
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orientation independent vibrational dynamics of lipid-bound interfacial water†
Malte
Deiseroth
 a,
Mischa
Bonn
a and
Ellen H. G.
Backus
*ab
a,
Mischa
Bonn
a and
Ellen H. G.
Backus
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: backus@mpip-mainz.mpg.de
bDepartment of Physical Chemisty, University of Vienna, Währinger Straße 42, 1090 Wien, Austria
First published on 2nd April 2020
Abstract
Zwitterionic phospholipids are one of the main constituents of biological membranes. The electric field associated with the two opposite headgroup charges aligns water molecules in the headgroup region. Here, we study the role of water alignment on the sub-picosecond vibrational dynamics of lipid-bound water. To this end, we compare the dynamics of oppositely oriented water associated with, respectively, a phosphocholine (PC) headgroup and an inverse-phosphocholine with non-ethylated phosphate groups (CP). We find that the dynamics are independent of the water orientation, implying that the vibrational dynamics report on the local properties of the water molecules.
1 Introduction
The main constituents of biological membranes are zwitterionic phosphatidylcholine (PC) lipids, using amphiphilic interactions with the adjacent water molecules to form the naturally occurring bilayer structure.1 Interestingly, the naturally occurring zwitterionic phosphatidylcholine lipid only exists in the PC conformation. The CP, or CPe, conformation, containing a reversed headgroup structure, has to be synthesized and does not occur naturally.2 This raises the question whether there are fundamental differences between the PC and CP(e) conformation making the latter biologically irrelevant. Perttu et al.2 discovered that DPPC and DPCPe liposomes release encapsulated anionic carboxyfluorescein as well as glucose at different rates, suggesting that the configuration of the headgroup charge plays a significant role in determining membrane permeability. Further, it was found that CPe liposomes do not appreciably interact with Ca2+ ions. It was hypothesized that the permeability for Na+ and Cl− might be altered as well. Another important but often overlooked feature of a lipid is its interaction with water itself. Dreier et al.3 have shown that the charge distribution throughout the zwitterionic headgroup determines the orientation of the water bound to the headgroup.4,5 Given the opposite orientation of water bound to PC- and CPe-based lipids, one might expect that the molecular structure of lipid-bound water is different for the two lipids.For oppositely oriented water in contact with, respectively, the negatively charged lipid DPPG and positively charged DPTAP, it has indeed previously been shown that both the structure and vibrational dynamics of water in contact with these headgroups is different. For negative (positive) lipids, the water dipole points towards (away from) the lipid layer, exhibiting an ultrafast relaxation path available only for the positively charged lipid.6–8 In the case of the negatively charged surfactant sodium dodecyl sulfate (SDS) water interface two types of water were reported.9 Given the ubiquitous presence of the lipid/water interface, these kinds of differences in vibrational dynamics – that reflect differences in structure10 – could be important for the functionality of lipid bilayers.
To investigate the role of interfacial water alignment on the water properties, we study the lipid water interaction using sum-frequency-generation (SFG) spectroscopy, an even-order nonlinear process that is almost exclusively interface-specific owing to its selection rules.5,11–13 We use an infrared beam in resonance with the OD-stretch vibration to probe the interfacial (heavy) water (D2O) in contact with the lipids. The OD-stretch vibrational frequency is a marker of the strength of the local hydrogen bond network and the intensity of the SFG signal provides information about the average orientation of the transition dipole moment.14 To study the vibrational dynamics of the water lipid interface we use pump–probe SFG (PP-SFG) spectroscopy, the interface-specific counterpart to infrared (IR) pump–probe spectroscopy.15 With PP-SFG it is possible to study vibrational dynamics at a sub ps time scale and thus the spectral diffusion.16,17 Here, we use this method to investigate vibrational dynamics and spectral diffusion of water interacting with PC and CP monolayers. Our results demonstrate, that despite the opposite orientation, the vibrational dynamics of CP- and PC-bound water molecules are indistinguishable.
2 Experimental section
A pulsed laser beam is produced by an oscillator (Mai Tai, Spectra Physics) with a central wavelength of 800 nm and roughly 50 nm bandwidth. Two Empowers (22 mJ pulse energy at central wavelength of 527 nm) pump a Spitfire Ace optical amplifier, providing a power output of about 10 W at a pulse length of 40 fs with a repetition rate of 1 kHz. The sum frequency signal is generated by overlapping a narrow-band 800 nm visible laser pulse (s-polarized, pulse energy of 20 μJ, central wavelength of 810 nm, incidence angle to surface normal of 70°) with a focused broadband IR beam generated by a Topas-c with DFG stage (p-polarized, pulse power of 7 μJ, FWHM of 350 cm−1, central frequency of 2500 cm−1, incidence angle of 40° to surface normal). The SFG signal has a central frequency of 674 nm and is sent into a spectrometer and detected using an Andor Newton electron-multiplied charged coupled device (EMCCD) camera. The accumulation time of a single spectrum is 100 s. To correct for the intrinsic spectrum of the IR beam, all SFG spectra are normalized to the non-resonant SFG spectrum of z-cut quartz.The narrowband pump beam (p-polarized, ≈90 cm−1 FWHM, 55° incidence angle to surface normal) is generated by doubling the frequency of the 1000 nm idler output of a second Topas-c within a BBO crystal, generating IR pulses at ≈2000 nm. These pulses are overlapped with 800 nm pulses within a LiNbO3 crystal generating the narrowband pump pulses. The infrared pump beam can be tuned between 2350 cm−1 and 2750 cm−1, by tuning the frequency of the doubled idler and adjusting the phase-matching condition at the LiNbO3 crystal. The resulting IR power varies from 5 μJ to 10 μJ depending on the central pump frequency.
