
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRigidly hydrogen-bonded water molecules facilitate proton transfer in photosystem II†
Naoki
Sakashita
a,
Hiroshi
Ishikita
 ab and
Keisuke
Saito
*ab
ab and
Keisuke
Saito
*ab
aDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8654, Japan
bResearch Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo, 153-8904, Japan. E-mail: ksaito@appchem.t.u-tokyo.ac.jp; Fax: +81-3-5452-5083; Tel: +81-3-5452-5056
First published on 2nd July 2020
Abstract
In the water-splitting enzyme photosystem II (PSII), the proton is released from the catalytic site and transferred to the protein bulk surface via the proton-relay mechanism. Proton transfer occurs in a proton-conducting channel consisting of a series of water molecules connected by hydrogen-bonded (H-bonded) chains. The water-transport protein aquaporin (AQP) also contains a water chain with structure similar to that of the PSII proton channel, although the water chain does not transport protons. We compared the PSII proton channel with the AQP water channel from the following standpoints: (1) the energetics of proton transfer based on crystal structures obtained from quantum mechanical/molecular mechanical calculations, and (2) fluctuations in water molecules obtained from molecular dynamics simulations. The results showed that residues facing the channel and acting as H-bonded partners of water molecules predominantly determined the proton-transfer ability. In PSII, the water chain is surrounded by H-bond acceptors (e.g., carbonyl groups), and the water chain transports protons where the water molecules are rigidly fixed. In AQP, the water chain is surrounded by hydrophobic sidechains or H-bond donors (e.g., NH2 groups), and it does not transport protons where the water molecules are flexible and fluctuating.
Introduction
The photosynthetic protein photosystem II (PSII) mediates a light-driven, water-splitting reaction (2H2O → 4H+ + O2 + 4e−),1,2 where PSII incorporates two water molecules and releases four protons. Protons are released in response to changes in the oxidation state (the Sn state, where n represents the number of oxidation steps).3 During the first step (the S0 → S1 transition), one proton is released from the Mn4CaO5 cluster and ejected to the outside of the protein.4 Around the Mn4CaO5 cluster, there exist water molecules forming hydrogen-bond (H-bond) networks. Among these water molecules, a water chain from the O4 site of the Mn4CaO5 cluster and oriented toward the protein bulk surface (Fig. 1a) has been suggested to play a role in a proton-conducting wire.5–8 Proton release in the S0 → S1 transition occurs very rapidly (i.e., it is not a rate-limiting process) as compared with other proton-release processes in the other (S2 → S3 and S3 → S0) transitions (observed to be rate-limiting).1,9 The rapid proton transfer can be explained as a consequence of the proton-relay mechanism (the so-called Grotthuss-like mechanism),10,11 which requires a single-file arrangement of properly H-bonded water molecules oriented unidirectionally along the channel.11 The proton-conducting wire from O4 consists of a series of water molecules connected by H-bonded chains (Fig. 1a and c).5,6 The Grotthuss-like proton transfer involves successive proton-transfer reactions between adjacent water molecules. Specifically, for each proton-transfer reaction, an O–H covalent bond is cleaved and transformed to an H-bonded O atom along with simultaneous formation of another O–H covalent bond (Fig. 1c). Because of proton transfer, the H-bond patterns of water molecules are converted from the pre-proton transfer (pre-PT) pattern to the post-proton transfer (post-PT) pattern (Fig. 1b and c).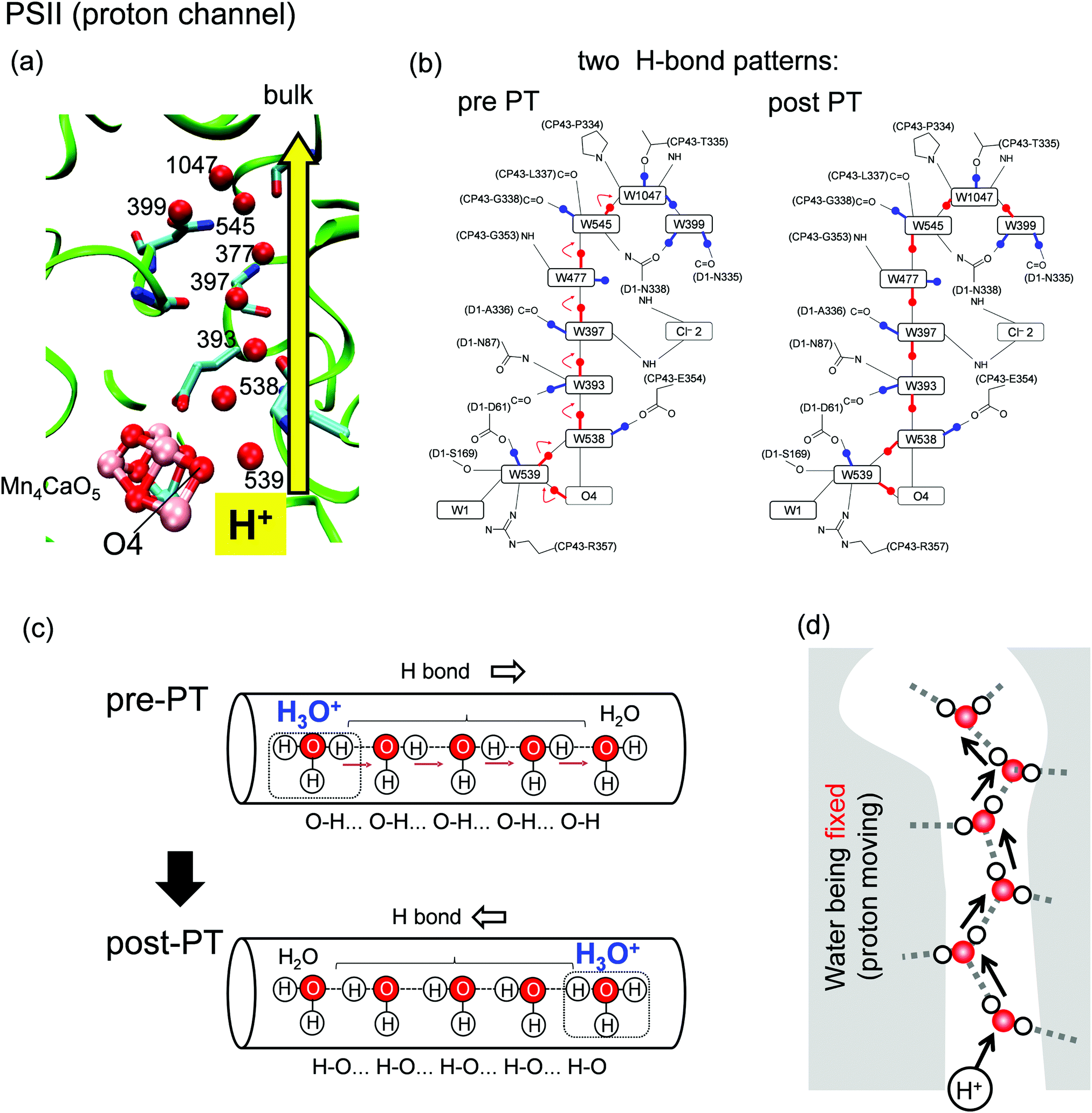 | ||
| Fig. 1 (a) Structure of the proton-transfer channel and (b) H-bond patterns5 before (pre-PT) and after (post-PT) proton transfer in PSII. (c) Schematics of a segment of a proton-conducting channel. The Grotthuss like proton-transfer mechanism transforms the H-bond pattern of water molecules from the pre-PT pattern to the post-PT pattern. (d) No water mobility in the proton-transfer channel. | ||
Not only protons but also water molecules are transferred in proteins via special channels. A number of studies reported multiple candidate proton/water channels surrounding the Mn4CaO5 cluster in PSII.12–15 Human water-transport proteins, aquaporins (AQPs) contain a water channel consisting of a water chain oriented along a transmembrane axis (Fig. 2a)16–18 and are capable of transporting water rapidly (the effective conductance rates are close to 109 s−1);16,17 however, the channel cannot transfer protons16,17 despite the single-file arrangement of H-bonded water molecules similar to that of the O4 proton channel in PSII.
