
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton transfer dynamics modified by CH-stretching excitation
Tim
Michaelsen
,
Björn
Bastian
 ,
Patrick
Strübin
,
Jennifer
Meyer
and
Roland
Wester
*
,
Patrick
Strübin
,
Jennifer
Meyer
and
Roland
Wester
*
Institut für Ionenphysik und Angewandte Physik, Universität Innsbruck, Technikerstraße 25/3, 6020 Innsbruck, Austria. E-mail: roland.wester@uibk.ac.at; Tel: +43 512 507 52620
First published on 22nd April 2020
Abstract
Gaining insight how specific rovibrational states influence reaction kinetics and dynamics is a fundamental goal of physical chemistry. Purely statistical approaches often fail to predict the influence of a specific state on the reaction outcome, evident in a great number of both experimental and theoretical studies. Most detailed insight in atomistic reaction mechanisms is achieved using accurate collision experiments and high level dynamics calculations. For ion–molecule reactions such experiments are scarce. Here we show the influence of symmetric CH-stretching vibration on the rate and dynamics of proton transfer in the reaction of F− + CH3I. We find a pronounced shift in the reaction dynamics for excited reactions from indirect to preferred direct dynamics at higher collision energy. Moreover, excited reactions occur at larger impact parameters. Finally, we compare vibrational excitation with collision energy and find that vibration is overall more efficient in promoting reactivity, which agrees with recent theoretical calculations.
1 Introduction
A long standing question in chemistry is how specific rovibrational states influence reactivity, especially in comparison with equal amounts of collision energy. Early on this led John Polanyi to explain this influence with the location of the transition state on the reaction's potential energy surface.1 With the development of suitable lasers, they became the dominant tool for experimental state-preparation,2 but other approaches such as thermally populating many states and later disentangling their individual contribution are feasible as well.3 There have been a great number of experimental and theoretical studies with counter-intuitive results on the influence of specific rovibrational states on reaction kinetics and dynamics. They range from atom di-atom reactions, such as the well-studied reaction of F + H2,4,5 to larger systems such as halide methane and water reactions,6–9 ion molecule reactions10,11 and state specific surface interactions.12 To gain insight into the atomic level mechanisms and how a specific quantum state influence them, experiments under single collision conditions are desired. The combination of crossed beams with velocity map imaging has proven a powerful tool in the study of reaction dynamics,13,14 especially in comparison with state-of-the-art trajectory calculations.15 For example the reactions of CH-stretching excited deuterated methane CHD3 with Cl, F and O nicely illustrate the range of results and the need for state-of-the-art theory to interpret them.16–18 For the F and O cases the Polanyi rules1 do not predict the influence of vibrational excitation. Instead, a stereodynamic effect leads to suppression in the reaction with fluorine and to an enhanced cone of acceptance for reactions with oxygen. The effect was corroborated using quasi-classical trajectory (QCT) calculations on an ab initio potential energy surface (PES).19–23In order to extend the Polanyi rules to poly-atomic systems and predict the influence of vibrations, the sudden vector projection model (SVP) has been developed by Guo and coworkers.24,25 It projects the different internal modes and translational coordinates onto the intrinsic reaction coordinate at the transition state and calculates the individual overlap. From these overlap integrals a qualitative ranking is obtained of how effective each mode is at promoting reactivity. The SVP method has been successfully applied to explain many different experimental observations.25
Here we present an experimental study about the influence of symmetric CH-stretching excitation on the proton transfer channel of F− reacting with CH3I. The dominant channel in this reaction is the bimolecular nucleophilic substitution (SN2), but three additional endothermic pathways exist (reaction enthalpies taken from ref. 26 and ref. 27).