To record pump–probe spectra, a chopper blocks every second laser pulse in the pump laser path, and a vibrating mirror separates the pumped and unpumped signal spatially on the CCD camera. With a motorized delay stage the time delay between the pump- and the probe pulse is changed using various step sizes between 50 fs and 6 ps. The total time range is −10 ps to 20 ps where the pump arrives after (before) the probe for negative (positive) times. In total 27 different pump–probe time delay spectra were recorded.
To minimize heating effects from the laser, the sample is rotated at 13.6 rpm with the laser spots on a 55 mm diameter cycle.18 A chiller is used to keep a constant temperature of 20 °C throughout the experiment and the water level is kept constant using a reservoir.
The lipids are obtained from Avanti Polar Lipids and dissolved in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of chloroform (>99%, stabilized with amylene, Fischer Scientific) and methanol (99.99%, Fischer Scientific) at a concentration of 0.43 mmol l−1. To avoid oxidation of the unsaturated lipid molecules, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 2-((2,3-bis(oleoyloxy)propyl)-dimethyl-ammonio)ethyl-ethyl-phosphate, the zwitterionic lipid with the inverted headgroup (DOCPe) are dissolved under a nitrogen atmosphere. D2O (99.9%) is obtained from Euriso-Top and used as received.
:1 mixture of chloroform (>99%, stabilized with amylene, Fischer Scientific) and methanol (99.99%, Fischer Scientific) at a concentration of 0.43 mmol l−1. To avoid oxidation of the unsaturated lipid molecules, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 2-((2,3-bis(oleoyloxy)propyl)-dimethyl-ammonio)ethyl-ethyl-phosphate, the zwitterionic lipid with the inverted headgroup (DOCPe) are dissolved under a nitrogen atmosphere. D2O (99.9%) is obtained from Euriso-Top and used as received.
Lipid monolayers are prepared by dropping DOPC and DOCPe on D2O and have a coverage of 64(6) Å2 per molecule and 77(2) Å2 per molecule at a surface pressure of 112 mN m−1 and 131 mN m−1 for DOPC and DOCPe respectively.3 Details of the monolayer preparation can be found in Table S1 (ESI†). To prevent the lipids from degradation, all sample preparation steps and measurements are performed under N2 atmosphere. The stability of the monolayer was monitored throughout the course of the 12 h data acquisition and an example of a surface pressure measurement is shown in Fig. S1 (ESI†)
3 Results and discussion
3.1 Static spectra
Fig. 1 shows SFG intensity spectra of the DOPC–D2O and the DOCPe–D2O interface and the molecular structure of DOPC and DOCPe in the inset. The broad peak from 2200 cm−1 to 2600 cm−1 originates from the OD-stretch vibration of the interfacial water.19,20 This peak is broadened by a wide distribution of different OD-bond strengths, intermolecular coupling as well as a Fermi resonance with the overtone of the water stretch vibration.21–25 No significant difference can be observed between DOPC and DOCPe in the OD-stretch region of the SFG spectrum. CH-Stretch vibrations of the lipid give rise to the SFG signal between 2800 cm−1 and 2950 cm−1.3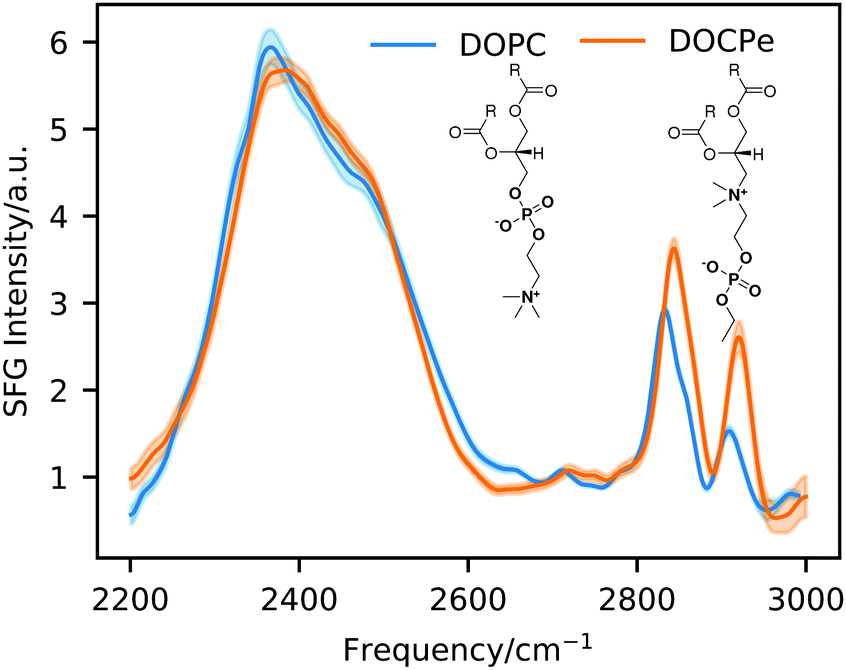 | ||
| Fig. 1 Static SFG intensity spectra of DOPC and DOCPe. The inset shows the molecular structure of DOPC and DOCPe. | ||
Phase-resolved SFG measurements were previously performed on the DOPC– and DOCPe–H2O interface, showing that the orientation of the interfacial water depends on the headgroup of the lipid.3 The CPe headgroup showed a net downwards orientation of the interfacial water dipole moment, indicative of the hydrogen atoms pointing down towards the bulk. For the PC headgroup the situation is reversed, meaning that the interfacial water hydrogen atoms point on average upwards, away from the bulk phase. The phase-resolved experiments showed as well a small blue shift of the Im(χ(2)) spectrum of DOPC compared to the Im(χ(2)) spectrum of DOCPe, indicating slightly weaker hydrogen bonding of water for DOPC.
