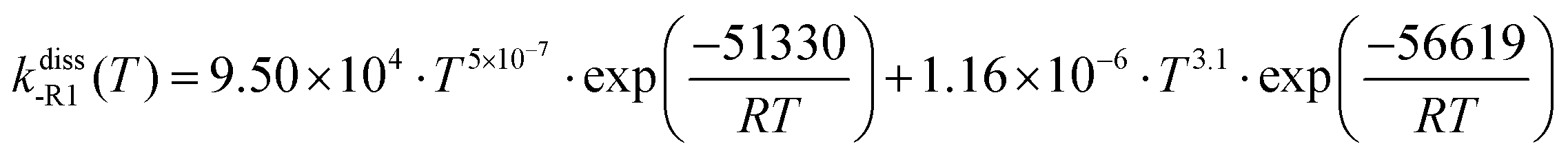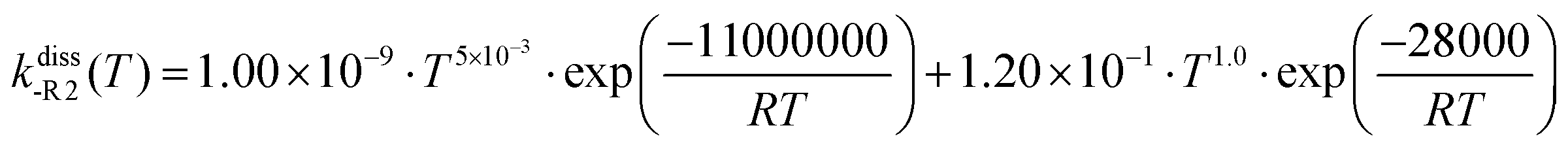Kinetics of the benzyl + HO2 and benzoxyl + OH barrierless association reactions: fate of the benzyl hydroperoxide adduct under combustion and atmospheric conditions†
Received
10th February 2020
, Accepted 27th March 2020
First published on 30th March 2020
Abstract
Radical–radical association reactions are challenging to address theoretically due to difficulties finding the bottleneck that variationally minimizes the reactive flux. For this purpose, the variable reaction coordinate (VRC) formulation of the variational transition state theory (VTST) represents an appropriate tool. In this work, we revisited the kinetics of two radical–radical association reactions of importance in combustion modelling and poly-aromatic hydrocarbon (PAH) chemistry by performing VRC calculations: benzyl + HO2 and benzoxyl + OH, both forming the adduct benzyl hydroperoxide. Our calculated rate constants are significantly lower than those previously reported based on VTST calculations, which results from a more efficient minimization of the reactive flux through the bottleneck achieved by the VRC formulation. Both reactions show different trends in the variation of their rate constants with temperature. We observed that if the pair of single occupied molecular orbitals (SOMOs) of the associating radicals show a similar nature, i.e. similar character, and thereby a small energy gap, a highly stabilized transition state structure is formed as the result of a very efficient SOMO–SOMO overlap, which may cancel out the free energy bottleneck for the formation of the adduct and result in large rate constants with a negative temperature dependence. This is the case of the benzoxyl and OH radical pair, whose SOMOs show O2p nature with an energy gap of 20.2 kcal mol−1. On the other hand, the benzyl and HO2 radical pair shows lower rate constants with a positive temperature dependence due to the larger difference between both SOMOs (a 28.9 kcal mol−1 energy gap) as a consequence of the contribution of the multiple resonance structures of the benzyl radical. The reverse dissociation rate constants were also calculated using multi-structural torsional anharmonicity partition functions, which were not included in previous work, and the results show a much slower dissociation of benzyl hydroperoxide. Our work may help to improve kinetic models of interest in combustion and PAH formation, as well as to gain further understanding of radical–radical association reactions, which are ubiquitous in different environments.
1. Introduction
Toluene (C7H8) and benzyl alcohol (C7H7OH) are common aromatic species in the combustion of diesel, gasoline and jet fuels, as well as in the formulation of surrogate fuels for combustion modeling.1,2 They can, respectively, generate benzyl (C7H7) and benzoxyl radicals (C7H7O) by hydrogen abstraction reactions of their benzyl and hydroxyl hydrogens, as they are weaker than the phenyl hydrogens.3
The radicals C7H7 and C7H7O can later either undergo unimolecular decomposition or bimolecular reactions with other species. At low and intermediate temperatures, when decomposition is not likely, the association reaction with other species might be prominent. Da Silva and Bozzelli found, in a theoretical study, that benzyl and benzoxyl radicals react with hydroperoxyl (HO2) and hydroxyl (OH) radicals, respectively, to form the adduct benzyl hydroperoxide (C7H8O2) at temperatures below 800 K (reactions (R1) and (R2));4 at higher temperatures (800–2000 K) they found that the oxygen transfer reaction between benzyl and HO2 radicals is the most prominent reaction in the consumption of the benzyl radical (reaction (R3)),
| | | C7H7 + HO2 → C7H7O + OH | (R3) |
Other species, such as O
25–7 and O,
8–10 are also involved in the removal of C
7H
7 and C
7H
7O, although to a minor extent.
Reactions (R1) and (R2), which take place on the singlet PES, are barrierless and very exothermic, and therefore are expected to be fast and important in combustion systems. In addition, the radical C7H7 is an important intermediate in the formation and growth of poly-aromatic hydrocarbons (PAHs), which are known to be soot precursors. For these reactions, da Silva and Bozzelli obtained,4 based on variational transition state theory (VTST) and electronic structure calculations at the G3B311 level, very large rate constants which vary within the 9.02 × 10−10–5.82 × 10−12 and 5.73 × 10−5–1.57 × 10−10 cm3 molecule−1 s−1 ranges, respectively, at temperatures between 300 and 2000 K.
The adduct benzyl hydroperoxide formed in the radical–radical association reactions (R1) and (R2) might be ro-vibrationally excited, as both reactions are highly exothermic with reaction enthalpies of −60.44 and −45.01 kcal mol−1, respectively4 (values calculated at the G3B3 level of theory). This ro-vibrationally excited adduct can be also formed by the reactions of the benzylperoxy radical (C7H7O2) with H and HO2 radicals,12,13
| | | C7H7O2 + HO2 → C7H8O2 + O2 | (R5) |
Reaction (R4) has been previously investigated by da Silva
et al. as an important step in the oxidation of the benzyl radical,
12 who reported quick formation of the adduct benzyl hydroperoxide with 87 kcal mol
−1 excess energy over the ground state. These authors performed a VTST study based on G3B3 electronic structure calculations and found that, under conditions of interest in combustion, benzyl hydroperoxide is not collisionally stabilized if formed through the radical–radical barrierless association
reaction (R4). The lifetime of the adduct toward decomposition to C
7H
7O + OH (reaction (-R2)), and to a lesser extent to C
7H
7 + HO
2 (reaction (-R1)), is very short; the former decomposition channel, with a reaction enthalpy of 45.01 kcal mol
−1, dominates at temperatures higher than 800 K and any pressure, while the latter, with a reaction enthalpy of 60.44 kcal mol
−1, becomes a minor decomposition channel at temperatures beyond 1500 K. Only at pressures equal to or higher than 10 atm does the collisional stabilization of the adduct C
7H
8O
2 become significant, but only if the temperature is not too high (below 800 K). As a result, da Silva
et al.12 concluded that benzyl hydroperoxide formed
viareaction (R4) is not likely to play a role in toluene oxidation as it will quickly dissociate
via reactions (-R2) and (-R1). This conclusion was also extended to the formation of benzyl hydroperoxide
viareaction (R1).
4
Nevertheless, the role of the reactions (R1), (R2), (-R1) and (-R2), and therefore that of benzyl hydroperoxide, in combustion of aromatics might be still uncertain due to two limitations of the former study by da Silva and Bozzelli.4 The first is the low level of theory used for the electronic structure calculations, G3B3, which can be nowadays upgraded with more sophisticated methods and basis sets using more advanced computational resources; G3B3 uses geometries and frequencies obtained with the B3LYP hybrid functional,14 which may not be appropriate to describe the investigated radical–radical association reactions. This second is the kinetic theory used for the calculations, that is, VTST. It is well known that VTST may fail to accurately describe the bottleneck to barrierless reactions such as these radical–radical association ((R1) and (R2)) and dissociation ((-R1) and (-R2)) reactions. Instead, variable reaction coordinate transition state theory, VRC-TST,15,16 is recommended as it variationally minimizes the reactive flux through that bottleneck more efficiently.
Therefore, in this work we revisited the previously calculated rate constants by da Silva and Bozzelli for the reactions (R1), (R2), (-R1) and (-R2) in order to obtain more accurate values over the wide temperature and pressure range 200–2500 K and 0.001–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atm, respectively, by using a higher level of theory for the electronic structure calculations and the VRC-TST methodology. Our goal is to shed light on the fate of the benzyl hydroperoxide adduct under combustion and atmospheric conditions; in addition, our calculated rate constants are useful to update kinetic models of fuels containing aromatic species, in order to obtain more accurate predictions on their combustion kinetics.
000 atm, respectively, by using a higher level of theory for the electronic structure calculations and the VRC-TST methodology. Our goal is to shed light on the fate of the benzyl hydroperoxide adduct under combustion and atmospheric conditions; in addition, our calculated rate constants are useful to update kinetic models of fuels containing aromatic species, in order to obtain more accurate predictions on their combustion kinetics.
2. Computational methods
2.1 Electronic structure calculations
The most computationally expensive step in the VRC-TST calculations is the electronic structure calculations. Hundreds of thousands of energy calculations need to be performed to explore different configurations of the system and obtain accurate rate constants. Since the rate constants are very sensitive to the level of theory used, one needs to carefully choose an appropriate level of theory. This is especially complicated if the system under investigation is large and contains several heavy atoms, as is the case of the title reactions.
Coupled cluster calculations, such as the widely used CCSD(T) method,17 are prohibitive for our VRC-TST calculations and thus we opted for density functional theory calculations. We tested several functionals in conjunction with the aug-cc-pvtz basis set18 by comparing their performance against the CCSD(T)/aug-cc-pvtz level, which was used as a benchmark. The functionals we tested were M06-2X19 for its good performance in kinetics and thermochemistry, M06-L20 for its good computational performance, MN12-L21 for its favorable computational performance and accuracy ratio for energetic purposes, and WB97XD22 with the aim of including dispersion corrections. Therefore, not only accuracy but also computational performance was considered in the functional selection for our computationally expensive VRC-TST calculations. The electronic structure calculations were carried out with the Gaussian09 package.23
2.2 VRC-TST formalism
The location of the dividing surface that minimizes the reactive flux in barrierless reactions is not straightforward because the separation between the two reacting fragments used to define that surface is very sensitive to the energy (E) and angular momentum (J), and also because the motion of one fragment relative to the other can be of large amplitude. The VRC formulation of TST15,16 overcomes these aspects by optimizing not only the value of the reaction coordinate s, but also its definition, which is now given by the distance between two pivot points which are located on each of the two reactants, as will be explained later. We minimized the number of transitional states N(E,J,s) as a function of E, J and s, yielding an E and J resolved reactive flux, N(E,J,s)E,J-μVT, within the VRC-E,J-μVT scheme.
The VRC-TST formalism sorts the normal modes into two different kinds, conserved and transitional modes. The conserved modes are the vibrational modes of the reacting fragments; their character barely changes during the reaction. The translational modes are the rotation of the reacting fragments as well as their relative motion; they are converted into vibrations and overall rotations along the course of the reaction as the association product is formed.
