
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
What is the trigger for the hydrogen evolution reaction? – towards electrocatalysis beyond the Sabatier principle†
Aleksandar R.
Zeradjanin
 *abc,
George
Polymeros
b,
Cigdem
Toparli
d,
Marc
Ledendecker
b,
Nejc
Hodnik
e,
Andreas
Erbe
f,
Michael
Rohwerder
b and
Fabio
La Mantia
a
*abc,
George
Polymeros
b,
Cigdem
Toparli
d,
Marc
Ledendecker
b,
Nejc
Hodnik
e,
Andreas
Erbe
f,
Michael
Rohwerder
b and
Fabio
La Mantia
a
aUniversität Bremen, Energiespeicher- und Energiewandlersysteme, Bibliothekstr. 1, 28359, Bremen, Germany. E-mail: aleksandar.zeradjanin@uni-bremen.de; Tel: +49-(0)421-2246-7336
bMax-Planck-Institut für Eisenforschung GmbH, Department of Interface Chemistry and Surface Engineering, Max-Planck-Strasse 1, 40237 Düsseldorf, Germany. E-mail: zeradjanin@mpie.de
cMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: aleksandar.zeradjanin@cec.mpg.de
dLaboratory for Electrochemical Interfaces, Departments of Nuclear Science and Engineering, and Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
eDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
fDepartment of Materials Science and Engineering, NTNU, Norwegian University of Science and Technology, 7491 Trondheim, Norway
First published on 17th March 2020
Abstract
The mechanism of the hydrogen evolution reaction, although intensively studied for more than a century, remains a fundamental scientific challenge. Many important questions are still open, making it elusive to establish rational principles for electrocatalyst design. In this work, a comprehensive investigation was conducted to identify which dynamic phenomena at the electrified interface are prerequisite for the formation of molecular hydrogen. In fact, what we observe as an onset of the macroscopic faradaic current originates from dynamic structural changes in the double layer, which are entropic in nature. Based on careful analysis of the activation process, an electrocatalytic descriptor is introduced, evaluated and experimentally confirmed. The catalytic activity descriptor is named as the potential of minimum entropy. The experimentally verified catalytic descriptor reveals significant potential to yield innovative insights for the design of catalytically active materials and interfaces.
Introduction
Electrolytic splitting of water to molecular oxygen and hydrogen has attracted the attention of researchers for over two centuries.1 Nowadays, water electrolysis is intensively investigated as an essential technology in a future energy landscape (e.g. PEM electrolyzers).2,3 In that context, the efficiency of water electrolysis appears to be one of the bottlenecks for a hydrogen-based economy.4,5 The efficiency of water electrolysis relies on the ability to identify catalytic structure–performance descriptors.6–8 Of particular importance are the mechanisms of inner-sphere electrocatalytic reactions, where the electron transfer is coupled to the adsorption/desorption of intermediates at the solid/liquid electrified interface.9–12 While interest in the HER has grown intensively over the last few decades,7,13 unifying guidelines for active catalyst design are still missing.14 Lack of a sufficiently comprehensive picture of the reaction mechanism of the two-electron reaction, like the HER, suggests why mechanistic understanding of more complex reactions (e.g. oxygen reduction/evolution reaction) will probably remain a challenge for a long time.12 As postulated by Trasatti: “A true theory of electrocatalysis will not be available until activity can be calculated a priori from some known properties of the materials”.15 An important contribution in this direction has come from density functional theory (DFT), which has evolved into a powerful tool for predicting reaction rates of heterogeneous gas-phase catalysis.16 In electrocatalysis, however, the application of DFT to obtain adsorption energies of intermediates often over-simplifies the phenomena at the electrified interface.7 Nevertheless, the obtained activity trends, based on computational adsorption energies, to a large extent overlapped with experimental activity trends.17 In the case of transition metals, DFT has rationalized trends through a linear relation between the position of the d-band center relative to the Fermi level and the adsorption energies of the key intermediates.18 The proposed model resulted in a volcano-type dependence where the maximum in kinetics responds to the optimal adsorption energy.18 Consequently, the Sabatier principle was suggested to be an underlying principle on which the theory of electrocatalysis is grounded.19 Till today, this remains a widely accepted conventional view.19–21However, solvent effects are often not considered7,10 and their impact on the reaction rate is still largely unknown.8,9 In fact, the solvent strongly influences bond formation, bond breaking, the existence of adsorbed intermediates and electron transfer.22,23 Based on this, three questions arise: (i) what are adequate catalytic descriptors for the HER, which also include the role of the solvent, (ii) can we confirm them experimentally and (iii) can these descriptors contribute in forming an accurate picture of the activation process?
Many physico-chemical parameters were correlated as descriptors with the HER reaction rate.13 The majority of the proposed descriptors lacked an obvious link to the structure of the interface and the rate law.23 However, one of these correlations deserves special attention. Namely, Trasatti observed a linear relationship between the work function and the logarithm of the exchange current density of HER.24 The observed relation was understood as a secondary effect, where the high work function responds to more positive potentials of zero charge (Epzc).25 Due to the more positive Epzc, the excess proton concentration at the interface close to the reversible potential of the HER will be large, which in turn should influence the HER initiation through a facilitated Volmer step (reaction (1)).26 The Volmer step is followed by the Heyrovsky or Tafel step or, in other words, the intermediate formation is followed by the recombination that requires one or two active sites that are dependent on the intermediate coverage.26
| H+ + e− ⇌ Had Volmer step | (1) |
| Had + H+ + e− ⇌ H2 θmax ≤ 0.3 Heyrovsky step | (2a) |
| 2Had ⇌ H2 θmax ≤ 1.0 Tafel step | (2b) |
In this work, we set out to reinterpret the phenomena occurring at the electrified interface during HER and offer a new perspective on the origins of electrocatalytic activity. In our approach, we propose a relation between the nature of the electrode material, the structural properties of the interface and the reaction rate,23 by introducing some dynamic aspects of the reaction mechanism. In order to gain in-depth insights into the electrified interface, we combine the existing mechanistic understanding with new experimental insights using a combination of classic and advanced electroanalytical techniques: specifically, we utilized electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV), in situ spectroscopic ellipsometry (SE) and electrochemical surface force apparatus (EC-SFA) to probe the electrode interface, under dynamic conditions of HER.
Results and discussion
Potential of maximum entropy vs. potential of minimum entropy – reorganization of the surface water layer as a promotor of proton reduction
Starting from the consideration that the activation entropy is manifested through double layer restructuring via interfacial water molecules and their noncovalent interactions, an intuitive question arises: is there a counterpart to the Epzc, a potential of minimum entropy that might be relevant for the electrocatalytic activity? To find an answer to this question, it is essential to develop a reasonable hypothesis of the HER activation process, based on the experimental data available on the most active catalysts. These, including polycrystalline platinum (Pt-poly), exhibit the Volmer–Tafel pathway with Tafel's step as the rate determining step (reaction (2b)). Instead of trying to understand why the recombination of intermediates in the Tafel step is relatively slow, we will try to understand why the formation of intermediates in the Volmer step (reaction (1)) is so fast. The platform for our heuristic model is the visual representation of the potential-induced double layer changes on polycrystalline platinum in acidic electrolyte, in some aspects similar, but not identical, to the one previously shown by Jerkiewicz.28 The Pt-surface has a dipole moment (μsurf) that originates from the lower coordination number of surface atoms in comparison to bulk atoms and a surface charge (qpt) that is related to the applied potential.29 Dependent on applied potential, μsurf and qpt vary in magnitude and sign.30 At the same time, surface water molecules have a dipole moment (μwater), wherein spatial orientation is electric field-dependent, and interact with the Pt-surface via surface dipole–water dipole and surface charge–water dipole interactions.28 Four characteristic points during a potential sweep from the double layer region towards the reversible potential of HER are shown in Fig. 1, together with the corresponding cyclic voltammogram: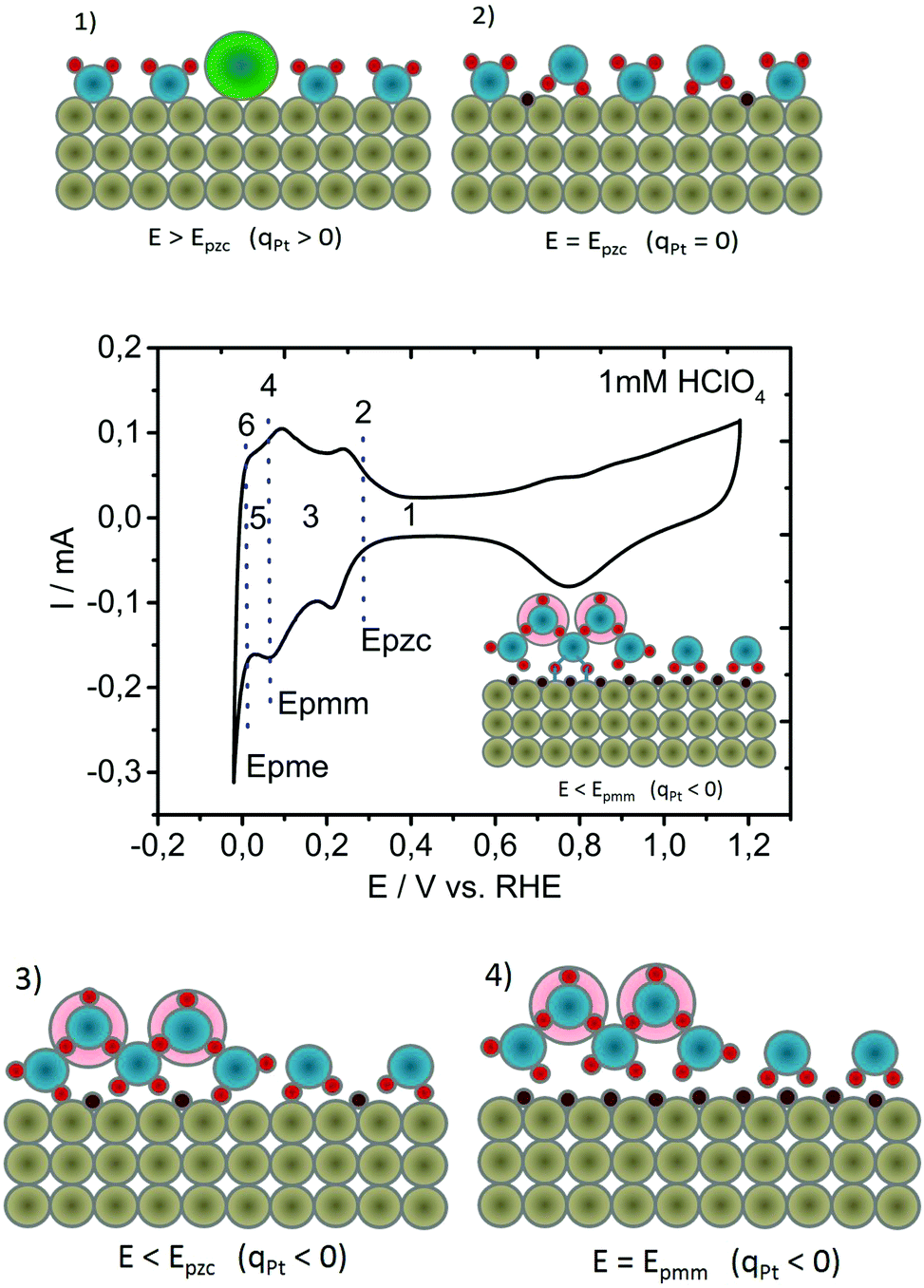 | ||