MD simulations support the hypothesis of the opposing water orientation at the DOPC– and DOCPe–water interface, showing that the orientation of the field in between the choline (+) and the phosphate group (−) determines the orientation of the interfacial water.26–28 Further, there exist an alternative interpretation based on phase-resolved SFG data and MD simulations of the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) water interface.12,29 There, three differently bound types of water were proposed to be responsible for the Im(χ(2)) spectrum of the POPC–water interface. However, the exact molecular mechanism causing the water orientation on zwitterionic headgroups is out of the scope of this work. Instead we study the effect of water orientation on the vibrational dynamics. To do so we use 2D-SFG spectroscopy of the DOPC and DOCPe–water interface.
3.2 2D-SFG Spectra
Fig. 2 shows 2D-SFG spectra of upwards (DOPC) and downwards (DOCPe) oriented interfacial water at four different pump–probe time delays. See Fig. S2 (ESI†) for details on the pump frequency spectrum. The t = 0 ps time axis is calibrated by measuring the bleach on a gold sample and setting t = 0 ps to the maximum bleach. The color (ΔI) denotes the difference of SFG intensity between the pump beam being on (pumped) and the pump beam being off (unpumped). The pumped and the unpumped spectra are corrected for heating effects and normalized to the specific pump power. Details of the heat correction are described in Fig. S3 (ESI†). The signal ΔI is dominated by a combination of bleach and stimulated emission.17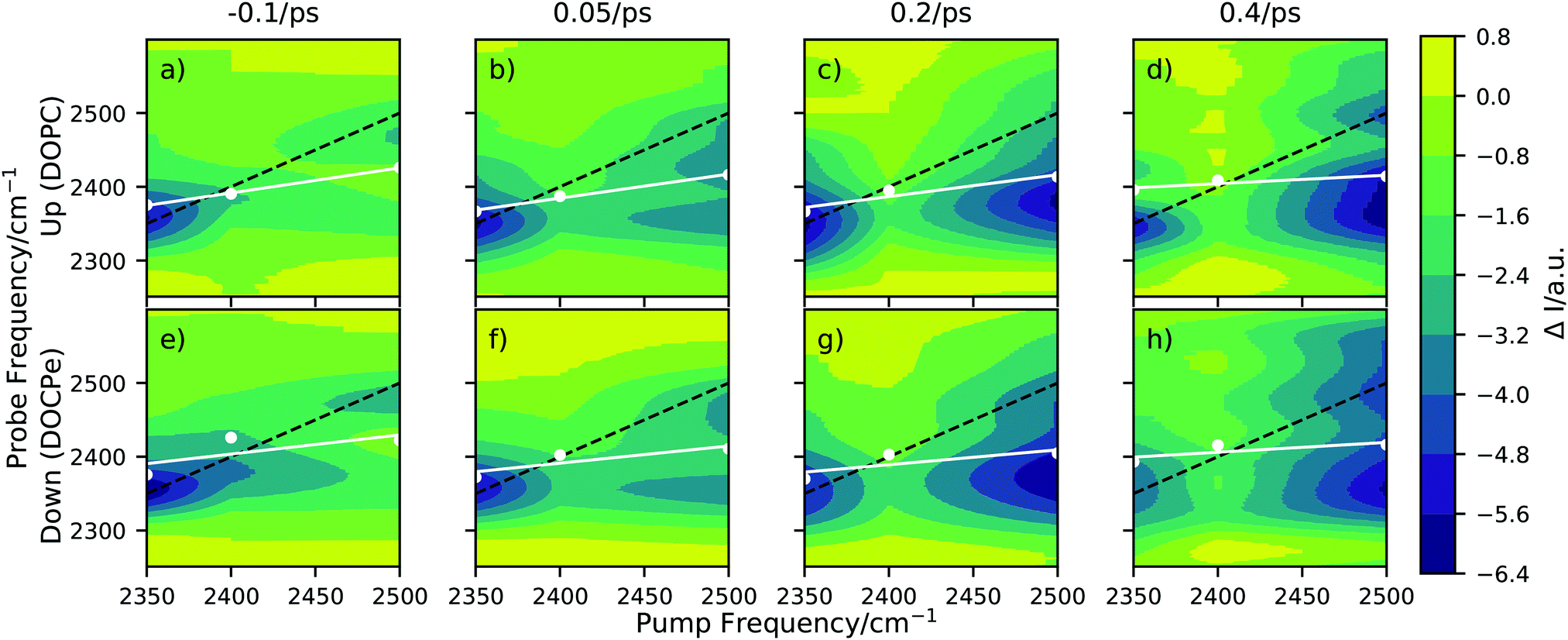 | ||
| Fig. 2 2D-SFG spectra of D2O in the hydrogen bond OD-stretch region for (a)–(d) the upwards oriented water (DOPC), and for (e)–(h) the downwards oriented water (DOCPe), at four different pump–probe time delays. The white dots correspond to a spectral weight frequency. (Details in Fig. S4, ESI†) The white line denotes a linear regression through the spectral weight. The black dotted line indicates the pump = probe frequency diagonal. | ||
A qualitative comparison of the 2D-SFG spectra of upwards (DOPC) and downwards (DOCPe) oriented water shows no significant differences between the two orientations. As such, we discuss in the following the common spectral features of both water orientations. We note that 2D-SFG and 2D phase-resolved SFG have been shown to contain virtually identical information in terms of vibrational dynamics and coupling.17
The amplitude of ΔI on the diagonal is comparable for a pump beam frequency of 2350 cm−1 and 2500 cm−1, but significantly smaller for a pump beam frequency of 2400 cm−1. At a pump beam frequency of 2350 cm−1, both systems show an instantaneous response at the same probe frequency. At a pump beam frequency of 2400 cm−1 the bleach appears at a lower probe frequency of about 2380 cm−1. Pumping at a central frequency of 2500 cm−1 results in an instantaneous signal at a probe frequency of 2500 cm−1, but after less then 0.2 ps additional signal is observable at a 2350 cm−1 probe frequency, hinting towards spectral diffusion or a cross-peak.