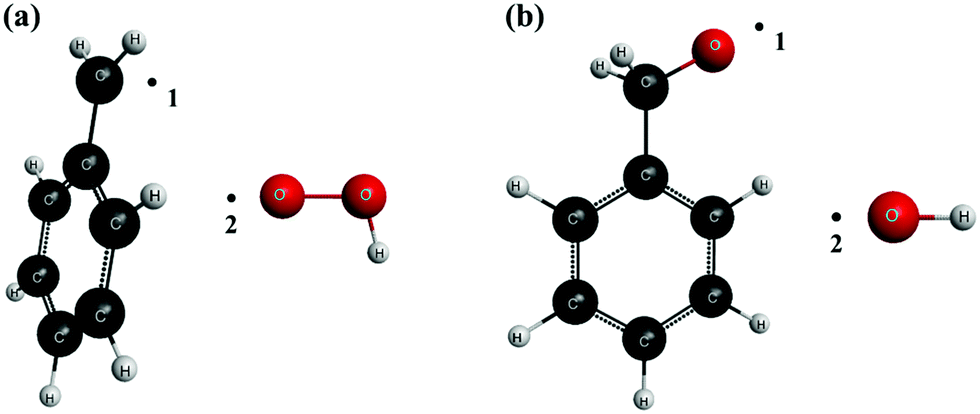
The variable reaction coordinate was defined by a single-faceted dividing surface which is determined by two pivot points, one for each reactant. The location of these pivot points is shown in Fig. 1 for reactions (R1)/(-R1) (panel a) and (R2)/(-R2) (panel b), respectively, as black dots. The distances d between pivot point 1 and the C atom of C7H7, and between pivot point 2 and the O atom of HO2 are, respectively, 0.3 Å and 0.35 Å; those between pivot point 1 and the O atom of C7H7O, and between pivot point 2 and the O atom of OH are also 0.3 Å and 0.35 Å, respectively. Pivot points 1 and 2 of the reactants are separated by a distance r12, so that s = r12, and the reactive flux was variationally minimized within the interval 1.5 Å ≤ s ≤ 5.0 Å; the location of the pivot points was also varied during the minimization of the flux from its original location, shown in Fig. 1, up to a distance d of 0.465 Å for C7H7 (pivot point linked to a C atom) and 0.515 Å for the rest of the species (pivot points linked to an O atom). The parameters s and d were scanned using intervals of 0.1 Å and 0.015 Å, respectively, determining, for each combination, N(E,J,s)E,J-μVT. We made sure that the reactive flux, and therefore the rate constant, was efficiently minimized at each temperature within the considered intervals for the parameters s and d.
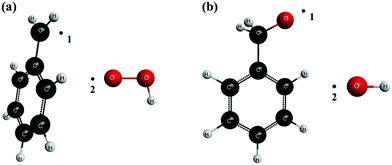 |
| | Fig. 1 Location of the pivot points that define the single-faceted dividing surface of reactions (R1,)/(-R1) (a) and (R2,)/(-R2) (b). | |
The high-pressure limit rate constant was determined as
| | 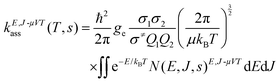 | (1) |
where
ge is the ratio of the electronic partition function of the transition state to the product of the electronic partition functions of the reactants,
Q1 and
Q2 are the rotational partition functions of the reactants without symmetry numbers,
σ≠,
σ1 and
σ2 are the symmetry numbers for the transition state, reactant 1 and reactant 2, respectively, and
ħ is the reduced Planck's constant. The
2Π
1/2 low-lying electronic excited state of the OH radical, with an energy
ε of 140 cm
−1,
24 was included in the calculations of the electronic partition function. For each combination of
d and
s, a set of 7000 configurations was explored by Monte Carlo integration to determine the flux
N(
E,J,s)
E,J-μVT, with a maximum angular momentum quantum number
J equal to 350. The calculated rate constants for
reaction (R1) were multiplied by two to account for the two equivalent pathways that can form C
7H
8O
2, that is, the approach of the HO
2 radical along one or the other side of the ring. The VRC-
E,J-μVT calculations were performed with the Polyrate
25 and Gaussrate
26 software. Additional details on these calculations can be found elsewhere.
15,16,25
For the definition of the pivot points, and therefore of the reaction coordinate, we tried to reproduce the most likely way both reactants approach each other. The pivot point of the benzyl species is located along the direction of its radical orbital, while those of the HO2, benzoxyl, and OH radicals were located along the direction of their O–O, C–O, and O–H bonds, respectively, that is, perpendicular to their radical orbitals. A multi-faceted divided surface that includes pivot points on both sides of the radical orbital of the species HO2, benzoxyl, and OH, that is, above and below their respective O–O, C–O, and O–H bonds, might be more appropriate to describe the way both reactants approach each other during the course of the reaction; however, this doubles the total number of electronic structure calculations for each of the two investigated reactions, which is not feasible given the size of the studied reactive system and its numerous heavy atoms.
The described VRC-E,J-μVT methodology was used to calculate the rate constants of reactions (R1) and (R2), kE,J-μVTass,R1 and kE,J-μVTass,R2 (cm3 molecule−1 s−1), respectively. The rate constants of the reverse dissociation reactions (-R1) and (-R2), kdiss-R1 and kdiss-R2 (s−1), respectively, were obtained by using kE,J-μVTass,R1 and kE,J-μVTass,R2 and the concentration equilibrium constants of the dissociation process, Kdissc,-R1 and Kdissc, -R2 (cm−3 molecule),
| | | kdiss-R1/-R2 = Kdissc,-R1/-R2·kE,J-μVTass,R1/R2 | (2) |
The calculation of the concentration equilibrium constants is described in Section 2.3.
2.3 Thermochemistry and NASA polynomials
The thermodynamic functions free energy (G), entropy (S), enthalpy (H) and heat capacity at constant pressure (Cp) of the species benzyl hydroperoxide were calculated by using the multi-structural torsional anharmonicity approximation with uncoupled torsions, MS-T(U), which includes the effect of the multiple conformers by means of multi-structural torsional partition functions;27 the software MSTor28 was used for the calculations. The term “conformers” refers to distinguishable structures that can be converted into each other by internal rotations. Under the MS-T(U) scheme, the conformational ro-vibrational partition function of a given species is calculated as| | 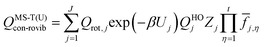 | (3) |
where J (this term should not be confused with the previously defined angular momentum quantum number) is the set of conformers. The conformer with the lowest potential energy (global minimum) is labeled as j = 1, and the potential energy of each conformer, Uj, is defined with respect to that of the global minimum; therefore, Uj=1 = 0. Qrot,j is the classical rotational partition function, the factor β is 1/kbT, kb is the Boltzmann's constant and Zj is a factor that makes the partition function reach the correct high temperature limit. The normal-mode harmonic oscillator partition function, QHOj, is given by| | | QHOj = exp(−hω/2kbT)[1 − exp(−hω/kbT)]−1 | (4) |
and it is adjusted to account for the torsional anharmonicity associated with the torsion η by the internal-coordinate torsional anharmonicity function ![[f with combining macron]](https://www.rsc.org/images/entities/i_char_0066_0304.gif) j,η (as well as by Zj). In eqn (4), h, ω, and T are Planck's constant, frequency and temperature, respectively.
j,η (as well as by Zj). In eqn (4), h, ω, and T are Planck's constant, frequency and temperature, respectively.
The function j,η is defined as
| | 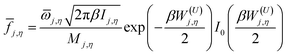 | (5) |
where
![[small omega, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0da.gif) j,η
j,η is an internal coordinate torsional frequency,
Ij,η is the torsional moment of inertia calculated by Pitzer's method without coupled torsions,
Mj,η is the local periodicity parameter for the torsion
η and
W(U)j,η is an uncoupled effective barrier height defined as
| | 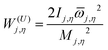 | (6) |
The thermochemistry data are given in the NASA polynomial format by determining the an fitting parameters
| | | Cp = (a1 + a2T + a3T2 + a4T3 + a5T4)·R | (7) |
| | 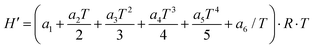 | (8) |
| | 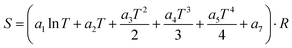 | (9) |
H′ in
eqn (8) is defined as
| | | H′(T) = ΔHf(298) + H(T) − H(298) | (10) |
where Δ
Hf(298) for benzyl hydroperoxide was determined with isodesmic reactions and
H is the multi-structural torsional thermodynamic function calculated with MSTor.
The change in the Gibbs free energy of the dissociation reactions (-R1) and (-R2), ΔGR, was also calculated from the multi-structural torsional thermodynamic functions G of the corresponding reactant and product species; this magnitude was used to calculate their dissociation equilibrium constants (unitless) as
| | 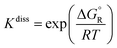 | (11) |
Then, the concentration equilibrium constant for reactions (-R1) and (-R2) (in cm
−3 molecule), which is used to determine the corresponding dissociation rate constants
kdiss-R1 and
kdiss-R2, as defined by
eqn (2) in Section 2.2, was calculated as
| | 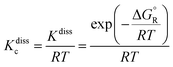 | (12) |
2.4 Pressure-dependent rate constants. SS-QRRK theory
Pressure-dependent rate constants of reactions (R1) and (R2) were calculated with the system-specific quantum RRK theory using the chemical activation mechanism for a bimolecular association reaction as implemented in the SS-QRRK utility code.29 This method, as opposed to other well-established and accurate methods to include pressure effects such as master equations, allows the user to include multi-structural anharmonicity effects, and has been successfully used to calculate rate constants for other radical–radical association reactions.30
The SS-QRRK method applies the steady-state approximation to the chemical activation mechanism, yielding the following pressure dependent association rate constant for reaction (R1) (and similarly for reaction (R2))
| | 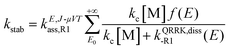 | (13) |
where [M] is the concentration of the bath gas (calculated using the ideal gas law),
E0 is the threshold energy of the dissociation of the adduct benzyl hydroperoxide to form the reactants benzyl and HO
2 radicals,
kc is the collisional deactivation rate constant for the activated adduct [benzyl hydroperoxide]*,
f(
E) is the fraction of energized species of [benzyl hydroperoxide]* at energy
E, and
kQRRK,diss-R1(
E) is a QRRK energy-resolved rate constant for the dissociation back to reactants of [benzyl hydroperoxide]*. They are calculated as
| | 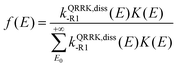 | (15) |
| | 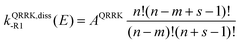 | (16) |
where
| | 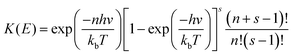 | (17) |
| | 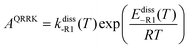 | (18) |
| | m = Ediss-R1/hc![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) | (19) |
AQRRK is the frequency factor,
Z and
βc are the Lennard-Jones collision rate constant and the collision efficiency, respectively,
n is the number of quanta excited at energy
E,
m is the number of quanta excited at energy
E0, and
s is the number of vibrational degrees of freedom of the adduct.
Ediss-R1 is the activation energy for the dissociation of the adduct into the reactants.
First, we obtained Ediss-R1 from the calculated kE,J-μVTass,R1 and the concentration equilibrium constant Kdissc,-R1 (eqn (2)), and, second, we fitted the high-pressure limit rate constants Kdiss-R1 to the following equation
| | 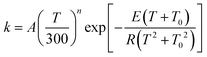 | (20) |
and determined the fitting parameters
A,
n,
E, and
T0. These fitting parameters were then used to determine the threshold energy
E0, which is set equal to the high-pressure limit Arrhenius activation energy for the dissociation reaction,
Ediss-R1| | 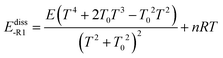 | (21) |
Using the values yielded by
eqn (21),
AQRRK can be calculated by using
eqn (18), so that
kstab can be also estimated.
