
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid†
Jari
Peltola
 ,
Prasenjit
Seal
,
Anni
Inkilä
and
Arkke
Eskola
*
,
Prasenjit
Seal
,
Anni
Inkilä
and
Arkke
Eskola
*
Department of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtasen aukio 1), FI-00014, Helsinki, Finland. E-mail: arkke.eskola@helsinki.fi
First published on 7th April 2020
Abstract
We present a time-resolved broadband cavity-enhanced UV-absorption spectrometer apparatus that we have constructed and utilized for temperature- and pressure-dependent kinetic measurements of formaldehyde oxide (CH2OO) reactions. We also introduce and utilize a new photolytic precursor, bromoiodomethane (CH2IBr), which photolysis at 213 nm in presence of O2 produces CH2OO. Importantly, this precursor appears to be free from secondary reactions that may regenerate CH2OO in kinetic experiments. The unimolecular decomposition rate coefficient of CH2OO has been measured over wide pressure (5–400 Torr) and temperature (296–600 K) ranges and master equation simulations of the decomposition kinetics have been performed using MESMER program. The MESMER simulations of the experimental data with the calculated zero-point energy corrected transition state energy 85.9 kJ mol−1 for decomposition required no adjustment and returned 〈ΔE〉down = 123.2 × (T/298 K)0.74 cm−1 for temperature-dependent exponential-down model of the collisional energy transfer in He. A very good agreement between results of simulations and experiments is obtained. The results are compared with the previously reported unimolecular decomposition study by Stone et al. (Phys. Chem. Chem. Phys., 2018, 20, 24940–24954). Current master equation simulations suggest about 61% decomposition yield for the predominant H2 + CO2 channel, whereas the yields of two other channels, H2O + CO, and HCO + OH, are sensitive on the parameters involved in the simulations. The kinetics of CH2OO reaction with formic acid has also been investigated as function of pressure (5–150 Torr) and temperature (296–458 K). The bimolecular rate coefficient for CH2OO + HCOOH reaction shows a negative temperature dependency, decreasing from (1.0 ± 0.03) × 10−10 cm3 molecule−1 s−1 at 296 K to (0.47 ± 0.05) × 10−10 cm3 molecule−1 s−1 at 458 K with an Arrhenius activation energy of −4.9 ± 1.6 kJ mol−1, where statistical uncertainties shown are 2σ. Estimated overall uncertainty in the measured rate coefficients is about ±20%. Current bimolecular rate coefficient at room temperature agrees with the previously reported rate coefficients from the direct kinetic experiments. The reaction is found to be pressure independent over the range between 5 and 150 Torr at 296 K in He.
Introduction
Alkenes are important non-methane hydrocarbon species in the Earth's atmosphere. Alkenes with natural origin (e.g. isoprene, monoterpenes, and sesquiterpenes) are released into the troposphere from vegetation whereas anthropogenic alkene emissions are often byproducts of combustion and are composed of smaller hydrocarbons (ethene, propenes, butenes, etc.).1 A major loss pathway of alkenes in the troposphere is reaction with ozone, i.e. ozonolysis. In ozonolysis a highly excited primary ozonide is formed in a very exothermic O3 + alkene reaction.2 In gas phase and at atmospheric pressure, any excited primary ozonide decomposes to a Criegee intermediate, also known as carbonyl oxide, and a carbonyl compound. In case of acyclic (e.g. trans-2-butene, isoprene) and exocyclic (e.g. β-pinene) alkenes, roughly 50% of Criegee intermediates decompose or isomerize further before stabilization, while other ∼50% are stabilized at atmospheric pressure and are consequently called stabilized Criegee intermediates, sCIs. sCIs can further react with atmospheric constituents (H2O, (H2O)2, SO2, acids, NO2, etc.) in bimolecular reactions or decompose and/or isomerize in unimolecular reactions.1,3Formaldehyde oxide, CH2OO, is the smallest and probably the most studied sCI.4–9 It is formed in the ozonolysis of ethene and any larger terminal alkene (e.g. isoprene).10 In breakthrough experiments using multiplex photoionization mass-spectrometer (MPIMS) utilizing synchrotron radiation for ionization, Welz et al.5 discovered 2012 that CH2I radical (an α-iodoalkyl radical) reaction with molecular oxygen produces the smallest sCI, CH2OO.
| CH2I + O2 → CH2OO + I | (R1) |
| → other products |
Since high [O2] conditions are easy to prepare, sCIs do not react with O2 at any known extent, reaction (R1) is fast11 and formaldehyde oxide yield is close to unity at low pressures,12,13reaction (R1) is the excellent source of CH2OO for direct kinetic measurements. Impetus to study reaction (R1) with MPIMS originated from this laboratory.11,14
Direct kinetics measurements of formaldehyde oxide have shown that CH2OO reacts fast with SO2,5 organic acids,15 and water dimer ((H2O)2).8 Bimolecular rate coefficients of the smallest sCI with the above and many other reactants, obtained from direct kinetic measurements in different laboratories, are consistent with each other.16 For example, results of direct kinetic experiments of CH2OO reaction with SO217 agree with each other and show significantly faster kinetics than now outdated estimates based on results of indirect measurements suggested.10 Especially CH2OO reactions with acids are very fast with bimolecular rate coefficients in excess of 1 × 10−10 cm3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) molecule−1 s−1. Welz et al.15 measured a bimolecular rate of (1.1 ± 0.1) × 10−10 cm3 molecule−1 s−1 for the reaction of CH2OO with HCOOH at room temperature (∼298 K) and pressure of 4 Torr.
molecule−1 s−1. Welz et al.15 measured a bimolecular rate of (1.1 ± 0.1) × 10−10 cm3 molecule−1 s−1 for the reaction of CH2OO with HCOOH at room temperature (∼298 K) and pressure of 4 Torr.
| CH2OO + HCOOH → products | (R2) |
In this work, we measure kinetics of CH2OO + HCOOH reaction over extended temperature (296–458 K) and pressure (5–150 Torr) ranges, report bimolecular rate coefficients, and discuss on the observed kinetics.
Stone et al.9 have recently measured unimolecular decomposition kinetics of CH2OO in the temperature 450–650 K and helium pressure 2–350 Torr ranges using a time-resolved broadband cavity enhanced absorption spectrometer (TR-BB-CEAS) and probing formaldehyde oxide at UV.
| CH2OO → products | (R3) |
Since the first study by Welz et al.,5 alkyl-substituted α-iodoalkyl radicals have also been shown to produce corresponding sCIs in presence of oxygen.24–27 Hitherto CH2I radical in the reaction (R1) has been prepared by photodissociation of CH2I2 at 248,5 266,7 or 35528 nm. Important for the current work, a reaction between CH2OO and CH2I2 precursor has been observed in many previous studies.9,29 This may be a significant problem in kinetic experiments, because only a few percent of the precursor is typically consumed in the photodissociation process and, most importantly, CH2OO + CH2I2 reaction may generate CH2I and thus CH2OO, resulting in chain-propagation and distorting information from kinetic measurements. For example, Liu et al.29 reported a bimolecular rate coefficient (5.2 ± 2.6) × 10−14 cm3 molecule−1 s−1 for the reaction at 298 K. The reaction becomes faster at higher temperatures. Stone et al.9 observed a bimolecular rate coefficient (8.2 ± 1.7) × 10−12 cm3 molecule−1 s−1 at 450 K in their unimolecular decomposition study of CH2OO. They also stated that the reaction might have caused a contribution to the measured decay data at the temperatures above 450 K. Buras et al.30 observed a baseline offset in the 375 nm absorption of CH2OO at high temperatures. They stated that it indicated an extra absorption by an unknown product formed in their system at high temperatures. In addition, CH2I2 has a relatively strong absorption at the 340 nm region, where the typical probing of CH2OO is conducted in UV-absorption-based experiments, leading to a negative (and often non-constant) measurement baseline in most cases.