| Fig. 1 Schematic illustration of four characteristic interfacial changes at the Pt/electrolyte boundary during indicated potential modulations, relevant for intermediate formation. Four characteristic potential-dependent stages in the reconstruction of the double layer are also indicated using the voltammogram recorded in 1 mM HClO4 at a scan rate of 100 mV s−1. Color code: Pt atoms – grey, Hupd – brown, oxygen – blue, hydrogen – red, hydronium ions – pale red background, and anions – green. | ||
(1) at potentials more positive than Epzc (Fig. 1(1)), the Pt-surface is positively charged with surface dipole pointed away from the Pt-surface, causing adsorption of anions as well as forcing water molecules to be preferentially oriented. Oxygen atoms point towards the surface, while hydrogen atoms point towards the electrolyte;31
(2) when more negative potentials are applied, Epzc is reached (Fig. 1(2)), where the net surface charge at the electrode is equal to zero (qpt = 0) and at which the entropy in the double layer is at its maximum.32 Despite the fact that the surface of the metal at Epzc is electroneutral, on Pt and several other noble metals, protons get specifically adsorbed forming a low coverage of underpotentially deposited hydrogen (Hupd). Here, hydrogen is adsorbed at potentials more positive than the reversible potential for hydrogen evolution, and it is considered not to be an intermediate of the HER (see the voltammogram in Fig. 1).33 The exact location of Hupd, including the consequent direction of the Pt–Hupd dipole,34 has been a matter of dispute for decades, with very contradictory spectroscopic data.35 However, independent of the exact locations where Hupd is formed (more probably interatomic holes in the Pt outer layer36 or even sub-surface locations,28 instead of on-top locations), the bond between the water molecule and the metal surface will be significantly weakened.28 Namely, if a Pt–water noncovalent bond is established through hydrogen, a repulsive interaction between Pt–Hupd and surface water molecules is expected, thus causing partial detachment of water molecules from the surface, as shown by Osawa.37,38 The possibility that Hupd minimizes the adhesion of surface water molecules was also indicated by Jerkiewicz et al.28 The surface can be perceived as “hydrophobic”.
(3) applying a more negative potential, we arrive at a region where the Pt-surface is negatively charged (Fig. 1(3)). Consequently, water molecules now become preferentially oriented with the hydrogen atoms towards the surface32 while the concentration of protons in the interfacial region grows.39 Simultaneously, the Hupd coverage increases proportionally to the applied potential.7 An important consequence is, in comparison to Epzc, that the repulsion between water molecules and the Hupd is more emphasized. The result increases water detachment from the surface.
(4) Finally, with further negative polarization, full Hupd coverage is reached causing most of the water molecules to be detached from the surface (Fig. 1(4)). By means of an electrochemical quartz-crystal nanobalance (EQCN), Jerkiewicz detected a minimum mass at the electrode surface at full Hupd coverage, consequently introducing the potential of minimum mass (Epmm) as an interfacial parameter.28 Water detachment from the hydrophobic surface and the formation of a “hydrophobic gap” have been discussed in the literature intensively in the last ten years, although they still remain very controversial.37,38,40–43 Due to the existing electric field at Epmm, the water layer demonstrates a higher level of order than at Epzc. However, due to minimized Pt–H2O interaction and surface detached water molecules, Epmm could not be considered as a point of minimum entropy. When a water molecule is completely detached from the surface, the electric field increases due to the negative surface charge of Pt atoms and the drop in dielectric constant. The consequence is a local attraction of protons resulting in an increased surface concentration. At potentials more negative than Epmm, the probability to form an Had-intermediate becomes significant (in the inset of the CV in Fig. 1, hydrogen atoms are shown as linked to the surface with blue lines/bonds).
From the analyzed potential-induced interfacial structural changes (Fig. 1(1)–(4)), we can conclude that if we continue with negative polarization, we will reach a potential at which the surface is highly populated with Had, which is right before Had recombination starts. The potential region is marked as Nr.5 in the CV in Fig. 1. The concentration of protons in the interfacial region is now significantly enlarged. Simultaneously, at that potential, surface water layers exhibit the highest level of ordering. In the whole manuscript, we refer to it as the potential of minimum entropy (Epme) for the negatively charged surface. At potentials at which Had starts to recombine to form hydrogen molecules, the entropy has to increase again. Namely, generated hydrogen molecules diffuse out from the double layer via Brownian motion, creating a distortion in the interfacial structure of the hydrogen-bonding network that interconnects water molecules and protons. It becomes evident that in the region between Epzc and Epme, the entropy of the double layer is gradually reduced from a maximum to a minimum. Under the influence of the electric field, thermal motions are drastically minimized allowing surface water molecules to become ordered.44 The solvation shell of each proton in the double layer is now partially re-arranged, thus influencing the entire hydrogen bonding network.39 Our assumption is that Had in the transition state bonds to surface water molecules (cf. inset Fig. 1). To further understand intermediate Had formation and its recombination, it is essential to address the behavior of interfacial water under the influence of the electric field generated by the excess surface charge at the interface. The latter is connected to the potential difference between Epzc and Epme. Under the influence of an external electric field, interfacial water molecules have the tendency to orient themselves with the direction of the dipole moment opposite to the direction of the electric field, assuring a configuration with minimum energy. This configuration is achieved via a significant rearrangement of the hydrogen bonding network and the establishment of a more rigid structure in comparison to bulk water. It is suggested that the interfacial water layers have a structure similar to the structure of ice.45,46 Besides forming an ice-like structure, there are several logical consequences stemming from the exposure of interfacial water molecules to the electric field generated by the excess surface charge at the interface:
(1) the existing electric field and the negative electrode surface charge result in a positive excess surface concentration of protons;39
(2) a positive excess surface concentration of protons implies that protons in the double layer are only partially solvated,39 forming rather transition forms between Zundel cations (H5O2+) and Eigen cations (H9O4+);47
(3) the activation energy for electron transfer from the negatively charged surface towards partially solvated protons is lower compared to fully solvated protons;39
(4) due to the strong polarization of water molecules at fields of 1 GV m−1, which is the typical field strength in the double layer, dielectric saturation could occur.48 Consequently, oriented and aligned water molecules can be also a proton source due to enhanced water autoprotolysis.49
The consequential result of this reasoning is that water should act as a promotor of the initial (Volmer) elementary step in the HER sequence, it is not just a passive solvent. The role of water as promotor for electrocatalytic reactions is a relatively unexplored research area.8,50 At and below Epme (Fig. 2a), interfacial water molecules behave as a proton source that will be adsorbed on-top of the Pt surface forming Had. Protons in the form of hydronium ions that are located above the first water layer mutually attract oxygen atoms from the water molecules of the first water layer and weaken their Had–O bond (Fig. 2(1)) and together with the negatively polarized Pt-surface, they favour Pt–Had formation. When the bond between Pt–Had and the surface water molecule is cleaved, Pt–Had is not particularly stable due to the repulsion from strongly adsorbed Hupd, thus neighboring Had will have the tendency to recombine and form molecular H2 (Fig. 2(2)). From this analysis, it seems that the Epme or a potential that is slightly more negative could be the potential that triggers the HER.