To compute the Lennard-Jones collision rate constant we used the Lennard-Jones parameters σ = 6.000 and ε/kb = 410.0 K for benzyl hydroperoxide, which have been used in other work for toluene.31 We used N2 as the bath gas in our calculations, whose Lennard-Jones parameters are σ = 3.798 and ε/kb = 71.4 K.32
The collision efficiency βc calculated by the SS-QRRK utility code29 depends on the value of the thermal fraction of unimolecular states above the threshold energy, FE,
| |  | (22) |
| | 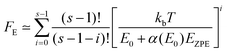 | (23) |
The definition given by
eqn (23) for
FE was derived by Troe,
33 where
α(
E0) is a correction factor and
EZPE is the zero point energy of the activated complex. It is well known that
FE adopts too large values for molecules with more than 8 atoms at high temperatures, leading to an unphysical underestimation of
βc and therefore to an underestimation of
kstab. The net result is an overestimation of pressure effects at high temperatures in large systems; we have highlighted this behavior in previous work,
34 and fixed it in a recent study.
35 In our approach, we used a more accurate collision efficiency parameter by introducing the correction factor
Δ proposed by Gilbert
et al.36| |  | (24) |
Eqn (24) allowed us to derive a more accurate
FE parameter, called
FE′, which was used instead in our SS-QRRK calculations as input. This corrected
FE′ parameter is not overestimated, adopting significantly lower values than the former one, and therefore correcting the described underestimation of the rate constants as the temperature increases and pressure decreases.
The de-activation averaged energy transferred value was determined in our calculations by using the following temperature-dependent function
| | 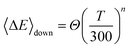 | (25) |
where
Θ = 200 cm
−1 and
n = 0.85.
37
3. Results and discussion
3.1 Benchmark of the DFT methods against CCSD(T)
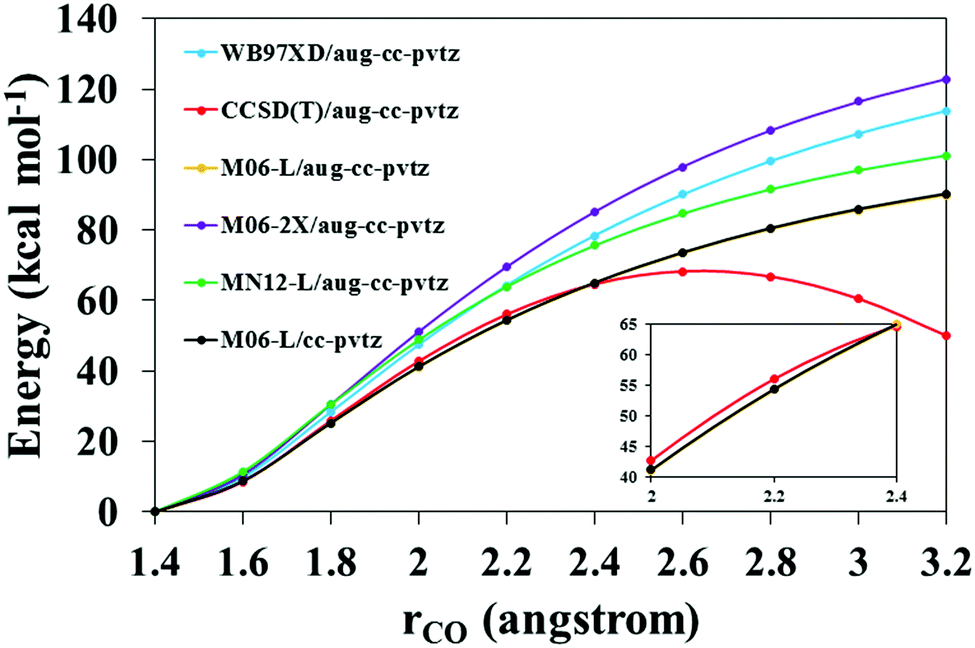
The reactive system C7H7 + HO2(R1) was used to test the performance of different functionals by comparing them to the benchmark CCSD(T)/aug-cc-pvtz level of theory. The comparison consisted of an evaluation of the potential energy profile as the association product C7H8O2 dissociates. We first used the previously described functionals with the same basis set as that used at the benchmark level (as described in Section 2.1), and selected the one with the best performance, which turned out to be the M06-L functional. However, the M06-L/aug-cc-pvtz level of theory was too computationally expensive to perform VRC-E,J-μVT calculations for a system with 9 heavy atoms. We therefore reduced the basis set, and analyzed the performance of the M06-L/cc-pvtz level as well. These results are shown in Fig. 2.
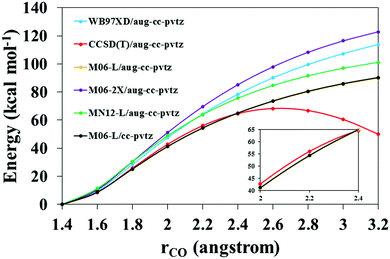 |
| | Fig. 2 Dissociation potential energy profile of the adduct C7H8O2 as a function of the C–O bond distance. Energies are defined with respect to that of the optimized C7H8O2 species. CCSD(T)/aug-cc-pvtz, M06-L/aug-cc-pvtz and M06-L/cc-pvtz levels are shown in the inset figure for a closer comparison. Orange and black lines overlap. | |
The M06-L/aug-cc-pvtz level (orange line) yields the best agreement with the CCSD(T)/aug-cc-pvtz benchmark level (red line). Using a smaller basis set, that is, the M06-L/cc-pvtz level (black line), we obtained essentially the same results; both the orange and black lines overlap. Therefore, we selected the M06-L/cc-pvtz level of theory for the VRC-E,J-μVT calculations as it is computationally cheaper and made our calculations feasible; the corresponding frequency scale factor 0.994 was also use in the rate constant calculations.38
It should be noted that those geometries with interatomic distances rCO > 2.4 Å show multi-reference character, with larger T1 values39 than those with rCO ≤ 2.4 Å (T1 < 0.025). This is the reason for the decrease in the potential energy predicted by the CCSD(T)/aug-cc-pvtz level beyond rCO = 2.4 Å (Fig. 2), leading to an unphysical bump. However, appropriate functionals, such as those used in the present work, can correct this unphysical behavior and lead to smooth potential energy curves. The higher level of theory CCSD(T)/aug-cc-pvtz should be thus considered as a benchmark in our calculations only for values of the reaction coordinate up to 2.4 Å; beyond that point, the abovementioned functionals perform better, predicting a physically correct behavior with a smooth asymptote at larger rCO distances which reaches the correct dissociation limit.
3.2 High-pressure limit rate constants of the association reactions (R1) and (R2): VRC-E,J-μVT calculations
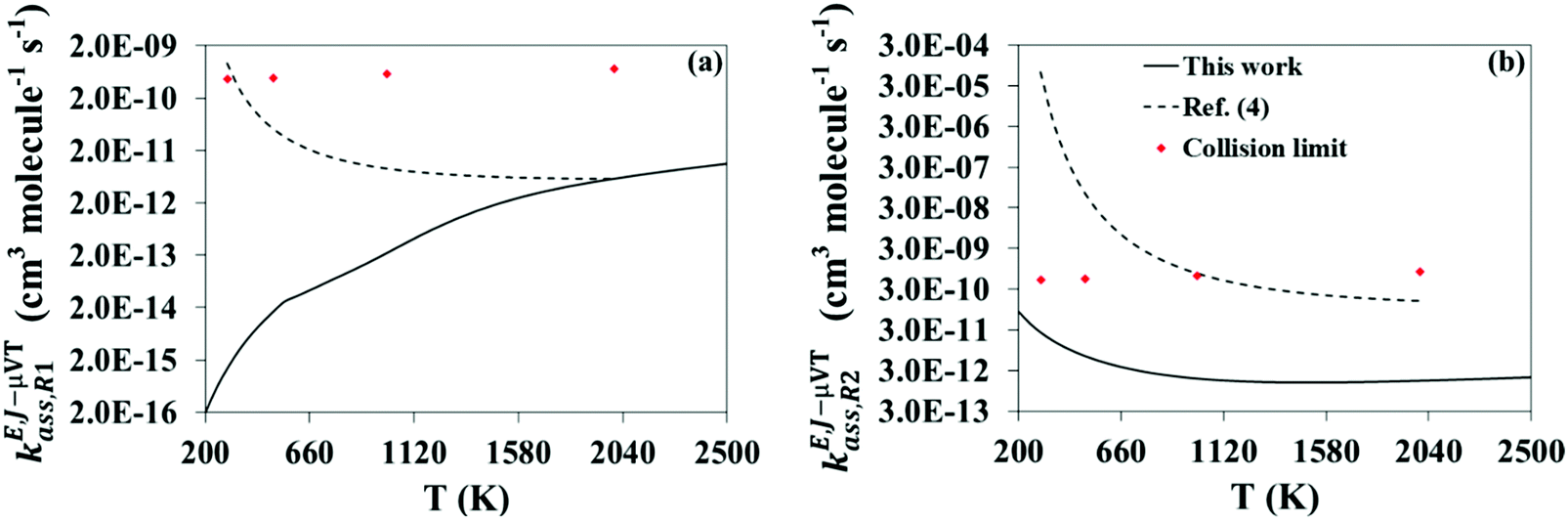
The calculated high-pressure limit association rate constants for (R1), kE,J-μVTass,R1, and (R2), kE,J-μVTass,R2, are plotted as a function of temperature in Fig. 3(a and b), respectively, together with those previously reported by da Silva and Bozzelli4 for comparison; the rate constants as determined by the collision limit40 are also plotted for further discussion. Our calculated values are also tabulated in Table 1, and fitted to the following two-term Arrhenius expression (in cm3 molecule−1 s−1)| | 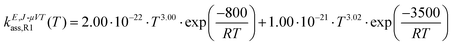 | (26) |
| | 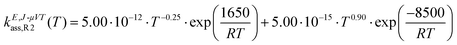 | (27) |
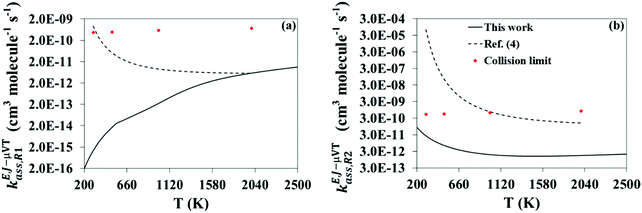 |
| | Fig. 3 Calculated high-pressure limit rate constants for reactions (R1) (a) and (R2) (b) using the VRC-E,J-μVT scheme (black solid line). Those calculated by da Silva and Bozzelli4 (black dashed line) with the VTST method and estimated by the collision limit40 (red markers) are also shown for comparison. | |
Table 1 VRC-E,J-μVT high-pressure limit rate constants for reactions (R1), kE,J-μVTass,R1, and (R2), kE,J-μVTass,R2
|
T (K) |
k
E,J-μVTass,R1 (cm3 molecule−1 s−1) |
k
E,J-μVTass,R2 (cm3 molecule−1 s−1) |
| 200 |
1.96 × 10−16 |
8.38 × 10−11 |
| 250 |
5.91 × 10−16 |
4.32 × 10−11 |
| 298 |
1.40 × 10−15 |
2.61 × 10−11 |
| 350 |
3.14 × 10−15 |
1.67 × 10−11 |
| 400 |
5.99 × 10−15 |
1.17 × 10−11 |
| 450 |
1.04 × 10−14 |
8.66 × 10−12 |
| 500 |
1.69 × 10−14 |
6.70 × 10−12 |
| 600 |
3.29 × 10−14 |
4.44 × 10−12 |
| 700 |
5.19 × 10−14 |
3.25 × 10−12 |
| 800 |
8.27 × 10−14 |
2.57 × 10−12 |
| 900 |
1.33 × 10−13 |
2.15 × 10−12 |
| 1000 |
2.22 × 10−13 |
1.89 × 10−12 |
| 1100 |
3.77 × 10−13 |
1.72 × 10−12 |
| 1200 |
6.23 × 10−13 |
1.61 × 10−12 |
| 1300 |
9.77 × 10−13 |
1.55 × 10−12 |
| 1400 |
1.44 × 10−12 |
1.52 × 10−12 |
| 1500 |
2.00 × 10−12 |
1.51 × 10−12 |
| 1600 |
2.65 × 10−12 |
1.51 × 10−12 |
| 1700 |
3.37 × 10−12 |
1.54 × 10−12 |
| 1800 |
4.14 × 10−12 |
1.57 × 10−12 |
| 1900 |
4.96 × 10−12 |
1.62 × 10−12 |
| 2000 |
5.83 × 10−12 |
1.67 × 10−12 |
| 2300 |
8.87 × 10−12 |
1.87 × 10−12 |
| 2500 |
1.13 × 10−11 |
2.02 × 10−12 |
Large discrepancies were observed between both series of data, especially in kE,J-μVTass,R1 at low temperatures. We believe that these discrepancies are mainly due to the different methodologies used in both studies to calculate the rate constants, VRC-E,J-μVT and VTST. The former (used in this work) is the methodology recommended for barrierless association reactions as the latter (used in ref. 4) may fail to locate the correct bottleneck or transition state. At 300 K, da Silva and Bozzelli located the transition state of reaction (R1) at rCO = 2.9 Å, but our calculations reveal that a transition state located at 2.6 Å (Fig. S1 in the ESI†) minimizes the rate constants much more efficiently. As the temperature increases, the opposite trend was found. For instance, at 2000 K da Silva and Bozzelli found the transition state at shorter interatomic distances, 2.4 Å, while that found by our VRC-E,J-μVT calculations is located at a greater distance, 2.9 Å.