In this work, we introduce and utilize a new photolytic precursor, bromoiodomethane (CH2IBr), which photolysis at 213 nm and in presence of O2 produces CH2OO. This precursor is more stable against secondary reaction chemistry, which may regenerate CH2OO in kinetic experiments. We report a detailed study of thermal unimolecular decomposition kinetics of CH2OO over wide pressure (5–400 Torr) and temperature (296–600 K) ranges using new CH2IBr precursor. We also introduce and utilize our new time-resolved broadband cavity enhanced absorption spectrometer (TR-BB-CEAS) apparatus that locates in the University of Helsinki and probes in the ultraviolet (UV) region. We perform master equation simulations and compare obtained outcome with the current experimental results as well as with the results of unimolecular study by Stone et al.9
Experimental
A schematic figure of the new TR-BB-CEAS apparatus utilizing UV absorption to probe CH2OO is shown in Fig. 1. The design of our TR-BB-CEAS apparatus is a modified version of the experimental setup developed by L. Sheps et al.31 The gas mixture flowing through a quartz tube reactor contained the radical precursor (CH2IBr or CH2I2), O2, and HCOOH (for the bimolecular reaction) diluted in helium or nitrogen carrier gas. Calibrated mass flow controllers (Omega Engineering) were used to deliver gases to a mixing manifold before flowing into the reactor. The CH2I radicals were homogeneously generated along the flow reactor by the 5th harmonic (213 nm) or the 4th harmonic (266 nm) of a pulsed Nd:YAG laser (Quantel Q-smart 850). The photolysis laser beam is expanded by a 2:1 telescope giving an 18 mm diameter spot inside the reactor. Typical pulse fluences at 213 nm and 266 nm were about 4 mJ cm−2 and 18 mJ cm−2 inside the reactor, which were sufficient to dissociate approximately 2.5% and 3% of the CH2IBr and CH2I2 precursors, respectively. The inner diameter of the reactor tube is 36 mm and linear gas flow speed about 1 m s−1, which ensured that the gas mixture was completely replaced between laser pulses with the repetition rate of 1 Hz. The reactor tube is combined with a 2 m long confocal cavity formed by two highly reflecting concave mirrors (Layertec). The radius of curvature and the diameter of the mirrors are 2 m and half an inch (12.7 mm), respectively. The mirror holders are coupled to the reactor construction using flexible stainless-steel bellows. A beam from a continuous-wave laser-driven plasma light-source (Energetiq, EQ-99X) is used to probe the transient absorption of CH2OO. The output of the light-source covers wavelengths from 180 to 2100 nm. UV portion of the light is coupled into the cavity using a high-pass filter (with a cut-off wavelength of ∼300 nm). The reflectivity of the mirrors as stated by the manufacturer is 99.6 ± 0.3% across the λ = 300–450 nm wavelength range, giving together with the cavity length the total effective optical path length (OPL) 30–160 m. Details of the calibration procedure of the effective OPL are given in the ESI.†
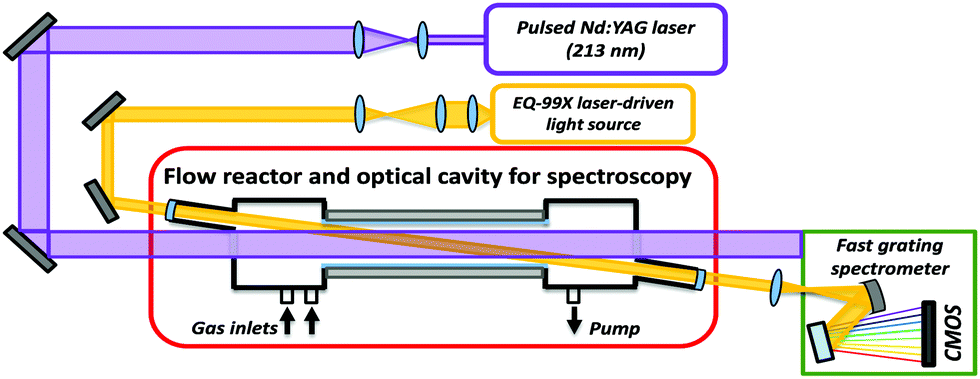 | ||
| Fig. 1 Schematic figure of the time-resolved broadband cavity-enhanced absorption spectrometer. The sCI is produced along a heated quartz flow tube reactor by a single-pass photolysis laser pulse at 213 or 266 nm. The sCI is probed by overlapping incoherent laser-driven broadband light source. The sensitivity of the detection is enhanced using an optical cavity formed by two highly reflecting concave mirrors between 300 and 450 nm. The time-dependent broadband absorption spectrum of sCI is measured by a grating spectrometer combined with a fast CMOS line array camera. | ||
The probe beam leaking out of the cavity is guided to a grating spectrometer and dispersed in wavelength horizontally by a ruled grating. The transient spectrum of CH2OO is recorded by focusing the dispersed light onto an image sensor of a fast CMOS line array camera (JAI SW-2000M-CL-80) in vertical and horizontal directions with 100 mm and 30 mm cylindrical UV fused silica lenses, respectively. The camera (1 × 2048 pixels, 12-bit) has a maximum line rate of 80 kHz, however, to gain good enough signal-to-noise ratio (SNR), the line rate was reduced to 20 kHz leading to time resolution of about 50 μs. The use of the fast line array camera enables the transient spectrum measurements without moving parts (e.g., a spinning mirror31,32) inside the grating spectrometer. All the transient absorption traces of CH2OO measured in the current work were probed at 340 nm region, where the strong absorption band of CH2OO has its maximum and the effective path length about 80 m. This region is also free from unwanted interferences of byproducts, e.g. from absorption of IO radical.
The signal from the CMOS line array camera is processed and digitized by a fast 12-bit image acquisition card (National Instruments, PCIe-1427). Single-exponential function At = A0 × exp(−k′t) is subsequently fitted to a background-subtracted CH2OO time-trace by the least squares method using the Levenberg–Marquardt algorithm in a custom-made LabVIEW program. Here k′ is the pseudo-first-order decay rate coefficient, and At is the absorbance at time t, and A0 is the initial absorbance (at time t = 0). The wavelength scale of the spectrometer is calibrated with atomic (Hg and Ar) emission lamps. For the experiments described here, we averaged signal between 600 and 3000 shots for each decaying experimental time-trace. Fig. S4 in the ESI† presents a temporal absorption profile of CH2OO measured at 340 nm with the presence of HCOOH. The estimated initial concentration of CH2OO was ∼8.5 × 1010 molecule cm−3 and the observed SNR of the time-trace was ∼35 for an averaging time of 1800 s (1800 shots). This yielded a minimum detectable [CH2OO] of ∼2.4 × 109 molecule cm−3.
The temperature control of the flow tube reactor is achieved by two temperature-controlled custom-made aluminum blocks, which are placed around the quartz-glass reactor tube. Each aluminum block has four 300 W cartridge heaters, which are regulated using a PID-controller program from LabVIEW. The temperature of the system can be heated anywhere between 296 K and 600 K. Temperature of the gas flow was measured continuously in the middle of the reactor just outside of the photolysis beam by a K-type thermocouple. Complete axial temperature profile within the overlap volume of the probe and the photolysis beams were measured separately for all experimental conditions (temperature, pressure and flow rate) used in this work. The observed temperature uncertainty was ±3 K. The gases were pre-heated close to the setpoint temperature before entering to the reactor.
The CH2I radicals were principally generated from CH2IBr at 213 nm.
| CH2IBr + hν(213 nm) → CH2I + Br | (R4a) |
| → other products | (R4b) |
 | ||
| Fig. 2 The UV absorption cross-section of gaseous CH2IBr as a function of wavelength.33 | ||
A few measurements in this work were done with CH2I2 precursor for comparison.
| CH2I2 + hν(266 nm) → CH2I + I | (R5a) |
| → other products | (R5b) |
Theoretical methods
In this work we accompanied the same potential energy surface (PES) for the thermal unimolecular decomposition of the smallest Criegee intermediate, CH2OO, as Stone et al.9 utilized in their study. Their PES was to significant extent based on Nguyen et al. calculations.36 The geometry optimization along with the vibrational frequency calculations of the stationary points on the PES, viz., CH2OO, transition states, and intermediates were performed using complete active space self-consistent field (CASSCF) method taking into account 8 electron and 8 orbital combination in the active space, i.e., CASSCF(8,8). The products and one transition state leading to the formation of H2O + CO, were optimized with MN1537 density functional. In all these cases, def2-TZVP basis set was used.The thermal unimolecular decomposition of CH2OO is believed to proceed via the formation of a cyclic dioxirane and the transition state leading to its formation determines the rate of reaction from CH2OO to different products. Therefore, accurate estimation of the barrier height and the energy of the cyclic dioxirane is crucial. We performed the T1 diagnostics for the CH2OO reactant, the transition state leading to the formation of the cyclic dioxirane, and the dioxirane intermediate. For the reactant and the TS, the T1 diagnostic has a value of 0.043 and 0.051, respectively, which is an indication of the possibility of multireference character in those systems. Hence, we chose CASSCF method to carry out the electronic structure calculations. All above methods have been implemented in Gaussian 16 suite of programs.38
In order to get reliable energies, we performed single-point energy calculations with domain based local pair natural orbital coupled cluster methods or the DLPNO-CCSD(T) approach as employed in ORCA code.39 The DLPNO calculations were then extrapolated to the complete basis set limit using correlation consistent Dunning's augmented basis sets, i.e., aug-cc-pVXZ (X = 5 and 6)40,41 following the extrapolation scheme as given below42
| ΔECBS = ΔEaug-cc-pV6Z − 0.694(ΔEaug-cc-pV5Z − ΔEaug-cc-pV6Z) | (1) |
To account for the effect of helium bath-gas pressure on unimolecular decomposition rate coefficient of CH2OO, we performed master equation simulations using MESMER 5.1 (Master Equation Solver for Multi-Energy well Reactions) program.43 For well-defined transition states Rice–Ramsperger–Kassel–Marcus (RRKM) theory was used along with Eckart tunneling corrections to calculate microcanonical rate coefficients. For collisional energy transfer a temperature-dependent single-exponential-down model was used.