| F− + CH3I → I− + CH3F ΔH = −1.96 eV (SN2) | (1) |
| → CH2I− + HF ΔH = 0.69 eV (proton tr.) | (2) |
| → FI− + CH3 ΔH = 0.7 eV (halide abstr.) | (3) |
| → FHI− + CH2 ΔH = 0.9 eV (halide abstr.) | (4) |
We have recently shown that for the SN2 channel the symmetric stretching mode acts as a spectator, which is confirmed by SVP and QCT calculations.28 For the proton transfer reaction the situation is different and more similar to neutral halide reactions with CHD3,19 as we excite the bond that is broken during the reaction. This leads to interesting questions: Is vibration more efficient in promoting the reaction compared to collision energy and if so, does the enhancement depend on the collision energy? Is there a difference in the reaction dynamics for ground and excited state? Are stereodynamic effects important? To answer these questions we have obtained differential cross sections with and without vibrational excitation and compare our results to recently published QCT calculations by the Czakó group.26
2 Methods
We use a combination of crossed beams and 3D velocity map imaging to obtain angular- and energy-dependent differential cross sections for ion molecule reactions. The experiment has been described in more detail in previous publications.29,30 The F− ions are produced in a pulsed plasma discharge of NF3 in argon, thermalized in an octupole rf ion trap and crossed with a neutral molecular beam of CH3I seeded in helium under an angle of 60° in the center of the velocity map imaging spectrometer. We use a pulsed Laservision infrared OPO/OPA laser to excite the symmetric CH-stretching mode of CH3I prior to the reactive collision. A cylindrical lens is used to focus it into the neutral beam in one dimension to excite a large volume in the direction of expansion. The laser is pulsed at 10 Hz while the whole experiment runs at 20 Hz resulting in alternating laser on and off events under identical conditions.We obtain the fraction of excited molecules experimentally using a combination of spatial map imaging (SMI),31 electron photo-detachment of the F− in the ion beam and a photo-dissociation/REMPI scheme for CH3I adapted from Hu et al.32 This requires knowledge of the overlap of the two reactant beams and the IR laser. Fig. 1a shows a sketch of this overlap inside the bottom two VMI electrodes (beams scaled up for better visibility). The size of the ion and neutral beam is measured using SMI, photo-detachement and photo-dissociation/REMPI. The neutral beam was considerably smaller compared to the ion beam in all measurements, which reduces the overlap problem to the IR laser beam and the neutral beam. The excitation can be directly mapped using REMPI of the stretch-excited state (v = 1) of the CH3 fragment. By moving the spherical lens that focuses the photo-dissociation and REMPI UV lasers vertically through the neutral beam, as shown by the black arrow in the sketch in Fig. 1a, we obtain the spatial profile shown in green without and in red with the IR laser present in Fig. 1b. To quantify the excitation we use the depletion of the ground state at the center of the neutral beam (marked by shaded area in Fig. 1b) and extrapolate this result to the entire beam. The excited fraction was determined regularly and the mean value is used to scale our results to a fully excited beam. It is important to note that the vibrational mode stays for relatively long times (> 4 μs) without intramolecular vibrational redistribution (IVR), as tested using delayed REMPI.
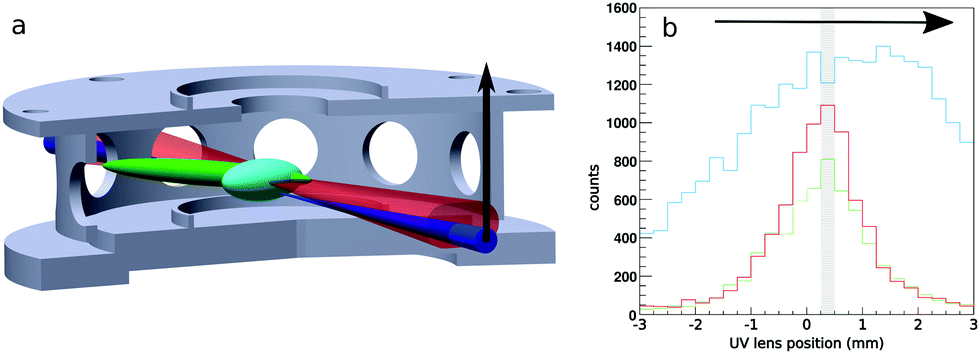 | ||
| Fig. 1 (a) Sketch of the ion (turquoise), neutral (green) and infrared laser beam (red) overlap with the photo-dissociation/REMPI ultraviolet laser (blue) in the interaction volume of the VMI spectrometer. (b) Vertical spatial profile of the neutral beam with (red) and without IR laser (green) along with the ion beam profile (turquoise). Each bin contains counts from either a photo-dissociation (v = 1) REMPI measurement of the CH3I beam with and without IR laser present or from photo-detached electrons from the F− beam. The x axis shows the position of the UV lens that focuses the two parallel UV beams into the chamber which is stepped through to obtain the spatial profile relative to the center of the vacuum window. A background is subtracted from the ion beam measurement for easier comparison. The shaded bin marks where the ground state depletion REMPI measurement was recorded. | ||
We record the velocity and arrival time for each product ion, which allows us to obtain branching ratios between competing product channels and differential cross sections for each product in a single measurement. To obtain sufficient statistics (roughly 1 million counts in the SN2 channel) at the three investigated collision energies a total measurement time of approx. 650 hours was necessary. Regular systematic checks of the collision energy (approx. every 6 h), the excited fraction and the beam overlaps (approx. every 30 h) were performed to ensure stable experimental conditions. The product velocity distributions are represented in the center of mass frame, where we choose the neutral beam direction as forward (see illustration at the top of Fig. 2). We project the 3D velocity distribution onto a 2D representation of the velocity along the collision axis vx and the radial velocity vr perpendicular to the collision axis.