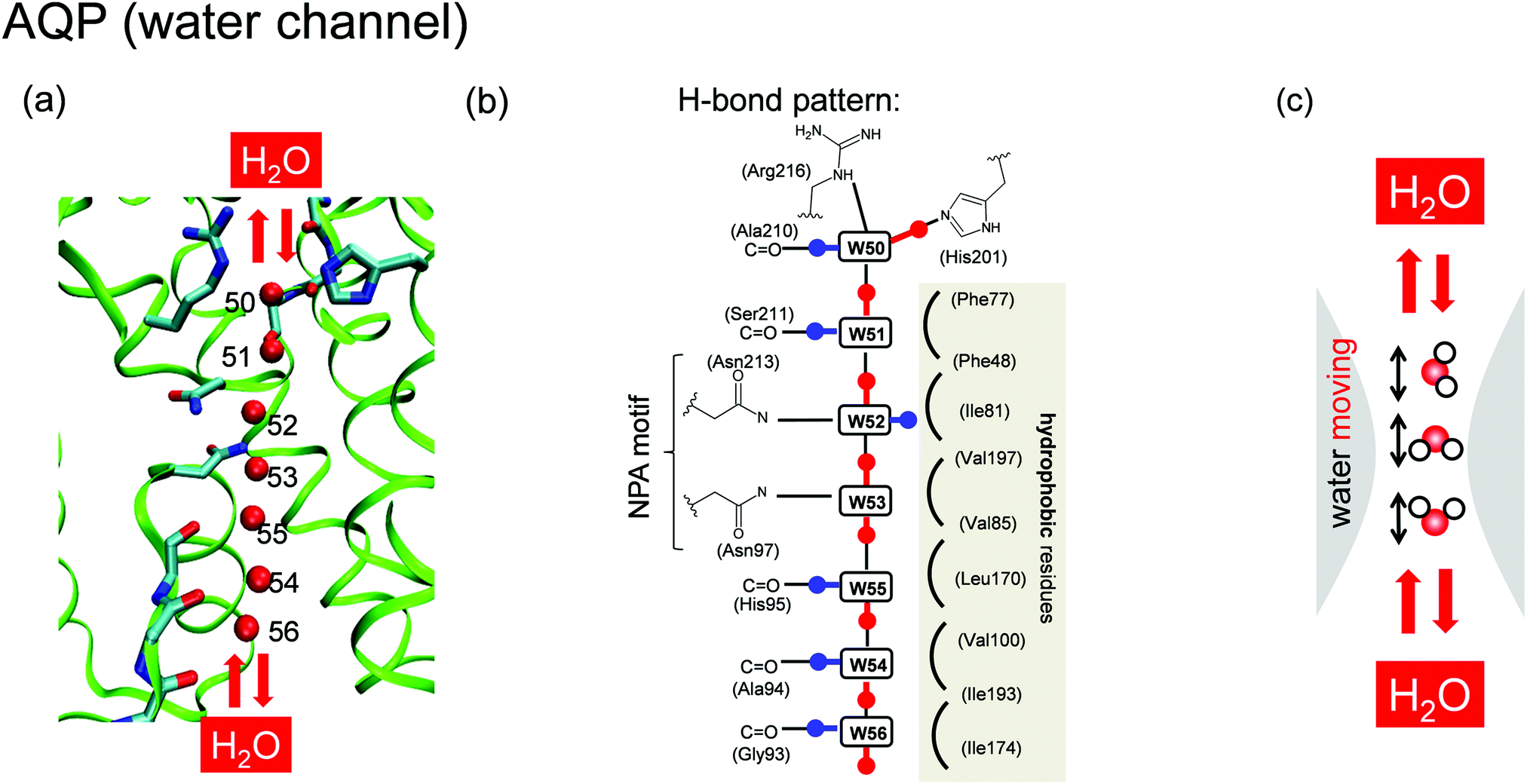 | ||
| Fig. 2 Structure of the (a) water-transfer channel and (b) proposed H-bond pattern18,19 in AQP. (c) Water mobility in the water-transfer channel. | ||
The mechanism preventing proton transfer in AQP has been well described. Early studies19,20 suggested that blockage of proton transfer is attributable to water-orientational effects.10,11 On the basis of both structural interpretations18 and molecular-dynamics (MD) simulation results,19,20 it was suggested that a specific H-bond pattern forms in the channel, and the orientation of the H-bonds switches at the center of the pore, resulting in orientation of the H atoms of the water molecules toward the channel exits (Fig. 2b).18,19 This H-bond orientation inhibits a proton from entering the channel19 and disrupts proton transfer via the Grotthuss-like mechanism. AQP contains two conserved asparagine-proline-alanine (NPA) motifs located at the center of the channel and constituting the termini of two transmembrane half α-helices (M3 and M7).19 The opposite orientations of the two water molecules at the center are caused by H-bonds to the oxygen atoms from the NH2 groups of two asparagines (Asp213 and Asp97) on the NPA motifs (Fig. 2b). Additionally, blockage of proton transfer was attributed to electrostatic effects between the positive charge of the proton and the protein structural environment (e.g., the NPA motif,21–24 the aromatic/arginine constriction-selectivity filter,25–27 ionized residues,23 and dipoles generated by α-helices M3 and M728,29). It was also claimed that the barrier preventing proton transfer originates from the loss of solvation upon moving the proton charge from the bulk of the protein to the central channel.17,30,31 Another study suggested the charge-delocalization effect of the Zundel cation on blockage of proton transfer,26 which was subsequently validated in studies of AQP mutants.25,27,32
The water-transfer channel in AQP and the proton-transfer channel in PSII contain similar water chains that exhibit different functions. The origin of the difference in function can be related to H-bond networks and the mobility of water molecules, as implied by the following facts: (1) the water channel has only one H-bond pattern (Fig. 2b),18,19 whereas the proton channel has two patterns (pre- and post-PT) (Fig. 1b);5 (2) in the water channel, water molecules move (Fig. 2c), whereas in the proton channel, they are fixed, although the protons move (Fig. 1d); and (3) in the water channel, the water chain is surrounded by both hydrophilic and hydrophobic residues (Fig. 2b),18,19 whereas in the proton channel, it is surrounded by only hydrophilic residues (Fig. 1b).5 Here, we identify factors differentiating the functions of the AQP water channel and the PSII proton channel in regard to water chains, using (1) quantum mechanical/molecular mechanical (QM/MM) calculations, and (2) MD simulations. The advantage of QM/MM calculation is that it can describe bond cleavage and formation, and it can draw the energy profile of the proton transfer (Fig. 3a). On the other hand, the advantage of MD simulation is that it can investigate molecular structural fluctuations (Fig. 3b).