This cross-peak-like structure, visible at a probe frequency of 2350 cm−1 if pumped at 2500 cm−1 and small-time delays (0.05 ps), at the lipid water interface has previously been observed for the cetyltrimethylammonium bromide (CTAB) water and SDS–water interfaces.9,30 However, the interpretations are very different in both cases. Inoue et al.30 showed in the case of CTAB that the cross-peak vanishes upon isotopic dilution, and assigned it to the coupling of the water stretch mode with the overtone of the bending mode, i.e. the Fermi resonance. Livingstone et al.9 proposed for the SDS case two types of water, coupling differently with the lipid headgroup and showed that the Fermi resonance cannot account for all observed effects. Work on DPTAP showed only a single broad peak when studied with D2O and HOD.6,8 Overall, the discussion of the origin of the cross-peak is out of the scope of this paper. Instead, the goal of this work is to investigate and highlight the possible effects of interfacial water orientation on interfacial vibrational water dynamics. Because isotopic dilution significantly reduces the signal intensity and thus the signal-to-noise ratio, and does not completely resolve the complication induced by the Fermi resonance, we perform the experiments with D2O and not HOD to ensure best possible sensitivity regarding differences induced by the interfacial water orientation.
On the downside of this, we must deal with complex coupling dynamics, which we account for by analyzing the data in two ways. At first, we investigate the time evolution for the spectral weight frequency. Afterward, we treat the system as a set of coupled oscillators, with central vibrational frequencies at 2350 cm−1 and 2500 cm−1, giving rise to two cross-peaks at their respective frequencies.
Fig. 3 shows the time dependency of the SWL modeled with the convolution of Gaussian function and an exponential decay (eqn (S4), ESI†). Here the Gaussian function accounts for the instrument response function and has a width of σ = 0.16 ps.32 The exponential decay describes the time scale of the spectral diffusion and shows comparable decay times of 0.47 ps ± 0.07 ps for DOPC and 0.5 ps ± 0.1 ps for DOCPe.
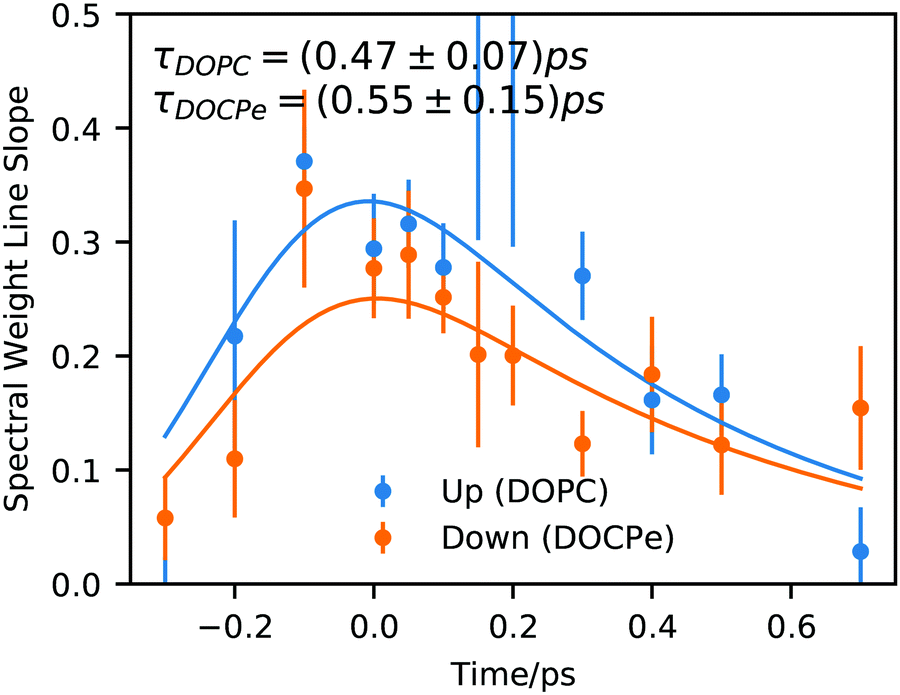 | ||
| Fig. 3 Spectral weight line slope of the interfacial OD-stretch vibration of D2O covered with a monolayer of DOPC and DOCPe as a function of pump–probe delay time. Solid lines show fits of eqn (S4) (ESI†). | ||
Previous studies of DPTAP6–8 showed spectral diffusion of the timescale of 0.35 ps for D2O, 1.1 ps for HOD within the OD-stretch region and 0.75 ps ± 0.35 ps for HOD within the OH-stretch region. Interestingly it seems that a timescale of about 0.5 ps is characteristic for the interfacial spectral diffusion of vibrational energy and is almost independent of the chemical structure of the lipid as well as the isotopic content of the water.
