
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Four resonance structures elucidate double-bond isomerisation of a biological chromophore†
Evgeniy V.
Gromov
 * and
Tatiana
Domratcheva
*
* and
Tatiana
Domratcheva
*
Max-Planck Institute for Medical Research, Jahnstraße 29, 69120 Heidelberg, Germany. E-mail: tatjana.domratcheva@mpimf-heidelberg.mpg.de; evgeniy.gromov@mpimf-heidelberg.mpg.de
First published on 9th April 2020
Abstract
Photoinduced double-bond isomerisation of the chromophore of photoactive yellow protein (PYP) is highly sensitive to chromophore–protein interactions. On the basis of high-level ab initio calculations, we scrutinise the effect of hydrogen bonds on the photophysical and photochemical properties of the chromophore. We identify four resonance structures – two closed-shell and two biradicaloid – that elucidate the electronic structure of the ground and first excited states involved in the isomerisation process. Changing the relative energies of the resonance structures by hydrogen-bonding interactions tunes all photochemical properties of the chromophore in an interdependent manner. Our study sheds new light on the role of the chromophore electronic structure in tuning in photosensors and fluorescent proteins.
1. Introduction
Photoactive yellow protein (PYP)1 is a remarkable model system for studying double-bond isomerisation in photoreceptor proteins, as this photoreceptor has amply been characterised using a wide variety of advanced methods such as time-resolved X-ray crystallography,2–5 neutron crystallography,6 NMR,7 and ultrafast spectroscopy.8–12 These studies provided important insights into photoinduced double-bond isomerisation of the PYP chromophore that triggers the PYP photoresponse. The chromophore is derived from the anionic phenolate form of the p-coumaric thioester (pCTM−), which is in the E(trans)-configuration and stabilised by hydrogen bonds (H-bonds) with the protein (Fig. 1a).13 The excited-state dynamics of the pCTM− anion in the protein is different from that in solution and the gas phase.14,15 Despite numerous studies, the exact mechanism of the protein control of the chromophore's isomerisation remains elusive. In particular, it is not clear how to describe the impact of the protein on the chromophore at the electronic structure level. | ||
| Fig. 1 (a) Chromophore binding pocket (active site) of PYP featuring the anionic E(trans)-p-coumaroyl chromophore bound to PYP via a thioester linkage of the Cys69 residue and hydrogen-bonded to the backbone of Cys69 and side chains of Tyr42 and Glu46. (b) Three models considered here: the anionic p-coumaric thiomethyl (pCTM−) chromophore (1), pCTM− hydrogen-bonded with one water molecule at the carbonyl oxygen (2), mimicking the H-bond with the backbone of Cys69, and pCTM− hydrogen-bonded with two water molecules at the phenolic oxygen (3), mimicking the H-bonds with the Tyr42/Glu46 residues. | ||
The moderate molecular size and high sensitivity to intermolecular interactions render the pCTM− chromophore (model 1 in Fig. 1b) particularly suitable for accurate quantum-chemistry calculations addressing the chromophore's tuning by intermolecular interactions.16–22 Similarly to other ionic chromophores of photoresponsive proteins, pCTM− undergoes a pronounced charge-transfer, from the phenolic moiety to the carbonyl fragment, upon photoexcitation.23 Twisting the chromophore's central double bond (C7![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C8) or single bond (C4–C7) results in localisation of the negative charge either on the phenolic or carbonyl moiety, respectively, in the excited (S1) state.17,20,24,25 The localisation of the negative charge in the ground state (S0) is reversed to that in the S1 state at the twisted geometries. Polar interactions of pCTM− with the protein or solvent profoundly influence the ratio of the S1-state trajectories returning to the S0 state via the single- and double-bond twists.16,18,24,26 Experimentally it has been established that PYP photoactivation is triggered by the double bond isomerisation yielding a Z(cis) isomer with a quantum yield of 0.35,27 which proceeds via a one-bond flip (OBF) mechanism destabilizing the carbonyl H-bond.2,3,9 Activation of the single-bond rotation has been reported for a mutated variant with weakened phenolic H-bonds.3 The mutation leads to a slight red shift of the absorption maximum,28 reduced charge-transfer character of the S0–S1 transition,23 and a so-called hula-twist (HT) isomerisation combining rotations around the single and double bonds.3 In contrast to the OBF isomerisation, the HT isomerisation preserves the carbonyl H-bond in the resulting cis photoproduct, which impairs the signal transduction.3
C8) or single bond (C4–C7) results in localisation of the negative charge either on the phenolic or carbonyl moiety, respectively, in the excited (S1) state.17,20,24,25 The localisation of the negative charge in the ground state (S0) is reversed to that in the S1 state at the twisted geometries. Polar interactions of pCTM− with the protein or solvent profoundly influence the ratio of the S1-state trajectories returning to the S0 state via the single- and double-bond twists.16,18,24,26 Experimentally it has been established that PYP photoactivation is triggered by the double bond isomerisation yielding a Z(cis) isomer with a quantum yield of 0.35,27 which proceeds via a one-bond flip (OBF) mechanism destabilizing the carbonyl H-bond.2,3,9 Activation of the single-bond rotation has been reported for a mutated variant with weakened phenolic H-bonds.3 The mutation leads to a slight red shift of the absorption maximum,28 reduced charge-transfer character of the S0–S1 transition,23 and a so-called hula-twist (HT) isomerisation combining rotations around the single and double bonds.3 In contrast to the OBF isomerisation, the HT isomerisation preserves the carbonyl H-bond in the resulting cis photoproduct, which impairs the signal transduction.3
Here, by performing quantum-chemistry calculations for models 1–3 (Fig. 1b) we study the impact of H-bonds on the S0 and S1 energy at the geometries of pCTM− constituting the single and double bond isomerisation pathways, including the geometries of the S0/S1 conical intersections (CoIns). The accurate description of the models is attained by using a high-level ab initio method, the extended multi-state multi-configurational quasi-degenerate perturbation theory of second order (XMCQDPT2),29 which accounts for both static and dynamical electron correlation. In this context, the present work critically extends the previous studies performed using the complete-active-space self-consistent field (CASSCF) method21,24,30 and the single-reference second-order approximate coupled-cluster (CC2) method.17,20,22,25 To explain the trends revealed from contrasting the three models, we deduce four resonance structures, two closed-shell (CS) and two biradicaloid (BR), that describe charge delocalisation and charge transfer upon the S0–S1 excitation and the bond twists. Stabilisation and destabilisation of these structures by H-bonds account for interdependent changes of all photochemical properties of the pCTM− chromophore. We compare our findings to previously published results on PYP, fluorescent proteins and rhodopsin photoreceptors noting multiple parallels among the properties of ionic chromophore in these proteins.