 | ||
| Fig. 3 Theoretical approaches to investigate the water channel. (a) QM/MM calculation draws the proton-transfer potential by moving a proton from H3O+ to the H-bonded water molecule (b) MD simulation investigates the structural fluctuation of water molecules at equilibrium. | ||
Methods
MD simulations of AQP
The atomic coordinates of AQP were obtained from the X-ray structure of aquaporin-4 from Homo sapiens determined at 1.8 Å resolution (PDB: 3GD833). The AQP assembly was embedded in a lipid bilayer consisting of 90 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) and 110 cholesterol molecules and then soaked in 5874 flexible water models (SPC-Fw).34 The MD simulations were based on the AMBER-ff14SB force field for protein residues. After structural optimization with position restraints on heavy atoms of the protein and channel water molecules, the system was quenched from 0.001 K to 1.0 K over 0.2 ps, with a 0.01 fs time step, followed by gradual release of the restraints over 1.0 ns at 300 K, with a 2 fs time step and the SHAKE algorithm used for hydrogen constraints.35 After an equilibrating MD run for 60 ns, with an MD time step of 0.5 fs (because of flexible water models), a production run was conducted over 10 ns for statistical analysis. Simulations were conducted using the MD engine AMBER 14.36 For temperature and pressure control, a Berendsen thermostat and barostat37 were employed to maintain consistency with earlier work on the AQP.20 We confirmed that the results were unchanged when we used the Langiven piston instead of the Berendsen thermo/barosta.MD simulations of PSII
The simulations for PSII were performed using the X-ray crystal structure determined at 1.9 Å resolution (PDB: 3ARC)12 and based on the same procedure described previously,15,38 except for the following points. The heavy atoms of the Mn4CaO5 cluster with its ligands (side chains: D1-Asp61, -Asp170, -Glu189, -His332, -Glu333, -His337, -Asp342, CP43-Glu354, and -Arg357; backbone: D1-Ala344; and water molecules ligated to the W1–W4 cluster) were treated as harmonic constraints, with force constants set to 5 kcal mol−1. Their partial charges were derived from the calculated structure from our previous QM/MM studies5,39 for the O4-protonated S0 state.After structural optimization with positional restraints on heavy atoms of the PSII assembly, the system was heated from 0.001 to 300 K over 5.0 ps. The positional restraints on heavy atoms were gradually released over 16.5 ns. After an equilibrating MD run for 40 ns, with an MD time step of 0.5 fs (because of flexible water models), a production run was conducted over 10 ns for statistical analysis. For temperature and pressure control, a Berendsen thermostat and barostat37 were employed to assure consistency with the AQP simulation.