 | ||
| Fig. 2 Differential cross sections, internal energy and angular distributions for ground and vibrationally excited proton transfer reactions in the reaction of F− + CH3I. The rows give the results for the three investigated collision energies 0.7, 1.2 and 2.3 eV. The first column shows the velocity distributions of CH2I− products for ground state reactions. The black circle marks the kinematic cutoff. The second column shows the difference of velocity distributions with and without vibrational excitation, corresponding to excited CH3I reactions. The red circle again marks the kinematic cutoff, taking the quanta of vibrational excitation into account. The final two columns show the internal energy of the two molecular products and the angular distribution as the cosine of scattering angle θ for the ground and excited state reactions in black and red respectively. The green circle and histograms illustrate the ground state indirect mechanism at 0.7 eV. The illustration at the top shows the Newton diagram in the chosen center-of-mass frame. The histograms are area-normalized. | ||
3 Results
In Fig. 2 we present measured differential cross sections for proton transfer, reaction (2), and the derived internal energy and angular distributions. The data have been obtained using the crossed beam velocity map imaging technique (see Methods section). Each row contains results for one of the experimental collision energies, 0.7, 1.2 and 2.3 eV respectively. The first two columns contain the two-dimensional velocity distributions in the scattering plane and in the center-of-mass frame of CH2I− products. The first column shows products without laser interaction, corresponding to the reaction of CH3I in the vibrational ground state. The black circles marks the maximum available kinetic energy for the product ions, calculated from conservation of momentum and energy as well as the endothermicity of reaction (2), which is referred to as the kinematic cutoff. The second column depicts the difference of the velocity distribution with and without infrared laser interaction and therefore of products stemming from reactions with vibrationally excited CH3I. The red circle again marks the kinematic cutoff, for which now also the energy put into the vibrational mode is taken into account (+0.37 eV). The other two columns show the internal energy and angular distributions of the aforementioned products from ground state and excited reactants in black and red respectively. The internal energy is calculated as the difference of the obtained velocities from the kinematic cutoff. The grey shaded area in the internal energy histogram gives a one Sigma interval around zero internal energy, which has been determined from the experimental uncertainties in forward direction. A detailed description of how we obtain this uncertainty is given in a previous publication.293.1 Energy-differential change in the reaction dynamics
At the lowest investigated collision energy of 0.7 eV the proton transfer is just opening up energetically. Yet, we obtain a significant contribution of the proton transfer to the total product flux. As can be seen from the velocity distribution in Fig. 2a, most of the product ion velocities exceed the kinematic cutoff (black circle). This is due to the finite width of the relative energy distribution (shown as a grey bar in the Eint histogram in Fig. 2c). The high energy tail of this distribution has a higher reaction probability, as the cross section rises quickly at the threshold. These high energy reactions lead to product ions several hundred m/s above the calculated maximum velocity. The excess energy in these events also leads to negative values for the internal energy, because it is calculated relative to the kinematic cut-off.The velocity distribution without vibrational excitation reveals two mechanisms leading to proton transfer (see Fig. 2a). A direct, forward scattered mechanism, often referred to as stripping and an indirect, isotropically scattered mechanism around zero product velocity. We know from previous studies,27 that the general trend with additional collision energy in this reaction leads to more forward, direct scattering. For the vibrationally excited reaction, the additional energy instead leads only to an indirect scattering signature (see Fig. 2b). The direct mechanism is suppressed. The indirect mechanism shows high internal excitation, in fact the entire additional energy seems to be efficiently transferred into internal excitation, which is evident in the Eint distribution shown in red in Fig. 2c. Taking a closer look at the angular distribution of the indirect mechanism for vibrationally excited reactions shown in red in Fig. 2d reveals