In a study using DPPG as negative and DPTAP as positive lipid, the centerline slope of DPPG and DPTAP were found to be significantly different.6,7 This difference was attributed to an ultrafast, but invisible process occurring on a sub-0.1 ps timescale and was attributed to water orientation in the direct vicinity of water in contact with the lipids. The DPPG/DPTAP study used phase-resolved 2D-SFG, but as explained in ref. 17 the two methods probe fundamentally the same dynamic properties. Instead, our results suggest that the orientation of water in the direct vicinity of DOPC and DOCPe seems to be unimportant for spectral diffusion of the OD-Stretch vibration, because both amplitude and temporal evolution of the SWL are comparable within the uncertainty of the experiment. Hence, a comparison between the results of Tahara and co-workers6,7 and the present work, highlights the importance of the detailed chemical environments that different lipids constitute for the water. The orientational arrangement of water for the DPPG and DPTAP headgroups are caused by the net charge on the head groups. In contrast, the reorientation of water for DOPC and DOCPe is the result of the change of the electric field direction in the headgroup region, and not of a sign change of the net charge of the lipid headgroups. In the following, we switch to the interpretation of Fig. 2 and analyze the 2D-SFG spectra by assuming two coupled oscillators at 2350 cm−1 and 2500 cm−1.
3.3 Dynamics
The traces shown in Fig. 4 are obtained by averaging vertical areas of probe frequencies at 2350 cm−1 ± 30 cm−1, 2400 cm−1 ± 30 cm−1 and 2500 cm−1 ± 30 cm−1 at a given pump frequency and plotted vs. the pump–probe time delay. 30 cm−1 corresponds roughly to the 1σ spectral width of the pump pulse (see Fig. S2, ESI†). If the central pump frequency equals the probe frequency, the trace is labeled diagonal. At a central pump frequency of 2350 cm−1 the cross up labeled trace is centered around a probe frequency of 2500 cm−1 and describes an uphill energy transport process. At the 2500 cm−1 pump frequency, the cross down labeled trace is centered around a probe frequency of 2350 cm−1 and describes a downhill energy transport process. Note that the traces in Fig. 4 contain no heat correction, unlike the 2D-SFG spectra in Fig. 2. Heating effects are accounted for within the fitting model and thus do not need to be subtracted beforehand.8,9,32–34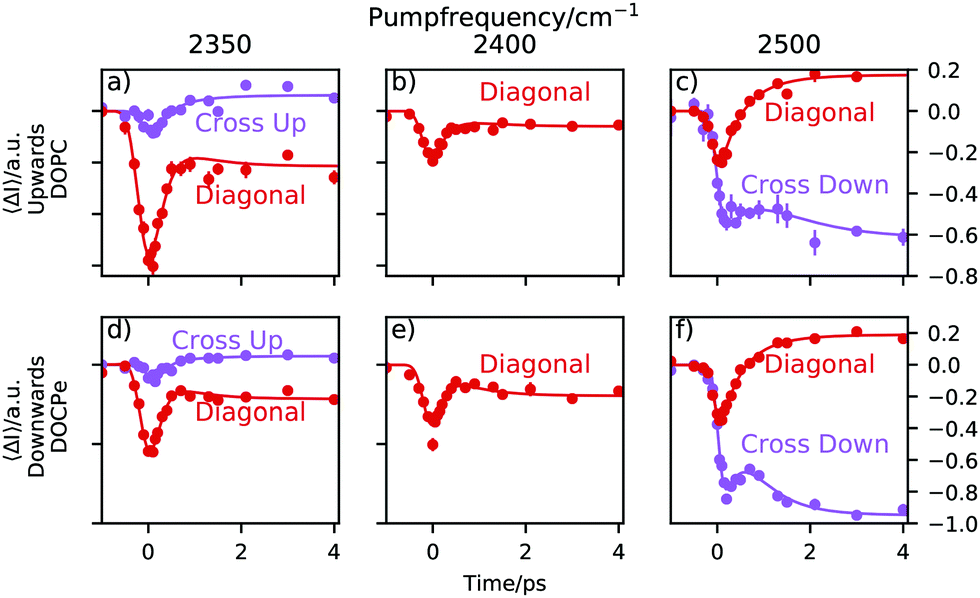 | ||
| Fig. 4 Traces of three different central pump frequencies. The “Diagonal” labeled traces are obtained by averaging areas of Fig. 2, where the central pump frequency equals the central probe frequency. The “Cross” labeled traces have a central probe frequency of 2500 cm−1 if pumped at 2350 cm−1 and vice versa if pumped at 2500 cm−1. The direction of energy transfer is labeled with “Up” for uphill and “Down” for downhill energy transfer. | ||
A trace consists of three sections. For negative times, e.g. the probe pulse arrives at the sample before the pump pulse and consequently, the trace is a flat line at 0. As the pump and the probe start to overlap, the transient signal appears. The point in time, where the signal has reached about 50% of its amplitude is called the lag time and the duration of the signal rise is determined by the instrument response function of the setup. Following, one observes the relaxation of the system. This is due to the relaxation of the pump-induced vibrational excitation and reflects the lifetime of the excited vibrational state. Due to heating effects, the final state after >4 ps is not necessarily the initial state but can be different.
