Ar+ and O2+ (Ecom = 4.4 eV) is found to be centred at ∼4.7 eV, with a full width at half maximum (FWHM) of 2.1 eV. The experiments performed at a lower TOF-MS source field, to give a higher energy resolution, revealed no new structures in this exoergicity spectrum.
To interpret the exoergicity spectrum for Reaction A (Fig. 2) we need to determine the relative energies of the reactant and product states that could be involved in the reaction. For this collision system this energetic data is readily accessible. Previous studies have shown that Ar2+ beams, generated in a similar manner to those in our experiments, are composed of ions in all three electronic states (3P, 1D and 1S) arising from the Ar2+ p4 configuration;48 the relative abundance of these states appears approximately statistical in these previous investigations.49 The reactant O2 molecule, admitted as an effusive beam, will be in its ground vibronic state, 3Σ−gv = 0. The accessible states of the O2+ product are well studied.50 The ground state of O2+ (X2Πg) lies 12.07 eV above the ground state of the neutral molecule and requires an additional 6.7 eV to break the O–O bond to form O+ + O.51 All O2+ states lying above this O+ + O asymptote are unstable to dissociation within the lifetime of our experiment and therefore cannot contribute to the formation of the O2+ observed in this non-dissociative SET channel.52 For example, metastable minima in the O2+ c4Σ−u state, and levels of the b4Σ−g state above v = 3 are expected to dissociate before they are detected in our experiments.53–56 There are three energetically accessible electronic levels for the product Ar+ ions (2P, 2S and 4D).57
From the above energetic considerations (see Table SI 1 in the ESI†) we find that there are three possible reaction pathways that match the favoured 3–6.5 eV range of exoergicities we see in Fig. 2: reactions (2)–(4). These three pathways all involve the formation of O2+ in its ground electronic state, X2Πg, which is stable to dissociation. The exoergicities of these three channels are indicated in Fig. 2. The match of the calculated exoergicities with the experimental spectrum (Fig. 2) is good but not perfect due to the neglect of product vibrational excitation and the distribution of centre of mass collision energies.
| Ar2+(1S) + O2(3Σ−g) → Ar+(4D) + O2+(X2Πg) ΔE = 3.3 eV | (2) |
| Ar2+(1D) + O2(3Σ−g) → Ar+(2S) + O2+(X2Πg) ΔE = 3.8 eV | (3) |
| Ar2+(1S) + O2(3Σ−g) → Ar+(2S) + O2+(X2Πg) ΔE = 6.2 eV | (4) |
3.2 Formation of Ar+ and O+
The most intense product channel we observe following the reaction of Ar2+ with O2 is dissociative single-electron transfer (DSET), in agreement with Ascenzi et al.38 The well-established mechanism of such dicationic DSET reactions, in this collision energy regime, is that initial Landau–Zener (LZ) style electron transfer occurs, at significant interspecies separations, forming one or both of the nascent product ions in dissociative states. These dissociative state(s) subsequently fragment to yield the detected products.28 These dynamics, given the LZ curve crossing is favoured in a “reaction window” centred at an inter-reactant separation of around 4 Å, result in strong forward scattering. Such strong forward scattering has been observed in several different collision systems.28,46,58,59 In this specific reaction channel, the nascent O2+* ion formed in the initial electron transfer would subsequently dissociate, resulting in the formation of Ar+ + O+ + O.Fig. 3a shows the CM scattering diagram for the Ar+ and O+ product ions from this DSET channel. The Ar+ is primarily forward scattered but has a significant tail to higher scattering angles. Such a tail is indicative of a short-lived association, a collision complex, between the O2 and Ar2+: [ArO2]2+. Clearly, this