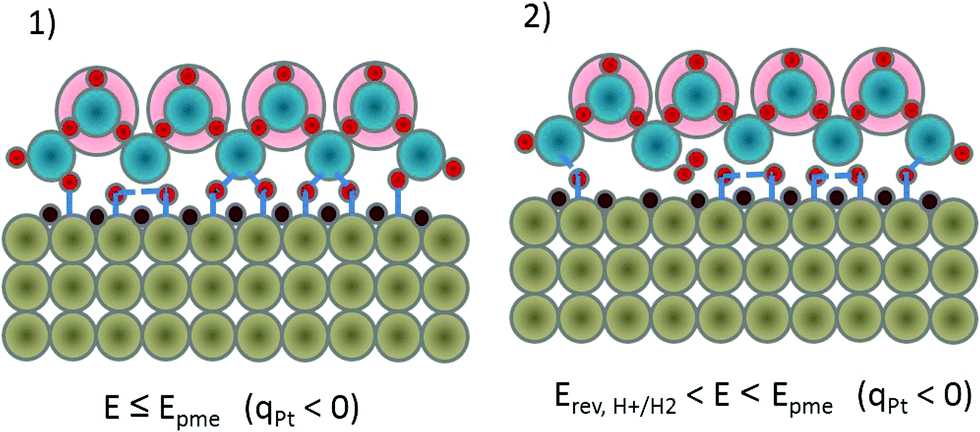 | ||
| Fig. 2 Schematic illustration of interfacial changes at the Pt/electrolyte boundary during indicated potential modulations, relevant for Had-intermediate recombination/hydrogen molecule formation. Color code: Pt atoms – grey, Hupd – brown, oxygen – blue, hydrogen – red, hydronium ions – pale red background and anions – green. | ||
Dynamic interfacial structural changes at the potential of minimum entropy
At potentials close to Epme, the interfacial structure goes through dynamic transformation including: (1) an increase in Pt–Had coverage that should take place rapidly, reaching coverages of at least 0.5 in the potential range of only a few millivolts,37 and (2) the fast establishment of Had that will intensify the high arrangement of the complex hydrogen bonding network in the ice-like bilayer, where some rotational modes/motions of water molecules should be drastically limited.Both mentioned phenomena are expected to result in changes in the capacitance of the double layer. Namely, the potential-induced increase in Had coverage results in an initial increase in the adsorptive pseudo-capacitance as well as in a drop in the double-layer capacitance. The high level of arrangement of the ice-like bilayer should result in a drop in dielectric constant and in the double-layer capacitance, respectively. In order to confirm our hypothesis, the double layer capacitance was monitored as a function of the potential, starting from the pure double-layer region (see the cyclic voltammogram in Fig. 1) and going towards the reversible potential for HER. This was done using electrochemical impedance spectroscopy (EIS) in the so-called Mott–Schottky operational mode. As the presence of specifically adsorbed anions could alter the double layer structure and a large amount of protons could hinder expected transient changes in the interfacial region, EIS measurements were done in 1 mM HClO4. While changes in surface charge originating from (non-)specific adsorption could be easily monitored with EIS (e.g. Epzc is usually analyzed in the literature at very low frequencies of a few Hz), water molecule re-orientation is expected to take place at frequencies in the GHz-range. However, in the case of the Epme, it is expected that due to the highly arranged hydrogen-bonding network in the double layer, the frequency at which water molecules in the ice-like bilayer will change orientation due to thermal fluctuations can be orders of magnitude lower than the one of bulk water. This assumption is well-grounded, as shown in Fig. 3a.51 Specifically, the dielectric constant of pure ice is shown to be a function of the frequency of the applied alternate electric field. In the region of 1–100 kHz, the dielectric constant of ice drops significantly from the so-called static value (for ice ε = 120) towards the so-called dynamic value that is more than an order of magnitude lower (the opposite happens with the conductivity).51 The result of this experiment in the case of Pt is shown in Fig. 3b, where a drastic drop in the capacitance at the potential of 0.04 V vs. RHE can be observed.
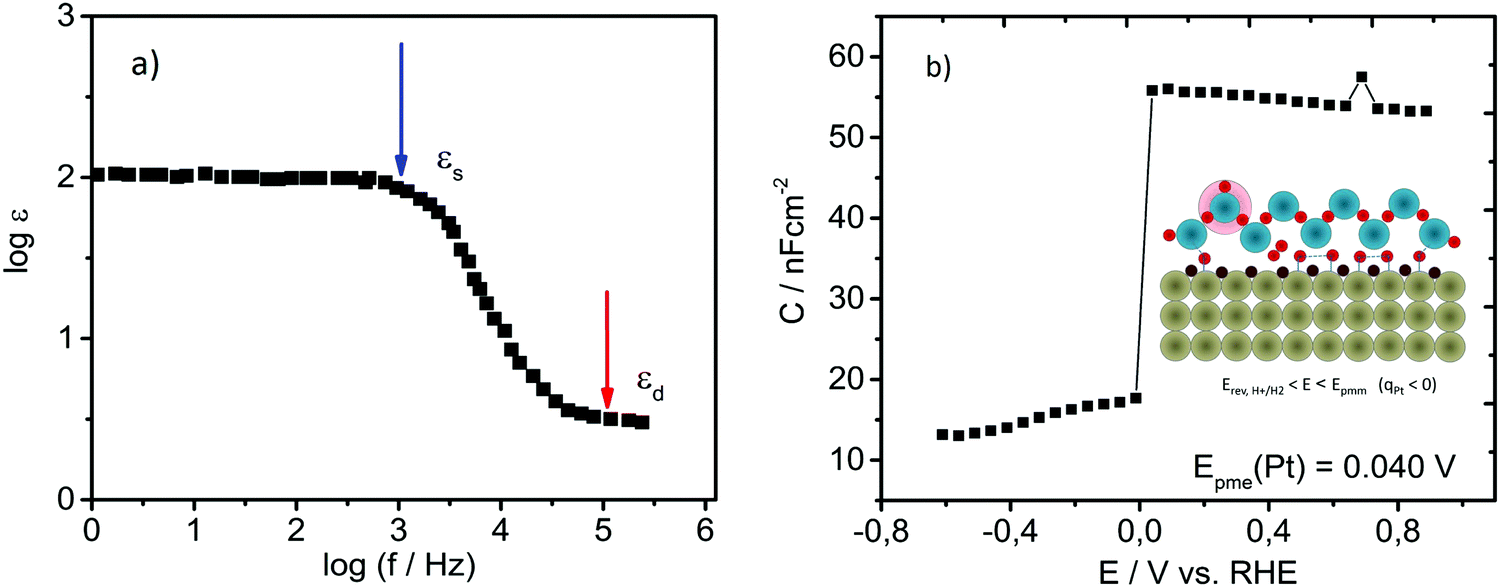 | ||
Fig. 3 (a) Illustration of the Debye dispersion (frequency dependence on relative permittivity) for pure ice. Replotted using data from ref. 51. (b) Detection of potential of minimum entropy (Epme) for Pt using electrochemical impedance spectroscopy in Mott–Schottky operational mode; the double layer capacitance was monitored as a function of applied potential using a linear sweep with imposed sinusoidal perturbation recorded at a frequency of at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz in 1 mM HClO4 with a step of 50 mV. Inset: Scheme of the double layer at Epme (similar to Fig. 2(1)) in diluted electrolyte, where protons from the interfacial region are depleted due to Had formation. 000 Hz in 1 mM HClO4 with a step of 50 mV. Inset: Scheme of the double layer at Epme (similar to Fig. 2(1)) in diluted electrolyte, where protons from the interfacial region are depleted due to Had formation. | ||
The lowest frequency at which this effect can be observed is 10 kHz. At such a moderately high frequency, the mass transport of protons into the double layer is inhibited. Consequently, the value of the double-layer capacitance is very low (approximately three orders of magnitude lower than the values recorded at low frequencies; check Fig. S1 in the ESI†). After discharge of protons that were already in the double layer, the capacitance of the double layer drops. It seems that Had intermediate formation and consequent coverage growth are detectable at the frequency at which the Debye relaxation of water molecules in real ice occurs, suggesting unequivocally that Had displays an integral character of the ice-like bilayer. It is difficult to distinguish whether the formation of the specific form of the ice-like bilayer is a prerequisite for Had formation or vice versa. In our understanding, it is more probable that restructuring of an ice-like layer with a complex hydrogen bonding network is a sluggish process in comparison to the transport of protons in the double layer including on-top adsorption at the Pt-surface. Results based on surface-enhanced infrared absorption spectroscopy (SEIRAS) expressed skepticism towards the existence of hydrogen bonds between Had and the ice-like bilayer,37 however EQCN results obtained by Jerkiewicz show an evident increase in mass on the Pt-surface at potentials more negatively than Epmm (around Epme), indicating clearly that water bonds to the surface. Due to steric reasons, these bonds are more likely to be established through Had instead of through the bare Pt-surface.
To test our hypothesis and to validate the general character of our descriptor, similar experiments were conducted for six other metals (Pd, Ir, Rh, Ru, Au and Cu), as shown in Fig. 4. For all studied metals, a similar behavior can be observed. It seems that the double layer capacitance drops quickly with potential, as a consequence of the facile increase in Had coverage. At approximately 50 mV more positive than Epme, the Had coverage spans from 0.44 for Cu up to 0.68 for Pt. Importantly, the coverage cannot be directly correlated with the adsorption energy, due to the unknown impact of non-covalent interactions on the interaction parameter in the adsorption isotherm. However, it is straightforward that Had formation proceeds in a very small potential range of a few tens of mV. Evidently, Epme changes depending on the electronic structure of the metal. A more positive Epme compared to Pt is only shown by Pd (0.090 V vs. RHE). Ir, Rh and Ru have very similar values between −0.010 and −0.012 V while Au and Cu have much more negative values of Epme, namely −0.31 and −0.41 V vs. RHE, respectively. From Fig. 4, it can be concluded that Epme is unique for each metal characteristic interfacial parameter.