The transition state of (R2) seems to be tighter and does not shift with temperature as that of reaction (R1). Our calculations predict this transition state to be located at values of the rOO distance of 2.3 Å at both temperatures, 300 K and 2000 K (Fig. S1, ESI†). This is in agreement with the results reported in ref. 4, whose authors also found a tighter transition state located at 2.5 Å at 300 K and 2000 K. The agreement between both methodologies, VRC-E,J-μVT and VTST, in the location of the transition state of (R2) is better than that found in reaction (R1), and results from the tighter nature of the transition state of the former compared to that of the latter. This might be the reason why the discrepancies between the results reported in both studies are smaller in reaction (R2) than in (R1). A potential energy profile diagram showing the stationary points of the reactions (R1)–(R5) considered in this work is shown in Fig. S2 of the ESI.†
It is worth noting the presence of a very shallow minimum at 1500 K in the rate constants for (R2), showing a non-Arrhenius behavior. This might be due to the presence of a shallow and non-bonded pre-reactive intermediate complex with van der Waals and/or hydrogen bond interactions located along the association reaction coordinate. These intermediate complexes sometimes induce a submerged barrier or transition state, resulting in rate constants with non-Arrhenius behavior as a consequence of a change in the sign of the activation energy.41,42 All our attempts to optimize and characterize such an intermediate complex failed, but, if present, it might have been detected by the electronic structure calculations, affecting the determination of the reactive flux and therefore the final VRC-E,J-μVT rate constant temperature dependence.
An interesting feature of our calculated rate constants is the different temperature dependence in reactions (R1) and (R2). The former, with a positive temperature dependence, and thus a positive activation energy, is in stark contrast to the results reported in ref. 4, which show the opposite trend. The latter, in agreement with the results in ref. 4, show a negative temperature dependence, and therefore a negative activation energy.
For a bimolecular reaction and in the absence of tunneling, as is the case of reactions (R1) and (R2), the activation energy Ea is given by
| | | Ea = ΔH≠° + RT = ΔG≠° + T(ΔS≠° + R) | (28) |
where Δ
H≠°, Δ
G≠° and Δ
S≠° are the activation enthalpy, free energy and entropy (barriers), respectively,
T is temperature and
R is the ideal gas constant. Although the studied reactions are barrierless in terms of potential energy (Δ
H≠°), they all are hindered by entropy (Δ
S≠°), as two molecules are being brought together to form an adduct. The value and sign of
Ea, and thus the temperature dependence of the rate constants, will be determined by the ability of the term Δ
H≠° to cancel out the hindering effect of Δ
S≠°. The more stabilizing the interaction between the two associating radicals, the lower is Δ
H≠° and therefore
Ea. This will be reflected in the evaluation of the flux
N(
E,
J,
s)
E,J-μVT used to calculate
kE,J-μVTass(
T,
s) in
eqn (1);
N(
E,
J,
s)
E,J-μVT is determined for each set of the parameters
s and
d by calculating the energy of the interacting system in thousands of configurations by means of electronic structure calculations, as explained in Section 2.2.
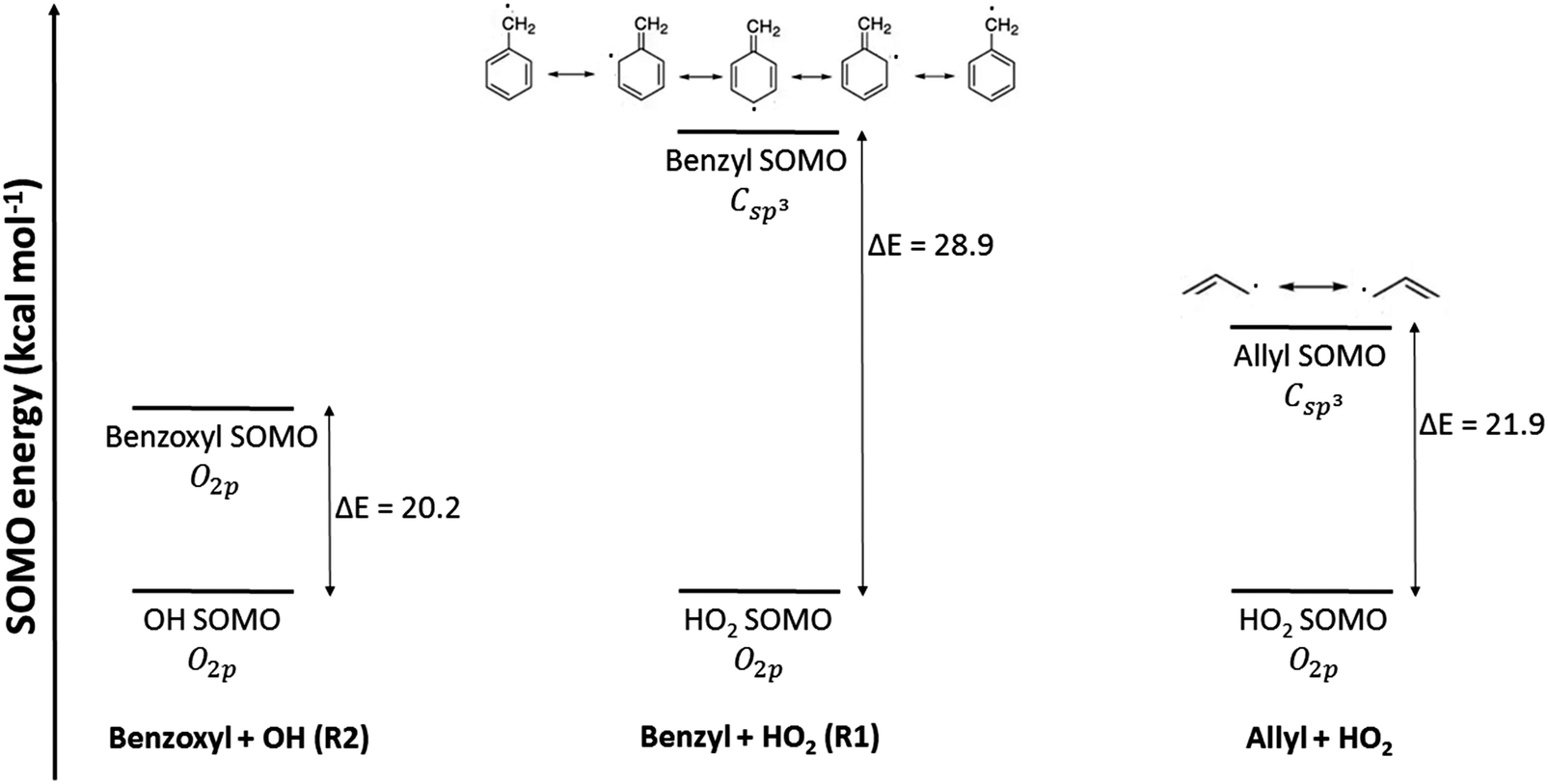
Therefore, we attempt to explain the observed temperature dependence of the calculated rate constants from a fundamental point of view. An important factor that should be considered to estimate the energy and stability of a configuration resulting from two associating radicals is the extent of the overlap between each of the single occupied molecular orbitals (SOMOs) of both radical fragments. These SOMOs are combined during the association reaction to form a new molecular orbital (MO) in the incipient/final association product. Two SOMOs, ψA(r) and ψB(r), that overlap very efficiently, as determined by the overlap integral,
| | 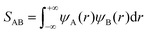 | (29) |
will yield highly stabilized configurations (which will eventually lead to an adduct) with respect to the separated radicals A and B, yielding low values of Δ
H≠° and therefore of
Ea, and thus favoring a negative temperature dependence of the rate constant. Similarly, low values of Δ
H≠° would give large values of the reactive flux
N(
E,
J,
s)
E,J-μVT and therefore large rate constants
kE,J-μVTass(
T,
s) (
eqn (1)). The degree of overlap between any two MOs, SOMOs in the case of radical–radical association reactions, will be largely determined by their nature and energy. In
reaction (R1), the SOMO orbital of the benzyl radical, C
sp3, does not overlap as efficiently with the SOMO orbital of the HO
2 radical, O
2p, as occurs between the two O
2p SOMOs in
reaction (R2). In the former and latter SOMO pairs, the differences in energy between both orbitals are respectively 28.9 and 20.2 kcal mol
−1 (using the same level of theory as that for the VRC-TST calculations). This is exemplified in
Fig. 4; SOMOs with similar nature and energy, as those in the benzoxyl (O
2p) and hydroxyl (O
2p) radicals (
reaction (R2)), lead to a larger overlap and more stabilized configurations, and this may explain the observed negative activation energy and negative temperature dependence of the rate constants, as opposed to the trend observed in
reaction (R1).
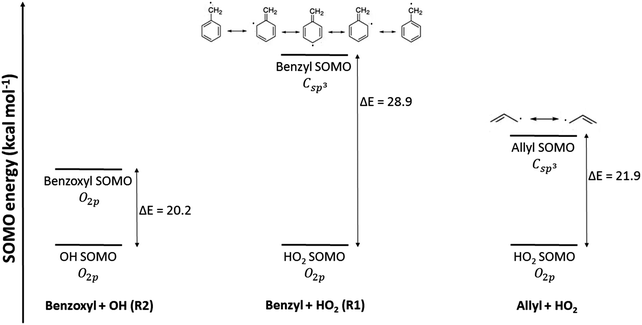 |
| | Fig. 4 Energy diagrams of the SOMO of the radicals involved in the studied reactions. Those of the radical–radical association reaction allyl + HO2 → allyl hydroperoxide investigated in ref. 43 are also shown for comparison. Energies were calculated at the M06-L/cc-pvtz level, and those of the OH and HO2 radicals were arbitrarily set as zero. | |
For comparison purposes, it is useful to look into a similar radical–radical association reaction between the allyl and the HO2 radicals, which forms allyl hydroperoxide. This reaction has been investigated by Goldsmith et al.,43 who also ran VRC-TST calculations to calculate the rate constants, and the energy gap between the SOMOs of the allyl and HO2 radicals is only 21.9 kcal mol−1 (Fig. 4), that is, much smaller than that of the benzyl and HO2 pair and similar to that of the benzoxyl and OH pair, and suggests that the radicals allyl and benzyl may not be so similar. Based on this analysis of the MOs, one could also expect a more similar trend between the Ea values of reactions (R2) and the association reaction between the allyl and HO2 radicals; in fact, the rate constants reported by Goldsmith et al.43 for the latter show a negative temperature dependence as those we calculated for reaction (R2), as opposed to those calculated for reaction (R1). These conclusions on the differences in the reactivity of the investigated radical species can be also inferred from the values of the rate constants; at low temperatures, when the rate constants are more sensitive to the ΔH≠° and Ea magnitudes, we find the following rate constants (in cm3 molecule−1 s−1) at 500 K: 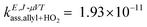 ,43kE,J-μVTass,R2 = 6.70 × 10−12 and kE,J-μVTass,R1 = 1.69 × 10−14.