Heat weakens the hydrogen bonds and shifts the spectrum towards higher frequencies. Consequently, the spectral intensity increases at around a probe frequency of 2500 cm−1 and decreases at a probe frequency of 2350 cm−1. This is the cause for a positive ΔI in Fig. 4 at a probe frequency of 2500 cm−1 and negative ΔI at a probe frequency of 2350 cm−1.
At a central pump frequency of 2350 cm−1, a relatively large bleach of the diagonal peak for both water orientations (DOPC and DOCPe) is observed. With a delay of about 0.2 ps we see a much weaker, but still significant bleach in the cross-peak region. After about 1 ps the dynamic processes have largely ended. At a central pump frequency of 2400 cm−1 it is not possible to define a cross-peak, and thus only the diagonal trace is shown. Tuning the pump frequency to 2500 cm−1 reveals an interesting structure in the dynamics. There is significant bleach visible for the diagonal peak, but also for the cross-peak. This hints towards spectral diffusion from the diagonal peak downhill towards the cross-peak. Note that no significant qualitative differences between the observed dynamics for the two opposite water orientations are found. The reproducible bump, visible in e.g. the purple trace in Fig. 4f, makes it clear that at least a two-lifetime model is needed to describe the data.
The traces are fitted with the four-level-model commonly used.8,35,36 The inset of Fig. 5 shows the energy levels postulated by the model. For further details see a previous publication, where the analytical solution to this model was presented.32 The lifetime of the vibrationally excited state amounts to τ1, with the system decaying into an intermediate state. The decay from the intermediate state into a heated ground state is characterized by a lifetime of τ2 for which 0.7 ps has been used.32,37 Using this model, we obtain the lifetime of the excited vibrational states for different interfacial water orientation.
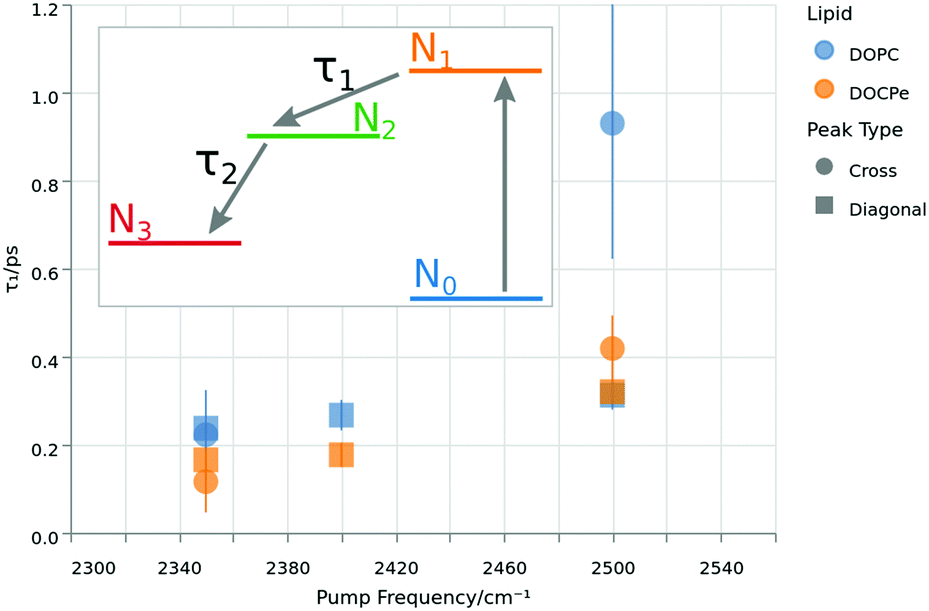 | ||
| Fig. 5 Lifetimes of the excited vibrational state τ1 as function of the pump frequency. The colors denote the lipid, while the shape corresponds to the selected peak. The inset shows the energy levels of the four-level-model used to extract τ1 by fitting the data of Fig. 4. | ||
Fig. 5 shows τ1 lifetimes extracted from the four-level-model. The vibrational dynamics are fastest at a pump frequency of 2350 cm−1 with lifetimes around 0.2 ps. Upon increasing pump frequencies, the lifetimes increase to τ1 greater than 0.4 ps at a central pump frequency of 2500 cm−1. A similar trend has already been reported for the neat water–air, as well as the water–DPTAP interface.8,32,38 At the lowest pump frequency, there is neither a significant difference between the cross and diagonal peaks, nor between the opposing water orientations. At 2400 cm−1 pump frequency, there is a 1σ difference between the two water orientations, and at the 2500 cm−1 pump frequency, there is no significant difference between the lipids for the diagonal peak, but a 1σ difference in τ1 at the cross-peak lifetime for the two water orientations. The overall impression of Fig. 5 could be that DOPC shows a slightly larger τ1 then DOCPe. However one has to keep in mind, that the error bars of Fig. 5 do not account for correlation effects of the fit parameters. A significant improvement in the signal to noise ratio would be needed to consolidate this observation.