forward–backward symmetric scattering. Such features are typically attributed to reactive collisions that occur at large impact parameters,33–36 where conservation of angular momentum constrains the leaving products into directions along the collision axis. In contrast, the indirect mechanism for the ground state reaction, marked in green in Fig. 2c and d, shows completely isotropic scattering (the maximum velocity chosen for this mechanism is marked in green in Fig. 2a). For isotropic scattering the mechanical rotation of the molecular reactant contributes significantly to the total angular momentum. Assuming the coupling of angular momentum does not change, we can attribute the shift from isotropic scattering to a forward–backward symmetric 1/cos(θ) distribution to an increase in initial orbital angular momentum and therefore larger reactive impact parameters.
At the intermediate collision energy of 1.2 eV, we can no longer clearly separate the two mechanisms in the ground state velocity distribution shown in Fig. 2e. The forward and isotropic parts overlap to a combined distribution. Interestingly, in the excited state image in Fig. 2f two contributions might be separable into a direct forward and an indirect, isotropic mechanism. This is also evident in the internal energy distribution (Fig. 2g), which is broader compared to the laser off distribution. The FWHM are 0.44 and 0.81 eV respectively and the difference is therefore very close the vibrational energy (0.37 eV). Instead of the forward–backward symmetric angular distribution at 0.7 eV, the excited reactions lead to more forward, direct scattering compared to the ground state (see Fig. 2h). This trend again fits the observations of larger impact parameters for excited reactions, as more forward scattering means less deflection due to short range collisions. This shows that there is a change from the observed dominantly indirect scattering at 0.7 eV to a mix of both indirect and direct dynamics at 1.2 eV for reactions of vibrationally excited reactants.
At 2.3 eV, the highest investigated collision energy, the indirect isotropically scattered mechanism is almost completely absent. Instead, direct forward stripping dominates (Fig. 2i). There is an additional small backward-scattered component. The forward scattered distribution shows lower velocities for larger angles. Lower velocities are equivalent to higher internal excitation and large scattering angles generally correspond to small impact parameters. Thus small impact parameter collisions lead to higher internal excitation. We have seen a similar effect in cation-neutral charge transfer reactions.37,38 At small impact parameters the short range repulsive part of the interaction potential is explored, which leads to more efficient coupling to vibration and thus to the observed higher internal excitation. The backward contribution is known from theory as a direct rebound mechanism,39 similar to the one known from SN2 reactions.
In contrast to the lower collision energies vibrationally excited reactions show more direct dynamics, compared to the ground state, at the highest collision energy. This can best be identified in the internal energy distribution (Fig. 2k). We find a shift towards lower internal energies, indicative of more direct dynamics. Furthermore the angular distribution is dominated almost exclusively by direct forward scattering. This observation is again interpreted as a larger contribution of large impact parameter collisions leading to stripping with very little angular deflection.
Larger impact parameters for vibrationally excited reactants have been seen before in the reactions of O + CHD3 and O + HCl.40,41 The authors argue that it is evidence for an enlarged cone of acceptance. For the reaction with CHD3, Wang et al. find a change from dominant backward scattering for the ground state to forward and sideways scattering with the CH-stretching vibration excited. They attribute this change to larger impact parameters and argue that the change of dipole moment due to the vibration modifies the long range interaction leading to an attraction at larger distances. This explanation is supported by quasi-classical trajectory calculations on an accurate potential energy surface.20,22,42 A similar steric effect can also explain the suppression of reactivity in the reaction of F + CHD3.21,43 Liu and coworkers refer to these effects as being analogous to an optical lens for approaching reactants steering them towards or away from the preferred transition state geometry.19 It is surprising that we find a similar effect despite the very different strength of the long range interaction due to the ion–dipole interaction in the title reaction.