| 〈ΔE〉down = 〈ΔE〉down,ref(T/298 K)n | (2) |
Results and discussion
CH2OO spectrum
The absorption spectrum of CH2OO measured in this work, produced using the CH2IBr precursor and 213 nm photolysis followed by reaction (R1), is shown with blue line in Fig. 3. The spectrum is averaged over 0–3 ms (with ∼67 μs time resolution) after the photolysis. The corresponding transient spectrum is presented in Fig. S5 in the ESI.† The absorption cross-section is estimated using the measured absorption coefficient at t = 0 ms, and the resulting initial CH2I concentration using the estimated yield of the channel (R4a) = 0.4. The estimated initial CH2OO concentration in the measurement is ∼2.0 × 1011 molecule cm−3. For comparison, the CH2OO absorption spectrum of Ting et al.44 is presented in Fig. 3 as black line, which was measured using the CH2I2 precursor and 248 photolysis, and much higher initial CH2OO concentration, about 5 × 1013 molecule cm−3.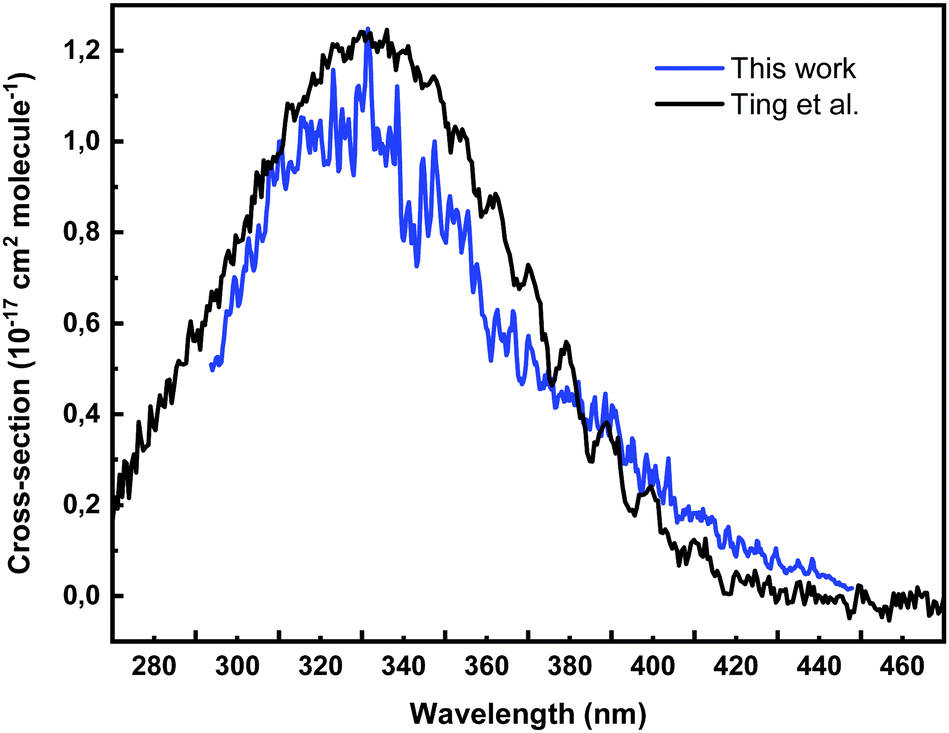 | ||
| Fig. 3 Absorption spectrum of CH2OO using CH2IBr as photolytic precursor and measured at 296 K and 10 Torr (N2). The spectrum is averaged over t = 0–3 ms. The initial CH2OO concentration was ∼2.0 × 1011 molecule cm−3. The O2 concentration was ∼3.8 × 1016 molecule cm−3. | ||
Bimolecular CH2OO + HCOOH reaction
The kinetics of CH2OO reaction with formic acid was measured as a function of temperature between 296 and 458 K at low pressure (5–15.5 Torr). Fig. 4 shows typical transient traces of CH2OO at varying concentrations of HCOOH at 458 K. All the CH2OO traces were fitted using a single-exponential decay function. In the absence of added acid reactant, the CH2OO signal follows a first-order decay loss, kloss (s−1), which originates from diffusion out of the measurement volume, from slow reaction of CH2OO with the precursor, and to some small extend from radical–radical reactions. Heterogeneous loss is negligible in our measurement system, since the radicals are generated and probed inside the same volume element in the middle of the flow reactor tube away from wall. In addition, the measurements were done in a temperature region where the unimolecular decomposition is negligible (below 475 K). The kloss was measured by reducing the precursor concentration until its value no longer depended on the precursor concentration. Once this condition was obtained, it was concluded that radical–radical reactions, especially CH2OO–CH2OO reactions, were suppressed. The initial CH2OO concentration in the measurements was typically below 1.0 × 1011 molecule cm−3, which was estimated from the laser fluence, the precursor absorption cross-section,33 and the effective OPL at the used wavelength (see ESI† for more details).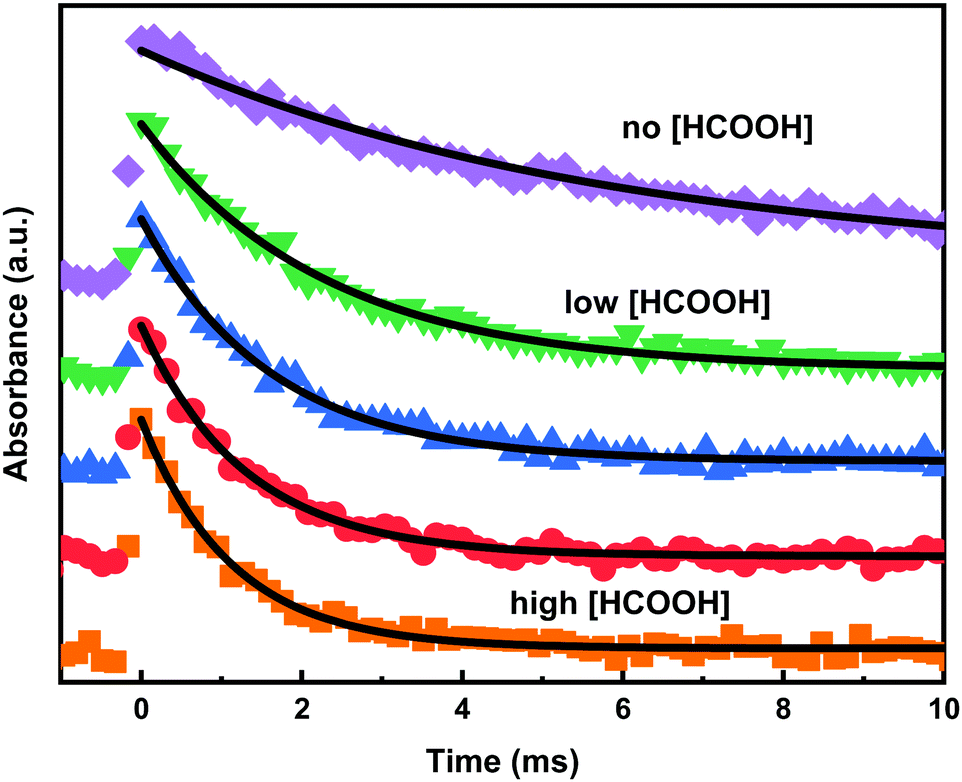 | ||
| Fig. 4 The decay rates of CH2OO as function of [HCOOH] at 458 K and total density of 3.3 × 1017 molecule cm−3 (15.5 Torr). The CH2OO traces were probed at 338 nm. | ||
By adding acid, the decay of CH2OO became faster. All the measurements were performed under pseudo-first-order conditions, i.e. [CH2OO] ≪ [HCOOH]. The photolysis of formic acid, HCOOH + hν(213 nm) → HCO + OH, was negligible (∼0.06%) in our measurements, because of low laser fluence (∼4 mJ cm−2) and small absorption cross-section of HCOOH at 213 nm (1.4 × 10−19 cm2 molecule−1).46 Even at the highest [HCOOH] used, 2.06 × 1013 molecule cm3, the resulting [OH] was only about 1.0 × 1010 molecule cm3, which could not have any important effect on the current measurements. Concomitant product of photolysis, HCO radical, would rapidly react with O2 and form HO2 radical that, due to its low reactivity, would have even smaller effect than the OH radical. In Fig. 5, the obtained pseudo-first-order decay rate coefficients (kobs) of CH2OO are shown as function of temperature and [HCOOH]. The bimolecular rate coefficient k(CH2OO + HCOOH) is obtained from the slope of the equation kobs = kloss + k(CH2OO + HCOOH) × [HCOOH] fitted to the data, while the intercept reflects the kloss. Obtained bimolecular rate coefficients were measured with the both precursor-photolysis wavelength combinations and the results are shown in Table 1 along with experimental conditions and statistical 2σ experimental uncertainties. Estimated overall uncertainties in the measured rate coefficients are about ±20%. The current bimolecular rate coefficient (1.0 ± 0.03) × 10−10 cm3 molecule−1 s−1 for CH2OO + HCOOH reaction measured at room temperature agrees with the previously reported rate coefficients (1.1 ± 0.1) × 10−10 cm3 molecule−1 s−1 measured by Welz et al.15