Depending on the nature of the coupling, the ingrowth of the cross-peak can be delayed relative to that of the diagonal peak. Indeed such a delay can be found in Fig. 4, if the lag times of the diagonal and cross-peak are compared. Here the lag time is obtained from the fits of Fig. 4, and corresponds to the point in time when the trace has reached 50% of its maximum height. The result is, that if pumped at 2350 cm−1, the uphill cross peak appears about 0.2 ps after the diagonal peak. The situation is similar if pumped at 2500 cm−1, where the diagonal peak appears about 0.1 ps prior to the downhill cross-peak. To exclude instrumental artifacts as the source of the lag time difference, the same analysis has been performed on gold, showing lag times of at most 40 fs ± 10 fs. See Fig. S5 (ESI†). This observation is consistent with near-resonant vibrational energy transfer. However for the upwards (DOPC) and the downwards (DOCPe) oriented water, there is no significant difference in the lag times, again showing that the vibrational dynamics are independent of water orientation for this system.
4 Conclusion
We have investigated the influence of the water orientation on the dynamics of the interfacial OD-Stretch vibration of D2O. 2D-SFG spectroscopy of oppositely oriented water interfaces reveal no significant differences of neither the time dependency of the SWL, nor the vibrational lifetimes of a potential cross or diagonal peak. These results contrast previous findings,6,7 where the water orientation, resulting from charged headgroups, was identified as the cause of dynamical changes. For the present results, the orientation is induced by the lipid headgroup dipolar field. Because the interfacial water vibrational response and dynamics are independent of water orientation at the DOPC– and DOCPe–water interfaces, we conclude that the properties of water cannot explain the biological differences between DOPC and DOCPe on a molecular level.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – BA 5008/3 and the ERC Starting Grant (no. 336679). The authors thank G. Florian and M. van Zadel for engineering, H. Menges for laser maintenance, J. Worm for programming, L. Dreier for the help with sample preparation and S. Das, G. Gonella, M. Grechko for fruitful discussions. This research made use of the MINUIT algorithm39via the iminuit40 Python interface, Matplotlib,41 Altair,42 Vegalite,43 and Numpy.44 Open Access funding provided by the Max Planck Society.Notes and references
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112 CrossRef CAS PubMed.
- E. K. Perttu, A. G. Kohli and F. C. Szoka, J. Am. Chem. Soc., 2012, 134, 4485–4488 CrossRef CAS PubMed.
- L. B. Dreier, A. Wolde-Kidan, D. J. Bonthuis, R. R. Netz, E. H. Backus and M. Bonn, J. Phys. Chem. Lett., 2019, 10, 6355–6359 CrossRef CAS PubMed.
- W. Sung, S. Seok, D. Kim, C. S. Tian and Y. R. Shen, Langmuir, 2010, 26, 18266–18272 CrossRef CAS PubMed.
- X. Chen, W. Hua, Z. Huang and H. C. Allen, J. Am. Chem. Soc., 2010, 132, 11336–11342 CrossRef CAS PubMed.
- P. C. Singh, K.-i. Inoue, S. Nihonyanagi, S. Yamaguchi and T. Tahara, Angew. Chem., Int. Ed., 2016, 55, 10621–10625 CrossRef CAS PubMed.
- K.-i. Inoue, P. C. Singh, S. Nihonyanagi, S. Yamaguchi and T. Tahara, J. Phys. Chem. Lett., 2017, 8, 5160–5165 CrossRef PubMed.
- R. A. Livingstone, Z. Zhang, L. Piatkowski, H. J. Bakker, J. Hunger, M. Bonn and E. H. G. Backus, J. Phys. Chem. B, 2016, 120, 10069–10078 CrossRef CAS PubMed.
- R. A. Livingstone, Y. Nagata, M. Bonn and E. H. G. Backus, J. Am. Chem. Soc., 2015, 137, 14912–14919 CrossRef CAS PubMed.
- J. D. Cyran, E. H. G. Backus, Y. Nagata and M. Bonn, J. Phys. Chem. B, 2018, 122, 3667–3679 CrossRef CAS.
- Y. Shen, Nature, 1989, 337, 519 CrossRef CAS.