collision complex does not live long enough for a complete loss of the correlation between the Ar+ velocity and the initial velocity of the Ar2+ reactant before fragmenting into O2+* and Ar+. The observed scattering shows that this DSET reaction does not follow the standard (direct) mechanism of dicationic electron transfer outlined above. Dynamics supporting the standard (direct) model of dication SET have been revealed, using the PSCO experiment and other angularly resolved techniques, for SET in many other collision systems.28,46,58,59 However, other isolated cases of dicationic electron transfer involving significant complexation, as we see for this reaction, have also been previously reported.60,61
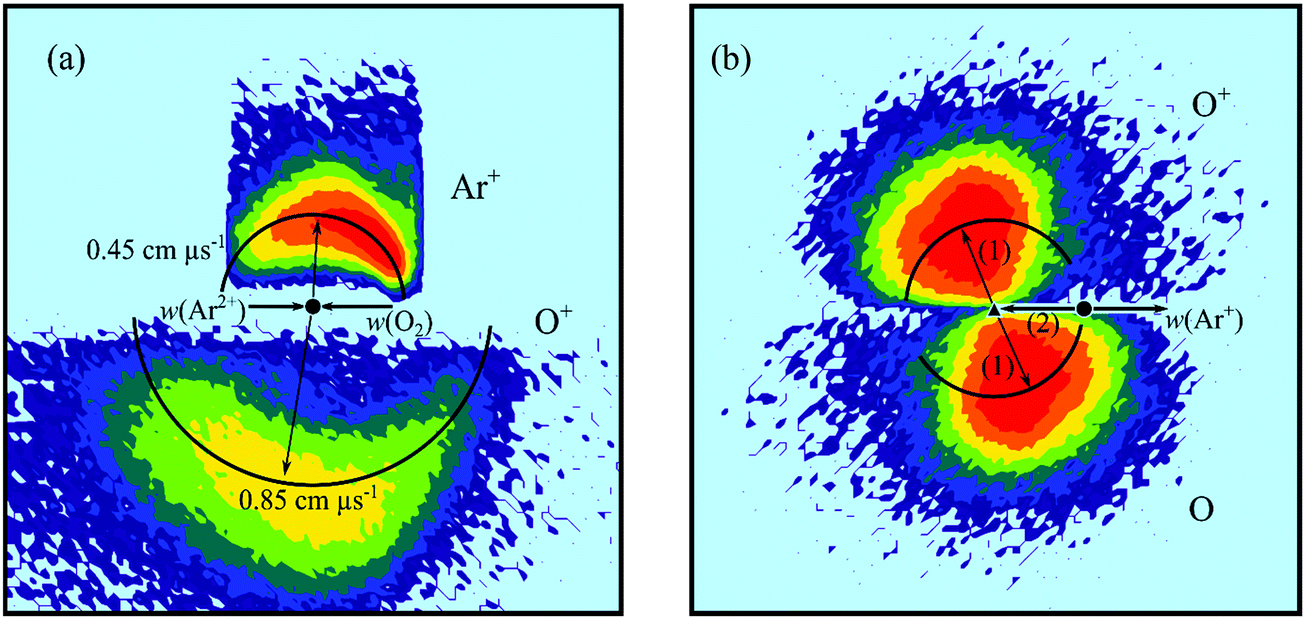 | ||
| Fig. 3 Scattering diagrams for the reaction Ar2+ + O2 → Ar+ + O+ + O at a CM collision energy of 4.4 eV. (a) CM scattering diagram showing the scattering of O+ and Ar+ relative to the incident dication velocity, w(Ar2+). (b) Internal frame scattering diagram showing the scattering of O+ and O relative to the velocity of the Ar+ product ion. In part (b) the labelled vectors represent: (1) 0.55 cm μs−1; (2) the precursor velocity, w(O2+*). | ||
Fig. 3b shows the internal frame scattering of O+ and O, relative to the Ar+ ion. The diagram clearly shows the O and O+ fragments are both backward scattered relative to the Ar+ ion, clearly indicating that the [ArO2]2+ complex dissociates into O2+* and Ar+ before the molecular ion subsequently fragments. A similar case of DSET via a collision complex was observed following collisions between Ne2+ and N2.60 In the Ne2+/N2 collision system the dissociative SET reaction proceeded along two competing routes, via a collision complex, [NeN2]2+, and via a fast-sequential pathway. The two different pathways could be distinguished by distinct peaks in the product ions’ angular distributions. In the current Ar2+/O2 collision system, such peaks are not observed, implying a single mechanism, involving short-lived complexation, is responsible for the DSET reaction.
The internal frame scattering (Fig. 3b) has a symmetrical character, but the O+ is slightly more strongly backward scattered, relative to the Ar+, than the O. This additional acceleration of the O+ product is very likely due to Coulombic repulsion with the Ar+. That is the scattering indicates that the O2+ dissociates relatively rapidly, before it leaves the electric field of its Ar+ partner. Dissociation of the O2+ within the electric field of the Ar+ gives the O+ a larger velocity away from the Ar+ than the O fragment. By looking at the extra velocity of the O+ ion, the point of O2+* dissociation can be estimated. The difference in velocities between the O+ and O fragments corresponds to an energy difference of 3 ± 0.2 eV. The extra kinetic energy of the O+ fragment would result from O2+* dissociation at an interspecies separation of about 5 Å between the Ar+ and O2+. This distance corresponds to an O2+* lifetime of approximately 200 fs, which is comparable to lifetimes of O2+ dissociative states determined from the rotational bandwidths of photoelectron spectra,54,62 and calculated potential curves.63 DSET reactions where the dissociation of one of the product ions occurs within the field of the other have been previously observed, for example in the reaction between Ar2+ and C2H2.28 Here the C2H2+* fragment formed from the initial electron transfer dissociates into CH+ and CH, and the CH+ fragment is backscattered with a larger velocity than the CH fragment relative to the Ar+ ion. In this earlier work dissociation of C2H2+* was estimated to occur at a distance of about 20 Å from the Ar+ ion, involving a C2H2+* lifetime of about 500 fs, values comparable with those derived for the current collision system.