2. Computational details
The energies and geometries for models 1–3 in the S0 and S1 states were obtained from the XMCQDPT2 calculations. As a zero-order wave function employed in the XMCQDPT2 calculations, we used a state-averaged CASSCF wave function, SA2-CASSCF(12,11). The active space comprised 12 electrons over 11 π molecular orbitals (MOs), as previously used by Boggio-Pasqua and Groenhof.21 The state-averaging was done for the two lowest electronic states, S0 and S1, with equal weights. The subsequent XMCQDPT2 calculations using the SA2-CASSCF(12,11) zero-order wave function further accounted for dynamical electron correlation, up to the second order of perturbation theory, and interaction of the S0 and S1 states, via their mixing in a 2 × 2 Hamiltonian. The resolution of the identity (RI) approximation31–33 implemented in Firefly version 8.2 afforded a substantial speed-up of the XMCQDPT2 computations and a decrease of memory requirements, significantly facilitating numerical evaluation of the energy gradients utilizing supercomputer resources. An intruder state avoidance (ISA) energy shift34 of 0.02 Hartree was applied throughout. In all our calculations, the correlation consistent double-zeta basis set of Dunning (cc-pVDZ) was used,35,36 resulting in 231 to 277 MOs depending on the model. All MOs were included in the MCSCF procedure, whereas the chemical-core MOs (1s of carbon and oxygen and 1s, 2s and 2p of sulphur) were not correlated in the XMCQDPT2 calculations.The search of stationary points on the S1 and S0 PESs and points of S1/S0 minimum energy conical intersections (CoIn) was performed using numerical XMCQDPT2 energy gradients computed with finite differencing of second order. In the search, internal coordinates were used without imposing constraints, i.e., the full geometry optimisation was performed. The root mean square (RMS) gradient for optimisation convergence was 10−3 Hartree per Bohr. In the CoIn geometry optimisation, a penalty function method was used, with a penalty function as proposed by Martinez and co-workers.37 The threshold of the energy gap in the CoIn optimisation was 10−3 Hartree. Because of exceedingly high computational demands for numerical evaluation of the Hessians at the XMCQDPT2 level, vibrational analysis at the stationary points was not performed. The type of the stationary points was inferred based on previously published computations.17,20 For the saddle points, the existence of a negative curvature was confirmed by calculating relaxed energy scans along the coordinate for which the negative curvature was expected. The planar structures of models 1 and 2 (E-S1-Sad) and the single-bond twisted structure of model 3 (α-S1-Sad) were demonstrated to correspond to saddle points by computing relaxed-energy scans along the single-bond twisting coordinate (α-torsion). The double-bond twisted barrier (β-S1) in all the models was demonstrated to correspond to a saddle point by computing relaxed-energy scans along the double-bond twisting coordinate (β-torsion).
For all the stationary points, the basis set superposition error (BSSE) corrections were computed for the S0 and S1 energies of models 2 and 3 using the protocol described in ref. 38. Overall, the BSSE corrections for the relative/excitation energies are small, not exceeding 0.1 eV, with the largest correction (−0.085 eV) found for the α-S1-Sad saddle point of model 3. Hereby, the corrections for the twisted structures are larger than that for the planar structures, which obviously relates to the different charge distribution for the planar and twisted configurations. Importantly, accounting for the BSSE corrections does not lead to changes in the relative order of the stationary points, either within a particular model or the different models. In the following, we therefore neglect the BSSE corrections, presenting and discussing the plain XMCQDPT2 results.
For analysis of results for models 1–3, we also considered two protonated isomers of model 1, which are obtained from 1 by protonating either the phenolate or enolate oxygen. The isomers are denoted HPhCS (protonation of phenolate) and HQnCS (protonation of enolate), with the meaning of such a notation becoming clear in Section 3.2. We are interested in the ground-state equilibrium geometries of HPhCS and HQnCS, which were obtained from full geometry optimisations at the XMCQDPT2/SA2-CASSCF(12,11) level of theory. In addition, the ground-state optimised geometries of HPhCS and HQnCS, as well as models 1–3, were obtained with the Møller–Plesset second-order (MP2) method.
Oscillator strengths, Mulliken atomic charges and free valences on atoms (number of unpaired electrons)39 were computed using the S0 and S1 zero-order QDPT electron densities. Here, the obtained zero-order QDPT densities were essentially the CASSCF S0- and S1-state densities. For analysis of the S0 and S1 PESs in the vicinity of CoIn, an approach suggested by Olivucci and co-workers was used.40,41 A series of points circumscribing a CoIn point (loops) lying in the branching plane were computed. The branching plane is defined by the gradient difference (x1) and derivative coupling (x2) vectors.42 We computed the x1 and x2 vectors with the SA2-CASSCF(12,11) method using Gaussian09,43 with solving the coupled-perturbed equations in the MCSCF cycle being skipped (“nocpmcscf” approximation).44 The error introduced by the latter decreases with the decrease of the S0–S1 energy gap; therefore it might be important at the Frank–Condon region and negligible at CoIn. Except computations of the branching vectors, all calculations were performed using version 8.2 of the Firefly quantum chemistry package,45 which is partially based on the Gamess-US source code.46 The computations utilised resources of the high performance computing cluster JURECA.47
3. Results and discussion
3.1 Characterisation of single- and double-bond twists
Fig. 2 depicts cross-sections through the S0 and S1 potential energy surfaces (PESs) computed for models 1–3 along the single- and double-bond twisting pathways starting from the E(trans) S0 minimum (E-S0-Min). For each model, the cross-section is composed of several stationary points on the S0 and S1 PESs and points of S1/S0 minimum-energy conical intersections (CoIns). Specifically, there are the E(trans) and Z(cis) planar minima in the ground S0 state, two minima and two saddle points of planar and twisted geometries in the S1 state, and two twisted CoIn points. All the points were obtained from a full geometry optimisation using the XMCQDPT2 method. In the following, we refer to the rotation around the C4–C7 bond as α-twist and around the C7–C8 bond as β-twist (Fig. 1b) according to the previously adopted notation.17,20,25 The geometries of the models at the stationary points are depicted in Fig. S1 in ESI.† The energies given in Fig. 2 are not corrected for the basis set superposition error (BSSE), which according to our estimation introduces only marginal changes in the cross-section energies (Table S3 in ESI†). | ||
| Fig. 2 Cross-sections of S0 and S1 PESs for models 1 (blue), 2 (green) and 3 (red) for the photoinduced α and β twisting. The following notations are used to indicate the geometry and type of the stationary points. Planar ground-state minima featuring E(trans) and Z(cis) configurations with respect to the central –C7C8– double bond, E-S0-Min and Z-S0-Min, respectively; planar excited-state saddle point and minimum, E-S1-Sad and E-S1-Min, respectively; α-twisted excited-state minimum and saddle point, α-S1-Min and α-S1-Sad, respectively; α-twisted CoIn, α-CoIn; β-twisted saddle point, β-S1-Sad; β-twisted minimum, β-S1-Min; β-twisted CoIn, β-CoIn. All geometries are shown in Fig. S1 (ESI†). The total and relative energies of the points are listed in Tables S1 and S2 in ESI.† | ||
At first, we consider the results for model 1, which relate to the intrinsic properties of the pCTM− chromophore (blue cross-section in Fig. 2). At the trans configuration E-S0-Min minimum, the S0–S1 vertical excitation energy (VEE) is 2.60 eV and the corresponding oscillator strength is close to unity. From the Franck–Condon (FC) point, the chromophore first undergoes a bond-length alteration (BLA) geometry relaxation toward the planar E-S1-Sad saddle point followed by the α-twist relaxation toward the 90°-twisted α-S1-Min minimum. Structurally close to the α-S1 minimum but higher in energy, there is a S1/S0 α-CoIn, mediating decay of the excited-state population back to the S0 state. The excited-state α-twist was previously implicated in internal conversion.20,22,24 Activation of the β-twist involves passing a small barrier, β-S1-Sad, which is 0.12 eV above VEE, and is associated with the change of the BLA coordinate and β torsion. Beyond the barrier, the chromophore relaxes by undergoing further β-twist toward the 90°-twisted β-S1-Min minimum. Structurally close to the β-S1 minimum but higher in energy, there is a S1/S0 β-CoIn, through which the chromophore decays back to the S0 state either forward to the Z-S0-Min minimum or backward to the E-S0 minimum. The ratio of the Z and E products depends on many factors, one of which is the topology of the S0 PES in the vicinity of β-CoIn. Both the α- and β-CoIns demonstrate a sloped topology, as each of them is found in the vicinity of a 90°-twisted S1 energy minimum.