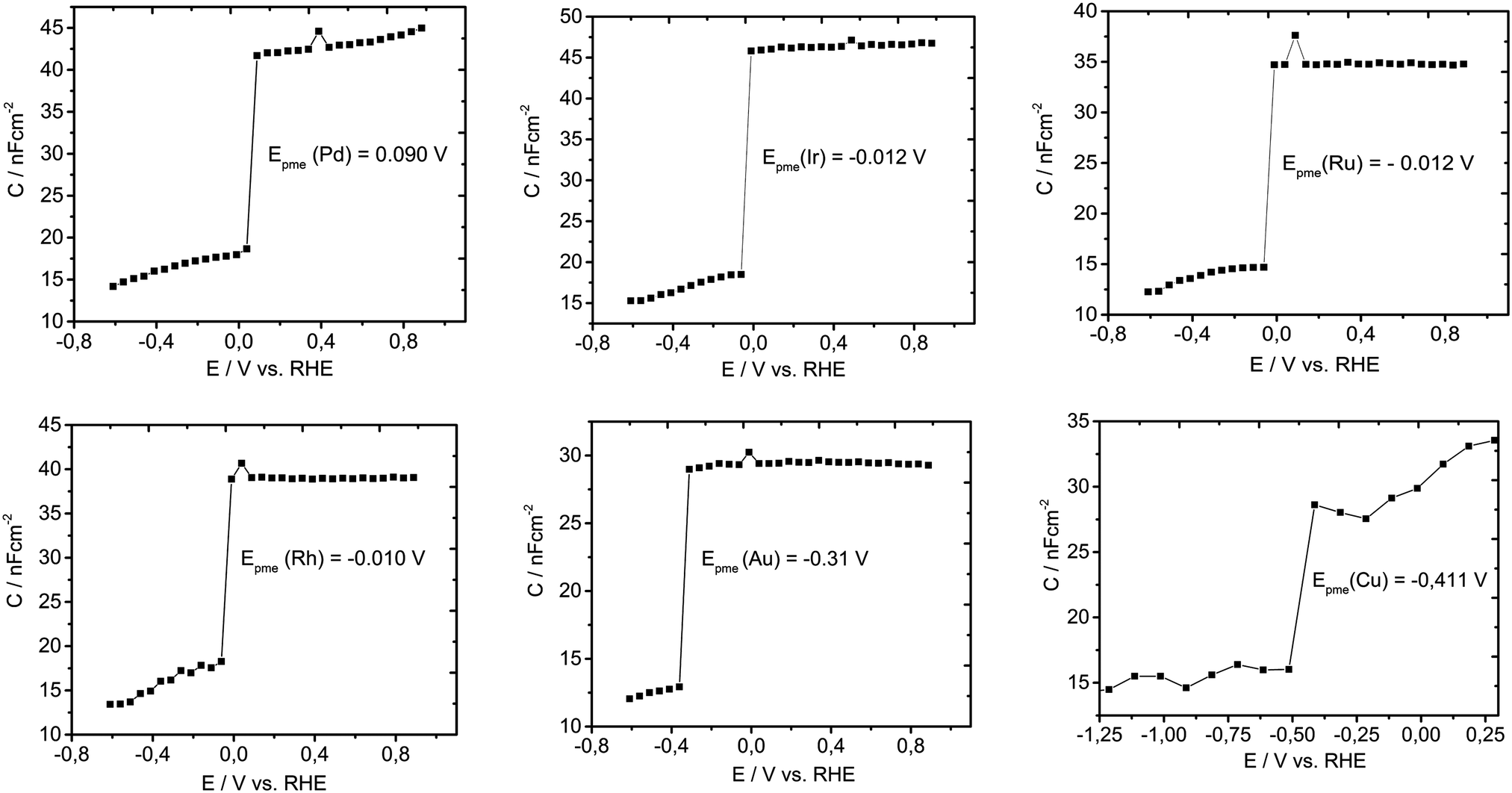 | ||
| Fig. 4 Detection of the potential of minimum entropy (Epme) for six different metals (Pd, Ir, Ru, Rh, Au, and Cu) using electrochemical impedance spectroscopy in Mott–Schottky operational mode; double layer capacitance was monitored as a function of the applied potential using a linear sweep with the imposed sinusoidal perturbation of 10 mV recorded at the frequency of 10000 Hz in 1 mM HClO4 with a step of 50 mV, with the exception of Cu where the step was 100 mV. | ||
Entropy driven reaction – a pathway for minimizing the activation energy
If we re-assess the widely accepted phenomenology of the “volcano” plot for the HER, it seems that the best catalysts are those for which the Gibbs free energy of adsorption is zero (ΔGads = 0).20,21 This implies that at the top of the “volcano,” the adsorption energy (enthalpy) is equal to the adsorption entropy. Putting this into relation with a computational study of Norskov,18 where ΔGads = 0 corresponds to the enthalpy of adsorption of −0.24 eV, we conclude that for an optimal catalyst, adsorption is driven by an entropic change of −0.24 eV or more precisely by a drop in the entropic term of 0.24 eV. For example, for Pt, the enthalpy of adsorption was accurately estimated to be around −0.33 eV in a computational study by Norskov et al.18 and −0.34 eV in an experimental study by Zeradjanin et al.23 The Gibbs free energy of adsorption was found to be around −0.1 eV while the entropic contribution to adsorption was −0.24 eV. A key contribution to the adsorption energy comes from the adsorption entropy. Therefore, it is suggested that reordering/restructuring in the double layer that happens more positively from the reversible potential of hydrogen evolution could reduce the activation energy. Therefore, the reaction rate is related to a change in the entropy of the double layer determined by a potential difference between Epzc and Epme. Consequently, Epme should be an indicator of the HER onset and should be related to the exchange current for the HER, as shown in Fig. 5a. Intriguingly, the slope of the Epme – logj0 dependence is around 104 mV dec−1, which is very similar to the slope of the ΔGact – logj0 dependence plotted by Schmickler et al.36 (Fig. 5b), where ΔGact was calculated by taking into consideration the solvent reorganization energy during HER. This was surprising due to the fact that on the experimentally tested metals, the rate determining steps vary (e.g. Volmer discharge step vs. Tafel chemical recombination step) or even different mechanisms could appear (the Heyrovsky electrochemical recombination step instead of the Tafel step).26,52 From Fig. 5b, it can be observed that Cu, Au, Ir, Rh, and Pt almost perfectly fit the plotted linear dependence while Ru and Pd slightly deviate. Pd has a more positive Epme than Pt, which suggests that Pd should have a higher exchange current than Pt, which was indeed reported in the literature.53 At the same time, Ru has a slightly lower exchange current than Ir and Rh, even though the latter both have an almost identical Epme. This can be a consequence of the thin oxide formed on Ru, which is highly challenging to remove.
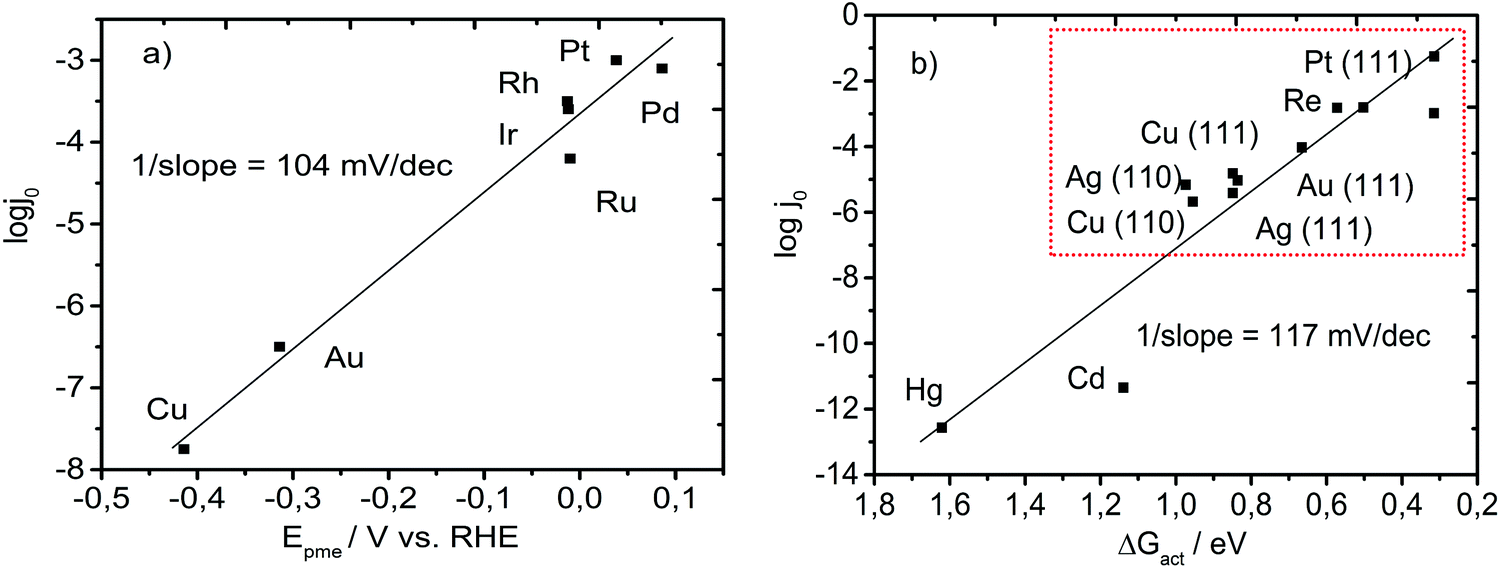 | ||
| Fig. 5 (a) Logarithm of the exchange current density taken from ref. 24 as a function of Epme obtained experimentally in our laboratory. (b) Logarithm of the exchange current density as a function of the theoretically calculated free energy of activation taking into consideration the reorganization energy of the solvent (replotted using data from ref. 36). The red rectangle in (b) indicates transition metals (noble and coinage metals) that were used for the experimental determination of Epme and further analysis. The value of the inverse slope in (b) is obtained by dividing the activation energy with the Faraday constant with an assumption of one electron exchanged in the rate determining step. | ||
It seems that Epme captures very well the terminal point in the entire activation process, or in other words, it seems that at the potential at which an intermediate Had is formed, most of the energy necessary for the activation process is supplied. Importantly, it can be noticed that Epme is easily obtained using transient electrochemical measurements (Fig. 4). Epme is a new interfacial catalytic descriptor that indicates a novel and more realistic view of HER, compared with the conventional one based only on the mere catalyst/intermediate bond energy.