The modal total exoergicity of the DSET reaction (Rxn. B) determined experimentally is ∼9.5 eV with a FWHM of 6–15 eV (see Fig. SI 1b in the ESI†). This range of exoergicities encompasses a large number of accessible combinations of reactant and product electronic states: permutations of the Ar2+ (3P, 1D and 1S) states and the first five dissociation limits of O2+.57,64,65
Considering the DSET reaction to be stepwise, an approximation given the above analysis of the O and O+ velocities, we can derive the O2+* precursor velocity from the Ar+ velocity. Using this approximation, we find an exoergicity distribution for the initial electron transfer step (Ar2+ + O2 → Ar+ + O2+*) centred at ∼5.7 eV with a FWHM of ∼1.5–7.7 eV (Fig. 4). The broad distribution is indicative of a number of product and reactant electronic states being involved in this initial electron transfer. Considering the accessible reactant and product electronic states, specifically the dissociative states of O2+, and excluding some transitions due to poor Frank–Condon overlap, pathways (5)–(7) nicely match the peak of the exoergicity distribution for the initial electron transfer step (Fig. 4).66
| Ar2+(3P) + O2(3Σ−g) → Ar+(2P) + O2+(B2Σ−g) ΔE = 7.3 eV | (5) |
| Ar2+(3P) + O2(3Σ−g) → Ar+(2P) + O2+(c4Σ−u) ΔE = 3.1 eV | (6) |
| Ar2+(1D) + O2(3Σ−g) → Ar+(2P) + O2+(c4Σ−u) ΔE = 4.8 eV | (7) |
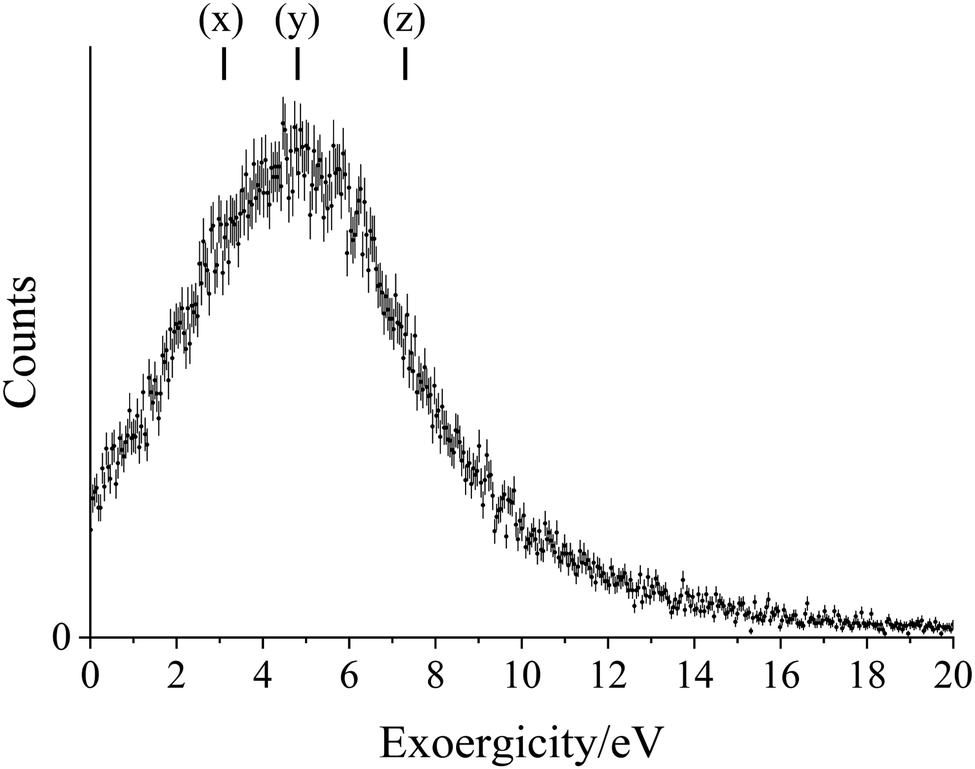 | ||
| Fig. 4 Exoergicity spectrum for the initial electron transfer reaction in the DSET channel, Ar2+ + O2 → Ar+ + O2+*. The exoergicities for potential electron transfer pathways calculated from literature values are also shown: (x) Ar2+(3P) + O2(3Σ−g) → Ar+(2P) + O2+(c4Σ−u), (y) Ar2+(1D) + O2(3Σ−g) → Ar+(2P) + O2+(c4Σ−u), (z) Ar2+(3P) + O2(3Σ−g) → Ar+(2P) + O2+(B2Σ−g). The error bars represent two standard deviations of the counts. | ||
When the PSCO data was recorded at a lower source field, to achieve higher energy resolution, peaks at lower energies in the total exoergicity spectrum were revealed. Specifically, a distinct peak at around 4 eV is visible (Fig. SI 1(a), ESI†), with another broad peak at 5–8 eV. It is important to remember, in this higher energy resolution experiment, sideways-scattered ions from reactions with higher exoergicities are not detected. The exoergicity for the initial electron transfer step, determined from the precursor velocity of Ar+ in this low source field experiment, shows a broad peak centred at 4 eV and a FWHM from 1.8–5.7 eV. This distribution is in good general agreement with that derived from the high source field experiments (Fig. 4). This high energy resolution experiment supports the population of O2+(c4Σ−u), shown by the exoergicities of reactions (6) and (7); these pathways have exoergicities of 3.1 eV and 4.8 eV respectively.
To summarise, there are multiple pathways for the initial electron transfer reaction in the DSET channel. The major pathways are likely eqn (5)–(7), however there may be contributions from higher energy O2+* states. The higher energy resolution experiments confirm the presence of pathways (6) and (7).
Using the O2+* precursor velocity, the KER for the dissociation of O2+* into O+ + O can also be extracted from our data.69 This exoergicity for the O2+ dissociation, from the high source field experiment, is shown in Fig. 5. The broad maximum of the exoergicity distribution is at 1.5–6.5 eV, with the FWHM from 0.2–10.3 eV. Previous work studying the dissociation of O2+* formed from neutral O2via electron transfer with O22+, in a similar experiment, revealed a dominant peak in the exoergicity spectrum at 1.5–3.0 eV, this overlaps with the lower end of the maximum exoergicity observed in this study.69 Dissociation energies for O2+* of up to ∼5.8 eV have been previously observed, resulting from dissociation of O2+(c4Σ−u) to the lowest energy dissociation limit of O+ + O.52,53,66,70 In order to explain the higher energy exoergicities, higher lying states of O2+ must be involved. Higher lying states of O2+ have been observed, including one at ∼27.5 eV, potentially accounting for the higher exoergicities we observe.67,68 The broad maximum observed in the current study results from the presence of multiple dissociation pathways, derived from the range of O2+ states, identified above, formed in the initial electron transfer step which fragment to the first dissociation limit, O(3P) + O+(4S) (L1), and second dissociation limit, O(1D) + O+(4S) (L2). The pathways, shown in Fig. 5, are (v) B2Σ−g → L2, (w) B2Σ−g → L1, (x) c4Σ−u → L2, (y) c4Σ−u → L1, (z) O2+* (E = 27.5 eV) → L1. The range of exoergicities attributable to each pathway is estimated by considering the possible vibrational excitation of the O2+ product, and these ranges are indicated in Fig. 5. As well as the first two dissociation limits, there are many additional minor O2+ dissociation pathways to higher dissociation limits that have been reported.52,53,70,71 The possibility of dissociation to higher energy O + O+ asymptotes, and the array of dissociative states of O2+ available, further broaden the kinetic energy release observed from the O2+* dissociation, shown in Fig. 5.