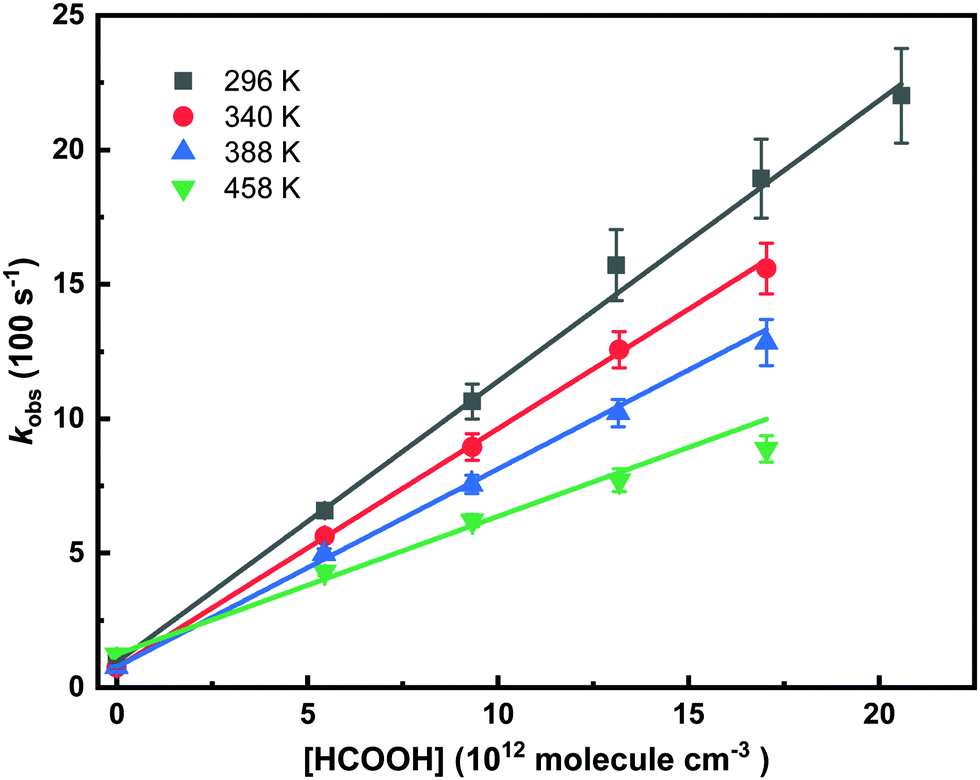 | ||
| Fig. 5 Determination of the bimolecular rate coefficients from pseudo-first-order decay rate coefficients at 296 K at total density of 1.6 × 1017 molecule cm−3, and 340 K, 388 K and 458 K at total density of 3.3 × 1017 molecule cm−3. | ||
| T (K) | [He] (×1017 molecule cm−3) | p (Torr) | [HCOOH]c (×1013 molecule cm−3) | k loss (s−1) | k (×10−10 cm3 molecule−1 s−1) |
|---|---|---|---|---|---|
| a Precursor concentrations used: 7.2 × 1012 molecule cm−3 for CH2IBr and 2.5 × 1012 molecule cm−3 for CH2I2. Estimated initial CH2OO concentration <1.0 × 1011 molecule cm−3. b O2 concentrations used: ∼3.8 × 1016 molecule cm−3 for CH2IBr and ∼2.8 × 1016 molecule cm−3 for CH2I2. c HCOOH reactant concentration in the reactor is calculated with accounting for the dimerization of the dilute HCOOH in helium mixture prepared in a bulb.45 The experiments in He and N2 were conducted with different HCOOH samples. d The statistical uncertainties shown are 2σ. Estimated overall uncertainty in the measured rate coefficients is about ±20%. e The linear gas flow velocity was ∼1.5 ms−1. | |||||
| Precursor: CH2IBra | |||||
| 296 | 1.6 | 5 | 0.55–2.06 | 95 | 1.0 ± 0.03 |
| 340 | 3.3 | 11.5 | 0.55–1.70 | 74 | 0.89 ± 0.01 |
| 388 | 3.3 | 13.1 | 0.54–1.70 | 77 | 0.74 ± 0.03 |
| 458 | 3.3 | 15.5 | 0.55–1.70 | 123 | 0.51 ± 0.05 |
Theoretical studies suggest that CH2OO + HCOOH reaction proceeds through a barrierless addition of the reactants leading to hydroperoxymethylformate (HPMF).18,19 In study by Vereecken,19 the rate coefficient (1.0 ± 0.1) × 10−10 cm3 molecule−1 s−1 is predicted at room temperature with very minor negative temperature dependency (∼8%) between 250 and 350 K. Current bimolecular rate coefficient at 458 K is about factor of two slower than the rate coefficient at room temperature, giving negative temperature dependency ∼50%. Fig. 6 shows an Arrhenius plot of the measured bimolecular rate coefficients of CH2OO + HCOOH reaction. The least squares fit to the data gives an Arrhenius expression, k = (1.5 ± 0.8) × 10−11 exp[(−4.9 ± 1.6) kJ mol−1/RT] cm3 molecule−1 s−1, with 2σ standard fitting uncertainties.
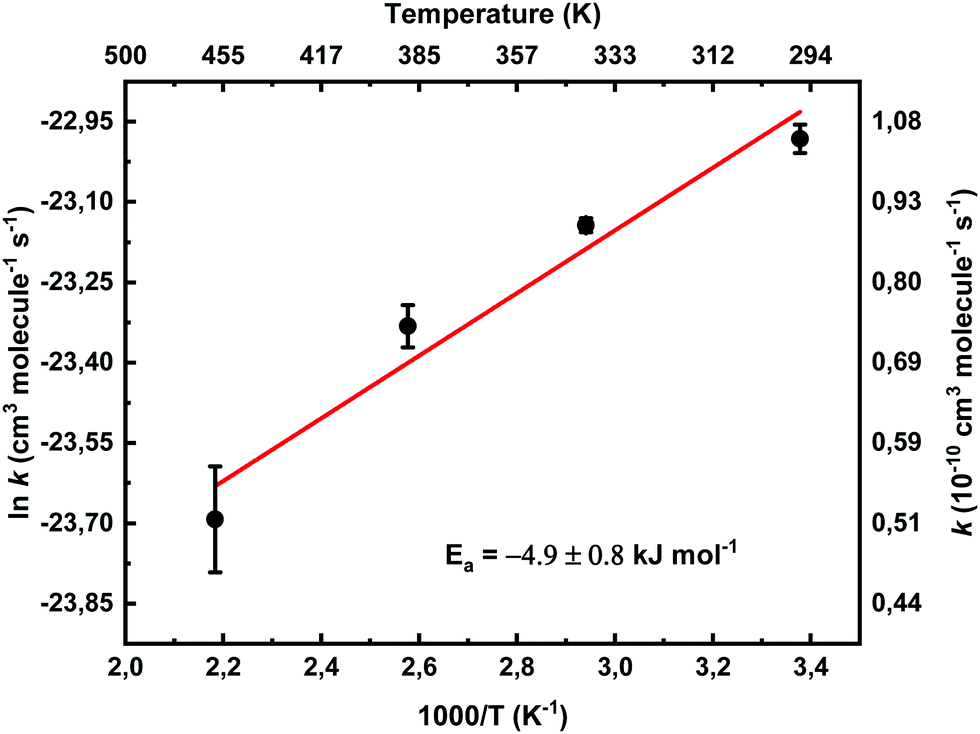 | ||
| Fig. 6 Arrhenius plot of the measured bimolecular rate coefficients utilizing CH2IBr photolytic precursor. The statistical uncertainties shown are 2σ. | ||
The bimolecular rate coefficient of the reaction was measured also as a function of helium density at 296 K. Fig. 7 presents the obtained pseudo-first-order decay rate coefficients (kobs) of CH2OO as function of pressure and [HCOOH]. A complete set of the results and experimental conditions is shown in Table S1 (in the ESI†). The reaction appears to be pressure independent over the range between 5 and 150 Torr, especially once considering the uncertainty of the measurements. An increase in the baseline of the measured absorption signal was observed at high pressures (with added HCOOH), which is possibly due to an enhanced stabilization of ICH2OO at higher pressure (see more details in the ESI†).
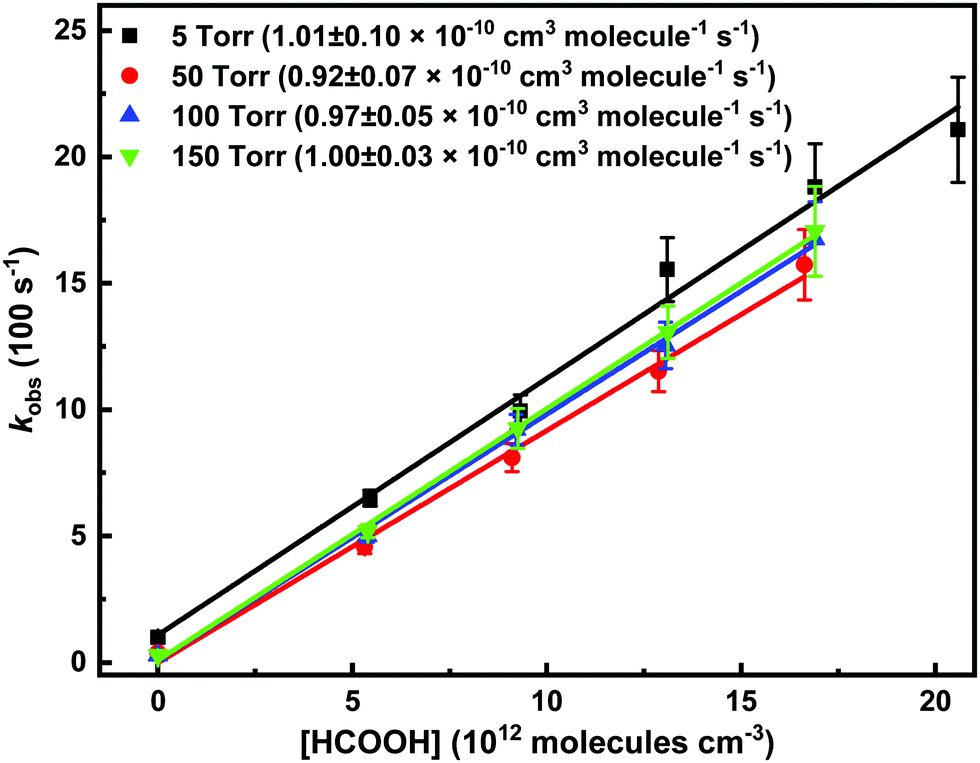 | ||
| Fig. 7 Bimolecular plots of the pseudo-first-order decay rate coefficients measured at 5, 50, 100, and 150 Torr total pressures versus [HCOOH] at constant temperature of 296 K. The obtained bimolecular rate coefficients are presented in the parentheses. The statistical uncertainties shown are 2σ. | ||
For comparison, the temperature dependency of the reaction was also measured using diiodomethane (CH2I2) precursor with 266 nm photolysis. The results are given in Table 1, which show that outcome of both precursor-photolysis wavelength combinations are in agreement with each other. The obtained Arrhenius expression for the CH2I2 study was k = (1.1 ± 0.4) × 10−11 exp[(−5.9 ± 0.8) kJ mol−1/RT] cm3 molecule−1 s−1, with 2σ standard fitting uncertainties. The inert buffer-gas in the measurements was nitrogen (N2).