- J. A. Mondal, S. Nihonyanagi, S. Yamaguchi and T. Tahara, J. Am. Chem. Soc., 2012, 134, 7842–7850 CrossRef CAS PubMed.
- A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103–145 CrossRef CAS.
- R. Rey, K. B. Møller and J. T. Hynes, J. Phys. Chem. A, 2002, 106, 11993–11996 CrossRef CAS.
- P. Hamm and M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, 2011 Search PubMed.
- P. C. Singh, S. Nihonyanagi, S. Yamaguchi and T. Tahara, J. Chem. Phys., 2012, 137, 094706 CrossRef PubMed.
- E. H. G. Backus, J. D. Cyran, M. Grechko, Y. Nagata and M. Bonn, J. Phys. Chem. A, 2018, 122, 2401–2410 CrossRef CAS PubMed.
- E. H. G. Backus, D. Bonn, S. Cantin, S. Roke and M. Bonn, J. Phys. Chem. B, 2012, 116, 2703–2712 CrossRef CAS.
- Q. Du, R. Superfine, E. Freysz and Y. R. Shen, Phys. Rev. Lett., 1993, 70, 2313–2316 CrossRef CAS.
- N. Ji, V. Ostroverkhov, C. S. Tian and Y. R. Shen, Phys. Rev. Lett., 2008, 100, 096102 CrossRef CAS.
- M. Sovago, R. K. Campen, G. W. H. Wurpel, M. Müller, H. J. Bakker and M. Bonn, Phys. Rev. Lett., 2008, 100, 173901 CrossRef.
- J. Schaefer, E. H. G. Backus, Y. Nagata and M. Bonn, J. Phys. Chem. Lett., 2016, 7, 4591–4595 CrossRef CAS.
- V. Buch, J. Phys. Chem. B, 2005, 109, 17771–17774 CrossRef CAS PubMed.
- H. Torii, J. Phys. Chem. A, 2006, 110, 9469–9477 CrossRef CAS.
- B. M. Auer and J. L. Skinner, J. Chem. Phys., 2008, 128, 224511 CrossRef CAS.
- A. Magarkar, T. Róg and A. Bunker, J. Phys. Chem. C, 2014, 118, 19444–19449 CrossRef CAS.
- T. Ohto, E. H. G. Backus, C.-S. Hsieh, M. Sulpizi, M. Bonn and Y. Nagata, J. Phys. Chem. Lett., 2015, 6, 4499–4503 CrossRef CAS PubMed.
- T. Ishiyama, D. Terada and A. Morita, J. Phys. Chem. Lett., 2016, 7, 216–220 CrossRef CAS PubMed.
- S. Re, W. Nishima, T. Tahara and Y. Sugita, J. Phys. Chem. Lett., 2014, 5, 4343–4348 CrossRef CAS.
- K.-i. Inoue, S. Nihonyanagi, P. C. Singh, S. Yamaguchi and T. Tahara, J. Chem. Phys., 2015, 142, 212431 CrossRef PubMed.
- K. Kwak, S. Park, I. J. Finkelstein and M. D. Fayer, J. Chem. Phys., 2007, 127, 124503 CrossRef PubMed.
- M. Deiseroth, M. Bonn and E. H. G. Backus, J. Phys. Chem. B, 2019, 123, 8610–8616 CrossRef CAS.
- T. Steinel, J. B. Asbury, J. Zheng and M. D. Fayer, J. Phys. Chem. A, 2004, 108, 10957–10964 CrossRef CAS PubMed.
- J. A. McGuire and Y. R. Shen, Science, 2006, 313, 1945–1948 CrossRef CAS PubMed.
- A. Ghosh, M. Smits, J. Bredenbeck and M. Bonn, J. Am. Chem. Soc., 2007, 129, 9608–9609 CrossRef CAS.
- J. Lindner, P. Vöhringer, M. S. Pshenichnikov, D. Cringus, D. A. Wiersma and M. Mostovoy, Chem. Phys. Lett., 2006, 421, 329–333 CrossRef CAS.
- K. Ramasesha, L. De Marco, A. Mandal and A. Tokmakoff, Nat. Chem., 2013, 5, 935–940 CrossRef CAS PubMed.
- S. T. van der Post, C.-S. Hsieh, M. Okuno, Y. Nagata, H. J. Bakker, M. Bonn and J. Hunger, Nat. Commun., 2015, 6, 8384 CrossRef CAS PubMed.
- F. James and M. Roos, Comput. Phys. Commun., 1975, 10, 343–367 CrossRef.
- Iminuit team, Iminuit – A Python Interface to Minuit, https://github.com/iminuit/iminuit, accessed 2019-02-28.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- J. VanderPlas, B. Granger, J. Heer, D. Moritz, K. Wongsuphasawat, A. Satyanarayan, E. Lees, I. Timofeev, B. Welsh and S. Sievert, J. Open Source Software Process, 2018, 3, 1057 CrossRef.
- A. Satyanarayan, D. Moritz, K. Wongsuphasawat and J. Heer, IEEE Trans. Visual. Comput. Graph., 2017, 23, 341–350 Search PubMed.
- T. Oliphant, NumPy: A Guide to NumPy, USA, Trelgol Publishing, 2006, http://www.numpy.org/, accessed 2019-02-28.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01099e |
| This journal is © the Owner Societies 2020 |