 | ||
| Fig. 5 Experimental exoergicity spectrum for the dissociation of O2+ to form O+ and O. The exoergicities for the potential dissociation pathways, calculated from literature values, are shown: (v) O2+(B2Σ−g) → O(1D) + O+(4S), (w) O2+(B2Σ−g) → O(3P) + O+(4S), (x) O2+(c4Σ−u) → O(1D) + O+(4S), (y) O2+(c4Σ−u) → O(3P) + O+(4S), (z) O2+(E = 27.5 eV) → O(3P) + O+(4S). The arrows indicate the approximate range of exoergicities from the potential vibrational excitation of O2+ in a vertical transition. The error bars represent two standard deviation of the counts. See text for details. | ||
Whilst DSET resulting from the reaction of Ar2+ with O2 has previously been observed,33,38 the current work provides much additional information on the dynamics and energetics of the reaction. The tail to higher scattering angles in the CM scattering diagram of Ar+ and O+ (Fig. 3a) is evidence for the formation of a short-lived [ArO2]2+ complex. This collision complex then dissociates into the nascent electron transfer products Ar+ and O2+* (B2Σ−g and c4Σ−u), with the O2+* subsequently fragmenting within the field of the Ar+ ion.
3.3 Formation of ArO+ and O+
Fig. 6 shows the CM scattering for ArO+ + O+, the products of the bond forming reaction we detect in this collision system. Scattering that is distinctly forward is apparent, where the velocity of the ArO+ ion is broadly oriented with the velocity of the incident dication, w(Ar2+). This form of scattering suggests a stripping-style mechanism, where an O− is transferred between the O2 and Ar2+ at a relatively large interspecies separation. Such dynamics for bond-forming reactions involving atom transfer have been observed before.28 In their earlier work, where this unusual reaction was first observed, Ascenzi et al.38 suggest that this bond-forming reaction proceeds via a collision complex, ArO22+. The scattering data we report here shows little evidence for any long-lived association between the reactants. If the channel proceeded via a long-lived collision complex, which survived for several rotational periods, prominent forward scattering would not be observed, and the scattering would have a more isotropic nature. Such a signature of long-lived collision complexes has been reported in other dicationic collision systems.43,72 | ||
| Fig. 6 CM scattering diagram for the reaction Ar2+ + O2 → ArO+ + O+ at a CM collision energy of 4.4 eV. The scattering of O+ and ArO+ are shown relative to the incident dication velocity, w(Ar2+). See text for details. | ||
The exoergicity distribution of the bond forming reaction, which is determined from the velocities of the product ions, is shown in Fig. 7. The exoergicity distribution has a maximum at 10.5 eV, with a FWHM of 5 eV. As above, the exoergicity spectrum can be rationalised by considering the possible electronic states of the products. Energetic calculations involving ArO+ usually consider the two lowest-bound electronic states: the ground state (4Σ−) and the first excited state (2Π).73,74 The minimum of the 2Π state lies ∼0.4 eV higher in energy than the 4Σ− state minimum, but at a markedly different equilibrium geometry.
 | ||
| Fig. 7 Experimental exoergicity spectrum for the reaction producing ArO+ and O+ from Ar2+ and O2. The range of exoergicities indicated by (x) is determined from literature values for the pathway: Ar2+(3P) + O2(3Σ−g) → ArO+(2Π) + O+(4S). The arrow represents the possible range of exoergicity from the vibrational excitation of ArO+.57,64,65,73 The error bars represent two standard deviations of the counts. See text for details. | ||
The exoergicity for forming ArO+(4Σ−) + O+(4S) from the ground states of Ar2+ + O2 is 11.4 eV, whilst populating ArO+(2Π) + O+(4S) would have an exoergicity of 11.0 eV. Both of these exoergicities coincide well with the peak in the experimental exoergicity distribution (Fig. 7). We note access to both these asymptotes is spin-allowed from Ar2+(3P) + O2(3Σ−g).