Unimolecular decomposition of CH2OO
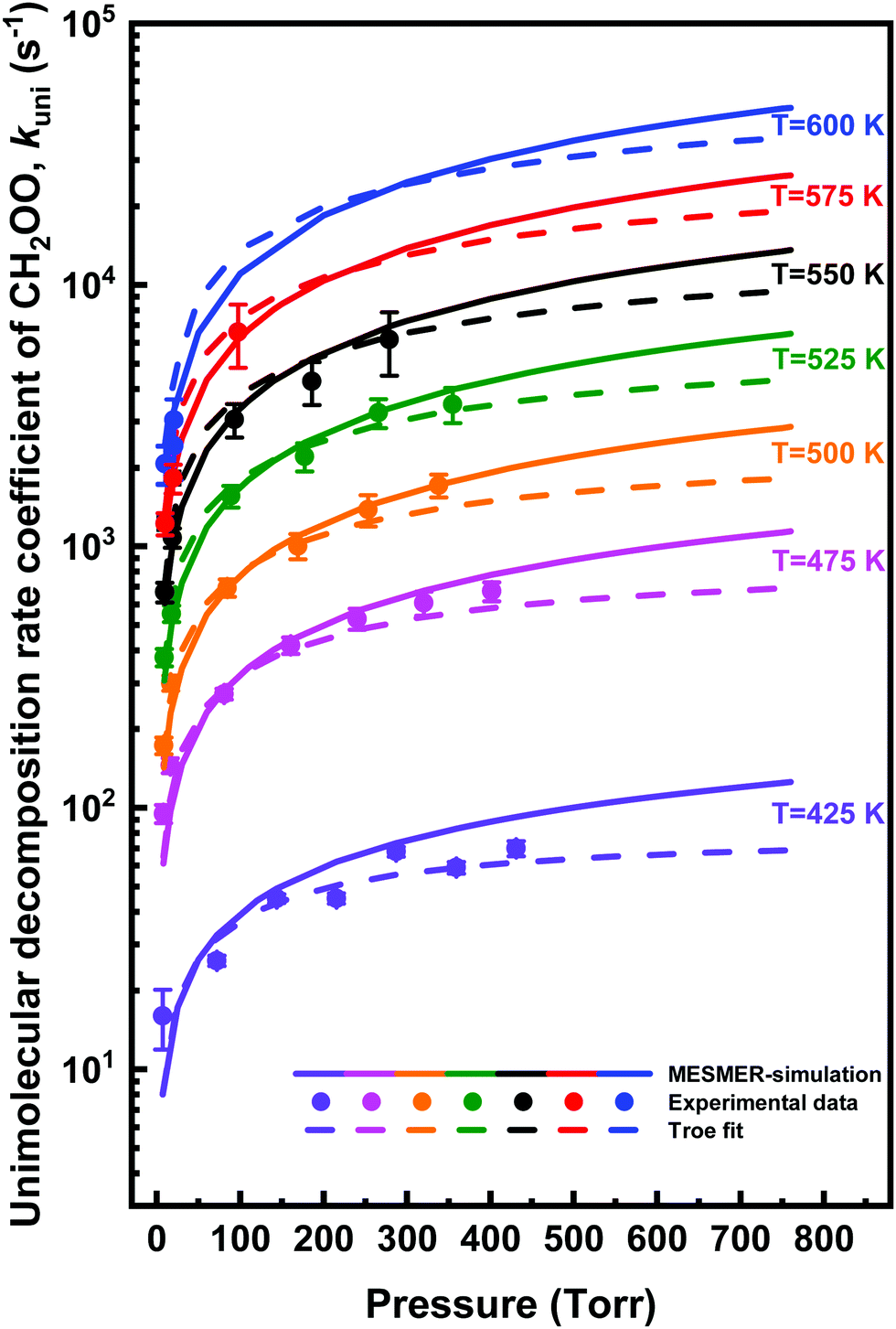 | ||
| Fig. 8 Measured thermal unimolecular decomposition rate coefficient of CH2OO as function of total helium density at different temperatures (coloured points). The statistical uncertainties shown are 2σ. Results of MESMER simulations (solid lines) and Troe equation fits (dashed lines) are also shown. | ||
| T (K) | [He] (×1018 molecule cm−3) | p (Torr) | k obs (s−1) | k loss (s−1) | k uni (s−1) | k uni,Troe (s−1) | k uni,MESMER (s−1) |
|---|---|---|---|---|---|---|---|
| a The fixed O2 concentration was ∼3.9 × 1016 molecule cm−3. | |||||||
| 296 | 0.16 | 5 | 99 ± 3.3 | 100 | −1 | 0 | 0 |
| 0.33 | 10 | 55 ± 1.3 | 62 | −7 | 0 | 0 | |
| 1.6 | 50 | 31 ± 0.7 | 29 | −2 | 0 | 0 | |
| 3.3 | 100 | 26 ± 0.7 | 24 | −2 | 0 | 0 | |
| 4.9 | 150 | 26 ± 0.8 | 26 | 0 | 0 | 0 | |
| 6.5 | 200 | 22 ± 0.5 | 19 | −3 | 0 | 0 | |
| 8.2 | 250 | 20 ± 0.6 | 19 | −1 | 0 | 0 | |
| 9.8 | 300 | 21 ± 0.7 | 18 | −3 | 0 | 0 | |
| ∞ | 0 | ||||||
| 325 | 0.16 | 5.5 | 104 ± 2.60 | 100 | −4 | 0 | 0 |
| 0.33 | 11 | 61 ± 1.2 | 62 | −1 | 0 | 0 | |
| 1.6 | 55 | 25 ± 0.4 | 29 | −4 | 0 | 0 | |
| 3.3 | 110 | 23 ± 0.4 | 24 | −1 | 0 | 0 | |
| 4.9 | 164.6 | 27 ± 0.6 | 26 | −1 | 0 | 0 | |
| 6.5 | 219.5 | 18 ± 0.3 | 19 | −1 | 0 | 0 | |
| 8.2 | 274.7 | 19 ± 0.4 | 19 | 0 | 0 | 0 | |
| 9.8 | 329.5 | 14 ± 0.3 | 18 | −4 | 0 | 0 | |
| ∞ | 0 | ||||||
| 375 | 0.16 | 6.35 | 96 ± 2.5 | 100 | −4 | 0 | 0 |
| 0.33 | 12.7 | 70 ± 1.6 | 62 | −8 | 0 | 0 | |
| 1.6 | 63.5 | 32 ± 0.5 | 29 | −3 | 0 | 0 | |
| 3.3 | 126.6 | 23 ± 0.4 | 24 | −1 | 0 | 0 | |
| 4.9 | 190 | 25 ± 0.4 | 26 | −1 | 0 | 0 | |
| 6.5 | 253.5 | 16 ± 0.3 | 19 | −3 | 0 | 0 | |
| 8.2 | 317 | 17 ± 0.3 | 19 | −2 | 0 | 0 | |
| 9.8 | 380.5 | 19 ± 0.4 | 18 | −1 | 0 | 0 | |
| ∞ | 0 | ||||||
| 425 | 0.16 | 7.2 | 115 ± 4.10 | 100 | 15 | 8 | 8 |
| 1.6 | 72 | 57 ± 1.2 | 29 | 28 | 32 | 33 | |
| 3.3 | 143.5 | 71 ± 1.7 | 24 | 47 | 43 | 49 | |
| 4.9 | 215.5 | 71 ± 2.0 | 26 | 45 | 51 | 62 | |
| 6.5 | 287 | 90 ± 3.0 | 19 | 71 | 55 | 73 | |
| 8.2 | 359.5 | 79 ± 3.0 | 19 | 60 | 59 | 83 | |
| 9.8 | 431 | 91 ± 4.7 | 18 | 73 | 62 | 92 | |
| ∞ | 347 | ||||||
| 475 | 0.16 | 8.05 | 194 ± 7.6 | 100 | 94 | 65 | 61 |
| 0.33 | 16.05 | 200 ± 9.5 | 62 | 138 | 110 | 97 | |
| 1.6 | 80.5 | 303 ± 13 | 29 | 274 | 287 | 281 | |
| 3.3 | 160.5 | 444 ± 30 | 24 | 420 | 402 | 438 | |
| 4.9 | 240.5 | 554 ± 48 | 26 | 528 | 480 | 566 | |
| 6.5 | 320.5 | 629 ± 65 | 19 | 610 | 537 | 677 | |
| 8.2 | 401.5 | 693 ± 56 | 19 | 674 | 580 | 778 | |
| ∞ | 3601 | ||||||
| 500 | 0.16 | 8.45 | 272 ± 13 | 100 | 172 | 156 | 143 |
| 0.33 | 16.9 | 355 ± 20 | 62 | 293 | 270 | 230 | |
| 1.6 | 84.5 | 725 ± 52 | 29 | 696 | 730 | 691 | |
| 3.3 | 169 | 1029 ± 111 | 24 | 1005 | 1031 | 1097 | |
| 4.9 | 253.5 | 1407 ± 188 | 26 | 1381 | 1240 | 1431 | |
| 6.5 | 338 | 1732 ± 172 | 19 | 1713 | 1397 | 1724 | |
| ∞ | 9589 | ||||||
| 525 | 0.16 | 8.9 | 475 ± 29 | 100 | 375 | 344 | 306 |
| 0.33 | 17.75 | 608 ± 40 | 62 | 546 | 605 | 500 | |
| 1.6 | 89 | 1587 ± 149 | 29 | 1558 | 1696 | 1553 | |
| 3.3 | 177.5 | 2231 ± 269 | 24 | 2207 | 2411 | 2497 | |
| 4.9 | 266 | 3271 ± 407 | 26 | 3245 | 2917 | 3282 | |
| 6.5 | 355 | 3518 ± 536 | 19 | 3499 | 3306 | 3977 | |
| ∞ | 23029 |
||||||
| 550 | 0.16 | 9.3 | 767 ± 58 | 100 | 667 | 706 | 606 |
| 0.33 | 18.6 | 1133 ± 910 | 62 | 1071 | 1255 | 1004 | |
| 1.6 | 93 | 3083 ± 451 | 29 | 3054 | 3641 | 3205 | |