The results of models 2 and 3 (green and red cross-sections, respectively, in Fig. 2) demonstrate that the H-bonds lead to considerable energy changes and, moreover, to a change of the S1 PES topology in the case of model 3. Specifically, the carbonyl H-bond in 2 reduces VEE, stabilises the α-pathway and destabilises the β-pathway. In contrast, the phenolic H-bonds in 3 increase VEE, destabilise the α-twist and stabilise the β-twist. Notably, the α-twist energies are more affected by the H-bonds than that of the β-twist. In 3, destabilisation of the α-twist results in appearance of the planar E-S1-Min minimum, whereas the α-S1 α-twisted geometry becomes a saddle point (red cross-section in Fig. 2).
3.2 Four resonance structures rationalizing effect of the H-bonds
In the previous studies, the effect of the H-bonds on the S0 and S1 energies has been explained by stabilisation and destabilisation of the negative charge either on the phenolic or carbonyl groups of pCTM−.17,20,24,25 At the planar E-S0 minimum, the charge de-localisation is explained by the resonance of two closed-shell (CS) anionic forms.17,25 At the same time, the origin of the reversed negative charge localisation at the α- and β-twisted S1 minima has not been linked explicitly to any resonance forms so far.17,20,25 To elucidate this issue, we analysed distribution of the negative charge and the number of unpaired electrons (NUE)39 at the different geometries (stationary points) in the S0 and S1 state (Fig. 3 and Tables S5–S7 in ESI†). The analysis indicates that the S0–S1 electronic excitation results in a significant charge transfer and increase of NUE, which is consistent with a biradicaloid (BR) structure of the S1 state (Table S8 in ESI†). The key result of our analysis is realising that the changes of the phenolic charge and NUE at all the stationary points (Fig. 3) can be accounted for by changing contributions of four resonance structures – two CS structures, characterizing the S0 state as mentioned above, and two BR structures, characterizing the S1 state (Scheme 1). The resonance structures are denoted according to the presence of the phenolic (Ph) or quinonic (Qn) π-system. Noteworthy, QnBR relates to PhCS, and PhBR relates to QnCS via an electron excitation. QnBR describes a single-electron charge-transfer excitation of PhCS, whereas PhBR describes a single-electron charge-transfer excitation of QnCS. To the best of our knowledge, the QnBR and PhBR structures have never been explicitly considered for the planar pCTM− chromophore before.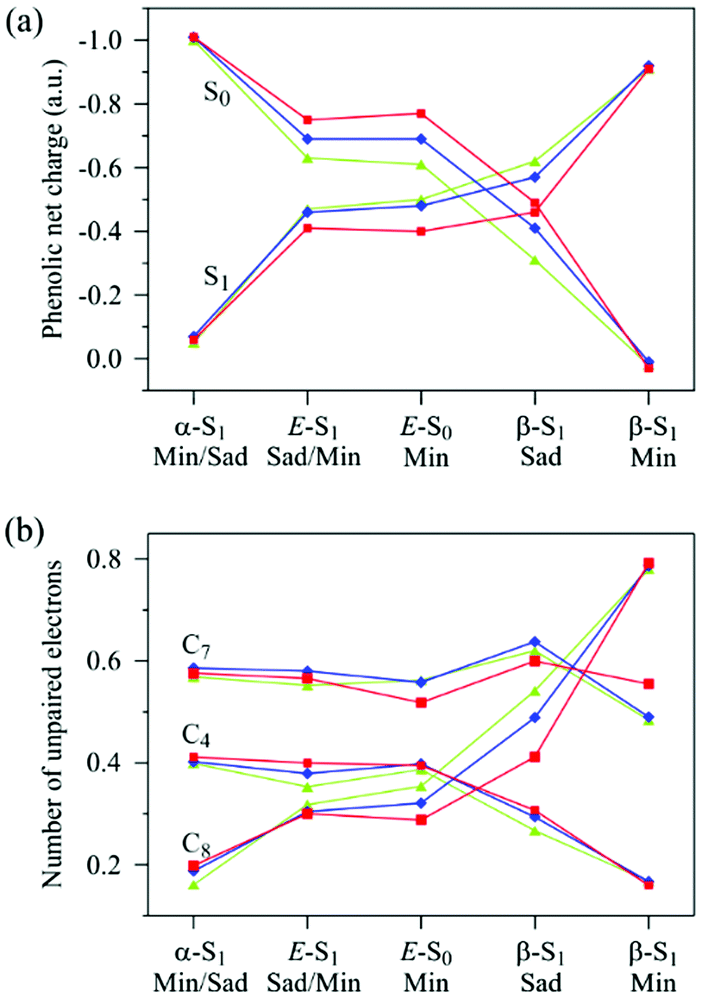 | ||
| Fig. 3 Distribution of (a) the net negative charge on the phenolic moiety in the S0 and S1 states and (b) the number of unpaired electrons (NUE) at the C4, C7 and C8 atoms in the S1 state in models 1–3 at the different stationary points along the α- and β-twisting pathways. The colour code is the same as in Fig. 2. The atomic and net charges on the phenolic and carbonyl moieties, as well as NUE are given in Tables S5–S7 in ESI.† | ||
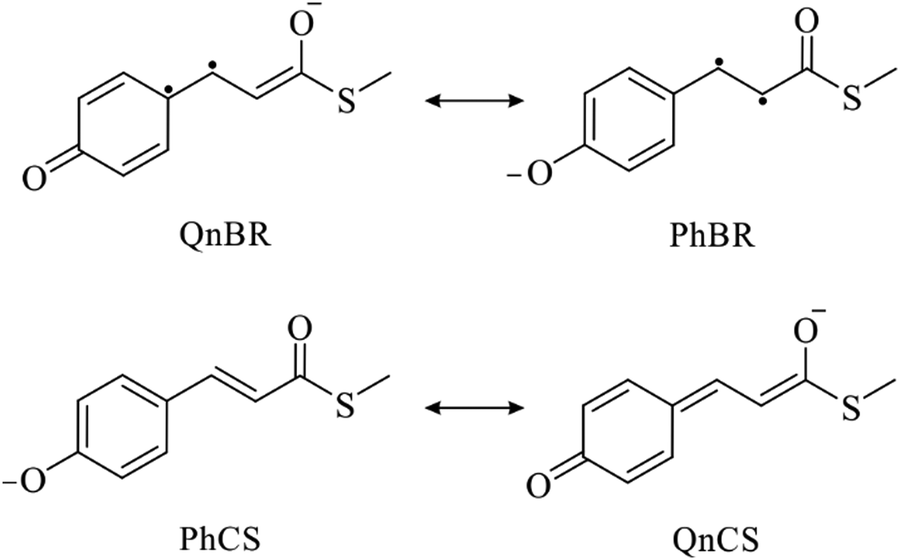 | ||
| Scheme 1 Resonance of two closed-shell (CS), phenolic (PhCS) and quinonic (QhCS), and two biradicaloid (BR), qiononic (QnBR) and phenolic (PnBR) structures elucidating photophysical and photochemical properties of the anionic PYP chromophore. | ||
It is worth noting that the four resonance structures are consistent with those introduced in the biradicaloid theory of Bonačić-Koutecký et al.48,49 and theory of twisted intermolecular charge-transfer (TICT) of Rettig and co-workers.50,51 The TICT theory treats the “hole-pair” (hp) configuration of a polar chromophore as electron-acceptor and electron-donor moieties connected via a central single bond. The electron transfer from the donor to the acceptor upon the HOMO–LUMO excitation (typically corresponding to the S0–S1 transition) yields a “dot–dot” (dd) structure with a translocated molecular charge. By twisting the central single bond, the HOMO and LUMO involved in the charge-transfer transition become localised on the mutually orthogonal molecular fragments, and thus do not interact. Therefore, the twisting decreases the energy of the dd configuration and increases the energy of the hp configuration, which altogether significantly reduces the S0–S1 energy difference and even leads to S0/S1 state crossing.48,51
In Scheme 1, PhCS and QnCS correspond to conjugated hp configurations featuring the phenolate and enolate negatively charged electron-donor moieties, respectively. Electron excitation from the PhCS hp configuration populates the QnBR dd configuration, whereas that from the QnCS hp configuration populates the PhBR dd configuration. At the same time, the α- (β-) twist leads to the QnBR (PhBR) structure in the S1 state, and to the PhCS (QnCS) structure in the S0 state. This picture is confirmed by Fig. 4, which displays the difference of the S1 and S0 total electron densities at the α-S1 and β-S1 twisted minima. As can be clearly seen, the negative charge at the α-S1 minimum resides on the alkene-carbonyl fragment in the S1 state and on the phenolic fragment in the S0 state (Fig. 4a), whereas it is vice versa at the β-S1 minimum (Fig. 4b). The same holds for models 2 and 3.