In interpreting the results of their experiments, Ascenzi et al.38 suggest this reaction involves ArO+ formed in its ground 4Σ− state along with O+(4S). Considering the formation of ArO+ at these two lowest energy product asymptotes, it is perhaps unlikely that a significant proportion of any ArO+(4Σ−) products would be long-lived enough to be detected by our experiment. Specifically, the ArO+ product is likely to be formed with significant vibrational excitation due to the long-range O− abstraction from the O2 by the Ar2+. However, the ArO+(4Σ−) ground state has only a shallow potential well (∼0.4 eV)73 and is most likely to be formed at an unstable geometry due to the expected level of vibrational excitation. Conversely, the first excited state of ArO+, 2Π, has a well depth of ∼2.0 eV, thus would more readily accommodate the expected significant vibrational excitation. Given these considerations, we suggest pathway (8) as the dominant route for this bond-forming reaction:
| Ar2+(3P) + O2(3Σ−g) → ArO+(2Π) + O+(4S) ΔE = 11.0 eV | (8) |
Ascenzi et al.38 also proposed the operation of a markedly less exoergic channel, ArO+(2Π) + O+(2P), involving the formation of an excited state of the product oxygen monocation. A hint of the involvement of such a channel came from evidence of a threshold in their cross sections for the formation of ArO+. The pathway to ArO+(2Π) + O+(2P) (ΔE = 6.0 eV) was considered a possible candidate for such a threshold due to a Coulombic barrier in the exit channel.38 Our exoergicity spectrum (Fig. 7) shows that such a channel, if present at this collision energy, makes only a minor contribution to the ArO+ yield.
PSCO experiments using a lower source field to achieve a higher energy resolution in the exoergicity spectrum for this reaction are impractical as the lower source field dramatically reduces the collection efficiency for the translationally energetic O+ ions.
3.4 Formation of O+ and O+
Analysis of the dynamics of the product channel generating a pair of O+ ions shows these charged species are effectively isotropically scattered about the velocity of the O2 reactant. Such scattering dynamics confirm that the source of these ion pairs is double electron transfer (DET). That is, two electrons are transferred from the oxygen molecule to the argon dication, at a significant interspecies separation, resulting in the formation of neutral argon and O22+. The O22+ then dissociates to form O+ and O+. The dissociation of the resulting O22+ ion should be uniformly distributed in the molecular (O2) frame, resulting in the scattering of O+ ions we observe.A simple, one-dimensional, electrostatic model has previously been used to model dicationic DET.28 In this model (Fig. 8), the reactant and product potentials are represented by simple polarization-attraction forces. The previous studies indicate that dicationic DET seems to occur via a concerted (two-electron transfer) mechanism in which the product asymptote lies close in energy to the reactant asymptote (<1 eV difference), as shown in Fig. 8.28 Such small asymptotic energy differences between the reactant and the product channels can result in a two-electron curve crossing lying within the Landau–Zener reaction window centred on an interspecies separation of 4 Å. Previous work has shown that when a collision system exhibits a significant yield of DET then the reactant and product asymptotes conform to the above energetic constraints and appropriately favoured crossings in the Landau–Zenner window exist.
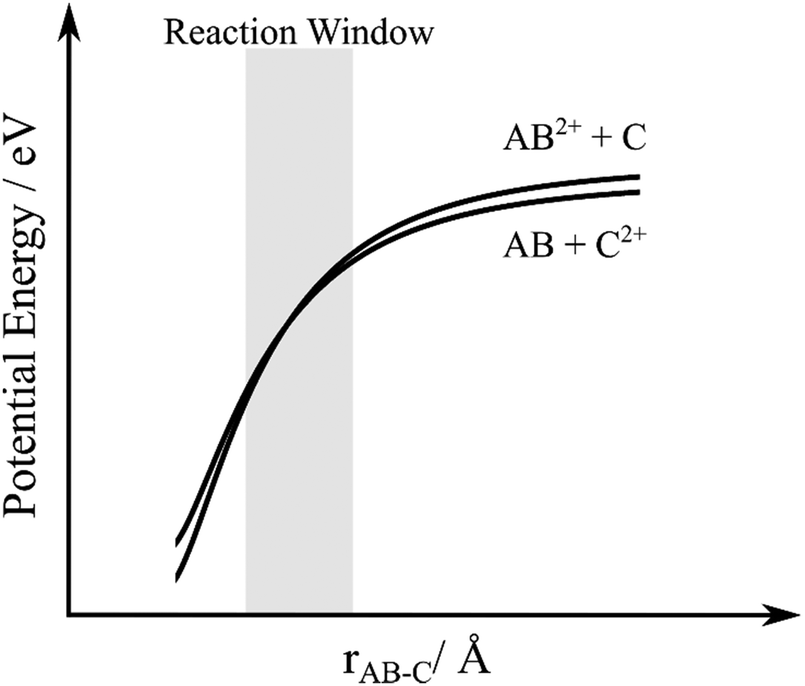 | ||
| Fig. 8 Schematic potential energy curves for double-electron transfer between a dication, AB2+, and neutral species, C. rAB–C is the interspecies separation between AB and C. | ||
To apply this model to the Ar2+/O2 collision system we note that the Ar2+ ground state (3P) and first two excited states (1D and 1S) have energies of 43.4, 45.1 and 47.5 eV above the ground state of Ar. Considering the products, there are several dissociative states of O22+ lying between 42 and 46 eV above the ground state of O2.75 From these simple energetic considerations we can immediately see that the Ar2+/O2 collision system is likely to involve DET as we have product and reactant asymptotes lying close in energy. However, since the polarizability of Ar (α = 1.6411 × 10−24 cm3) is slightly larger than the polarizability of O2 (α = 1.5689 × 10−24 cm3) if the DET process is exoergic overall, that is the Ar + O22+ limit lies below the Ar2+ + O2 asymptote, the reactant and product potentials will not cross in the simple polarization-attraction model outlined above.76 Of course, this simple model, with its additional assumption of spherical symmetry, will only provide an approximation to the true potentials, and of course no repulsive character is included. Indeed, there are potential Ar2+(1D) + O2 and Ar + O22+(11Πg and 11Δg) reactant and product asymptotes which lie very close in energy. Hence, it is quite possible, since the polarizabilities of O2 and Ar are very similar, that, given these close lying asymptotes, the potentials cross in the real collision system, particularly when the influence of differing repulsive terms, and the anisotropy, of the true interaction potentials are taken into account. Alternatively, it is possible that this DET reaction is an endothermic process, for which situation the differing polarizabilities will allow a simple curve crossing. Such an endothermic process would have to be facilitated by the collision energy. Such coupling of Ecom to the potential energy surface could, for example, allow the formation of O22+ B3Πg from Ar2+(3P) (ΔE = +0.2 eV).