| 3.3 | 186 | 4296 ± 800 | 24 | 4272 | 5212 | 5228 | |
| 4.9 | 279 | 6199 ± 1683 | 26 | 6173 | 6336 | 6928 | |
| ∞ | 50570 |
||||||
| 575 | 0.16 | 9.75 | 1319 ± 119 | 100 | 1219 | 1359 | 1123 |
| 0.33 | 19.45 | 1878 ± 233 | 62 | 1816 | 2438 | 1881 | |
| 1.6 | 97.5 | 6635 ± 1791 | 29 | 6606 | 7303 | 6181 | |
| ∞ | 102705 |
||||||
| 600 | 0.16 | 10.15 | 2170 ± 351 | 100 | 2070 | 2473 | 1956 |
| 0.33 | 20.3 | 3093 ± 610 | 62 | 3031 | 4473 | 3318 | |
| ∞ | 194777 |
||||||
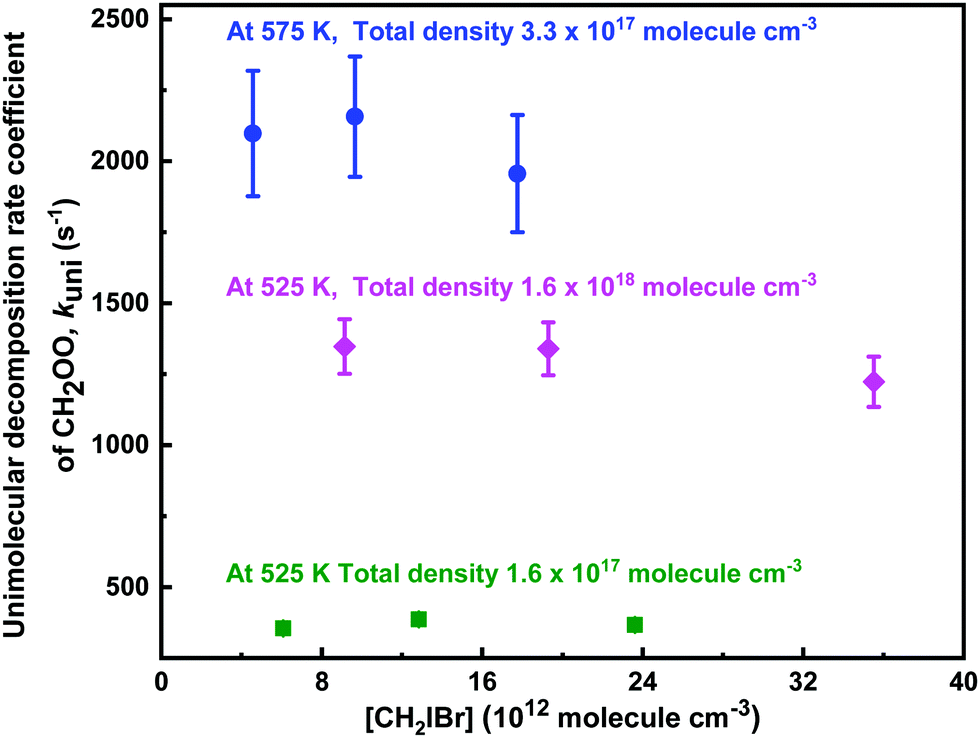 | ||
| Fig. 9 Measured unimolecular decomposition rate coefficients of CH2OO as function of CH2IBr concentration in different temperature and total density conditions. The statistical uncertainties shown are 2σ. | ||
The unimolecular reaction rate coefficient kuni at given temperature and total density is obtained by kuni = kobs − kloss, where both kobs and kloss are measured decay rate coefficients from single-exponential function fits to the experimental traces. The kloss is measured at a few temperatures below which any significant unimolecular decomposition reaction occurs, whereas the kobs is measured at a temperature where significant unimolecular reaction occurs, i.e., kobs > 3kloss. At low pressures (p < 20 Torr), the kloss depends strongly on total density, decreasing from ∼100 s−1 at low total density to ∼20 s−1 at high total density. However, values of kloss at each total density stay almost constant between the temperatures 296 K and 375 K, which is reasonable, since diffusion has a weak temperature dependency. The unimolecular decomposition reaction starts to become significant at temperatures only at and above 425 K (see Fig. S9 in the ESI†). Thus, mean of the measurements performed between 296 and 375 K was used as the kloss.
A positive baseline offset of the measured absorption signal was also present at high pressures in the thermal unimolecular decomposition measurements. The offset value increased as pressure increased indicating the possible enhanced stabilization of ICH2OO as observed in the bimolecular CH2OO + HCOOH reaction. However, in constant (high) density, the baseline offset decreased as the temperature increased indicating a possible decomposition of ICH2OO. Interestingly, at high temperatures, a small offset was present already at low pressures, which might indicate possible absorption by another formed product than ICH2OO. The baseline offset has been taken into account in the fitting of the first-order, single-exponential decays (see more details in ESI†).
The unimolecular decomposition rate coefficient data shown in Fig. 8 were fitted simultaneously (a global fit) using the following Troe expression47 employing the nonlinear least squares fitting method
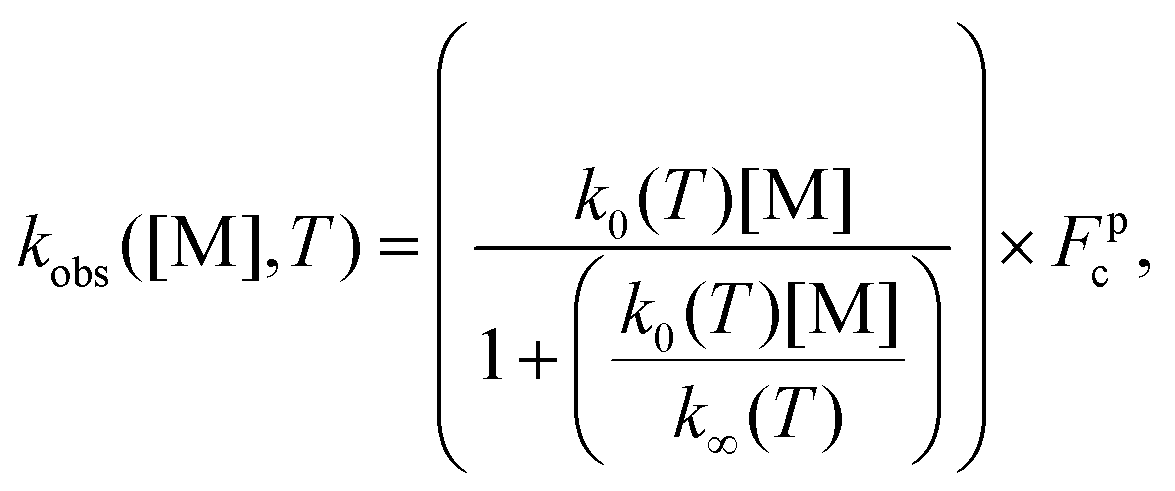 | (3) |
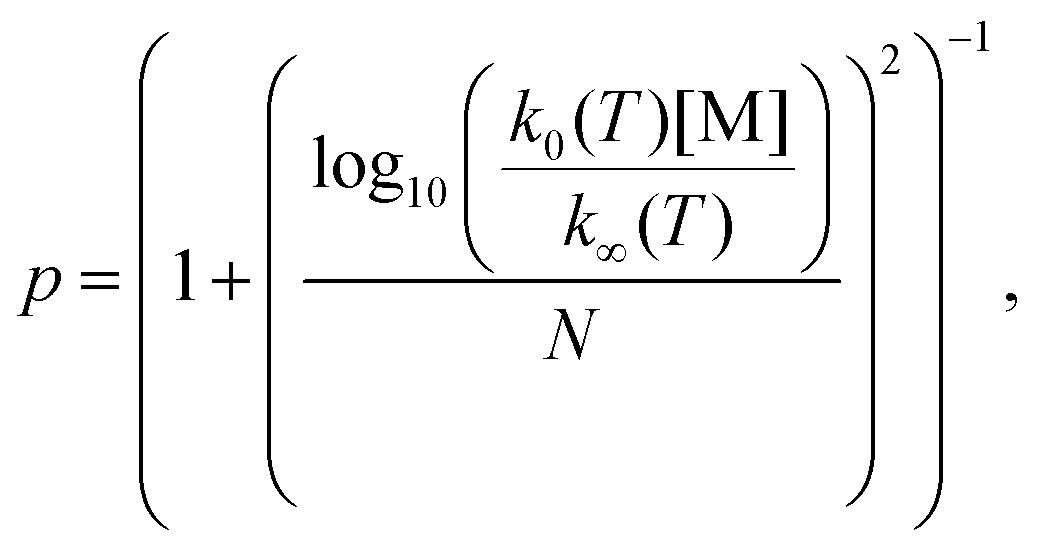 | (4) |
log10Fc is the width parameter. The global fits using eqn (3) were performed with Fc and N fixed to values of 0.648 and 1.0,47 respectively, or Fc allowed to float independently at each temperature. The fits with fixed Fc and N values gave lower χ2-value, although the parameterisations from each fit were in agreement within the fitting uncertainties. The fit with fixed Fc and N values gives k0(T) = (1.3 ± 3.0) × 10−8 exp[(−8065 ± 1170)/T] cm3 mol−1 s−1 and k∞(T) = (7.8 ± 17.8) × 1011 exp[(−9716 ± 1088)/T] s−1, where statistical uncertainties stated are 2σ.