Here, a note regarding the statistical analysis of the MD trajectory for PSII: to define the channel axis, we used the density of the oxygen atom of D1-Ser169, which donates an H-bond to the next water molecule (noted as P1 in the present study) to O4 on the Mn4CaO5 cluster, for the analysis of the PSII proton channel.
QM/MM potential of proton transfer in AQP
On the basis of the 1.8 Å crystal structure (PDB: 3GD8), hydrogen atoms were generated and energetically optimized using CHARMM,40 whereas the positions of all non-hydrogen atoms were fixed, and all titratable groups were maintained in their standard protonation states. For QM/MM calculations, we added additional counter ions to neutralize the entire system. Atomic partial charges of the amino acids were adopted from the all-atom CHARMM2241 parameter set.We used the Qsite42 code as described in a previous PSII study5,43 and employed the restricted density functional-theory method with the B3LYP functional and LACVP**+ basis sets. The geometries were refined by constrained QM/MM optimization (for optimized geometries, see ESI†). To avoid uncertainty associated with the MM force field, we constrained most of the atoms in the surrounding MM region. Namely, the coordinates of the heavy atoms in the MM region were fixed to the original X-ray coordinates. In contrast, those of the H atoms in the MM region from the residues within 7.0 Å of the QM region were optimized using the OPLS2005 force field. All atomic coordinates in the QM region were fully relaxed (i.e., not fixed) in the QM/MM calculations. The QM region was defined as the side chains of Asn97, His201, Asn213, and Arg216, the backbones of Gly93, Ala94, His95, Ser211, Ala210, and Gly209, and water molecules within H-bond distances specific to these residues (namely, W50–W56). The proton-transfer potential profile of the H-bonds was obtained using a procedure described in a previous PSII study5 (see ESI† for the optimized geometries). First, we obtained the QM/MM-optimized geometry with H3O+ at W1 without constraints and used the resulting geometry as the initial geometry.
Proton-transfer potentials were evaluated by moving a proton from the target H3O+ to the H-bonded water molecule (Fig. 3a). When obtaining the H-bond potential between W1 and W2, the H atom under investigation was then moved from the H-bond-donor atom (OW1) toward the acceptor atom (OW2) by 0.05 Å, after which the geometry was optimized by constraining the OW1–H and H–OW2 distances. The energy of the resulting geometry was plotted as a function of the H–OW2 distance. This procedure was repeated until the H atom reached the OW1 atom. Similarly, we obtained H-bond potentials for all water molecule pairs (for intermediate geometries of proton transfer, see ESI†).
H-bond network