,43kE,J-μVTass,R2 = 6.70 × 10−12 and kE,J-μVTass,R1 = 1.69 × 10−14.
The reason for the differences between the SOMOs of the benzyl and allyl radicals (Fig. 4), and therefore for their different reactivity towards association with the HO2 radical, might be the lower p character of the Csp3 SOMO of benzyl, which actually shows some Csp2 character because three of its five resonant structures, also shown in Fig. 4, display a double bond in that carbon center. This is not the case with the vinyl radical. That lower p character of the SOMO of benzyl, compared to that of allyl, leads to poorer mixing with the p SOMO of the O2p center of the HO2 radical, as was previously discussed (eqn (29)).
Our findings indicate that the rate constants reported in ref. 4 for reactions (R1) and (R2) are overestimated, and our calculated rate constants represent a more accurate estimation. This conclusion is supported by the collision limit rate constants we estimated for the investigated reactions based on the work by Chen et al.,40 which are plotted in Fig. 3. Our calculated rate constants for reactions (R1) and (R2) are below that limit within the whole temperature range we considered; this is not the case of the rate constants reported in ref. 4, which are affected by a collision limit violation at low and intermediate temperatures. This violation is especially pronounced at 300 K in reaction (R2), with a reported rate constant that exceeds the collision limit by 5 orders of magnitude.
We hope that this fundamental analysis can help understand trends in rate constants of other radical–radical reactions involving similar species, as well as in the formulation of more accurate analogies in kinetic modeling, which is a widespread practice among modelers and can be a source of uncertainty. However, it should be also noted that molecular orbitals are delocalized, which may make the task of establishing similarities between different SOMOs non-straightforward.
3.3 Thermochemistry of the benzyl hydroperoxide adduct and high-pressure limit rate constants of the dissociation reactions (-R1) and (-R2)
Using the methodology described in Section 2.3 and the M06-L/cc-pvtz level for the electronic structure calculations, we obtained the thermodynamic functions G, entropy S, H and Cp of the species C7H8O2 which results from reactions (R1) and (R2). In our calculations, we considered the effect of the multiple conformers or multi-structural anharmonicity, which was missed in previous studies,4 and included torsional anharmonicity as well. We found 3 different conformers, each of them with a non-superimposable specular image, by rotating the dihedrals H–O–O–C, O–O–C–C and O–C–C–C. In total, a set of 6 distinguishable conformers was considered in our calculations, whose optimized geometries are shown in Fig. 5; the global minimum is shown in Fig. 5(a), and the others are 0.76 kcal mol−1 (b) and 1.44 kcal mol−1 (c) higher in energy.
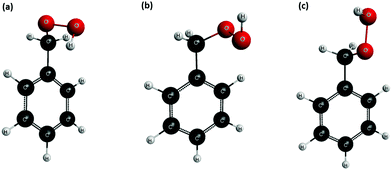 |
| | Fig. 5 Optimized geometries of the conformers found for C7H8O2 at the M06-L/cc-pvtz level. | |
The thermodynamic functions are given in the NASA polynomial format (eqn (7)–(9)), which is a common format in kinetic models. The fitted parameters of those polynomials are tabulated in Table 2 discerning two temperature intervals. The standard enthalpy of formation of the adduct C7H8O2 was determined by using the following isodesmic reactions, which involve the species methane (CH4), toluene (C7H8), methyl hydroperoxide (CH3OOH), propane (CH3CH2CH3), and 1-methylethyl hydroperoxide (CH3CH(HOO)CH3),
| | | C7H8O2 + CH4 → C7H8 + CH3OOH | (R6) |
| | | C7H8O2 + CH3CH2CH3 → C7H8 + CH3CH(HOO)CH3 | (R7) |
All the enthalpies of formation were obtained from the Active Thermochemical Tables
44 except that of the CH
3CH(HOO)CH
3 species, which was obtained from the NIST database.
38 At the M06-L/cc-pvtz level, the heat of reaction at 298 K of
reactions (R6) and (R7) is 5.39 and −4.44 kcal mol
−1, respectively, yielding an average value for the enthalpy of formation for the adduct C
7H
8O
2 of −5.87 kcal mol
−1; this value is in agreement with that calculated by da Silva and Bozzelli, who reported −5.00 kcal mol
−1.
Table 2 Fitted parameters of the NASA polynomials for C7H8O2
| Fitting parameter |
Low temperature (<1100 K) |
High temperature (1100–2500 K) |
|
a
1
|
4.00 |
8.38 × 10−11 |
|
a
2
|
4.80 × 10−2 |
4.32 × 10−11 |
|
a
3
|
−1.00 × 10−5 |
2.61 × 10−11 |
|
a
4
|
1.00 × 10−15 |
1.67 × 10−11 |
|
a
5
|
−5.00 × 10−12 |
1.62 × 10−12 |
|
a
6
|
−6.00 × 103 |
1.67 × 10−12 |
|
a
7
|
11.00 |
1.87 × 10−12 |
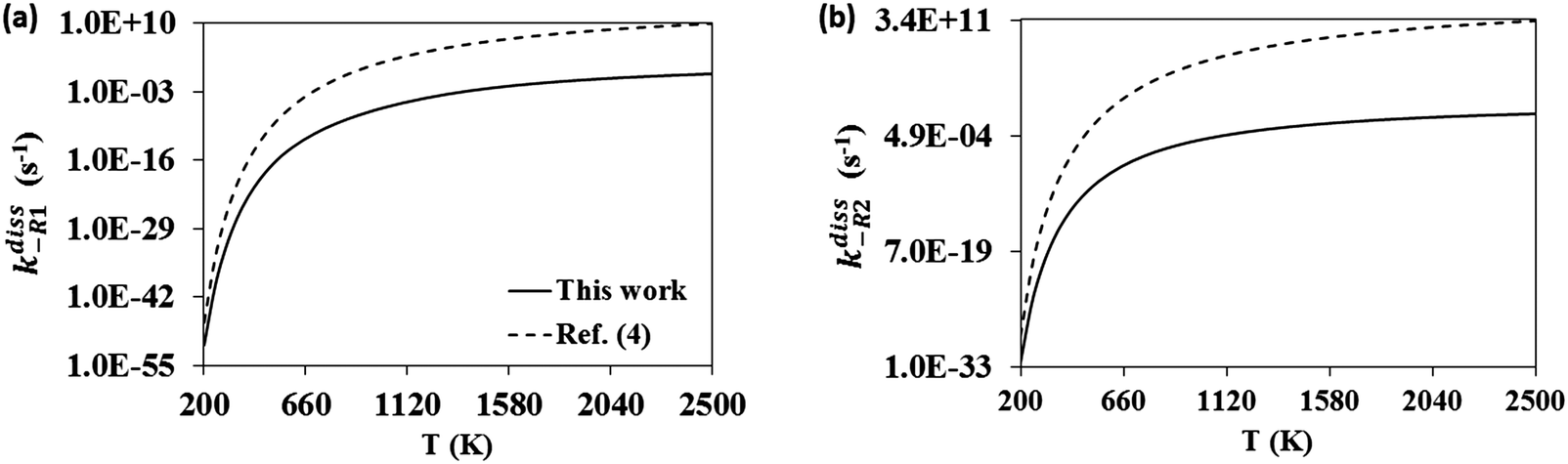
The calculated change in the Gibbs free energy, ΔGR, of the dissociation reactions (-R1) and (-R2) was used to calculate their concentration dissociation equilibrium constants Kdissc (eqn (12)) in order to determine the corresponding high-pressure limit dissociation rate constants (eqn (2)), kdiss-R1 and kdiss-R2. These rates are shown in Table 3 and plotted in Fig. 6(a and b), respectively. The results reported in ref. 4 are also plotted for comparison. The rate constants kdiss-R1 and kdiss-R2 were fitted to the following two-term Arrhenius expressions (s−1) within the 200–2500 K and 400–2000 K temperature intervals, respectively
| | 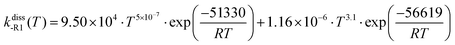 | (30) |
| | 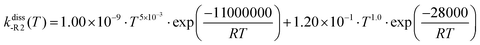 | (31) |
Table 3 High-pressure limit rate constants for reactions (-R1), kdiss-R1, and (-R2), kdiss-R2
|
T (K) |
k
diss-R1 (s−1) |
k
diss-R2 (s−1) |
| 200 |
5.22 × 10−52 |
2.84 × 10−33 |
| 250 |
1.07 × 10−40 |
1.03 × 10−25 |
| 298 |
2.18 × 10−33 |
6.51 × 10−21 |
| 350 |
1.02 × 10−27 |
2.94 × 10−17 |
| 400 |
1.18 × 10−23 |
1.11 × 10−14 |
| 450 |
1.68 × 10−20 |
1.04 × 10−12 |
| 500 |
5.65 × 10−18 |
3.72 × 10−11 |
| 600 |
2.90 × 10−14 |
7.07 × 10−9 |
| 700 |
1.14 × 10−11 |
2.69 × 10−7 |
| 800 |
1.05 × 10−9 |
3.82 × 10−6 |
| 900 |
3.71 × 10−8 |
2.86 × 10−5 |
| 1000 |
6.95 × 10−7 |
1.38 × 10−4 |
| 1100 |
8.15 × 10−6 |
4.86 × 10−4 |
| 1200 |
6.48 × 10−5 |
1.37 × 10−3 |
| 1300 |
3.72 × 10−4 |
3.22 × 10−3 |
| 1400 |
1.62 × 10−3 |
6.66 × 10−3 |
| 1500 |
5.64 × 10−3 |
1.24 × 10−2 |
| 1600 |
1.63 × 10−2 |
2.11 × 10−2 |
| 1700 |
4.05 × 10−2 |
3.37 × 10−2 |
| 1800 |
8.87 × 10−2 |
5.06 × 10−2 |
| 1900 |
1.75 × 10−1 |
7.26 × 10−2 |
| 2000 |
3.20 × 10−1 |
9.98 × 10−2 |
| 2300 |
1.35 × 100 |
2.15 × 10−1 |
| 2500 |
2.85 × 100 |
3.16 × 10−1 |
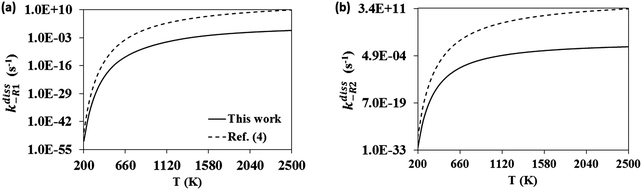 |
| | Fig. 6 Calculated high-pressure limit rate constants for reactions (-R1) (a) and (-R2) (b). Those calculated by da Silva and Bozzelli4 (dashed line) are also shown for comparison. | |
Large discrepancies are also observed between the two series of high-pressure limit dissociation rate constants we are comparing. Our rate constants for (-R1) and (-R2) are much smaller than da Silva's (also calculated with the VTST methodology at the G3B3 level), especially at intermediate and high temperatures, predicting a much slower dissociation back to reactants of the adduct C7H8O2 and probably a significantly larger lifetime towards decomposition. In addition to the limitations of the VTST methodology to tackle the studied reactions, the inclusion of multi-structural anharmonicity in the calculation of the partition functions of C7H8O2 may be also responsible to some extent for the observed discrepancies. This slower decomposition of C7H8O2 predicted in our study may change the picture drawn by da Silva and Bozzelli,4,12 who concluded that reaction (R4) may not play any role in toluene oxidation due to a quick decomposition of the adduct through reactions (-R1) and (-R2), supporting the exclusion of the adduct as an intermediate from kinetic models.