| 〈ΔE〉down = 123.2 × (T/298 K)0.74, | (5) |
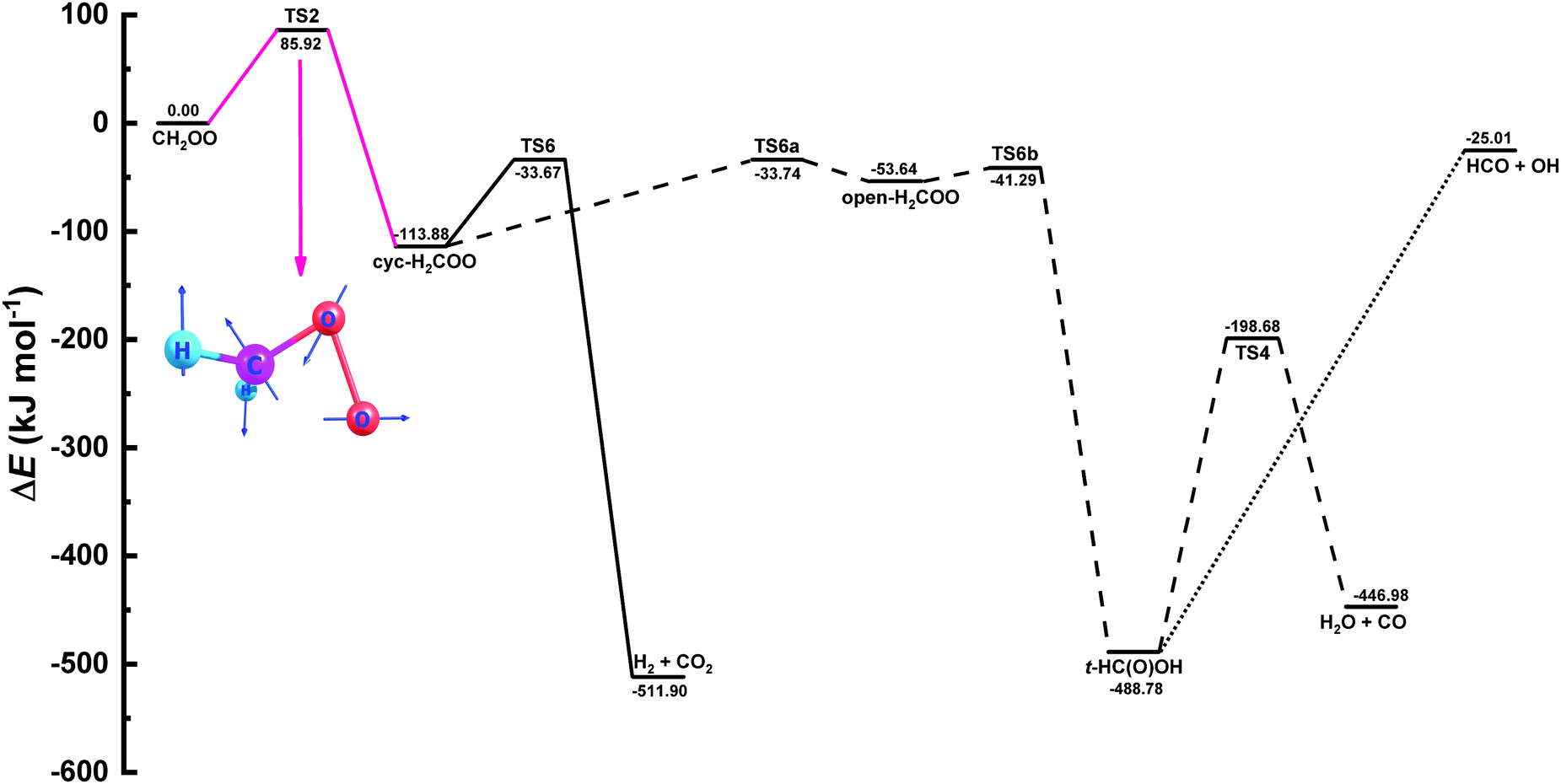 | ||
| Fig. 10 Zero-point inclusive potential energy surface (PES) for the unimolecular decomposition of CH2OO in kJ mol−1. The names of the species involved in the PES were taken from the works of Stone et al.9 and Nguyen et al.36 The portion of the PES used in MESMER calculations to fit the experimental rate coefficient data is shown in pink along with the structure of the crucial transition state. | ||
In literature several values are reported for the decomposition barrier of CH2OO to cyc-H2COO, ranging from 76 to 100 kJ mol−1.49 Our calculated barrier 85.92 kJ mol−1 lies well within this range. To further test compatibility between the current computational and experimental results, we performed MESMER simulations by fitting simultaneously not only the collisional energy transfer parameters but also the barrier height so as to observe any change in the barrier height. We noticed that using Eckart tunneling, the decomposition barrier increases slightly by 1.6 kJ mol−1, when fitting all three parameters simultaneously. On the other hand, when no tunneling is used, the barrier goes down by 1.8 kJ mol−1. A comparison between the current MESMER simulation and the simulations with floating all three parameters is provided in Fig. S10 of the ESI† along with the experimental rate coefficients.
Product yields for the three different channels, viz., H2 + CO2, H2O + CO, and HCO + OH presented in Fig. 10 were also estimated. According to MESMER simulations, H2 + CO2 is the predominant channel with 60.8% yield, whereas yields of the other two channels, H2O + CO, and HCO + OH, can differ depending on the parameters involved in the simulations. The formation of HCO + OH is believed to proceed via irreversible decomposition of t-HC(O)OH without a clear transition state. Hence, in the present work, we have used a measured value k(HCO + OH) = 1.83 × 10−10 cm3 molecule−1 s−1, as reported by Temps et al.,51 for the pre-exponential factor in the modified Arrhenius expression, without any temperature dependency, to obtain the yield of the OH-radical formation channel. Our results indicate the formation of H2O + CO channel with an overall yield of 30.9%, while the least dominant OH + HCO channel was having a percentage yield of 8.3.
In order to facilitate the utilization of current MESMER results in combustion or atmospheric chemistry models, we provide modified Arrhenius representations in ChemKin PLOG format (see ESI†) for the PES illustrated in Fig. 10. Also a MESMER input file is given in ESI.†
 | ||
| Fig. 11 Comparison of the current measurements (filled circles) and MESMER simulation results (solid lines) with the results of previous experiments (hollow triangles) of Stone et al.9 for the unimolecular decomposition rate coefficients of CH2OO as function of pressure at different temperatures. | ||
Conclusions
In this work we have presented a new TR-BB-CEAS apparatus for kinetic studies of reactive intermediates, which are probed utilizing their UV-absorption. We show the capability of and use the new set-up for temperature- and pressure-dependent measurements of formaldehyde oxide (CH2OO) reactions. The spectrometer is able to simultaneously measure transient absorption spectra over the wavelength range 300–450 nm with the time resolution of 50 μs using a fast CMOS line array camera-based grating spectrometer. The line array camera enables the transient spectra to be measured without any moving parts inside the grating spectrometer. For CH2OO, we could reach a detection limit ∼2.4 × 109 molecule cm−3 at 340 nm region with an averaging time of 30 min. This work also reveals a new photolytic precursor for formaldehyde oxide, bromoiodomethane (CH2IBr), which photolysis at 213 nm in presence of O2 produces CH2OO. This new precursor was found to be free from secondary reactions that may regenerate CH2OO in kinetic experiments. In addition, depletion of CH2IBr in a photolysis does not produce a negative baseline shift for the CH2OO absorption trace signal because of much smaller absorption cross-section of bromoiodomethane at 340 nm region.The bimolecular rate coefficient for CH2OO + HCOOH reaction was found to have a negative temperature dependency, decreasing from (1.0 ± 0.03) × 10−10 cm3 molecule−1 s−1 at 296 K to (0.47 ± 0.05) × 10−10 cm3 molecule−1 s−1 at 458 K with an Arrhenius activation energy −4.9 ± 1.6 kJ mol−1. The obtained bimolecular rate coefficient at room temperature agrees with the previously reported rate coefficients from the direct kinetic experiments by Welz et al.15 Photodissociation of HCOOH reactant was found to be negligible and, consequently, did not have any important effect on the bimolecular kinetics of the CH2OO + HCOOH reaction. However, if a reactant absorbs strongly at 213 nm, its photolysis could be a problem in bimolecular reaction kinetic measurements. This may well be the case with SO2 reactant, which absorbs radiation strongly at 213 nm.52 In the current study, the CH2OO + HCOOH reaction was also found to be pressure independent over the range between 5 and 150 Torr of helium at 296 K.