Optical properties of the electrified interface during metal–proton interaction
If we make a simple analogy to pure ice, it would be expected that the dielectric constant of the surface water bilayer increases while the density and the refractive index should decrease. However, from the theory of Booth, the dielectric constant of water exposed to the electric field strength of around 109 V m−1 drastically drops.54,55 The density of pure water exposed to an electric field, according to molecular dynamics studies, should decrease proportionally to the decrease in dielectric constant.56 Nevertheless, we operate in an acidic electrolyte and in a potential range that causes the excess surface concentration of protons. Results of Danielewicz-Ferchmin indicate that for water molecules whose distance from the ion/proton is less than 2 Å, the dielectric constant drops almost an order of magnitude,48 while the density increases proportionally to the excess surface charge.57 It should not be surprising that the “ice”-like structure, induced by an external electric field and multitude of dipoles, behaves quite distinctly from pure ice, induced by the low temperature. It is interesting that for both structures, due to the highly ordered arrangement of a complex hydrogen bonding network in the double layer, similar rotational modes/motions of water molecules are drastically inhibited in the same range of AC frequencies. What remains unknown is how the refractive index of an “ice”-like structure changes under potential control in the potential region of interest.In order to get a better indication of the interfacial structural changes, including changes in refractive index, we employed in situ ellipsometry where a potential was applied and an AC field was superimposed. The measured signal originates from the change in polarization as the incident light interacts with the electrified interfacial structure. The result is shown in Fig. 6a, where the ellipsometric parameter Δ is monitored as a function of electrode potential at various AC frequencies. The ellipsometric parameter Δ (phase shift of incident light) at 1 and 100 Hz appears to be nearly constant in the entire potential range. However, at 100 kHz, the ellipsometric parameter Δ changes by almost −0.8° compared to the open circuit potential (OCP), when Pt is polarized down to −100 mV vs. RHE. This indicates that the formed interfacial layer has a higher refractive index compared to the reference OCP, but only under dynamic conditions (100 kHz). Noticeable is a sudden change of Δ at potentials of approximately 0.35 V vs. RHE, which responds to the initiation of the Hupd formation as well as to Epzc. The very small absorption coefficient of Hupd58 suggests that the signal originates rather from the reorientation of surface water molecules at Epzc. Despite the existence of Hupd at this potential, we did not expect the presence of an ice-like bilayer structure. However, the behaviour at 100 kHz could suggest that the ice-like structure in the double layer at room temperature exists to a certain extent even at potentials close to Epzc, what was also postulated by computational models.59 Interestingly, after a sudden change, the ellipsometric parameter Δ remains relatively constant, despite the continuous increase in the Hupd coverage. This behaviour suggests that around Epzc, the water orientation on Pt changes fast, as previously reported by Iwasita et al.60 At the same time, the formed water layer has a structure with a higher refractive index than the structure that existed in the double layer region at potentials more positive than Epzc. From this perspective, it seems that the origin of the increase in refractive index at the interface at potentials of 0.35–0.40 V vs. RHE can be ascribed to water reorientation. Changes in proton concentrations in the double layer influence the refractive index only slightly. It is intriguing that the change in Δ is observable only at 100 kHz, which corresponds to the frequency at which the dielectric constant of pure ice drops by more than an order of magnitude (see Fig. 3a). As stated previously, this suggests that the ice-like structure and real ice have some similar rotation modes, which freely rotate/reorient up to the frequencies of the alternate electric field of 10–100 kHz. In this frequency range, they predominantly “freeze”. Importantly, while the “freezing” of the rotation/reorientation modes in the case of pure ice causes an increase in the conductivity of 2–3 orders of magnitude, it seems that in the case of an ice-like structure, the “freezing” of the rotation/reorientation modes keeps the double layer region in a configuration that favors interfacial proton and electron transfer. The exact structure of the ice-like bilayer that allows facile proton and electron transfer remains unknown.
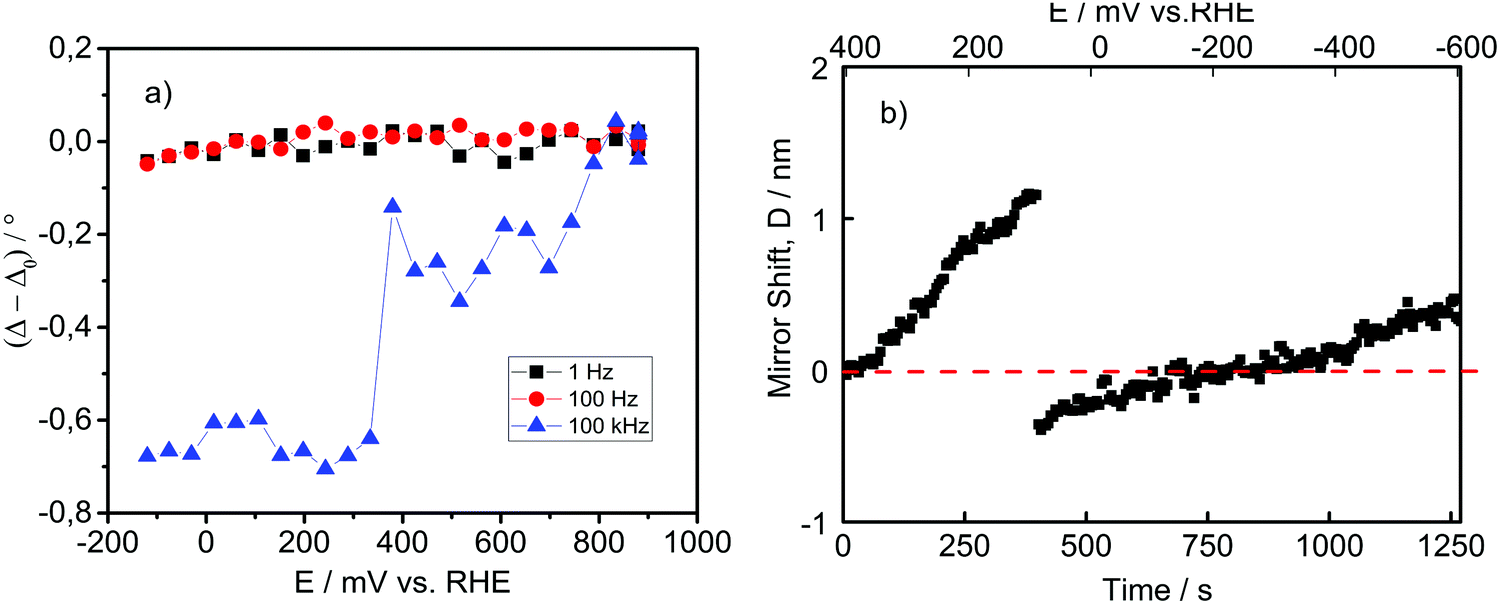 | ||
| Fig. 6 (a) Relative changes in ellipsometric parameter Δ as a function of applied potential using a linear sweep with imposed sinusoidal perturbation recorded at various frequencies in 1 mM HClO4 with a step of 50 mV and (b) EC-SFA (electrochemical surface force apparatus), where the mirror shift is monitored as a function of the applied potential step of 50 mV over a duration of 60 s. | ||
A method that is complementary to spectroscopic ellipsometry that can supply valuable information on potential-dependent alterations at the electrode/electrolyte interface is surface force apparatus (SFA). In general terms, SFA measures the interaction force between surfaces using multiple beam interferometry by monitoring surface separation and surface deformations. One surface is held by a cantilevered spring, and the deflection of the spring is used to calculate the force being exerted.61 Practically, during the electrochemical measurement, white light is transmitted through an interferometer after the interaction with the electrode surface. In this way, it is possible to measure the so-called mirror shift between the working electrode and the reference point as well as nonequilibrium forces due to electrochemical processes. The mirror-shift can be caused by changes in distance between surface and reference points (e.g. dissolution or deposition of metals), by changes in optical properties (e.g. change in reflectivity due to electrode potential-altered electron density of surface atoms) or a combination of changes in distance and changes in optical properties (e.g. oxide growth etc.).62 In our case, we do not expect a change in the position of the outer Pt surface, because we apply a potential in the direction of the HER. In Fig. 6b, the experimental results using EC-SFA are shown. After 70 s at OCP, potential steps of 60 s from the beginning of the Hupd region (around 400 mV vs. RHE) down to −600 mV vs. RHE were applied. The experiment was focused on the mirror shift (D) after the end of each pulse. The temporal changes of D were not on this occasion the focus of the investigation, but the focus was strictly on the changes in D as a function of the applied potential. Interestingly, while in the case of in situ ellipsometry, AC perturbation was necessary to observe a change in spectral features, EC-SFA did not give a response to AC perturbation, but rather it gave a response to the DC potential pulses. Fig. 6b shows the change in mirror shift with the applied potential, which was corrected for the thermal drift.