 | ||
| Fig. 4 Difference of the S1 and S0 electron densities at the α-twisted (a) and β-twisted (b) minima of pCTM− (model 1). Orange and purple correspond to S1 and S0 electron densities, respectively. | ||
Resonance of the PhCS and QnCS structures determines the single- and double-bond lengths at the planar E-S0-Min minimum and charge distribution in the S0 state. Fig. 5 shows the BLA of the C4–C7–C8 methine bridge in the anionic models in comparison to that in the protonated phenol and enol isomers of pCTM. Obviously, protonation of an electron-donating group, phenolate or enolate, favours the corresponding resonance structure, phenolic or quinonic. The shorter C7–C8 distance in comparison to C4–C7 (Fig. 5) and the amount of the negative charge on the phenolate (Fig. 3a) indicate the dominance of PhCS over QnCS in the three models. The prevailing phenolic character of the anionic models suggests that PhCS has a lower energy than QnCS in all the models under consideration. The decrease of the phenolic charge and increase of the C7–C8 bond length in the order 3–1–2 indicate the increase of the QnCS weight, and hence the increase of the PhCS and QnCS mixing in the same order.
 | ||
| Fig. 5 Bond length alteration (BLA) of the C4–C7–C8 methine bridge in models 1–3 in comparison to the protonated isomers of pCTM, HPhCS and HQnCS. In addition to the XMCQDPT2 results (red filled diamonds, red line), the MP2 results (green points, green line) are also shown. | ||
At the FC point in the S1 state, QnBR dominates over PhBR as it follows from the low negative charge on the phenolate (Fig. 3a) and higher NUE at C4 and C7 than at C8 (Fig. 3b). The weight of QnBR increases in the order 2–1–3, as indicated by a simultaneous decrease of the C8 NUE and phenolic negative charge in this order. In the same order, the S0–S1 charge-transfer character increases, as demonstrated by the increase of the S1–S0 phenolate charge difference (Fig. 3a) and the permanent dipole moment difference (Table S1 in ESI†). The increase of VEE in the same order of models (Fig. 2) is explained by the higher VEE of PhCS in comparison to that of QnCS.
Thus, mixing of PhCS and QnCS determines de-localisation of the molecular charge in the S0 state, the extent of BLA at the S0 equilibrium geometry, the S0–S1 VEE and the extent of the charge transfer upon S0–S1 excitation. Obviously, the more PhCS prevails in the S0 state (small PhCS/QnCS mixing), the larger the BLA and phenolic charge in the S0 state and the larger the S0–S1 VEE and the extent of the charge-transfer. In contrast, the less the PhCS prevails in the S0 wave function (large PhCS/QnCS mixing), the smaller the BLA and phenolic charge in the S0 state, and the smaller the S0–S1 VEE and the charge-transfer upon excitation.
3.3 Energies of the resonance structures – impact on the S1-state PES topology and S1/S0 conical intersections
The energies of the resonance structures can be estimated and compared for the 90°-twisted geometries.52 Indeed, the four resonance structures are practically uncoupled at the α-S1 and β-S1 twisted minima, with the negative charge being completely localised either on the phenolic or carbonyl fragment, as evident in Fig. 3a. At the α-S1 minimum, the S0 state corresponds to the PhBR structure, whereas the S1 state is represented by the QnBR structure, with the negative charge being localised on the phenolic moiety and carbonyl fragment in the S0 and S1 states, respectively (Fig. 3a and 4a), and the C8 NUE has the smallest value (Fig. 3b). At the β-S1 minimum, the S0 state is represented by the QnCS structure, while the S1 state is associated with the PhBR structure, with the negative charge being localised on the carbonyl fragment and phenolic moiety in the S0 and S1 states, respectively (Fig. 3a and 4b), and the C8 NUE has the largest value (Fig. 3b).By comparing the energies at the 90°-twisted geometries (Fig. 2), we infer that PhCS has the lowest energy among the four resonance structures in all the models, which is in agreement with the analysis of the resonance mixing at the planar geometries described above. The carbonyl H-bond stabilises QnCS and QnBR and destabilises PhCS and PhBR, whereas the phenolic H-bonds stabilise PhCS and PhBR and destabilise QnCS and QnBR. Among the four resonance forms, the energy of QnBR demonstrates the largest variation due to the H-bonding interactions. Explaining this observation would require further analysis, e.g., by invoking the valence bond picture.53,54
As the geometries of the stationary points demonstrate, the BLA, α-twist and β-twist are the main coordinates driving the relaxation of the pCTM− chromophore in the S1 excited state. If the QnBR energy is lower than VEE, the initial relaxation from the FC point involves the BLA coordinate followed by the α-twist. If that condition is not fulfilled, which is the case for model 3 (see Fig. 2), the planar E-S1 minimum appears on the S1 PES.