3.2 Enhancement of reactivity by vibration versus collision energy
In Table 1 we present the change of the proton transfer reactivity due to vibrational excitation as a function of collision energy along with the achieved excited fraction for each collision energy. The data show that proton transfer is enhanced by 26 ± 2% at the appearance threshold with the infrared excitation laser present. Considering that about 4.5 ± 0.5% of the CH3I molecules in the beam were excited, this corresponds to an enhancement by a factor of almost 7, if we scale our result to 100% excited molecules in the beam. Such a large effect is expected, as we are close to the endothermicity of the channel and extra energy is put into the bond that is broken during the reaction. At the two higher collision energies (1.2 and 2.3 eV) we still find an enhancement by a factor of 3 and 1.5, respectively. These values are compared with QCT calculations below.| Collision energy | PT counts IR off | PT counts IR on | Change (%) | Excited fraction (%) | Scaled factor | Theory non/soft/hard ZPE constraint |
|---|---|---|---|---|---|---|
| 0.7 eV | 5673 | 7156 | +26 ± 2.0 | 4.5 ± 0.5 | 6.9 ± 0.8 | 4.2/870/∞ |
| 1.2 eV | 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 713 713 |
83224 |
+5.7 ± 0.5 | 2.7 ± 0.4 | 3.0 ± 0.4 | 2.3/4.7/6.7 |
| 2.3 eV | 88551 |
89907 |
+1.5 ± 0.5 | 2.9 ± 0.3 | 1.5 ± 0.2 | — |
To gain insight how the enhancement depends on the collision energy, we have calculated the branching ratios for proton transfer relative to SN2 products at similar total energies, i.e., collision energy plus energy in the vibrational mode. Assuming no change in the SN2 channel due to vibrational excitation,28 the branching ratios yield insight whether translation or vibration is more efficient in promoting reactivity. At the highest collision energy the assumption of spectator mode SN2 dynamics is not strictly correct, as we recently found a small but significant enhancement of the SN2 channel, which will be discussed in a separate publication. Here, we have calculated the branching ratio relative to the ground state SN2 counts. The results are shown in Fig. 3. The results from the current study are shown in blue for ground state and red for vibrationally excited reactions. The same symbol is chosen for identical collision energies. The grey symbols connected by a line are results from a previous study over a large energy range on the same system by our group27 to illustrate the trend over the investigated energy range. Two additional points taken from ref. 28 are shown in light blue for the ground state and pink with vibrational excitation.
 | ||
| Fig. 3 Proton transfer to SN2 branching ratios versus total energy (Etotal = Ecoll + Evib). The points for excited reactions are scaled to 100% excitation in the neutral beam. The grey points27 and the light blue (ground state) and pink points (vibrationally excited)28 are from previous studies by our group. Points shown in blue are for ground state and in red for vibrationally excited reactions. The same symbol marks the same collision energy. Etotal uncertainties are calculated from the relative energy distributions. The branching ratio uncertainty is dominated by the error on the excited fraction for the excited points and is otherwise smaller than the symbols. | ||
Our ground state results are in good agreement with the branching ratios of the previous studies. We can now use points of similar total energy to compare whether vibration or translation is more efficient in promoting reactivity. It appears that there is a clear difference between the lowest collision energy and the two higher energy points. Comparing the two excited branching ratios (red and pink) at 1.07 eV with the ground state results at 1.2 eV (grey and blue) suggests that collision energy is more effective in enhancing the proton transfer. At the two higher collision energies we find the opposite effect. The effect that at lower collision energies translation is more efficient in promoting reactivity has been mentioned previously.19 It even occurs in ‘late’ barrier reactions.16,17 Guo and coworkers propose that this occurs, because a minimum of momentum transfer is required for the reaction to proceed, which leads to a preference for translation at low collision energies.24
Comparing the branching ratio without infrared excitation at 1.2 and 1.5 eV collision energy (grey points), we find that the additional collision energy leads to an enhancement by a factor of 2 due to 0.3 eV of additional collision energy. In contrast, vibrational excitation by a similar amount of energy enhances the branching ratio by a factor of 3 (red circle at 1.5 eV total energy). This amounts to a relative vibrational enhancement over translation by a factor of 1.5.
