
Soft XAS as an in situ technique for the study of heterogeneous catalysts

Abstract
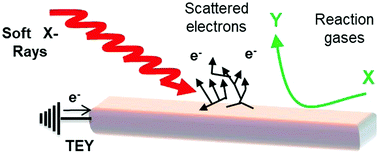
Soft X-ray absorption in situ studies of heterogeneous catalysts have been applied to areas such as copper methanol oxidation catalysts and cobalt Fischer–Tropsch type catalysts over a period of around two decades. The technique has the potential to offer several advantages for studying heterogeneous catalysts against hard X-ray XAS in: the systems that can be studied (includes elements such as C, N, O), the potential for surface sensitivity (crucial for catalysts, where reactions occur at surfaces) and the information content of the resulting spectra. Nevertheless, it is technically challenging and the necessary hardware has only been developed and evolved in a few specific groups worldwide. This perspective will introduce the technique in the context of other competing spectroscopies, summarise the development of hardware and the challenges that have been overcome in experimental terms, along with the outcome and impact on different fields within catalysis. Additionally, anticipated future trends and directions will be discussed.
- This article is part of the themed collections: PCCP Perspectives, 2020 PCCP HOT Articles and Synchrotron Radiation Techniques in Catalytic Science
 Please wait while we load your content...
Please wait while we load your content...