On first sight, it is unexpected that the Pt-surface exhibits a positive mirror shift with respect to the reference point (no transparent oxide growth, dissolution etc.). We assigned this observation to the low absorption coefficient of Pt–Hupd. From the qualitative point of view, it is possible that protons are adsorbed/absorbed beneath Pt surface atoms,28 causing strain in the subsurface region of Pt.63 At the same time, surface compression for more than 1 nm is not likely. The mirror-shift increase is almost linear from 400 mV vs. RHE, where Hupd formation starts, to a potential of approximately 50 mV vs. RHE, which is similar to Epme. At approximately 50 mV vs. RHE, the mirror shift drastically changes. In fact, after 50 mV vs. RHE, where the maximum positive value of 1.2 nm is reached, the mirror shift suddenly drops to −0.2 nm. Thereafter, the mirror shift again starts to increase linearly, but this time with a lower slope. This sudden change in mirror shift suggests a drastic dynamic change of the Pt surface. One of the plausible explanations for this could be that Hupd is actually transformed into Had by surface diffusion.
Important to notice is that after 400 mV vs. RHE, where we enter the Hupd region, atomic hydrogen gets adsorbed, which is something that reduces the work function of Pt-surface atoms.23 A drop in the work function coupled with Hupd formation results in a higher electron concentration in the conductive band of Pt surface atoms. In the past, IR experiments on Pt during Hupd formation revealed a baseline shift, attributed to an increase in reflectivity of the electrode surface.35 This should influence the detection on the interferometer due to altered reflectance. The continuous change in light reflection during cathodic polarization in the Hupd region in comparison to the “pure metal” part in the double layer region can have as a consequence a mirror shift. Taking this into consideration, we expected to see spectral changes inside the Hupd region also during ellipsometric measurements, but we observed spectral changes only at the initiation of Hupd, as explained previously. It seems that ellipsometric measurements indicate a dynamic structural change caused by water layer configuration, while the EC-SFA signal originates more from adsorption at the Pt-surface. Namely, a change in the ellipsometric signal is detectable at potentials close to Epzc where Hupd is initiated at an AC frequency of at least 100 kHz, evidently influencing interfacial water reorientation. On the other hand, EC-SFA exhibits a potential-dependent linear mirror shift in the entire Hupd region, indicating the formation of an interfacial layer whose reflectivity is continuously altered with potential. The disruptive point in the EC-SFA mirror-shift is approximately at a potential where Hupd formation was completed and Had formation is favoured. A plausible explanation for these mirror shift changes could originate from the previous explanation of Epme. Namely, a gradual completion of the Hupd layer is coupled with an increase in reflectance. As a consequence, the apparent mirror shift, caused by a change in reflectance, reaches a maximum around 50 mV vs. RHE. At that point, due to repulsive interactions between Pt–Hupd and interfacial water molecules (see Fig. 1(3) and (4)), a significant detachment of water molecules from the surface is expected. Knowing that this occurs at a potential substantially more negative than Epzc, the negative surface charge of Pt-atoms is very high. Therefore, protons will be rapidly attracted by Pt-atoms via on-top adsorption and neutralize this charge in the process of Had intermediate (or Hopd) formation. This will cause a drastic change in the reflection manifested through a sudden change in mirror shift. After this point, with further cathodic polarization, the coverage of Had will continuously grow, which will lead to a further linear increase in mirror shift.
Rationalizing electrocatalytic activity trends beyond the Sabatier principle
Considering that metals with the same adsorption intermediate energy do not necessarily give similar HER rates, we are now able to rationalize and delineate why Sabatier's principle is not sufficient to explain the electrocatalytic activity. In the very beginning of the reaction sequence, the Pt–Hupd bond strength dictates to what extent Hupd will fill the Pt hollow-sites. The potential-dependent completion of the Pt–Hupd layer, which neutralizes the partially negative surface charge at the Pt surface, causes simultaneous repulsion of water molecules from the Pt surface. Water molecules detached from the Pt-surface will again expose the negatively charged Pt-surface, which will attract protons. As a result, protons will be transported rapidly towards the surface. This is the reason why after the detachment of water molecules from the Pt-surface, the on-top Had intermediate will be formed almost instantly. The repulsion between M–Had and strongly bound M–Hupd dipoles as well as weakening of the Had–O bond between surface water molecules and Had at the metal surface will finally cause the recombination of two Had into one H2 molecule. Important to notice is that the exact location of Hupd remains still unknown while the possibility that Had is formed by surface diffusion of Hupd from Pt hollow sites towards on-top locations cannot be excluded. Similarly, recombination of two neighbouring Had and the formation of hydrogen molecules possibly require a weakening of the Had–O bond between surface water molecules and Had at the metal surface, although spectroscopic proof for this is lacking.In the entire activation process, there is an interplay and fine tuning between several effects. From Sabatier's principle, it is known that the ideal M–Had bond strength should be around half the H–H bond energy of molecular hydrogen. In the moment of recombination of neighboring Had intermediates and under the influence of repulsive M–Hupd surroundings, the M–Had bond energy should be around 220 kJ mol−1, assuming that the Had–O bond between surface water molecules and M–Had at the metal surface is cleaved. The cleavage of Had–O depends, besides the M–Had bond strength, on the ice-like bilayer structure, which is determined by (1) metal–water interactions, (2) the electric field strength and (3) the concentration of protons in the double layer. According to Conway,64 the proton recombines with a non-bonding electron pair of the water molecule forming H3O+ with an enthalpy of formation of −754 kJ mol−1. The H3O+ cation attracts three additional H2O molecules in the hydration shell and forms a time-averaged H9O4+ cation with an additional stabilization enthalpy of −356 kJ mol−1. Therefore, the high concentration of protons in the double layer, as well as the strong external electric field, will promote partial desolvation of protons in the double layer along with promoting water autoprotolysis. This will be the case especially for those water molecules that are in direct contact with the metal surface, where enhanced cleavage of the Had–O bond will result in M–Had bond formation. From here, it can be stated that the electrocatalytic HER activity is a function of: (1) the M–Hupd bond, which has the role of repulsing the surface water molecules, thus allowing the protons to access the surface, and the role of repulsing the intermediate, and should be as strong as possible; (2) the M–interfacial water interaction, which dictates the initial interfacial water structure and weakens the O–H bond of surface water molecules; (3) the proton–interfacial water interaction at Epzc, which additionally weakens O–H bond strength in surface water molecules; (4) Epzc–Epme potential difference, which dictates the electric field-induced water dipole orientation and controls the proton concentration in the interfacial region, causing additional partial desolvation of protons and deformation of the solvation sphere with larger exposure of positive charge towards the surface, weakening the O–H bond in water molecules that are not in direct contact with the metal surface and weakening of the Had–O bond; and (5) the balance between all four previous parameters will allow the optimal M–Had bond strength. It is evident that the HER mechanism is too complex if one intends to come up with a quantitative model that should explain the impact of the interfacial structure on the reaction rate. At the same time, from a qualitative point of view, it is straightforward that the existence of strongly bonded Hupd (not bonded with optimal strength as classical DFT predicts), a high work function (very positive value of Epzc) and strong to intermediate M–H2O bonds are the reasons why platinum group metals (PGMs) are the most active catalysts for the HER. To replace PGMs with less expensive materials and equally or even more active materials, we will probably need ternary alloys. One component will have to manifest strong adsorption of hydrogen (early transition metals/oxides), the second component will have to manifest strong to intermediate M–H2O bonds to enhance water dissociation, but not to form nonconductive bulk oxide (triad of iron) and a third component will be required that will have a high work function (coinage metals). What remains unknown is how the interfacial water structure depends on the nature of the electrode material and how exactly the structure of interfacial water will promote proton and electron transfer.
EIS experiments in this work indicate that intermediates are formed rapidly at potentials close to Epme. However, intermediate formation is detectable only at an AC frequency that responds to Debye relaxation of the ice-structure. At the same time, in situ spectroscopic ellipsometry indicates that from Epzc and towards more negative potentials, the refractive index of interfacial water molecules is higher than for bulk water. Interestingly, this behaviour was also detectable at the AC frequency that responds to Debye relaxation of the ice-structure. Finally, EC-SFA measurements suggest that after the Hupd layer is completed, an intermediate is formed after the disruption of the double layer structure at Epme. Having all this in mind, we believe that in the potential range from Epzc up to Epme, the double layer entropy is gradually reduced by altering gradually one form of the ice-like bilayer structure into another one. At Epme, this transition is completed along with the formation of intermediates. The proposed concept extends some of the old experimental observations by Trasatti25 as well as some of our recent analysis.7,23 However, it seems to be quite opposite to the concept recently proposed by Koper et al.,65 who postulated that the presence of a strong electric field makes interfacial water molecules more rigid and more difficult to reorganize during the charge transfer, finally inhibiting HER kinetics. Although Koper's notion65 cannot explain the well-established experimental correlation of Trasatti25 between the Epzc and the exchange current density in acidic media, it is not necessarily wrong. It might be possible that an optimal surface charge at the catalytic electrode, which establishes a delicate balance between the activation entropy and the activation enthalpy, is a prerequisite for a minimum in free activation energy, as postulated by Schmickler et al.39 To provide a full response on this matter, it would be necessary to make a detailed comparative study of d-metals in acidic and alkaline media.