Once formed, the O22+ dissociates to O+ + O+. Lundqvist et al.75 studied the formation and dissociation of O22+ and found O22+ states that result in dissociation to the first and second dissociation asymptotes (O+ + O+), with kinetic energy releases of 6.9–12.7 eV. From analysis of the O+ ion velocities, the exoergicity of the dissociation of the O22+ formed in the DET channel (Rxn. D) was determined to have a broad maximum centred at ∼8.5 eV with a FWHM from 6.3–11.6 eV. This KER is in good agreement with that determined by Lundqvist et al.75
4 Conclusion
The collisions of Ar2+ and O2 have been studied using coincidence methods at a collision energy of 4.4 eV. Four bimolecular reaction channels generating pairs of product ions are observed forming: Ar+ + O2+, Ar+ + O+, ArO+ + O+ and O+ + O+.The formation of Ar+ + O2+ is a minor channel involving strong forward scattering and generates O2+ in its ground electronic state. This single electron transfer process is expected to be facile by Landau–Zener arguments but the relative intensity of this channel is low because the electron transfer pathways involve multi-electron processes.
The formation of Ar+ + O++ O, is the most intense channel resulting from the reaction of Ar2+ with O2, in agreement with previous experiments. Many different combinations of Ar2+ and product electronic states contribute to the flux in this reaction. Major dissociation pathways of the nascent O2+ ion involve the first (O(3P) + O+(4S)), and second (O(1D) + O+(4S)) dissociation limits. Unusually, the experimental results clearly show the involvement of a short-lived collision complex [ArO2]2+ in this channel.
The formation of O+ and ArO+ involves direct abstraction of O− from O2 by Ar2+. Scant evidence of the involvement of a collision complex in this bond forming pathway is apparent. The formation of the first excited state of ArO+(2Π), accompanied by O+(4S) is the likely product channel.
The formation of O+ + O+ results from dissociative double electron transfer via the formation of O22+. The exoergicity of the O22+ dissociation, which we can extract from our data, is in good agreement with previous work investigating the unimolecular dissociation of this dication.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the EPSRC, the Leverhulme Trust and UCL.References
- C. Simon, J. Lilensten, O. Dutuit, R. Thissen, O. Witasse, C. Alcaraz and H. Soldi-Lose, Ann. Geophys., 2005, 23, 781–797 CrossRef CAS
.
- H. S. Bridge, J. W. Belcher, A. J. Lazarus, J. D. Sullivan, R. L. McNutt, F. Bagenal, J. D. Scudder, E. C. Sittler, G. L. Siscoe, V. M. Vasyliunas, C. K. Goertz and C. M. Yeates, Science, 1979, 204, 987–991 CrossRef CAS PubMed
- S. Ghosh, K. K. Mahajan, J. M. Grebowsky and N. Nath, J. Geophys. Res., 1995, 100, 23983–23991 CrossRef CAS
- J. H. Hoffman, C. Y. Johnson, J. C. Holmes and J. M. Young, J. Geophys. Res., 1969, 74, 6281–6290 CrossRef CAS
- E. Dubinin, R. Modolo, M. Fraenz, J. Woch, G. Chanteur, F. Duru, F. Akalin, D. Gurnett, R. Lundin, S. Barabash, J. D. Winningham, R. Frahm, J. J. Plaut and G. Picardi, J. Geophys. Res.: Space Phys., 2008, 113, A10217 CrossRef
- J. Lilensten, O. Witasse, C. Simon, H. Soldi-Lose, O. Dutuit, R. Thissen and C. Alcaraz, Geophys. Res. Lett., 2005, 32, L03203 CrossRef
- J. Lilensten, C. Simon, O. Witasse, O. Dutuit, R. Thissen and C. Alcaraz, Icarus, 2005, 174, 285–288 CrossRef CAS
- O. Witasse, O. Dutuit, J. Lilensten, R. Thissen, J. Zabka, C. Alcaraz, P.-L. Blelly, S. W. Bougher, S. Engel, L. H. Andersen and K. Seiersen, Geophys. Res. Lett., 2002, 29, 104 CrossRef
- S. D. Price, J. D. Fletcher, F. E. Gossan and M. A. Parkes, Int. Rev. Phys. Chem., 2017, 36, 145–183 Search PubMed
- D. Ascenzi, J. Aysina, E. L. Zins, D. Schröder, J. Žabka, C. Alcaraz, S. D. Price and J. Roithová, Phys. Chem. Chem. Phys., 2011, 13, 18330–18338 RSC
