
Identifying the catalyst chemical state and adsorbed species during methanol conversion on copper using ambient pressure X-ray spectroscopies†
 *a
Jesús S.
Lacasa,
a
David
Grinter,
c
Georg
Held,
c
Cruz S.
Esconjauregui
d
and
Robert S.
Weatherup
*bce
*a
Jesús S.
Lacasa,
a
David
Grinter,
c
Georg
Held,
c
Cruz S.
Esconjauregui
d
and
Robert S.
Weatherup
*bce
Abstract
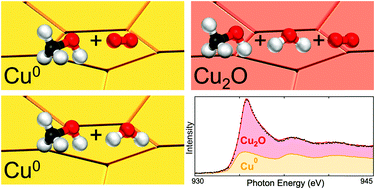
Methanol is a promising chemical for the safe and efficient storage of hydrogen, where methanol conversion reactions can generate a hydrogen-containing gas mixture. Understanding the chemical state of the catalyst over which these reactions occur and the interplay with the adsorbed species present is key to the design of improved catalysts and process conditions. Here we study polycrystalline Cu foils using ambient pressure X-ray spectroscopies to reveal the Cu oxidation state and identify the adsorbed species during partial oxidation (CH3OH + O2), steam reforming (CH3OH + H2O), and autothermal reforming (CH3OH + O2 + H2O) of methanol at 200 °C surface temperature and in the mbar pressure range. We find that the Cu surface remains highly metallic throughout partial oxidation and steam reforming reactions, even for oxygen-rich conditions. However, for autothermal reforming the Cu surface shows significant oxidation towards Cu2O. We rationalise this behaviour on the basis of the shift in equilibrium of the CH3OH* + O* ⇌ CH3O* + OH* reaction step caused by the addition of H2O.
- This article is part of the themed collections: 2020 PCCP HOT Articles and Synchrotron Radiation Techniques in Catalytic Science
 Please wait while we load your content...
Please wait while we load your content...