From all that was said above, it becomes clear that even without really resolving some old controversial issues (the exact location of Hupd, whether Hupd diffuses at the surface and becomes an intermediate, and whether Had is linked through hydrogen bonds to surface water molecules), we still obtained important insights. It has become evident why a fundamental understanding of catalysis that is based on a single material property (e.g. d-band center) did not provide a significant perspective for advanced catalyst design. A complex quantity like the reaction rate cannot be reduced only to the thermochemistry of adsorbent/adsorbate interactions, or in our case electrocatalyst/intermediate interactions. Somewhat relevant is the connection between material properties, interfacial properties, dynamic interfacial structural changes and the reaction rate.12,14,23
Conclusion
In this work, we set out to identify unifying guidelines to understand the origins of the electrocatalytic activity for the hydrogen evolution reaction. With the intention of resolving the puzzle of the HER mechanism, we asked new questions and we tackled some old controversial issues. On some aspects, it is still difficult to be conclusive; however, we definitely broaden the current perspective on the HER mechanism. It was shown for the first time that activity trends are influenced by several interrelated factors: (1) the M–Hupd bond strength, (2) the M–interfacial water interaction, (3) the proton–interfacial water interaction at Epzc, (4) the resulting Epzc–Epme potential difference and (5) the Pt–Had optimal bond strength. It was demonstrated that the reaction rate is rather linearly dependent on the potential at which these five factors interrelate. A definition was proposed for this potential, namely the potential of minimum entropy. We propose that at this potential, interfacial water bilayer undergoes dynamic transition from one ice-like structure to another at which the complex hydrogen bonding network is in a specific configuration allowing facilitated Had formation. The newly introduced interfacial catalytic descriptor is a multivariant parameter that gives a significant input in understanding the dynamic HER activation process. Importantly, we developed a concept and/or a theory that goes beyond the Sabatier principle and the resulting volcano plots. The given mechanistic insights represent an important advancement in the understanding of electrified interfaces, opening new avenues of thinking and may lead to a general theory of electrocatalysis providing a realistic platform for the design of new catalytic materials.Methods
Electrochemical characterization
To minimize the influence of anions, experiments were conducted in very diluted electrolytes with non-adsorbing or weakly adsorbing anions and in the absence of a supporting electrolyte. Therefore, the electrolyte was 1 mM HClO4 prepared from 70% HClO4 (supra pure, Merck, Darmstadt, Germany) and ultrapure deionized water (ELGA, PURELAB flex system with a resistivity of 18.2 MΩ cm, Celle, Germany). The electrolyte was pretreated by electrolysis over a duration of 30 min at 10 mA cm−2 to minimize the impact of impurities (especially chlorides, but also eventual organic impurities etc.). Electrochemical measurements were performed in a three-electrode home-made Teflon cell system using a Gamry Reference 600 potentiostat/galvanostat (Warminster, PA, USA) with an inbuilt impedance analyzer and rotator. Impedance data were collected using the Mott–Schottky operational mode where capacitance is measured as a function of the applied potential, in the potential range from 0.9 to −0.6 V vs. SHE. The employed AC (alternate current) perturbation was 10 mV using frequencies between 1 and 100000 Hz. All 7 working electrodes (platinum, palladium, rhodium, iridium, ruthenium, gold and copper) were polycrystalline metals (d = 5 mm, MaTecK, Juelich, Germany) with a geometric area of ca. 0.2 cm2, designed as rotating disc electrode (RDE) tips. A rotation speed of 1600 rpm was utilized to minimize diffusion limitations. The electrical equivalent circuit (EEC) for the potential region of interest (from the double layered region up to the reversible potential of HER) is constructed from an analytical model. Details were given previously.66 The counter electrode was a graphite rod while the reference electrode was commercial Ag/AgCl/3 M KCl (Metrohm AG, Herisau, Switzerland) with the potential of 0.210 V vs. standard hydrogen electrode (SHE). The reference electrode was kept in a separated compartment with a cation exchange membrane to minimize the influence of chloride ions on the working electrode.
In situ spectroscopic ellipsometry
In situ spectroscopic ellipsometry experiments were performed using an SE800 spectroscopic ellipsometer (Sentech Instruments, Krailling/Berlin, Germany) working in the wavelength range of 280–810 nm (1.5–4.4 eV). The measurements were carried out during potentiostatic pulses (t = 30 s) in the potential range from 0.9 down to −0.1 V vs. RHE and applying the same pulses with superimposed AC perturbation (see details about AC perturbation in Section 2.1). The electrode potential was controlled by a Compactstat potentiostat (Ivium Technologies, Eindhoven, the Netherlands). The details of the optical-electrochemical cell were described elsewhere.67,68 A Pt sample with a geometric area of 2.5 cm−2, prepared in-house using physical vapor deposition (PVD), was directly mounted in the optical-electrochemical cell, which was equipped with liquid flow connections. Cu tape was used to provide the electric contact to the surface of the sample. A Pt mesh counter electrode and a Ag|AgCl|3 M KCl micro reference electrode (DriRef-2SH, World Precision Instruments, Sarasota, FL, USA) were used to conduct the electrochemical part of the experiment. The electrode potential of the micro-reference electrode has been determined as +0.208 V vs. SHE. The electrolyte (see Section 2.1.) was externally purged with argon and flowed through the cell with a rate of 2 mL min−1 using a peristaltic pump (Ismatec IDEX Health and Science, Glattbrugg, Switzerland). The duration of a single ellipsometric measurement was 24 s. Analysis and interpretation are solely based on the experimentally determined ellipsometric parameter Δ. The complete set of Δ-spectra was first deionised by singular value decomposition filtering. Subsequently, three Δ-spectra recorded initially at the open circuit potential (OCP) were averaged. As layer formation frequently appears as a parallel shift of Δ, and subsequently Δ-values in the spectral region at 416–598 nm were averaged for all measurements. The spectral region was determined to exclude spectral features visible at lower wavelengths and a region with a larger noise level at higher wavelengths. Finally, the potential dependence of (Δ − Δ0), where Δ0 indicates the time-averaged OCP measurements, was plotted.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. Z. and F. L. M acknowledge EU initiative Horizon 2020, in the framework of the project “Eliont” (ERC Consolidator Grant Nr: 772579). The authors acknowledge Claudia Merola and Prof. Markus Valtiner for SFA experiments. Open Access funding was provided by the Max Planck Society.References
- J. Greeley and N. M. Markovic, The road from animal electricity to green energy: combining experiment and theory in electrocatalysis, Energy Environ. Sci., 2012, 5, 9246 RSC.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Oxygen Electrochemistry as a Cornerstone for Sustainable Energy Conversion, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Powering the planet: Chemical challenges in solar energy utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. O. M. Bockris, The hydrogen economy: Its history, Int. J. Hydrogen Energy, 2013, 38, 2579–2588 CrossRef CAS.
- J. O. Bockris, A Hydrogen Economy, Science, 1972, 176, 1323 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Energy and fuels from electrochemical interfaces, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- A. R. Zeradjanin, J.-P. Grote, G. Polymeros and K. J. J. Mayrhofer, A Critical Review on Hydrogen Evolution Electrocatalysis: Re-exploring the Volcano-relationship, Electroanalysis, 2016, 28, 2256–2269 CrossRef CAS.
- A. R. Zeradjanin, N. Menzel, P. Strasser and W. Schuhmann, Role of Water in the Chlorine Evolution Reaction at RuO2-Based Electrodes-Understanding Electrocatalysis as a Resonance Phenomenon, ChemSusChem, 2012, 5, 1897–1904 CrossRef CAS PubMed.
- E. Santos, P. Quaino and W. Schmickler, Theory of electrocatalysis: hydrogen evolution and more, Phys. Chem. Chem. Phys., 2012, 14, 11224 RSC.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Volcano plots in hydrogen electrocatalysis – uses and abuses, Beilstein J. Nanotechnol., 2014, 5, 846–854 CrossRef CAS PubMed.
- M. T. M. Koper, Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- A. R. Zeradjanin, Is a major breakthrough in the oxygen electrocatalysis possible?, Curr. Opin. Electrochem., 2018, 9, 214–223 CrossRef CAS.
- O. A. Petrii and G. A. Tsirlina, Electrocatalytic activity prediction for hydrogen electrode reaction: intuition, art, science, Electrochim. Acta, 1994, 39, 1739–1747 CrossRef CAS.
- N. Dubouis and A. Grimaud, The hydrogen evolution reaction: from material to interfacial descriptors, Chem. Sci., 2019, 10, 9165–9181 RSC.
- J. X. Wang, F. A. Uribe, T. E. Springer, J. Zhang and R. R. Adzic, Intrinsic kinetic equation for oxygenreduction reaction in acidic media: the double Tafel slope and fuelcell applications, Faraday Discuss., 2009, 140, 347–362 RSC.
- B. Hammer and J. K. Nørskov, Theoretical surface science and catalysis—calculations and concepts, Adv. Catal., 2000, vol. 45, 71–129 Search PubMed.
- J. K. Nørskov, et al., Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. K. Nørskov, et al., Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- M. T. M. Koper, Activity volcanoes for the electrocatalysis of homolytic and heterolytic hydrogen evolution, J. Solid State Electrochem., 2016, 20, 895–899 CrossRef CAS.
- H. Gerischer, Mechanismus der Elektrolytischen Wasserstoffabscheidung und Adsorptionsenergie von Atomarem Wasserstoff, Bull. Sociétés Chim. Belg, 2010, 67, 506–527 CrossRef.
- R. Parsons, The rate of electrolytic hydrogen evolution and the heat of adsorption of hydrogen, Trans. Faraday Soc., 1958, 54, 1053 RSC.
- E. Santos, A. Lundin, K. Pötting, P. Quaino and W. Schmickler, Model for the electrocatalysis of hydrogen evolution, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235436 CrossRef.