At the planar E-S1-Min minimum (in model 3), the S0–S1 oscillator strength is 0.9, and the computed radiative rate is ∼0.26 ns−1, suggesting a sizable emission with a Stokes shift defined by the extent of the BLA relaxation. At the twisted α-S1 minimum in models 1 and 2, the computed radiative rate is several orders of magnitude smaller, ∼0.51 s−1, because of the substantial decrease of the S0–S1 energy and especially the oscillator strength. Based on these results, we predict that the dynamics of the fluorescence signal in PYP correlates with the dynamics of the chromophore H-bonds that stabilise/destabilise the planar E-S1 minimum. It is conceivable that the protein folding and dynamics thus control the weak fluorescence of PYP. Furthermore, the smaller BLA variation at the planar S1 minimum as compared to that at the S0 minimum is caused by the chromophore instability with respect to the α-twist. In colour-tuned PYP mutants, the variation of the emission maximum should be significantly smaller than that of the absorption maximum as the α-twist favoured by stabilisation of QnBR virtually quenches the fluorescence. Thus, a decrease of both the fluorescence and Stokes shift is predicted for the red-shifted variants in comparison to the blue-shifted variants of PYP mutants. This does agree with the smaller variation of the emission maximum as compared to the absorption maximum in a series of single-point mutated PYP variants, reported by Hoff and co-workers.28
Finally, the energies of the resonance structures at the α-S1 and β-S1 minima correlate with the energies of α-CoIn and β-CoIn, which in turn define whether the excited chromophore returns to the S0 state via a single- or double-bond twist. As Fig. 2 demonstrates, the CoIn energies parallel the S0–S1 energy gap at the 90°-twisted geometries, i.e., the difference in energy of the corresponding CS and BR resonance structures. The smaller the energy gap at the 90°-twisted S1 minimum (or saddle point), the lower the CoIn energy and the smaller the distortion of the CoIn geometry in comparison to the geometry of the twisted minimum (or saddle point). As an example, we consider model 3, for which the PhCS and QnBR energy difference is rather high, and consistently the high-energy α-CoIn in this model demonstrates a substantial β-twist in addition to the α-twist (Fig. S1 in ESI†). In contrast, stabilisation of PhBR and destabilisation of QnCS by the phenolic H-bonds lowers the energy of the β-CoIn and reduces its distortion with respect to the β-S1 minimum. In models 1 and 2, the PhCS and QnBR energy difference is smaller than in model 3, the α-CoIn energy therefore decreases, whereas the PhBR and QnCS energy difference is larger, and hence the β-CoIn energy increases. The variation of the α-CoIn energy among the models suggests that accessibility of α-CoIn and therefore activation of the single bond rotation and internal conversion via the single-bond twist can be efficiently controlled by chromophore–protein interactions. Stabilisation of PhCS and PhBR by these interactions reduces the resonance mixing and favours the double-bond OBF isomerisation.
3.4 Double-bond isomerisation of the pCTM− chromophore
The β-CoIn provides the excited-state decay channel for the OBF double-bond isomerisation of pCTM−. We remind that a minimum energy CoIn geometry corresponds to a local energy minimum on the 3N-8 dimensional hypersurface (N is the number of atoms) where the S1 and S0 states are degenerate.42 The degeneracy is lifted in the first order along the gradient difference vector (x1) and the derivative coupling vector (x2), forming a so-called branching plane.55–57 Inspection of the energies in the branching plane unveils relaxation on the S0 PES after a non-adiabatic S1–S0 transition at the β-CoIn. We computed x1 and x2 at the β-CoIn geometries and constructed branching-plane loops following the procedure suggested by Olivucci and co-workers.58 In all our models, x1 is dominated by BLA, whereas x2 features the β-torsion and, in addition, hydrogen out-of-plane (HOOP)20,22,59 excursions, which mix with the BLA changes in models 1 and 2 (see Fig. S2 in ESI†). At the β-CoIn geometries, the HOOP value tends to be “ahead” of β-torsion, i.e., smaller than β-torsion, when β > 90° (in models 1 and 2), whereas it is “concerted” with β-torsion (close to β-torsion) when β < 90° (model 3) (Fig. S3 in ESI†). Fig. 6a presents the x1 and x2 vectors for model 3 (for the other models see Fig. S2 in ESI†). Fig. 6b demonstrates that β-CoIn has the β-torsion value slightly smaller than 90° in model 3, whereas it shifts toward a larger β-torsion value (i.e., closer to the initial E(trans) geometry) in models 1 and 2. The smaller β-torsion value at the β-CoIn, explained by stabilisation of PhBR and destabilisation of QnCS in model 3, is favourable for the E(trans)–Z(cis) isomerisation. The β-CoIn branching plane of pCTM− is similar to that previously reported for the double-bond isomerisation of the retinal protonated Schiff base (RPSB) chromophore and analogues in rhodopsins.60,61 | ||
| Fig. 6 Characterisation of the β-twisted CoIn: (a) the branching plane vectors x1 and x2 for model 3 (see also Fig. S2 (ESI†) presenting x1 and x2 for all three models). (b) Changes in the β-torsion and C7C8 bond length along the 0.005 Å loops for models 1–3. The values of the angular coordinate θ at some points of the loops, and the orientation of the x1 and x2 vectors are indicated. The colour code is the same as in Fig. 2. | ||
Fig. 7 shows the energy and property changes along the branching plane loops. In the x1 direction (θ = 90°, 270°), the S0 and S1 wave functions change from one resonance structure to another. Specifically, the charge distribution and NUE (Fig. 7b) indicate that the S0 wave function changes from QnCS to PhBR whereas the S1 wave function changes from PhBR to QnCS. The x2 direction (θ = 0°, 180°) corresponds to mixing of PhBR and QnCS and, to a lesser extent, of PhCS, as indicated by the changes of the phenolic charge and NUE (Fig. 7b). The presence of PhCS follows from the steeper phenolic charge curve in comparison to the NUE curve. The PhCS weight significantly increases as the chromophore becomes more planar.
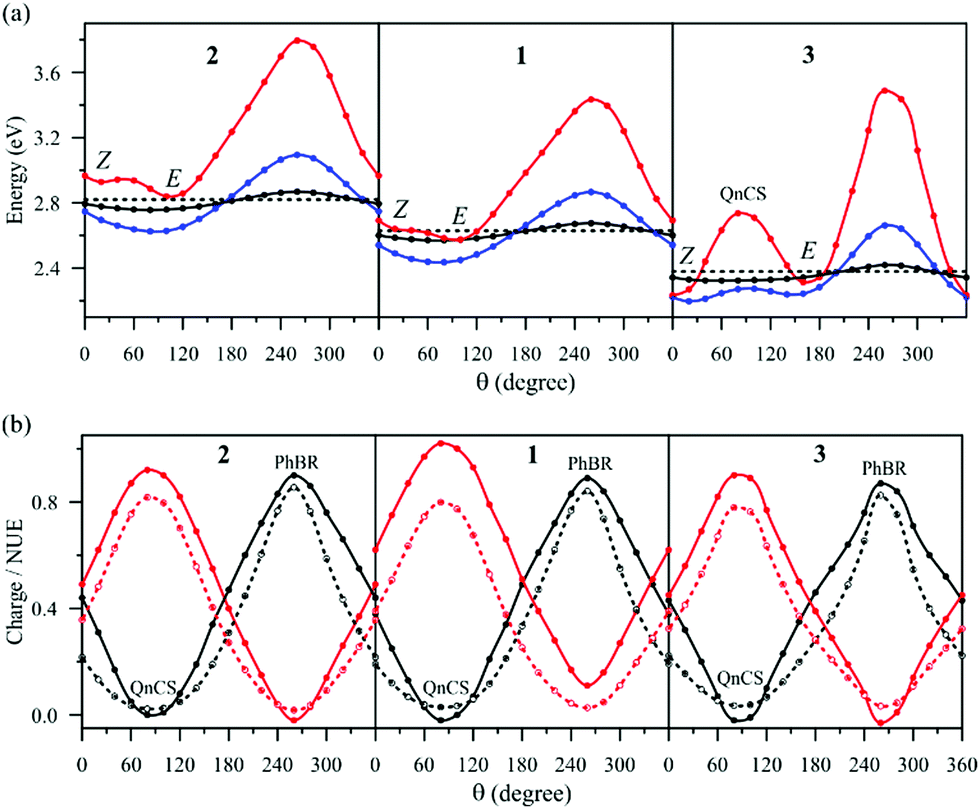 | ||
| Fig. 7 Energies and properties computed at the geometries constituting the loops circumventing the β-CoIn in the branching plane for models 1–3. θ represents the angular coordinate of the loops. θ = 0° and θ = 180° define the derivative coupling vector x2. θ = 90° and θ = 270° define the gradient difference vector x1. (a) S0 energies for the loops of radii 0.001 Å (black), 0.005 Å (blue) 0.0011 Å (red). The dashed line indicates the β-CoIn energy. (b) Net charge on the phenolic fragment (solid lines with filled symbols) and NUE at the C8 atom (dashed lines with empty symbols) in the S0 (black) and S1 (red) states. | ||
Two S0 energy minima emerge along x2 (Fig. 7a) as decay channels towards the Z(cis) and E(trans) minima. Notably, the Z(cis) decay channel lies below the β-CoIn energy only in 3 (at θ = 0°, Fig. 7a), correlating with the smaller β-torsion value at β-CoIn in this model. In addition, there is a high-energy barrier, associated with the QnCS structure, separating the two channels in model 3, which is explained by the higher QnCS energy in this model. Overall, the topology of the S0 PES along the loops suggests that the double-bond isomerisation quantum yield is higher in 3 as compared to 1 and 2.