Our kinetic measurements of thermal unimolecular decomposition of CH2OO over wide pressure (5–400 Torr) and temperature (296–600 K) ranges confirm the previously results that the thermal unimolecular decomposition is not an important atmospheric sink of CH2OO9,53 and the decomposition only becomes significant at temperatures above 425 K. Due to this reason, thermal unimolecular decomposition of CH2OO might play an important role under low-temperature combustion conditions.
A very good agreement between the experimental results and master equation simulations performed using MESMER code was obtained by fitting collision energy transfer parameters to the experimental data without tuning any transition state energy. This evidences high coherence between experiments and theory. The MESMER simulations suggest 60.8% decomposition yield for the predominant H2 + CO2 channel. Yields of two other channels, H2O + CO, and HCO + OH, are sensitive on the parameters involved in the simulations. Constraining simulations with an experimental value for (reverse) HCO + OH reaction, simulations return 30.9% and 8.3% yields for H2O + CO and HCO + OH reaction channels, respectively.
Appreciable differences observed between the current experimental thermal unimolecular decomposition kinetics of CH2OO and the results of Stone et al.9 measurements are suggested to originate from the use of different photolytic precursors.
Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
J. P., P. S., and A. I. acknowledge support from the Academy of Finland, Grants 298910 and 311967. A. E. acknowledges support from the Academy of Finland, Grant 288377. The financial support from the University of Helsinki and the Academy of Finland (Grant 298910) to construct the apparatus is gratefully acknowledged. The authors wish to acknowledge CSC – IT Center for Science, Finland, for computational resources. We also thank Dr Leonid Sheps, Combustion Research Facility, Sandia National Laboratories, California, US, for help in design of our TR-BB-CEAS apparatus.References
- L. Vereecken, A. Novelli and D. Taraborrelli, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- M. A. H. Khan, C. J. Percival, R. L. Caravan, C. A. Taatjes and D. E. Shallcross, Environ. Sci.: Processes Impacts, 2018, 20, 437–453 RSC.
- C. A. Taatjes, G. Meloni, T. M. Selby, A. J. Trevitt, D. L. Osborn, C. J. Percival and D. E. Shallcross, J. Am. Chem. Soc., 2008, 130, 11883–11885 CrossRef CAS PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204–207 CrossRef CAS PubMed.
- Y. Su, Y. Huang, H. A. Witek and Y. Lee, Science, 2013, 340, 174–176 CrossRef CAS PubMed.
- L. Sheps, J. Phys. Chem. Lett., 2013, 4, 4201–4205 CrossRef CAS PubMed.
- W. Chao, J. Hsieh, C. Chang and J. J. M. Lin, Science, 2015, 347, 751–754 CrossRef CAS PubMed.
- D. Stone, K. Au, S. Sime, D. J. Medeiros, M. Blitz, P. W. Seakins, Z. Decker and L. Sheps, Phys. Chem. Chem. Phys., 2018, 20, 24940–24954 RSC.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- A. J. Eskola, D. Wojcik-Pastuszka, E. Ratajczak and R. S. Timonen, Phys. Chem. Chem. Phys., 2006, 8, 1416–1424 RSC.
- H. Huang, A. J. Eskola and C. A. Taatjes, J. Phys. Chem. Lett., 2012, 3, 3399–3403 CrossRef CAS.
- H. Huang, B. Rotavera, A. J. Eskola and C. A. Taatjes, J. Phys. Chem. Lett., 2013, 4, 3824 CrossRef CAS.
- J. L. Miller, Phys. Today, 2012, 65, 17–19 Search PubMed.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. A. H. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., Int. Ed., 2014, 53, 4547–4550 CrossRef CAS PubMed.
- J. J. M. Lin and W. Chao, Chem. Soc. Rev., 2017, 46, 7483–7497 RSC.
- R. Chhantyal-Pun, A. Davey, D. E. Shallcross, C. J. Percival and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2015, 17, 3617–3626 RSC.
- B. Long, J. Cheng, X. Tan and W. Zhang, THEOCHEM, 2009, 916, 159–167 CrossRef CAS.
- L. Vereecken, Phys. Chem. Chem. Phys., 2017, 19, 28630–28640 RSC.
- A. Andersen and E. A. Carter, J. Phys. Chem. A, 2003, 107, 9463–9478 CrossRef CAS.
- A. Andersen and E. A. Carter, Mol. Phys., 2008, 106, 367–396 CrossRef CAS.
- B. Z. Chen, J. M. Anglada, M.-B. Huang and F. Kong, J. Phys. Chem. A, 2002, 106, 1877–1884 CrossRef CAS.
- R. A. Alvarez and C. B. Moore, J. Phys. Chem., 1994, 98, 174–183 CrossRef CAS.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Chem. Phys., 2013, 138, 20045 CrossRef PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. Lee, J. M. Dyke, D. K. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177–180 CrossRef CAS PubMed.
- Y. Chang, C. Chang and K. Takahashi, Chem. Phys. Lett., 2016, 653, 155–160 CrossRef CAS.
- F. Liu, J. M. Beames, A. M. Green and M. I. Lester, J. Phys. Chem. A, 2014, 118, 2298–2306 CrossRef CAS PubMed.
- D. Stone, M. Blitz, L. Daubney, N. U. M. Howes and P. Seakins, Phys. Chem. Chem. Phys., 2014, 16, 1139–1149 RSC.
- Y. Liu, F. Liu, S. Liu, D. Dai, W. Dong and X. Yang, Phys. Chem. Chem. Phys., 2017, 19, 20786–20794 RSC.
- Z. J. Buras, R. M. Elsamra, A. Jalan, J. E. Middaugh and W. H. Green, J. Phys. Chem. A, 2014, 118, 1997–2006 CrossRef CAS PubMed.
- L. Sheps and D. W. Chandler, Time-Resolved Broadband Cavity-Enhanced Absorption Spectroscopy for Chemical Kinetics, Sandia National Laboratories, Livermore, CA, 2013, SAND2013-2664 Search PubMed.
- J. J. Scherer, J. B. Paul, H. Jiao and A. O’Keefe, Appl. Opt., 2001, 40, 6725–6732 CrossRef CAS PubMed.
- J. C. Mössinger, D. E. Shallcross and R. A. Cox, J. Chem. Soc., Faraday Trans., 1998, 94, 1391–1396 RSC.
- L. Butler, E. Hintsa and Y. Lee, J. Chem. Phys., 1986, 84, 4104–4106 CrossRef CAS.
- L. Butler, E. Hintsa, S. F. Shane and Y. Lee, J. Chem. Phys., 1987, 86, 2051–2074 CrossRef CAS.
- T. Nguyen, R. Putikam and M. C. Lin, J. Chem. Phys., 2015, 142, 124312 CrossRef PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2016.
- F. Neese, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- A. K. Wilson, T. van Mourik and T. H. Dunning, THEOCHEM, 1996, 388, 339–349 CrossRef CAS.
- K. A. Peterson, D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 7410–7415 CrossRef CAS.
- T. T. Pekkanen, R. S. Timonen, G. Lendvay, M. P. Rissanen and A. J. Eskola, Proc. Combust. Inst., 2019, 37, 299–306 CrossRef CAS.
- D. R. Glowacki, C. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- W. Ting, Y. Chen, W. Chao and M. C. Smith, Phys. Chem. Chem. Phys., 2014, 16, 10438–10443 RSC.
- A. Winkler and P. Hess, J. Am. Chem. Soc., 1994, 116, 9233–9240 CrossRef CAS.
- D. Singleton, G. Paraskevopoulos and R. Irwin, J. Photochem., 1987, 37, 209–216 CrossRef CAS.
- J. Troe, J. Phys. Chem., 1979, 83, 114–126 CrossRef CAS.
- V. P. Barber, S. Pandit, A. M. Green, N. Trongsiriwat, P. J. Walsh, S. J. Klippenstein and M. I. Lester, J. Am. Chem. Soc., 2018, 140, 10866–10880 CrossRef CAS PubMed.
- T. Berndt, R. Kaethner, J. Voigtländer, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipilä, M. Kulmala and M. Olzmann, Phys. Chem. Chem. Phys., 2015, 17, 19862–19873 RSC.
- L. I. Stiel and G. Thodos, AIChE J., 1964, 10, 266–269 CrossRef CAS.
- F. Temps and H. G. Wagner, Ber. Bunsenges. Phys. Chem., 1984, 88, 415–418 CrossRef CAS.
- G. Stark, P. L. Smith, J. Rufus, A. Thorne, J. Pickering and G. Cox, J. Geophys. Res., 1999, 104, 16585–16590 CrossRef.
- T. A. Stephenson and M. I. Lester, Int. Rev. Phys. Chem., 2020, 39, 1–33 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00302f |
| This journal is © the Owner Societies 2020 |