- J. Roithová, H. Schwarz and D. Schröder, Chem. – Eur. J., 2009, 15, 9995–9999 CrossRef PubMed
- R. Thissen, O. Witasse, O. Dutuit, C. S. Wedlund, G. Gronoff and J. Lilensten, Phys. Chem. Chem. Phys., 2011, 13, 18264–18287 RSC
- O. Dutuit, N. Carrasco, R. Thissen, V. Vuitton, C. Alcaraz, P. Pernot, N. Balucani, P. Casavecchia, A. Canosa, S. Le Picard, J.-C. Loison, Z. Herman, J. Zabka, D. Ascenzi, P. Tosi, P. Franceschi, S. D. Price and P. Lavvas, Astrophys. J., Suppl. Ser., 2013, 204, 20 CrossRef
- C. L. Ricketts, D. Schröder, C. Alcaraz and J. Roithová, Chem. – Eur. J., 2008, 14, 4779–4783 CrossRef CAS PubMed
- E.-L. Zins and D. Schröder, J. Phys. Chem. A, 2010, 114, 5989–5996 CrossRef CAS PubMed
- J. Roithová and D. Schröder, Chem. – Eur. J., 2007, 13, 2893–2902 CrossRef PubMed
- J. Roithová and D. Schröder, Phys. Chem. Chem. Phys., 2007, 9, 731–738 RSC
- H. Sabzyan, E. Keshavarz and Z. Noorisafa, J. Iran. Chem. Soc., 2014, 11, 871–945 CrossRef CAS
- S. Falcinelli, F. Pirani, M. Alagia, L. Schio, R. Richter, S. Stranges, N. Balucani and F. Vecchiocattivi, Atmosphere, 2016, 7, 112 CrossRef
- S. Falcinelli, M. Rosi, P. Candori, F. Vecchiocattivi, J. M. Farrar, F. Pirani, N. Balucani, M. Alagia, R. Richter and S. Stranges, Planet. Space Sci., 2014, 99, 149–157 CrossRef CAS
- J. Lilensten, C. Simon Wedlund, M. Barthélémy, R. Thissen, D. Ehrenreich, G. Gronoff and O. Witasse, Icarus, 2013, 222, 169–187 CrossRef CAS
- M. Alagia, N. Balucani, P. Candori, S. Falcinelli, F. Pirani, R. Richter, M. Rosi, S. Stranges and F. Vecchiocattivi, Rend. Lincei, 2013, 24, 53–65 CrossRef
- D. F. Mark, F. M. Stuart and M. de Podesta, Geochim. Cosmochim. Acta, 2011, 75, 7494–7501 CrossRef CAS
- A. G. W. Cameron, Space Sci. Rev., 1973, 15, 121–146 CrossRef CAS
- S. A. Stern, Rev. Geophys., 1999, 37, 453–491 CrossRef CAS
- A. O. Nier, W. B. Hanson, A. Seiff, M. B. McElroy, N. W. Spencer, R. J. Duckett, T. C. Knight and W. S. Cook, Science, 1976, 193, 786–788 CrossRef CAS PubMed
-
D. D. Bogard, R. N. Clayton, K. Marti, T. Owen and G. Turner, in Space Science Reviews, ed. R. Kallenbach, J. Geiss and W. K. Hartmann, Springer, Dordrecht, 2001, vol. 12, pp. 425–458 Search PubMed
- M. A. Parkes, J. F. Lockyear and S. D. Price, Int. J. Mass Spectrom., 2009, 280, 85–92 CrossRef CAS
- G. Dupeyrat, J. B. Marquette, B. R. Rowe and C. Rebrion, Int. J. Mass Spectrom. Ion Processes, 1991, 103, 149–156 CrossRef CAS
- H. Störi, E. Alge, H. Villinger, F. Egger and W. Lindinger, Int. J. Mass Spectrom. Ion Phys., 1979, 30, 263–270 CrossRef
- B. Friedrich and Z. Herman, Chem. Phys. Lett., 1984, 107, 375–380 CrossRef CAS
- W. Lindinger, E. Alge, H. Störi, M. Pahl and R. N. Varney, J. Chem. Phys., 1977, 67, 3495–3499 CrossRef CAS
- E. Y. Kamber, P. Jonathan, A. G. Brenton and J. H. Beynon, J. Phys. B: At., Mol. Opt. Phys., 1987, 20, 4129–4142 CrossRef CAS
- G. C. Shields and T. F. Moran, J. Phys. B: At., Mol. Opt. Phys., 1983, 16, 3591–3607 CrossRef CAS
- P. Tosi, R. Correale, W. Lu, S. Falcinelli and D. Bassi, Phys. Rev. Lett., 1999, 82, 450–452 CrossRef CAS
- P. Tosi, W. Lu, R. Correale and D. Bassi, Chem. Phys. Lett., 1999, 310, 180–182 CrossRef CAS
- W. Lu, P. Tosi and D. Bassi, J. Chem. Phys., 2000, 112, 4648–4651 CrossRef CAS
- D. Ascenzi, P. Franceschi, P. Tosi, D. Bassi, M. Kaczorowska and J. N. Harvey, J. Chem. Phys., 2003, 118, 2159–2163 CrossRef CAS
- N. Lambert, D. Kearney, N. Kaltsoyannis and S. D. Price, J. Am. Chem. Soc., 2004, 126, 3658–3663 CrossRef CAS PubMed
- J. F. Lockyear, K. Douglas, S. D. Price, M. Karwowska, K. J. Fijalkowski, W. Grochala, M. Remeš, J. Roithová and D. Schröder, J. Phys. Chem. Lett., 2010, 1, 358–362 CrossRef CAS
- W.-P. Hu, S. M. Harper and S. D. Price, Meas. Sci. Technol., 2002, 13, 1512–1522 CrossRef CAS
- W.-P. Hu, S. M. Harper and S. D. Price, Mol. Phys., 2005, 103, 1809–1819 CrossRef CAS
- S. D. Price, Int. J. Mass Spectrom., 2007, 260, 1–19 CrossRef CAS