In the collision energy range above 1.5 eV, the branching ratios decrease slightly for the ground state reaction. This coincides with the opening of two halide abstraction channels.27 Vibrational excitation again leads to an increase in the proton transfer to SN2 ratio at 2.5 eV total energy, although this increase is not as strong compared to the data at 1.2 eV collision energy. We still observe a significant increase by a factor of 1.5. The ground and vibrationally excited state reactions therefore show opposite behaviour at the highest investigated collision energy. As the data at the appearance threshold is hardest to evaluate we can conclude from the other two, that the CH-stretching mode is indeed more effective in promoting reactivity towards the proton transfer in this system.
4 Comparison with theory
Quasi-classical trajectory calculations have been performed by the Czakó group for this system over a large range of collision energies and a variety of vibrational modes including 0.7 and 1.2 eV collision energy and the symmetric CH-stretching mode investigated in this work.26 Comparison at the threshold energy of 0.7 eV is difficult, because the experimental energy uncertainties underestimate the threshold behavior. On the theory side the introduction of different zero-point-energy (ZPE) constraints to correct for the quasi-classical nature of the method leads to significant changes in the results, i.e. the absolute cross sections, especially at low collision energies. Despite this, the agreement between the ground state angular and energy distributions from our measurements with the trajectory simulations is reasonable (see black histograms Fig. 2c, d and 5 in ref. 26). The calculations seem to overestimate the fraction of backward scattering. The change towards indirect dynamics for excited reactions, however, is not picked up and no significant shift towards larger impact parameters was found in the QCT calculations, while the stereodynamic effect was reproduced by QCT calculations in the case of the reaction F + CHD3. This makes a clear assignment of the origin of this effect difficult.At the intermediate collision energy, theory and experiment should compare better, as above threshold ZPE effects and the influence of experimental uncertainties have less relative contribution. Once again the angular and internal energy distribution agree well for the ground state, but the change in dynamics, evident most prominently in the experimental internal energy distribution (see Fig. 2g), is not reproduced. In the calculations, the Eint distribution is only shifted but is not significantly broadened.
Depending on the chosen ZPE constraint the trajectory calculations give an enhancement factor of the cross section at 1.2 eV collision energy due to symmetric stretching vibration of between 2 and 7 (Table 1). They also provide data at both 1.2 and 1.5 eV, which can be compared to our ground state results. Depending on the chosen ZPE constraint they find an increase of by a factor of 2–3 due to the additional collision energy. The trend that vibrational excitation is more efficient in promoting reactivity is therefore captured both in the quasi-classical trajectory calculations and our experiment. The experimental enhancement factors are slightly larger compared to the non-ZPE values (Table 1), but smaller than the soft-constraint values. It could be interesting to find a constraint that matches the experimental values. In both theory and experiment the vibrational enhancement is collision energy dependent and becomes smaller at high collision energies (Fig. 2 in ref. 26).
The preference for vibration is also seen in calculations using the sudden vector projection model by Guo and coworkers28 for this system. For the symmetric stretching mode they find a value of 0.30 and only a value of 0.03 for translation and therefore a clear preference for stretching vibration over translation, in qualitative agreement with our experimental results.