Finally, we discuss on some aspects related to the sloped character62 of the β-CoIn. There are two stationary points associated with the β-CoIn, the β-S1 minimum on the S1 PES, and a saddle point on the S0 PES, which are of the PhBR and QnCS electronic structure, respectively. The S0 saddle point is associated with the aforementioned QnCS energy barrier separating Z(cis) and E(trans) channel (Fig. 7a). Our preliminary calculations for model 3 have identified such a point. This is the only saddle point connecting the Z(cis) and E(trans) minima on the S0 PES, and hence it mediates the thermal isomerisation. The sloped type of β-CoIn implies that the aforementioned two stationary points lie on the same side from the β-CoIn along x1. This is different from the peaked CoIn found for the RPSB chromophore and its analogues, where two saddle points of different electronic structure lying on the S0 PES flank the CoIn along the BLA coordinate.63–66 Noteworthy, for the rhodopsin photoreceptor, the energy of the saddle point controlling the thermal isomerisation was found to correlate with VEE, such that the blue-shifted absorption maximum corresponds to a reduced thermal activation (higher energy of the saddle points).63 Despite different β-CoIn topology, a similar correlation as for the RPSB chromophore should be expected for the pCTM− chromophore, as the increase of VEE correlates with the increase of the QnCS energy.
3.5 Comparison to other computational studies
It is noteworthy that models 1–3 correctly grasp the major effects of intermolecular interactions in PYP previously reported by numerous computational studies. In particular, the energy changes presented in Fig. 2 are qualitatively consistent with those predicted by the hybrid quantum-mechanics/molecular mechanics (QM/MM) models of PYP16,18,19,26 and solvated chromophore.24 Specifically, the carbonyl H-bond lowers the α-CoIn energy, whereas the phenolic H-bonds lower the β-CoIn energy in the QM/MM models.24 In the surface-hopping molecular dynamics simulations of PYP,26 the presence of the protonated (positively charged) Arg52 stabilizing the phenolic negative charge, that is the PhCS and PhBR resonance structures with respect to the QnCS and QnBR ones, leads to stabilisation of the β-CoIn, and hence facilitates the OBF isomerisation.Comparison of the XMCQDPT2 energies of model 1 with the results obtained using the CASSCF(12,11) method,21 and of model 3 with the results obtained using the CC2 method17,20 indicates that the XMCQDPT2 treatment favours the double-bond isomerisation of pCTM−. It is instructive to compare the energies at the 90°-twisted geometries, which are associated with the energies of the resonance structures. Both CC2 and CASSCF clearly favour QnBR over PhBR when compared to the results of the XMCQDPT2 method. The β-CoIn is higher in energy than α-CoIn at the CASSCF level of theory, whereas it is vice versa at the XMCQDPT2 level. Thus, employing the CASSCF method to describe the pCTM− chromophore in surface-hopping dynamics of PYP16,26 underestimates the efficiency of the β-twist in the S1 state. For a similar anionic chromophore of the green fluorescent proteins (GFPs), it has indeed been reported that the CASSCF method underestimates the decay of the S1 state via the β-twist as compared to the more accurate XMSCASPT2 method.67 In the QM/MM simulations of PYP,26 this underestimation seems to be compensated by the effect of the positively charged residues, such as protonated Arg52 stabilizing, the PhCS and PhBR structures.
3.6 Similarity with other biological chromophores
Similar to pCTM−, the anionic p-hydroxybenzylideneimidazolinole (pHBDI−) chromophore of GFPs is described by resonance interactions of the PhCS and QnCS forms coupled along the BLA reaction coordinate.68,69 Recently, Boxer and co-workers69 suggested that the energy difference of the PhCS and QnCS forms can serve as a linear scale for quantitative prediction of the GFP photophysical properties using a small number of parameters intrinsic to the pHBDI− chromophore. This energy difference was implicated to explain all strong correlations of properties among a large number of systematically tuned GFP variants.69 We note that the charge-transfer GFP model of Boxer and co-workers69 is consistent with our computational results for the anionic pCTM− except that it does not invoke the QnBR and PhBR structures of pHBDI−. By analogy to pCTM−, we suggest that the energies and properties of the QnBR and PhBR resonance forms should be considered when addressing the S1 PES of pHBDI− in GFPs, in particular its photoactivation. Applying this prediction, stabilisation of PhBR in pHBDI− favours the OBF double-bond isomerisation, whereas stabilisation of QnBR gives a chance to the single-bond activation and HT isomerisation.70Mixing of the resonance forms describes charge delocalisation in protonated cationic chromophores, the RPSB chromophore of rhodopsins, and tetrapyrrole chromophores of phytochromes, and eventually determines the sensitivity of these chromophores to electrostatic effects. However, electrostatic effects are typically discussed without evoking the concept of chemical resonance (see e.g., a recent comparison of “electrostatic spectral tuning maps” of various biological chromophores).71 The difference in the electrostatic interaction energies of the S0 and S1 states explains the VEE tuning. Chemical resonance explains the energy and property tuning by changing contributions of the resonance forms, which depends on the relative energies of these forms and on the different stabilization of these forms by interactions and geometric constraints. Moreover, different sensitivities of ionic chromophores to electrostatic embedding relates to the extent of resonance mixing. For instance, a single dominant resonance form of RPSB features a positive charge localised on the Schiff-base fragment, giving rise to considerable charge-transfer character of the S0–S1 transition and high sensitivity to electrostatic interactions. These interactions in turn efficiently control the contributions of the resonance forms, as suggested by the correlations of the increased/decreased BLA and increased/decreased VEE.72–74 In contrast, the positive charge of the tetrapyrrole is redistributed among the four pyrrole rings, and hence the chromophore structure is described by at least four resonance forms.75 This charge delocalisation reduces the charge-transfer character of the S0–S1 transition and subsequently reduces the electrostatic effect of the protein on the chromophore.
4. Conclusions and outlook
Given the prominent role of ionic chromophores in light activation of naturally evolved photoreceptor proteins, it is important to rationalise the effect of chromophore–protein interactions that are crucial for the light sensory function. Here, by analysing how the anionic pCTM− chromophore of the PYP bacterial photoreceptor is influenced by phenolic or carbonyl H-bonds we derived a qualitative model explaining interdependent changes of all photochemical properties related to the PYP photoactivation. To account for electronic correlation effects, the state-of-the-art XMCQDPT2 method was employed to compute the S0 and S1 PES cross-sections. To rationalise the obtained trends, two pairs of resonance structures, each pair comprised of a CS structure and corresponding to it charge-transfer BR structure were invoked (Scheme 1). Delocalisation of the negative charge and other properties of the S0 and S1 states are described by chemical resonance of the structures with the opposite charge localisation. Contributions of the resonance structures depend on their energies that we compare at the 90° single- and double-bond twisted geometries where chemical resonance vanishes.From the computational results, the following picture of tuning pCTM− by intermolecular interactions emerges. Stabilisation of the phenolic CS and BR structures by the phenolic H-bonds leads to the larger difference in the lengths of the central single and double bonds (i.e. larger BLA) at the S0 minimum-energy geometry; there is also an increase of the excitation energy and stabilisation of the planar S1 minimum. Moreover, isomerisation around the central double bond becomes more favourable than the phenolic ring rotation around the single bond, and the energy barrier controlling the thermal double-bond isomerisation increases. In contrast, stabilisation of the quinonic structures by the carbonyl H-bond leads to delocalisation of the negative charge and a smaller BLA in the S0 state, smaller S0–S1 excitation energy and smaller charge transfer character, as well as activation of the single-bond rotation in the S1, increasing the probability of the HT photoisomerisation. Overall, our computational results predict that stabilisation of the pCTM− phenolic resonance structures, i.e. reduction of chemical resonance, is crucial for PYP photoactivation via the OBF double-bond isomerisation.