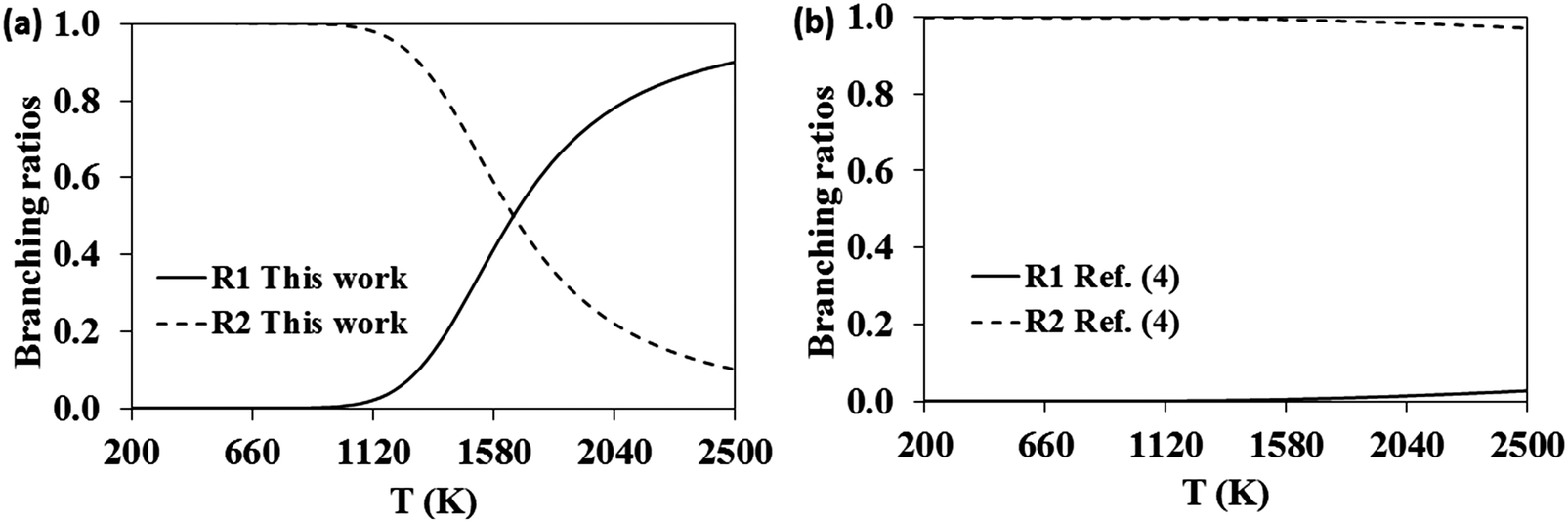
The relative importance of the pathways for the dissociation of the adduct in the high-pressure limit might be better addressed by analyzing their corresponding branching ratios, defined as kdiss-R1/(kdiss-R1 + kdiss-R2) and kdiss-R2/(kdiss-R1 + kdiss-R2) for reactions (-R1) and (-R2), respectively. These branching ratios and those calculated with the rate constants reported by da Silva and Bozzelli4 are plotted in Fig. 7. At low temperatures both studies predict similar results, and the adduct dissociates exclusively to the reactants C7H7O + OH, the dissociation channel to C7H7 + HO2 remaining unimportant. However, as the temperature increases, our results predict a larger role of the (-R1) dissociation channel, which becomes prominent at temperatures beyond 1700 K, as opposed to the behavior observed in ref. 4 in which (-R1) remains unimportant over the entire temperature range.
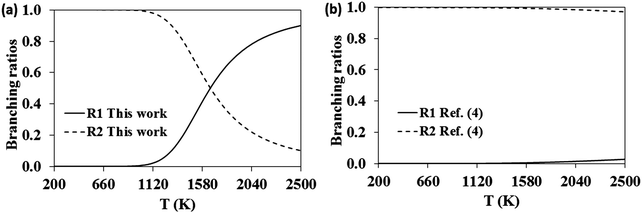 |
| | Fig. 7 High-pressure limit branching ratios for the dissociation reactions (-R1) (solid line) and (-R2) (dashed line). | |
3.4 Low-pressure rate constants of the association reactions (R1) and (R2): SS-QRRK calculations
We calculated the rate constants kE,J-μVTass,R1 and kE,J-μVTass,R2 at pressures below the high-pressure limit with SS-QRRK theory (Section 2.4), and observed no pressure effects even at pressures as low as 0.001 atm.
The reason for this behavior, which is in contrast to that found by da Silva and Bozzelli,4 is the much lower values we obtained for the dissociation rate constants kdiss-R1 and kdiss-R2, which make the frequency factor AQRRK (eqn (18)) and the microcanonical rate constant kQRRK,diss-R1(E) (eqn (16)) adopt very small values. This makes kstab (eqn (13)) insensitive to the collisional deactivation rate constant kc and the concentration of the bath gas M, and thus to pressure effects.
4. Conclusions
We conducted a theoretical kinetic study of the radical–radical barrierless association reactions benzyl + HO2 → benzyl hydroperoxide (R1,) and benzoxyl + OH → benzyl hydroperoxide (R2). These reactions are important in combustion of aromatic fuels and may play a role in poly-aromatic hydrocarbon (PAH) chemistry. The rate constants previously calculated for these reactions (R4) were revisited using higher levels of theory and more sophisticated methodologies.
We used the variable reaction coordinate (VRC) formulation of variational transition state theory (VTST) and obtained significantly lower rate constants than those previously reported. Pressure effects were estimated with the System-Specific Quantum Rice–Ramsperger–Kassel theory (SS-QRRK), and turned out to be negligible. In addition, rate constants for the dissociation of the adduct benzyl hydroperoxide were calculated by detailed balance using multi-structure torsional partition functions, also obtaining much lower values than those previously reported. Based on our calculated rate constants, we conclude that the adduct benzyl hydroperoxide is not as prone to dissociate back to either benzyl + HO2 (-R1) or benzoxyl + OH (-R2) as previously suggested, and may show larger lifetimes, and therefore a more important role during the combustion of aromatic fuels. At low temperatures dissociation to C7H7O + OH is the prominent dissociation channel, but beyond 1700 K dissociation to C7H7 + HO2 takes over.
Our calculations indicate a different trend of the rate constants of reactions (R1) and (R2) with temperature; the latter, with larger rate constants and negative temperature dependence, shows a similar trend to that reported in previous work43 for the radical–radical barrierless association reactions allyl + HO2 → allyl hydroperoxide, as opposed to the former with lower rate constant values and positive temperature dependence. These differences are explained by the degree of overlap between the SOMO orbitals of the associating radicals. The SOMOs of the radical pairs benzoxyl–OH and allyl–HO2 overlap very efficiently as a result of their similar p character, but this is not the case for those of the pair benzyl–HO2 due to the less pronounced p character of the SOMO of the radical benzyl as a result of its numerous resonant structures, which instead introduce some s character into the SOMO.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge funding from King Abdullah University of Science and Technology (KAUST), Clean Fuels Consortium and its member companies. For computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science and Technology (KAUST), Saudi Arabia. The work at LLNL was performed under the auspices of the U.S. Department of Energy (DOE), Contract DE-AC52-07NA27344, and was supported by the U.S. Department of Energy, Office of Basic Energy Sciences and Vehicle Technologies Office, program managers Wade Sisk, Kevin Stork, Mike Weismiller and Gurpreet Singh; it was conducted as part of the Co-Optimization of Fuels & Engines (Co-Optima) project sponsored by the DOE Office of Energy Efficiency and Renewable Energy (EERE), Bioenergy Technologies and Vehicle Technologies Offices.
References
- B. Chen, Z. Wang, J.-Y. Wang, H. Wang, C. Togbe, P. E. Alvarez Alonso, M. Almalki, M. Mehl, W. J. Pitz, S. W. Wagnon, K. Zhang, G. Kukkadapu, P. Dagaut and S. M. Sarathy, Fuel, 2019, 236, 1282–1292 CrossRef CAS.
- G. Kukkadapu, D. Kang, S. W. Wagnon, K. Zhang, M. Mehl, M. Monge-Palacios, H. Wang, S. S. Goldsborough, C. K. Westbrook and W. J. Pitz, Proc. Comb. Inst., 2019, 37, 521–529 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- G. da Silva and J. W. Bozzelli, Proc. Comb. Inst., 2009, 32, 287–294 CrossRef CAS.
- L. Elmaimouni, R. Minetti, J. P. Sawersyn and P. Devolder, Int. J. Chem. Kinet., 1993, 25, 399 CrossRef CAS.
- F. F. Fenter, B. Noziere, F. Caralp and R. Lesclaux, Int. J. Chem. Kinet., 1994, 26, 171 CrossRef CAS.
- Y. Murakami, T. Oguchi, K. Hashimoto and Y. Nosaka, J. Phys. Chem. A, 2007, 111, 13200 CrossRef CAS PubMed.
- K. Brezinsky, T. A. Litzinger and I. Glassman, Int. J. Chem. Kinet., 1984, 16, 1053 CrossRef CAS.
- M. Bartels, J. Edelbuttel-Einhaus and K. Hoyermann, Proc. Combust. Inst., 1989, 22, 1041 CrossRef.
- K. Hoyerman, J. Seeba, M. Olzmann and B. Viskolcz, Ber. Bunsen-Ges., 1997, 101, 538 CrossRef.
- A. G. Baboul, L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 1999, 110, 7650 CrossRef CAS.
- G. da Silva, M. R. Hamdan and J. W. Bozzelli, J. Chem. Theory Comput., 2009, 5, 3185–3194 CrossRef CAS PubMed.
- B. Du, W. Zhang, L. Mu, C. Feng and Z. Qin, Chem. Phys. Lett., 2007, 445, 17 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2003, 118, 5442–5455 CrossRef CAS.
- Y. Georgievskii and S. J. Klippenstein, J. Phys. Chem. A, 2003, 107, 9776–9781 CrossRef CAS.
- R. J. Barlett, J. Phys. Chem., 1989, 93, 1697–1708 CrossRef.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 13171–13174 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- M. W. Chase, C. A. Davis, J. R. Downey, D. J. Frurip, R. A. McDonald and A. N. Syverud, J. Phys. Chem. Ref. Data, 1985, 14, 1–383 CrossRef CAS.
-
J. Zheng, J. L. Bao, R. Meana-Pañeda, S. Zhang, B. J. Lynch, J. C. Corchado, Y.-Y. Chuang, P. L. Fast, W.-P. Hu and Y.-P. Liuet al., POLYRATE-version 2016-2A, University of Minnesota, Minneapolis, 2016 Search PubMed.
-
J. Zheng, S. Zhang, J. C. Corchado, R. Meana-Pañeda, Y. Y. Chuang, E. L. Coitiño, B. A. Ellingson and D. G. Truhlar, GAUSSRATE 2016, University of Minnesota, Minneapolis, 2016 Search PubMed.
- J. Zheng and D. G. Truhlar, J. Chem. Theory Comput., 2013, 9, 1356–1367 CrossRef CAS PubMed.
-
J. Zheng, S. L. Mielke, J. L. Bao, R. Meana-Pañeda, K. L. Clarkson and D. G. Truhlar, MSTor, version 2017-B, University of Minnesota, Minneapolis, 2017 Search PubMed.
-
J. L. Bao and D. G. Truhlar, SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory, version 2016-2A, Univeristy of Minnesota, Minneapolis, 2017 Search PubMed.