5 Conclusion
We have obtained differential scattering cross sections and branching ratios for the SN2 and proton transfer reaction channels with and without exciting the symmetric CH-stretching mode, and over a range of reactant collision energies. We have found that the vibrational excitation modifies proton transfer reaction dynamics in several ways. The change depends on collision energy over the investigated range from 0.7 to 2.3 eV. At the appearance threshold, direct dynamics are suppressed and only a single indirect scattering signature is observed. In contrast, at the highest investigated collision energy, the excitation instead leads to more direct dynamics compared to the ground state. We find a mixture of direct and indirect dynamics at the intermediate collision energy with vibrational excitation, confirming a trend towards more direct dynamics with additional collision energy. For all investigated collision energies we find evidence that reactions with vibrationally excited CH3I occur at larger impact parameters. Both effects are not reproduced by recent QCT calculations. In a comparison of adding kinetic versus vibrational energy to the system, we find that the latter is more efficient in promoting the reaction above 1.2 eV collision energy in agreement with both QCT and SVP calculations.This study shows that the atomistic reaction dynamics of polyatomic reactions are still far from being understood and that further theoretical work is needed. Our study also represents a step towards manipulating product branching ratios by quantum state preparation in halide anion reactions with methyl halides. The CH-bond cleavage necessary for proton transfer is also an essential step in elimination reactions.44 A similar enhancement by the CH-stretching mode could enable control of the competition between the elimination and nucleophilic substitution pathways.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Gabor Czako for fruitful discussions. This work was supported in parts by the Austrian Science Fund (FWF) through project No. P25956-N20. J. M. acknowledges support by a Hertha-Firnberg fellowship of the Austrian Science Fund (FWF), project No. T962-N34.Notes and references
- J. C. Polanyi, Acc. Chem. Res., 1972, 5, 161 CrossRef CAS.
- R. N. Zare, Science, 1998, 279, 1875–1879 CrossRef CAS PubMed.
- A. A. Viggiano, S. T. Arnold, R. A. Morris, A. F. Ahrens and P. M. Hierl, J. Phys. Chem., 1996, 100, 14397–14402 CrossRef CAS.
- T. Wang, J. Chen, T. Yang, C. Xiao, Z. Sun, L. Huang, D. Dai, X. Yang and D. H. Zhang, Science, 2013, 342, 1499–1502 CrossRef CAS PubMed.
- T.-g. Yang, L. Huang, Y.-r. Xie, T. Wang, C.-l. Xiao, Z.-g. Sun, D.-x. Dai, M.-d. Chen, D. H. Zhang and X.-m. Yang, Chin. J. Chem. Phys., 2015, 28, 471–475 CrossRef CAS.
- H. A. Bechtel, J. P. Camden, D. J. A. Brown, M. R. Martin, R. N. Zare and K. Vadopyanov, Angew. Chem., Int. Ed., 2005, 44, 2382 CrossRef CAS PubMed.
- R. Liu, F. Wang, B. Jiang, G. Czakó, M. Yang, K. Liu and H. Guo, J. Chem. Phys., 2014, 141, 074310 CrossRef PubMed.
- K. Liu, Annu. Rev. Phys. Chem., 2016, 67, 91–111 CrossRef CAS PubMed.
- A. W. Ray, J. Ma, R. Otto, J. Li, H. Guo and R. E. Continetti, Chem. Sci., 2017, 8, 7821–7833 RSC.
- R. D. Guettler, G. C. Jiones, L. A. Posey and R. N. Zare, Science, 1994, 266, 259–261 CrossRef CAS PubMed.
- J. B. Liu and S. L. Anderson, Int. J. Mass Spectrom., 2005, 241, 173–184 CrossRef CAS.
- P. R. Shirhatti, I. Rahinov, K. Golibrzuch, J. Werdecker, J. Geweke, J. Altschäffel, S. Kumar, D. J. Auerbach, C. Bartels and A. M. Wodtke, Nat. Chem., 2018, 10, 592 CrossRef CAS PubMed.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- J. Meyer and R. Wester, Annu. Rev. Phys. Chem., 2017, 68, 333–353 CrossRef CAS PubMed.
- J. Xie, R. Otto, J. Mikosch, J. Zhang, R. Wester and W. L. Hase, Acc. Chem. Res., 2014, 47, 2960–2969 CrossRef CAS PubMed.
- S. Yan, Y.-T. Wu, B. Zhang, X.-F. Yue and K. Liu, Science, 2007, 316, 1723–1726 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhou, D. H. Zhang, G. Czakó and J. M. Bowman, J. Phys. Chem. Lett., 2012, 3, 3416–3419 CrossRef CAS PubMed.