Comparison of our findings and conclusions to the previously published computational and theoretical results characterizing other biological chromophores identifies many common features. In particular, our model derived from the high-level quantum-chemistry calculations in its essence is similar to a model proposed by Boxer and co-workers69,76 explaining spectral properties of systematically mutated GFP proteins. Yet, our model extends Boxer's model to the twisted geometries and invokes BR structures to describe the S1 state properties. The broad similarities among the ionic chromophores imply that the concept of chemical resonance is instructive in experimental and especially computational studies addressing photochemical mechanisms of photosensory proteins operating via the double-bond isomerisation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to dedicate this paper to the memory of Alex A. Granovsky, an inspired scientist and the principal developer of the Firefly quantum chemistry program package, who developed and implemented the XMCQDPT2 method, and made other valuable contributions in quantum chemistry. The computing time on the supercomputer JURECA at Jülich Supercomputing Centre (JSC) granted by the John von Neumann Institute for Computing (NIC) is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- K. J. Hellingwerf, J. Hendriks and T. Gensch, J. Phys. Chem. A, 2003, 107, 1082–1094 CrossRef CAS.
- F. Schotte, H. S. Cho, V. R. Kaila, H. Kamikubo, N. Dashdorj, E. R. Henry, T. J. Graber, R. Henning, M. Wulff, G. Hummer, M. Kataoka and P. A. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19256–19261 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat, V. Srajer and H. Ihee, Nat. Chem., 2013, 5, 212–220 CrossRef CAS PubMed.
- J. Tenboer, S. Basu, N. Zatsepin, K. Pande, D. Milathianaki, M. Frank, M. Hunter, S. Boutet, G. J. Williams, J. E. Koglin, D. Oberthuer, M. Heymann, C. Kupitz, C. Conrad, J. Coe, S. Roy-Chowdhury, U. Weierstall, D. James, D. Wang, T. Grant, A. Barty, O. Yefanov, J. Scales, C. Gati, C. Seuring, V. Srajer, R. Henning, P. Schwander, R. Fromme, A. Ourmazd, K. Moffat, J. J. Van Thor, J. C. Spence, P. Fromme, H. N. Chapman and M. Schmidt, Science, 2014, 346, 1242–1246 CrossRef CAS PubMed.
- K. Pande, C. D. Hutchison, G. Groenhof, A. Aquila, J. S. Robinson, J. Tenboer, S. Basu, S. Boutet, D. P. DePonte, M. Liang, T. A. White, N. A. Zatsepin, O. Yefanov, D. Morozov, D. Oberthuer, C. Gati, G. Subramanian, D. James, Y. Zhao, J. Koralek, J. Brayshaw, C. Kupitz, C. Conrad, S. Roy-Chowdhury, J. D. Coe, M. Metz, P. L. Xavier, T. D. Grant, J. E. Koglin, G. Ketawala, R. Fromme, V. Srajer, R. Henning, J. C. Spence, A. Ourmazd, P. Schwander, U. Weierstall, M. Frank, P. Fromme, A. Barty, H. N. Chapman, K. Moffat, J. J. van Thor and M. Schmidt, Science, 2016, 352, 725–729 CrossRef CAS PubMed.
- K. Yonezawa, N. Shimizu, K. Kurihara, Y. Yamazaki, H. Kamikubo and M. Kataoka, Sci. Rep., 2017, 7, 9361 CrossRef PubMed.
- P. A. Sigala, M. A. Tsuchida and D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9232–9237 CrossRef CAS PubMed.
- D. S. Larsen and R. van Grondelle, ChemPhysChem, 2005, 6, 828–837 CrossRef CAS PubMed.
- L. J. van Wilderen, M. A. van der Horst, I. H. van Stokkum, K. J. Hellingwerf, R. van Grondelle and M. L. Groot, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15050–15055 CrossRef CAS PubMed.
- L. Mendonca, F. Hache, P. Changenet-Barret, P. Plaza, H. Chosrowjan, S. Taniguchi and Y. Imamoto, J. Am. Chem. Soc., 2013, 135, 14637–14643 CrossRef CAS PubMed.
- M. Creelman, M. Kumauchi, W. D. Hoff and R. A. Mathies, J. Phys. Chem. B, 2014, 118, 659–667 CrossRef CAS PubMed.
- H. Kuramochi, S. Takeuchi, K. Yonezawa, H. Kamikubo, M. Kataoka and T. Tahara, Nat. Chem., 2017, 9, 660–666 CrossRef CAS PubMed.
- U. K. Genick, S. M. Soltis, P. Kuhn, I. L. Canestrelli and E. D. Getzoff, Nature, 1998, 392, 206–209 CrossRef CAS PubMed.
- M. Vengris, M. A. van der Horst, G. Zgrablic, I. H. van Stokkum, S. Haacke, M. Chergui, K. J. Hellingwerf, R. van Grondelle and D. S. Larsen, Biophys. J., 2004, 87, 1848–1857 CrossRef CAS PubMed.
- I. R. Lee, W. Lee and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 258–262 CrossRef CAS PubMed.
- G. Groenhof, M. Bouxin-Cademartory, B. Hess, S. P. De Visser, H. J. C. Berendsen, M. Olivucci, A. E. Mark and M. A. Robb, J. Am. Chem. Soc., 2004, 126, 4228–4233 CrossRef CAS PubMed.
- E. V. Gromov, I. Burghardt, J. T. Hynes, H. Koppel and L. S. Cederbaum, J. Photochem. Photobiol., A, 2007, 190, 241–257 CrossRef CAS.
- C. Ko, A. M. Virshup and T. J. Martinez, Chem. Phys. Lett., 2008, 460, 272–277 CrossRef CAS.
- P. B. Coto, S. Marti, M. Oliva, M. Olivucci, M. Merchan and J. Andres, J. Phys. Chem. B, 2008, 112, 7153–7156 CrossRef CAS PubMed.
- E. V. Gromov, I. Burghardt, H. Koppel and L. S. Cederbaum, J. Phys. Chem. A, 2011, 115, 9237–9248 CrossRef CAS PubMed.
- M. Boggio-Pasqua and G. Groenhof, Comput. Theor. Chem., 2014, 1040, 6–13 CrossRef.
- E. V. Gromov, J. Chem. Phys., 2014, 141, 224308 CrossRef PubMed.
- L. L. Premvardhan, M. A. van der Horst, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2003, 84, 3226–3239 CrossRef CAS PubMed.
- M. Boggio-Pasqua, M. A. Robb and G. Groenhof, J. Am. Chem. Soc., 2009, 131, 13580–13581 CrossRef CAS PubMed.