- J. L. Bao, X. Zhang and D. G. Truhlar, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13606–13611 CrossRef CAS PubMed.
- R. A. Eng, A. Gebert, E. Goos, H. Hippler and C. Kachiani, Phys. Chem. Chem. Phys., 2002, 4, 3989–3996 RSC.
-
B. Poling, The Properties of Gases and Liquids, McGraw-Hill Education, 5th edn, 2001 Search PubMed.
- J. Troe, J. Phys. Chem., 1979, 83, 114–126 CrossRef CAS.
- M. Monge-Palacios, E. Grajales-Gonzalez and S. M. Sarathy, J. Phys. Chem. A, 2018, 122, 9792–9805 CrossRef CAS PubMed.
- E. Grajales-Gonzalez, M. Monge-Palacios and S. M. Sarathy, submitted to J. Phys. Chem. A. Search PubMed.
- R. G. Gilbert, K. Luther and J. Troe, Ber. Bunsen-Ges., 1983, 87, 169–177 CrossRef CAS.
- J. P. Senosiain, S. J. Klippenstein and J. A. Miller, J. Phys. Chem. A, 2006, 110, 6960–6970 CrossRef CAS PubMed.
- NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, ed. D. Russell, Johnson III, 2016.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., Quantum Chem. Symp., 1989, 36, 199–207 CrossRef.
- D. Chen, K. Wang and H. Wang, Combust. Flame, 2017, 186, 208–210 CrossRef CAS.
- M. Monge-Palacios, M. P. Rissanen, Z. Wang and S. M. Sarathy, Phys. Chem. Chem. Phys., 2018, 20, 10806–10814 RSC.
- M. Monge-Palacios and S. M. Sarathy, Phys. Chem. Chem. Phys., 2018, 20, 4478–4489 RSC.
- C. F. Goldsmith, S. J. Klippenstein and W. H. Green, Proc. Comb. Inst., 2011, 33, 273–282 CrossRef CAS.