- F. Wang, J.-S. Lin and K. Liu, Science, 2011, 331, 900–903 CrossRef CAS PubMed.
- K. Liu, J. Chem. Phys., 2015, 142, 080901 CrossRef PubMed.
- G. Czakó and J. M. Bowman, J. Chem. Phys., 2009, 131, 244302 CrossRef PubMed.
- G. Czakó and J. M. Bowman, J. Am. Chem. Soc., 2009, 131, 17534–17535 CrossRef PubMed.
- G. Czakó and J. M. Bowman, Phys. Chem. Chem. Phys., 2011, 13, 8306 RSC.
- G. Czakó and J. M. Bowman, Science, 2011, 334, 343–346 CrossRef PubMed.
- B. Jiang and H. Guo, J. Chem. Phys., 2013, 138, 234104 CrossRef PubMed.
- H. Guo and B. Jiang, Acc. Chem. Res., 2014, 47, 3679–3685 CrossRef CAS PubMed.
- B. Olasz and G. Czakó, J. Phys. Chem. A, 2018, 122, 8143–8151 CrossRef CAS PubMed.
- E. Carrascosa, T. Michaelsen, M. Stei, B. Bastian, J. Meyer, J. Mikosch and R. Wester, J. Phys. Chem. A, 2016, 120, 4711–4719 CrossRef CAS PubMed.
- M. Stei, E. Carrascosa, A. Dörfler, J. Meyer, B. Olasz, G. Czakó, A. Li, H. Guo and R. Wester, Sci. Adv., 2018, 4, eaas9544 CrossRef PubMed.
- R. Wester, Phys. Chem. Chem. Phys., 2014, 16, 396–405 RSC.
- M. Stei, E. Carrascosa, M. A. Kainz, A. H. Kelkar, J. Meyer, I. Szabó, G. Czakó and R. Wester, Nat. Chem., 2016, 8, 151–156 CrossRef CAS PubMed.
- M. Stei, J. von Vangerow, R. Otto, A. H. Kelkar, E. Carrascosa, T. Best and R. Wester, J. Chem. Phys., 2013, 138, 214201 CrossRef PubMed.
- L. Hu, Z. Zhou, C. Dong, L. Zhang, Y. Du, M. Cheng and Q. Zhu, J. Phys. Chem. A, 2013, 117, 4352–4357 CrossRef CAS PubMed.
- R. N. Zare, Angular Momentum, John Wiley & Sons, 1988 Search PubMed.
- R. D. Levine, Molecular reaction dynamics, Cambridge University Press, 2009 Search PubMed.
- Z. Herman, Int. J. Mass Spectrom., 2001, 212, 413–443 CrossRef CAS.
- J. Meyer, E. Carrascosa, T. Michaelsen, B. Bastian, A. Li, H. Guo and R. Wester, J. Am. Chem. Soc., 2019, 141, 20300–20308 CrossRef CAS PubMed.
- S. Trippel, M. Stei, J. A. Cox and R. Wester, Phys. Rev. Lett., 2013, 110, 163201 CrossRef CAS PubMed.
- T. Michaelsen, B. Bastian, E. Carrascosa, J. Meyer, D. H. Parker and R. Wester, J. Chem. Phys., 2017, 147, 013940 CrossRef PubMed.
- J. Zhang, J. Xie and W. L. Hase, J. Phys. Chem. A, 2015, 119, 12517–12525 CrossRef CAS PubMed.
- I. Schechter and R. Levine, J. Phys. Chem., 1989, 93, 7973–7975 CrossRef CAS.
- F. Wang and K. Liu, Chem. Sci., 2010, 1, 126–133 RSC.
- G. Czakó and J. M. Bowman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7997–8001 CrossRef PubMed.
- W. Zhang, H. Kawamata and K. Liu, Science, 2009, 325, 303–306 CrossRef CAS PubMed.
- E. Carrascosa, J. Meyer, J. Zhang, M. Stei, T. Michaelsen, W. L. Hase, L. Yang and R. Wester, Nat. Commun., 2017, 8, 25 CrossRef PubMed.
| This journal is © the Owner Societies 2020 |