- A. R. Zeradjanin, et al., Balanced work function as a driver for facile hydrogen evolution reaction – comprehension and experimental assessment of interfacial catalytic descriptor, Phys. Chem. Chem. Phys., 2017, 19, 17019–17027 RSC.
- S. Trasatti, Work function, electronegativity, and electrochemical behaviour of metals, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- S. Trasatti, Work function, electronegativity, and electrochemical behaviour of metals, J. Electroanal. Chem. Interfacial Electrochem., 1971, 33, 351–378 CrossRef CAS.
- G. Jerkiewicz, Hydrogen sorption ATIN electrodes, Prog. Surf. Sci., 1998, 57, 137–186 CrossRef CAS.
- A. N. Frumkin and O. A. Petrii, Potentials of zero total and zero free charge of platinum group metals, Electrochim. Acta, 1975, 20, 347–359 CrossRef CAS.
- G. Jerkiewicz, G. Vatankhah, S. Tanaka and J. Lessard, Discovery of the Potential of Minimum Mass for Platinum Electrodes, Langmuir, 2011, 27, 4220–4226 CrossRef CAS PubMed.
- S. Trasatti and R. Parsons, Interphases in systems of conducting phases, J. Electroanal. Chem. Interfacial Electrochem., 1986, 205, 359–376 CrossRef.
- M. Rohwerder and F. Turcu, High-resolution Kelvin probe microscopy in corrosion science: Scanning Kelvin probe force microscopy (SKPFM) versus classical scanning Kelvin probe (SKP), Electrochim. Acta, 2007, 53, 290–299 CrossRef CAS.
- N. Garcia-Araez, V. Climent and J. M. Feliu, Determination of the entropy of formation of the Pt(111)∣ perchloric acid solution interface. Estimation of the entropy of adsorbed hydrogen and OH species, J. Solid State Electrochem., 2008, 12, 387–398 CrossRef CAS.
- N. García-Aráez, V. Climent and J. M. Feliu, Potential-dependent water orientation on Pt(111) stepped surfaces from laser-pulsed experiments, Electrochim. Acta, 2009, 54, 966–977 CrossRef.
- B. E. Conway and B. V. Tilak, Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- B. E. Conway and J. O. Bockris, Electrolytic Hydrogen Evolution Kinetics and Its Relation to the Electronic and Adsorptive Properties of the Metal, J. Chem. Phys., 1957, 26, 532 CrossRef CAS.
- G. Jerkiewicz, Electrochemical Hydrogen Adsorption and Absorption. Part 1: Under-potential Deposition of Hydrogen, Electrocatalysis, 2010, 1, 179–199 CrossRef CAS.
- E. Santos, et al., Hydrogen Electrocatalysis on Single Crystals and on Nanostructured Electrodes, ChemPhysChem, 2011, 12, 2274–2279 CrossRef CAS PubMed.
- M. Osawa, M. Tsushima, H. Mogami, G. Samjeské and A. Yamakata, Structure of Water at the Electrified Platinum–Water Interface: A Study by Surface-Enhanced Infrared Absorption Spectroscopy, J. Phys. Chem. C, 2008, 112, 4248–4256 CrossRef CAS.
- K. Ataka, T. Yotsuyanagi and M. Osawa, Potential-Dependent Reorientation of Water Molecules at an Electrode/Electrolyte Interface Studied by Surface-Enhanced Infrared Absorption Spectroscopy, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- O. Pecina and W. Schmickler, A model for electrochemical proton-transfer reactions, Chem. Phys., 1998, 228, 265–277 CrossRef CAS.
- F. Niu, R. Schulz, A. Castañeda Medina, R. Schmid and A. Erbe, Electrode potential dependent desolvation and resolvation of germanium(100) in contact with aqueous perchlorate electrolytes, Phys. Chem. Chem. Phys., 2017, 19, 13585–13595 RSC.
- F. Niu, M. Rabe, S. Nayak and A. Erbe, Vibrational spectroscopic study of pH dependent solvation at a Ge(100)-water interface during an electrode potential triggered surface termination transition, J. Chem. Phys., 2018, 148, 222824 CrossRef PubMed.
- M. Mezger, et al., On the Origin of the Hydrophobic Water Gap: An X-ray Reflectivity and MD Simulation Study, J. Am. Chem. Soc., 2010, 132, 6735–6741 CrossRef CAS PubMed.
- M. Mezger, et al., High-resolution in situ X-ray study of the hydrophobic gap at the water-octadecyl-trichlorosilane interface, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18401–18404 CrossRef CAS PubMed.
- M. F. Toney, et al., Voltage-dependent ordering of water molecules at an electrode–electrolyte interface, Nature, 1994, 368, 444–446 CrossRef CAS.
- M. F. Toney, et al., Distribution of water molecules at Ag(111)/electrolyte interface as studied with surface X-ray scattering, Surf. Sci., 1995, 335, 326–332 CrossRef CAS.
- J. Carrasco, A. Hodgson and A. Michaelides, A molecular perspective of water at metal interfaces, Nat. Mater., 2012, 11, 667–674 CrossRef CAS PubMed.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, The nature of the hydrated excess proton in water, Nature, 1999, 397, 601–604 CrossRef CAS.
- I. Danielewicz-Ferchmin, Phase Diagram of Hydration Shells in Ionic Solutions, J. Phys. Chem., 1995, 99, 5658–5665 CrossRef CAS.
- E. M. Stuve, Ionization of water in interfacial electric fields: An electrochemical view, Chem. Phys. Lett., 2012, 519–520, 1–17 CrossRef CAS.
- J. Staszak-Jirkovský, et al., Water as a Promoter and Catalyst for Dioxygen Electrochemistry in Aqueous and Organic Media, ACS Catal., 2015, 5, 6600–6607 CrossRef.
- F. Victor, Petrenko. Electrical Properties of Ice, 1993.
- A. Lasia, Hydrogen evolution reaction, in Handbook of Fuel Cells, ed. W. Vielstich, A. Lamm, H. A. Gasteiger and H. Yokokawa, John Wiley & Sons, Ltd, 2010 Search PubMed.
- A. T. Kuhn, C. J. Mortimer, G. C. Bond and J. Lindley, A critical analysis of correlations between the rate of the electrochemical hydrogen evolution reaction and physical properties of the elements, J. Electroanal. Chem. Interfacial Electrochem., 1972, 34, 1–14 CrossRef CAS.
- F. Booth, The Dielectric Constant of Water and the Saturation Effect, J. Chem. Phys., 1951, 19, 391 CrossRef CAS.
- F. Booth, Dielectric Constant of Polar Liquids at High Field Strengths, J. Chem. Phys., 1955, 23, 453–457 CrossRef CAS.
- D. I. Karpov and D. A. Medvedev, Density dependence of dielectric permittivity of water and estimation of the electric field for the breakdown inception, J. Phys.: Conf. Ser., 2016, 754, 102004 CrossRef.
- I. Danielewicz-Ferchmin and A. R. Ferchmin, Water Density in the Double Layer, J. Phys. Chem., 1996, 100, 17281–17286 CrossRef CAS.
- K. Kunimatsu, T. Senzaki, G. Samjeské, M. Tsushima and M. Osawa, Hydrogen adsorption and hydrogen evolution reaction on a polycrystalline Pt electrode studied by surface-enhanced infrared absorption spectroscopy, Electrochim. Acta, 2007, 52, 5715–5724 CrossRef CAS.
- R. Guidelli and W. Schmickler, Recent developments in models for the interface between a metal and an aqueous solution, Electrochim. Acta, 2000, 45, 2317–2338 CrossRef CAS.
- T. Iwasita and X. Xia, Adsorption of water at Pt(111) electrode in HClO4 solutions. The potential of zero charge, J. Electroanal. Chem., 1996, 411, 95–102 CrossRef.
- J. Israelachvili, et al., Recent advances in the surface forces apparatus (SFA) technique, Rep. Prog. Phys., 2010, 73, 036601 CrossRef.
- B. R. Shrestha, T. Baimpos, S. Raman and M. Valtiner, Angstrom-Resolved Real-Time Dissection of Electrochemically Active Noble Metal Interfaces, ACS Nano, 2014, 8, 5979–5987 CrossRef CAS PubMed.
- E. Gileadi, M. A. Fullenwider and J. O. Bockris, The Permeation of Electrolytic Hydrogen through Platinum, J. Electrochem. Soc., 1966, 113, 926 CrossRef CAS.
- G. Jerkiewicz, Comparison of Hydrogen Electroadsorption from the Electrolyte with Hydrogen Adsorption from the Gas Phase, J. Electrochem. Soc., 1996, 143, 1240 CrossRef CAS.
- I. Ledezma-Yanez, et al., Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- D. Koster, A. R. Zeradjanin, A. Battistel and F. La Mantia, Extracting the kinetic parameters of the hydrogen evolution reaction at Pt in acidic media by means of dynamic multi-frequency analysis, Electrochim. Acta, 2019, 308, 328–336 CrossRef CAS.
- Y. Chen, P. Schneider and A. Erbe, Investigation of native oxide growth on zinc in different atmospheres by spectroscopic ellipsometry, Phys. Status Solidi A, 2012, 209, 846–853 CrossRef CAS.
- Y. Chen and A. Erbe, In situ spectroscopic ellipsometry during electrochemical treatment of zinc in alkaline carbonate electrolyte, Surf. Sci., 2013, 607, 39–46 CrossRef CAS.
- M. Valtiner, K. Kristiansen, G. W. Greene and J. N. Israelachvili, Effect of Surface Roughness and Electrostatic Surface Potentials on Forces Between Dissimilar Surfaces in Aqueous Solution, Adv. Mater., 2011, 23, 2294–2299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01108h |
| This journal is © the Owner Societies 2020 |