- K. Yamasaki and S. R. Leone, J. Chem. Phys., 1989, 90, 964–976 CrossRef CAS
- S. M. Harper, W.-P. Hu and S. D. Price, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, 4409–4423 CrossRef CAS
- Z. Herman, Int. Rev. Phys. Chem., 1996, 15, 299–324 Search PubMed
- S. A. Rogers, S. D. Price and S. R. Leone, J. Chem. Phys., 1993, 98, 280–289 CrossRef CAS
- T. Nakamura, N. Kobayashi and Y. Kaneko, J. Phys. Soc. Jpn., 1985, 54, 2774–2775 CrossRef CAS
- J. F. Lockyear, M. A. Parkes and S. D. Price, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 145201 CrossRef
- O. Edqvist, E. Lindholm, L. E. Selin and L. Asbrink, Phys. Scr., 1970, 1, 25–30 CrossRef CAS
- P. M. Guyon, T. Baer, L. F. A. Ferreira, I. Nenner, A. Tabche-Fouhaile, R. Botter and T. R. Govers, J. Phys. B: At. Mol. Phys., 1978, 11, L141–L144 CrossRef CAS
- T. Akahori, Y. Morioka, M. Watanabe, T. Hayaishi, K. Ito and M. Nakamura, J. Phys. B: At. Mol. Phys., 1985, 18, 2219–2229 CrossRef CAS
- X. Tang, G. A. Garcia and L. Nahon, J. Chem. Phys., 2018, 148, 124309 CrossRef PubMed
- A. Padmanabhan, M. A. MacDonald, C. H. Ryan, L. Zuin and T. J. Reddish, J. Phys. B: At., Mol. Opt. Phys., 2010, 43, 165204 CrossRef
- A. Mitrushenkov, P. Palmieri, G. Chambaud and P. Rosmus, Chem. Phys. Lett., 2003, 378, 463–469 CrossRef CAS
- J. T. Moseley, P. C. Cosby, J. B. Ozenne and J. Durup, J. Chem. Phys., 1979, 70, 1474–1481 CrossRef CAS
-
A. Kramida, Y. Ralchenko, J. Reader and NIST ASD Team, NIST Atomic Spectra Database (version 5.6.1), National Institute of Standards and Technology, Gaithersburg, MD, 2018 Search PubMed
- M. A. Parkes, J. F. Lockyear, D. Schröder, J. Roithová and S. D. Price, Phys. Chem. Chem. Phys., 2011, 13, 18386–18392 RSC
- S. D. Price, J. Chem. Soc., Faraday Trans., 1997, 93, 2451–2460 RSC
- S. M. Harper, S. W.-P. Hu and S. D. Price, J. Chem. Phys., 2004, 120, 7245–7248 CrossRef CAS PubMed
- C. L. Ricketts, D. Schröder, J. Roithová, H. Schwarz, R. Thissen, O. Dutuit, J. Žabka, Z. Herman and S. D. Price, Phys. Chem. Chem. Phys., 2008, 10, 5135–5143 RSC
- M. Evans, S. Stimson, C. Y. Ng and C.-W. Hsu, J. Chem. Phys., 1998, 109, 1285–1292 CrossRef CAS
- K. Tanakaa and M. Yoshimine, J. Chem. Phys., 1979, 70, 1626–1633 CrossRef
- J. E. Sansonetti and W. C. Martin, J. Phys. Chem. Ref. Data, 2005, 34, 1559–2259 CrossRef CAS
- I. Velchev, W. Hogervorst and W. Ubachs, J. Phys. B At. Mol. Opt. Phys., 1999, 32, L511–L516 CrossRef CAS
- M. Richard-Viard, O. Dutuit, M. Lavollée, T. Govers, P. M. Guyon and J. Durup, J. Chem. Phys., 1985, 82, 4054–4063 CrossRef CAS
- P. Baltzer, B. Wannberg, L. Karlsson, M. Carlsson Göthe and M. Larsson, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 45, 4374–4384 CrossRef CAS
- K. Ellis, R. I. Hall, L. Avaldi, G. Dawber, A. McConkey, L. Andric and G. C. King, J. Phys. B: At., Mol. Opt. Phys., 1994, 27, 3415–3426 CrossRef CAS
- M. A. Parkes, J. F. Lockyear, S. D. Price, D. Schröder, J. Roithová and Z. Herman, Phys. Chem. Chem. Phys., 2010, 12, 6233–6243 RSC
- A. Lafosse, J. C. Brenot, A. V. Golovin, P. M. Guyon, K. Hoejrup, J. C. Houver, M. Lebech and D. Dowek, J. Chem. Phys., 2001, 114, 6605–6617 CrossRef CAS
- L. J. Frasinski, K. J. Randall and K. Codling, J. Phys. B: At. Mol. Phys., 1985, 18, 129–135 CrossRef
- Z. Herman, Int. J. Mass Spectrom., 2015, 378, 113–126 CrossRef CAS
- S. M. McIntyre, J. W. Ferguson and R. S. Houk, Spectrochim. Acta, Part B, 2011, 66, 581–587 CrossRef CAS
- G. Frenking, W. Koch, D. Cremer, J. Gauss and J. F. Liebman, J. Phys. Chem., 1989, 93, 3410–3418 CrossRef CAS
- M. Lundqvist, D. Edvardsson, P. Baltzer, M. Larsson and B. Wannberg, J. Phys. B: At., Mol. Opt. Phys., 1996, 29, 499–514 CrossRef CAS
-
CRC Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press/Taylor & Francis, Boca Raton, FL, 100th edn, 2019 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01194k |
| This journal is © the Owner Societies 2020 |