- B. Ruscic, R. E. Pinzon, G. von Laszewski, D. Kodeboyina, A. Burcat, D. Leahy, D. Montoya and A. F. Wagner, J. Phys. Chem. Ref. Data, 1985, 14, 1–383 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00752h |
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Edwing
Grajales-González
a,
Goutham
Kukkadapu
b and
S. Mani
Sarathy
*a,
Edwing
Grajales-González
a,
Goutham
Kukkadapu
b and
S. Mani
Sarathy
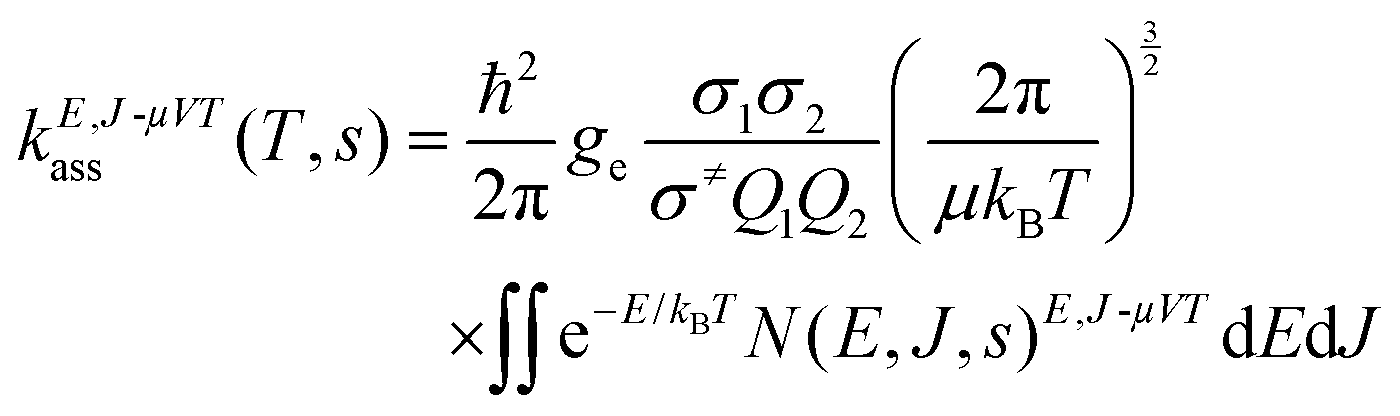
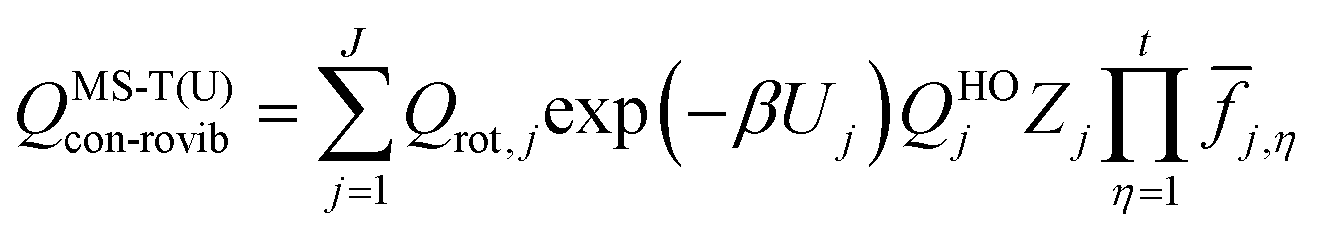
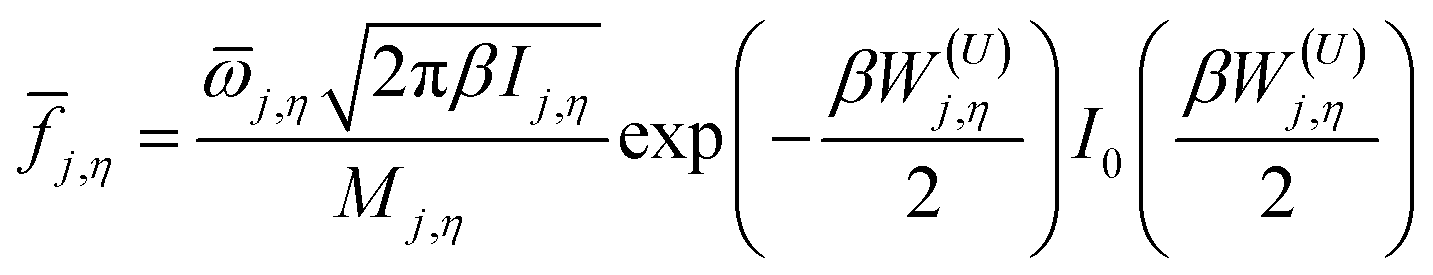
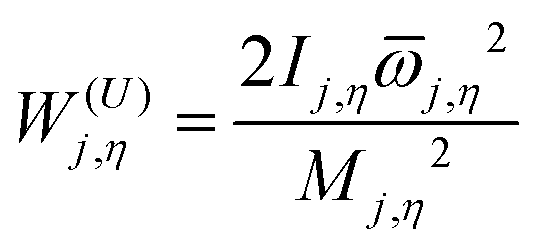
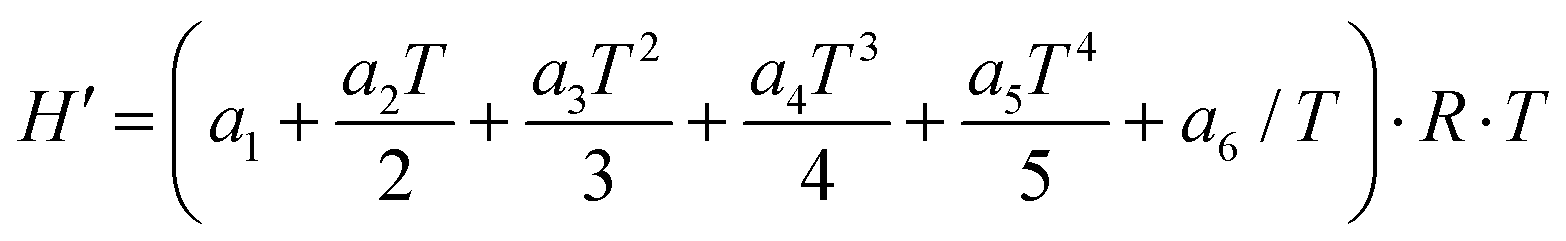
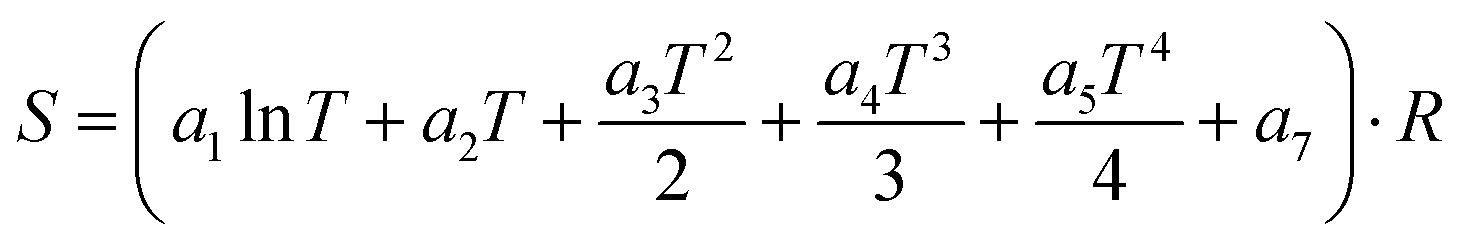
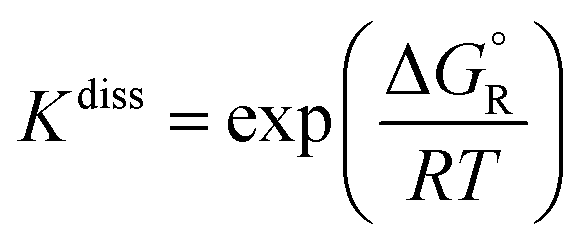
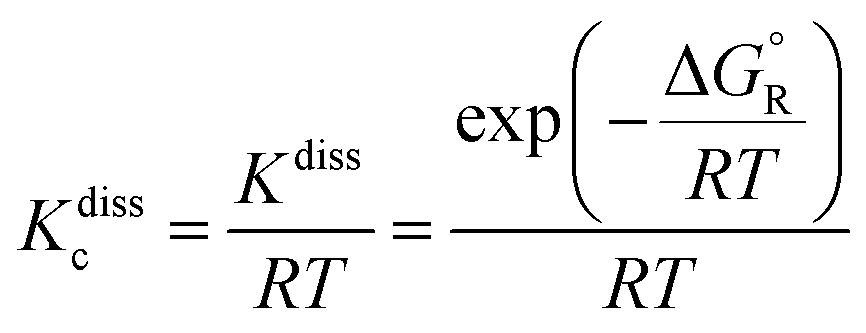
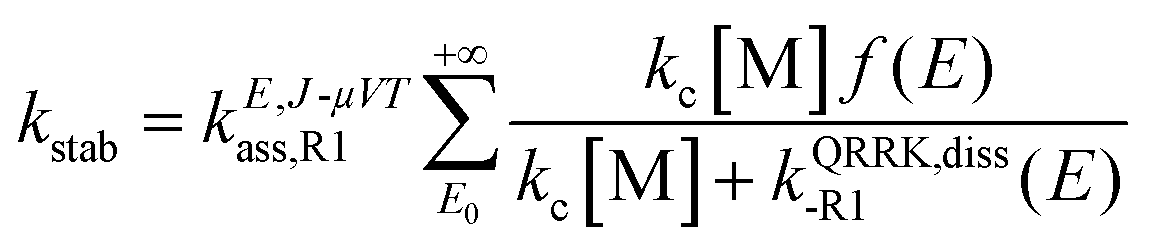
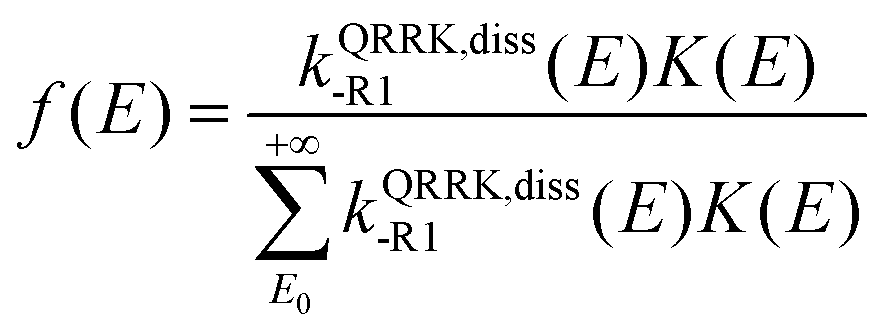
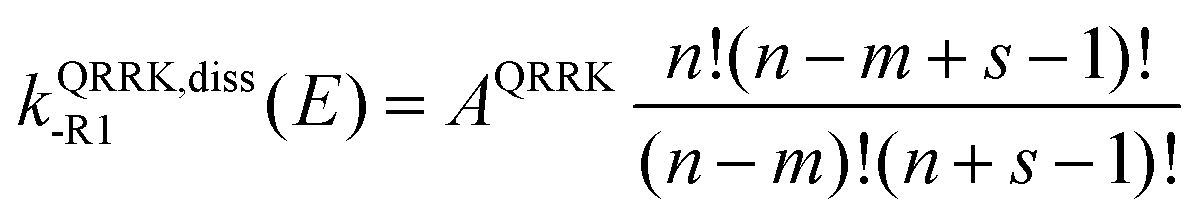
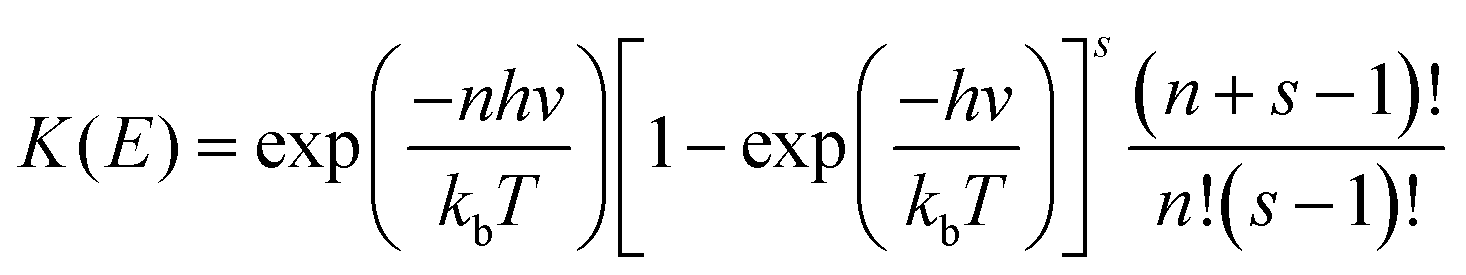
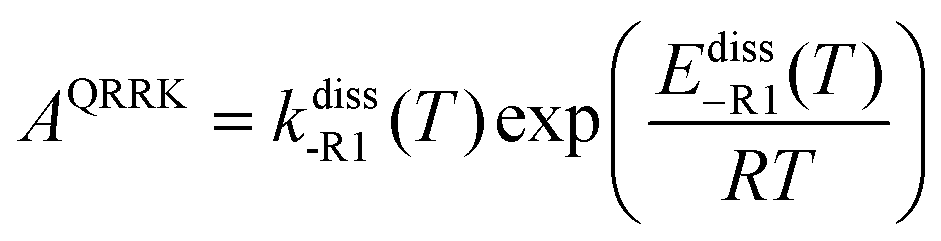
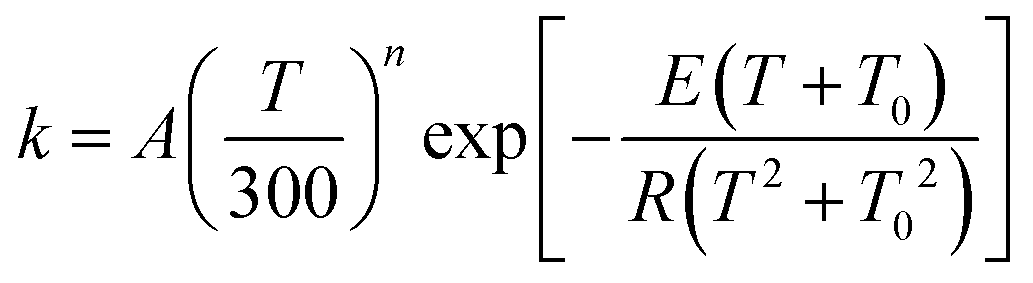
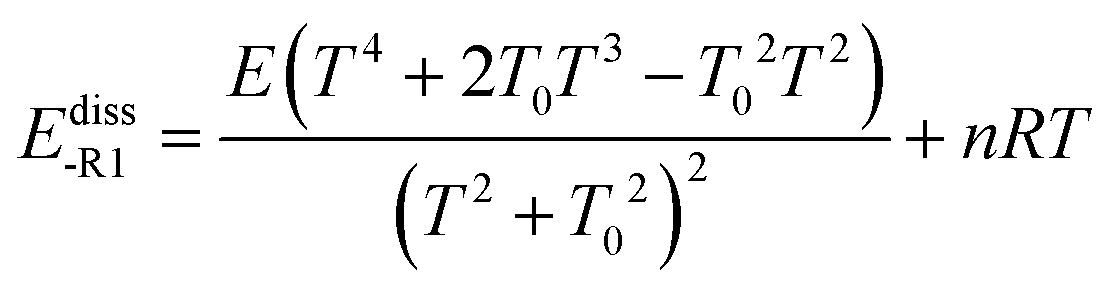
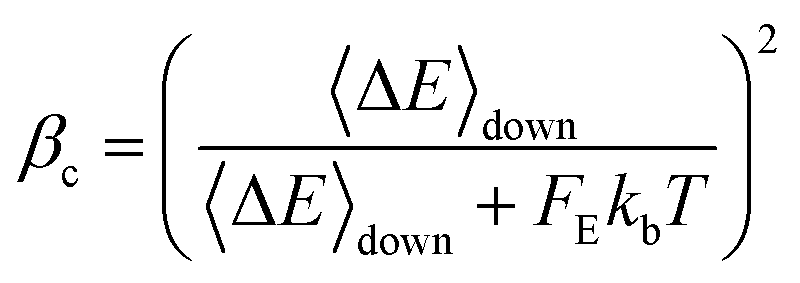
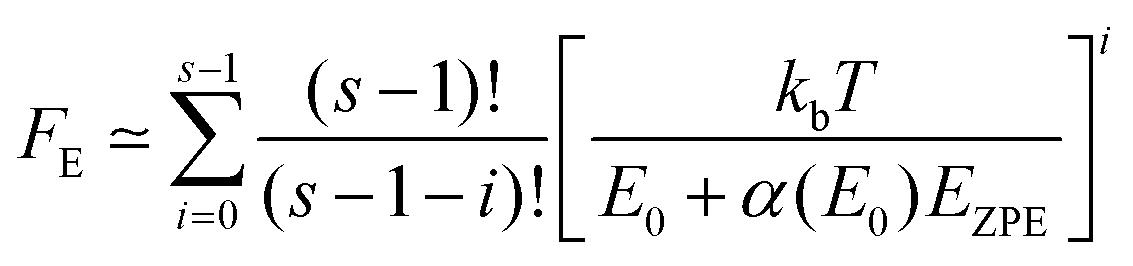
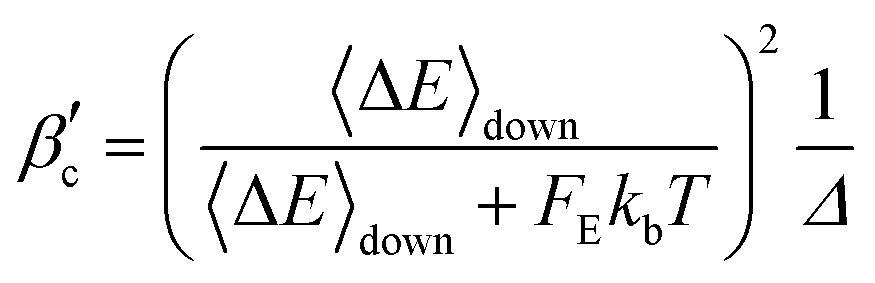
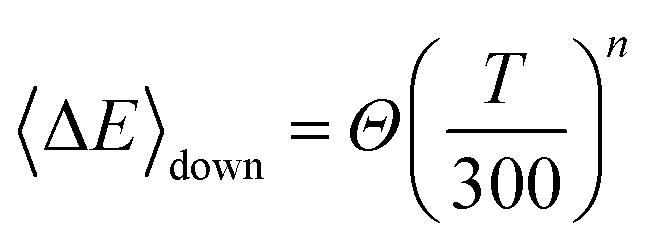
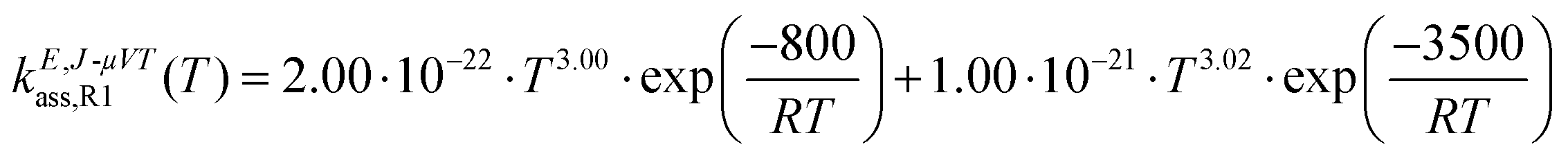
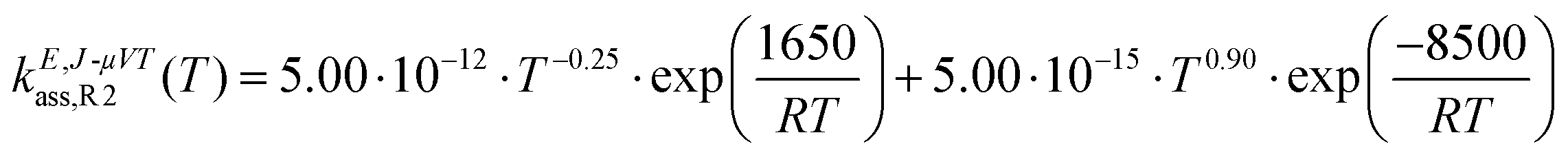
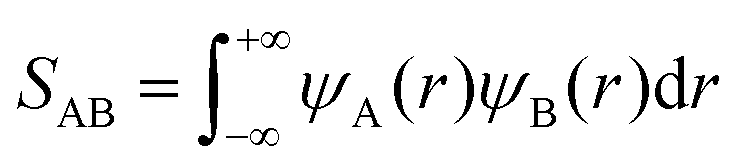
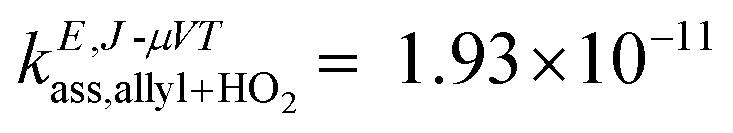 ,43kE,J-μVTass,R2 = 6.70 × 10−12 and kE,J-μVTass,R1 = 1.69 × 10−14.
,43kE,J-μVTass,R2 = 6.70 × 10−12 and kE,J-μVTass,R1 = 1.69 × 10−14.