- E. V. Gromov, I. Burghardt, H. Koppel and L. S. Cederbaum, J. Photochem. Photobiol., A, 2012, 234, 123–134 CrossRef CAS.
- G. Groenhof, L. V. Schafer, M. Boggio-Pasqua, H. Grubmuller and M. A. Robb, J. Am. Chem. Soc., 2008, 130, 3250–3251 CrossRef CAS PubMed.
- M. L. Groot, L. J. G. W. van Wilderen, D. S. Larsen, M. A. van der Horst, I. H. M. van Stokkum, K. J. Hellingwerf and R. van Grondelle, Biochemistry, 2003, 42, 10054–10059 CrossRef CAS PubMed.
- A. F. Philip, R. A. Nome, G. A. Papadantonakis, N. F. Scherer and W. D. Hoff, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5821–5826 CrossRef CAS PubMed.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- P. B. Coto, D. Roca-Sanjuan, L. Serrano-Andres, A. Martin-Pendas, S. Marti and J. Andres, J. Chem. Theory Comput., 2009, 5, 3032–3038 CrossRef CAS PubMed.
- O. Vahtras, J. Almlof and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Ohm, M. Haser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–289 CrossRef CAS.
- R. Ahlrichs, Phys. Chem. Chem. Phys., 2004, 6, 5119–5121 RSC.
- H. A. Witek, Y. K. Choe, J. P. Finley and K. Hirao, J. Comput. Chem., 2002, 23, 957–965 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- B. G. Levine, J. D. Coe and T. J. Martinez, J. Phys. Chem. B, 2008, 112, 405–413 CrossRef CAS PubMed.
- C. D. Sherill, Counterpoise Correction and Basis Set Superposition Error, 2010, http://vergil.chemistry.gatech.edu/notes/cp.pdf.
- I. Mayer, J. Comput. Chem., 2007, 28, 204–221 CrossRef CAS PubMed.
- S. Gozem, F. Melaccio, A. Valentini, M. Filatov, M. Huix-Rotllant, N. Ferre, L. M. Frutos, C. Angeli, A. I. Krylov, A. A. Granovsky, R. Lindh and M. Olivucci, J. Chem. Theory Comput., 2014, 10, 3074–3084 CrossRef CAS PubMed.
- D. Tuna, D. Lefrancois, L. Wolanski, S. Gozem, I. Schapiro, T. Andruniow, A. Dreuw and M. Olivucci, J. Chem. Theory Comput., 2015, 11, 5758–5781 CrossRef CAS PubMed.
- G. J. Atchity, S. S. Xantheas and K. Ruedenberg, J. Chem. Phys., 1991, 95, 1862–1876 CrossRef.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. W. Schmidt and M. S. Gordon, Annu. Rev. Phys. Chem., 1998, 49, 233–266 CrossRef CAS PubMed.
- A. A. Granovsky, Firefly version 8, http://classic.chem.msu.su/gran/firefly/index.html.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- D. Krause and P. Thörnig, Journal of large-scale research facilities, 2018, 4, A132 CrossRef.
- V. Bonacic-Koutecky, J. Koutecky and J. Michl, Angew. Chem., Int. Ed. Engl., 1987, 26, 170–189 CrossRef.
- V. Bonacic-Koutecky, K. Schoffel and J. Michl, Theor. Chim. Acta, 1987, 72, 459–474 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- M. Dekhtyar and W. Rettig, J. Phys. Chem. A, 2007, 111, 2035–2039 CrossRef CAS PubMed.
- G. J. M. Dormans, G. C. Groenenboom, W. C. A. Vandorst and H. M. Buck, J. Am. Chem. Soc., 1988, 110, 1406–1415 CrossRef CAS.
- E. Giner, C. Angeli, A. Scemama and J. P. Malrieu, Comput. Theor. Chem., 2017, 1116, 134–140 CrossRef CAS.
- S. Olsen, J. Phys. Chem. B, 2015, 119, 2566–2575 CrossRef CAS PubMed.
- M. A. Robb, F. Bernardi and M. Olivucci, Pure Appl. Chem., 1995, 67, 783–789 CAS.
- F. Bernardi, M. Olivucci and M. A. Robb, Chem. Soc. Rev., 1996, 25, 321–328 RSC.
- M. A. Robb, in Conical Intersections, Theory, Computation, and Experiment, ed. W. Domcke, D. R. Yarkony and H. Köppel, World Scientific, Singapore, 2011, vol. 17, pp. 3–50 Search PubMed.
- P. B. Coto, A. Sinicropi, L. De Vico, N. Ferre and M. Olivucci, Mol. Phys., 2006, 104, 983–991 CrossRef CAS.
- M. Boggio-Pasqua, C. F. Burmeister, M. A. Robb and G. Groenhof, Phys. Chem. Chem. Phys., 2012, 14, 7912–7928 RSC.
- F. Molnar, M. Ben-Nun, T. J. Martinez and K. Schulten, THEOCHEM, 2000, 506, 169–178 CrossRef CAS.
- S. Sen and I. Schapiro, Mol. Phys., 2018, 116, 2571–2582 CrossRef CAS.
- K. F. Hall, M. Boggio-Pasqua, M. J. Bearpark and M. A. Robb, J. Phys. Chem. A, 2006, 110, 13591–13599 CrossRef CAS PubMed.
- S. Gozem, I. Schapiro, N. Ferre and M. Olivucci, Science, 2012, 337, 1225–1228 CrossRef CAS PubMed.
- S. Rinaldi, F. Melaccio, S. Gozem, F. Fanelli and M. Olivucci, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1714–1719 CrossRef CAS PubMed.
- H. L. Luk, N. Bhattacharyya, F. Montisci, J. M. Morrow, F. Melaccio, A. Wada, M. Sheves, F. Fanelli, B. S. Chang and M. Olivucci, Sci. Rep., 2016, 6, 38425 CrossRef CAS PubMed.
- S. Gozem, H. L. Luk, I. Schapiro and M. Olivucci, Chem. Rev., 2017, 117, 13502–13565 CrossRef CAS PubMed.
- J. W. Park and T. Shiozaki, J. Chem. Theory Comput., 2017, 13, 2561–2570 CrossRef CAS PubMed.
- K. B. Bravaya, B. L. Grigorenko, A. V. Nemukhin and A. I. Krylov, Acc. Chem. Res., 2012, 45, 265–275 CrossRef CAS PubMed.
- C. Y. Lin, M. G. Romei, L. M. Oltrogge, I. I. Mathews and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15250–15265 CrossRef CAS PubMed.
- J. Chang, M. G. Romei and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15504–15508 CrossRef CAS PubMed.
- Y. Orozco-Gonzalez, M. P. Kabir and S. Gozem, J. Phys. Chem. B, 2019, 123, 4813–4824 CrossRef CAS PubMed.
- S. Sekharan, K. Katayama, H. Kandori and K. Morokuma, J. Am. Chem. Soc., 2012, 134, 10706–10712 CrossRef CAS PubMed.
- S. Sekharan, J. N. Wei and V. S. Batista, J. Am. Chem. Soc., 2012, 134, 19536–19539 CrossRef CAS PubMed.
- R. Pal, S. Sekharan and V. S. Batista, J. Am. Chem. Soc., 2013, 135, 9624–9627 CrossRef CAS PubMed.
- E. Maximowitsch and T. Domracheva, unpublished work.
- M. G. Romei, C. Y. Lin, I. I. Mathews and S. G. Boxer, Science, 2020, 367, 76–79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S8 and Fig. S1–S3. See DOI: 10.1039/d0cp00814a |
| This journal is © the Owner Societies 2020 |