H-Bond network was analyzed by MD simulation in the presence of only water molecules (without H3O+) (Fig. 3b). A method to analyze water-molecule dynamics and the H-bond networks of water chains from MD trajectories was developed for AQP by Tajkhorshid et al.19 However, their method is not directly applicable to PSII, because the water chain in the PSII proton channel is arranged in a winding orientation (Fig. 1a), whereas that in the AQP water channel is arranged along a straight line vertical to the membrane (Fig. 2a). Therefore, we developed a method to analyze dynamics applicable to winding water chains. In our method, we statistically analyzed H-bond networks from MD trajectories as follows (Fig. 4 and 5). We obtained the three-dimensional distribution of oxygen atoms for water molecules in the channel from the whole trajectory, followed by identification of the coordinates for the water-biding sites and at which the distribution takes the maximum value. We then defined the channel axis as the second-order spline curve interpolating the water-binding sites. For water molecules within 1.4 Å (corresponding to the van der Waals radius of a water molecule) of the channel axis, we obtained the coordinate, s, on the channel axis and the angle, θ, between the normal vector of the channel axis and the dipole vector (the C2-rotational axis) of the water molecule. We confirmed that the distribution of the water position and orientation was essentially independent from the simulation time, which indicate that the period of the production simulation is sufficiently long (Fig. S2, ESI†).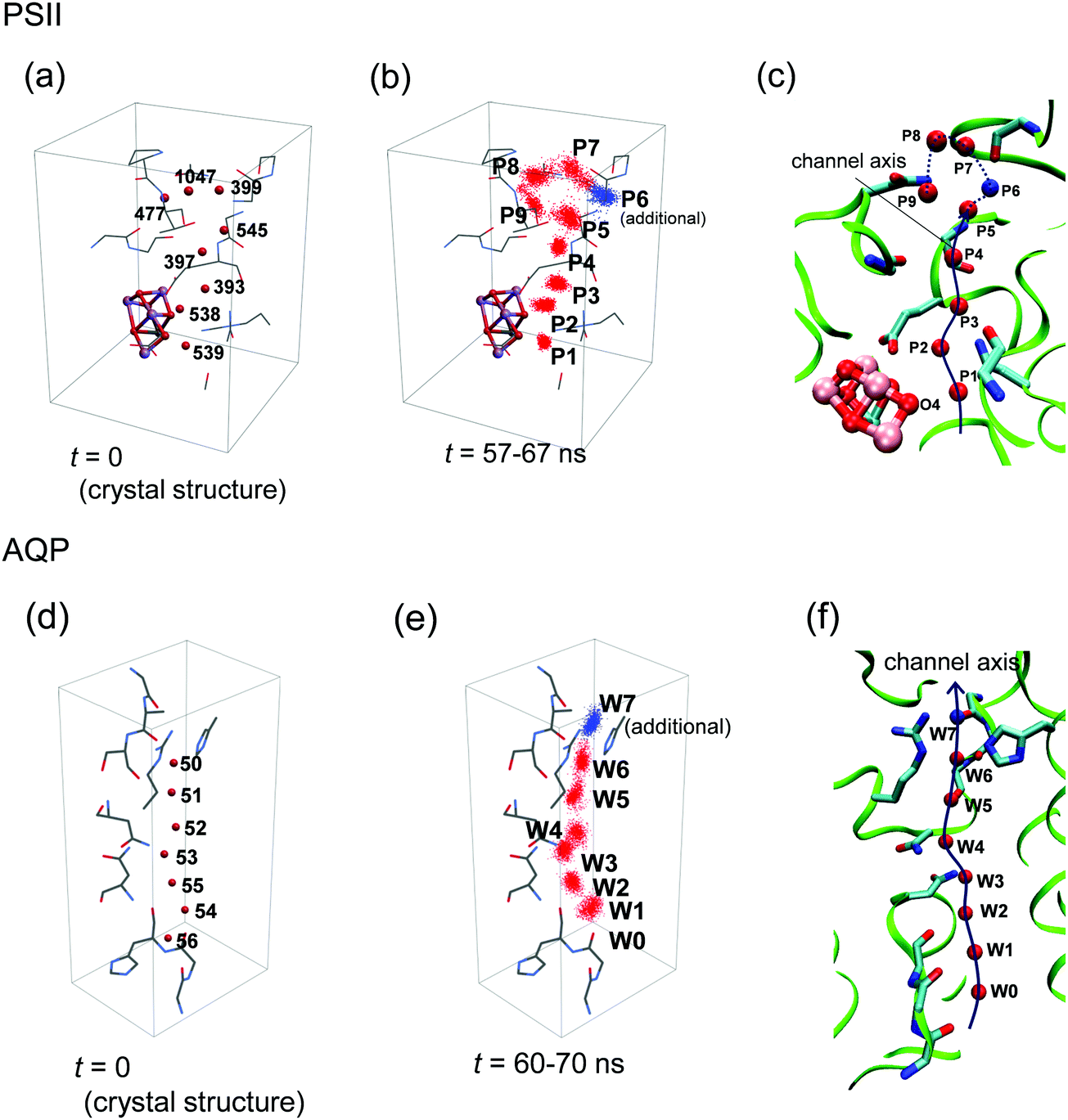 | ||
| Fig. 4 Definition of the channel axis in PSII and AQP. (a) Initial positions of the oxygen atoms of water molecules in PSII. (b) Distribution of water-molecule oxygen atoms in the MD simulation of PSII. The spots P1–P5 and P7–P9 correspond to W539, 538, 393, 397, 477, 545, 1047, and 399, respectively, in the original crystal structure (PDB: 3ARC),12 whereas the spot P6 has no corresponding water molecule in the crystal structure. (c) Channel axis in PSII defined by positions at the maximum densities of oxygen atoms (red or blue spheres). The axis of P5–P9 is represented by the dotted line, because we did not primarily focus on this region (see text). (d) Initial positions of the water-molecule oxygen atoms in AQP. (e) Distribution of water-molecule oxygen atoms in the MD simulation of AQP. The spots W0–W6 correspond to W57, 56, 55, 54 53, 52, and 51, respectively, in the original crystal structure (PDB: 3GD8).33 Spot W7 (blue) has no corresponding water molecule due to the presence of a glycerol molecule in the crystal structure. (f) Channel axis in AQP defined by positions at the maximum densities of the oxygen atoms (red or blue spheres). | ||
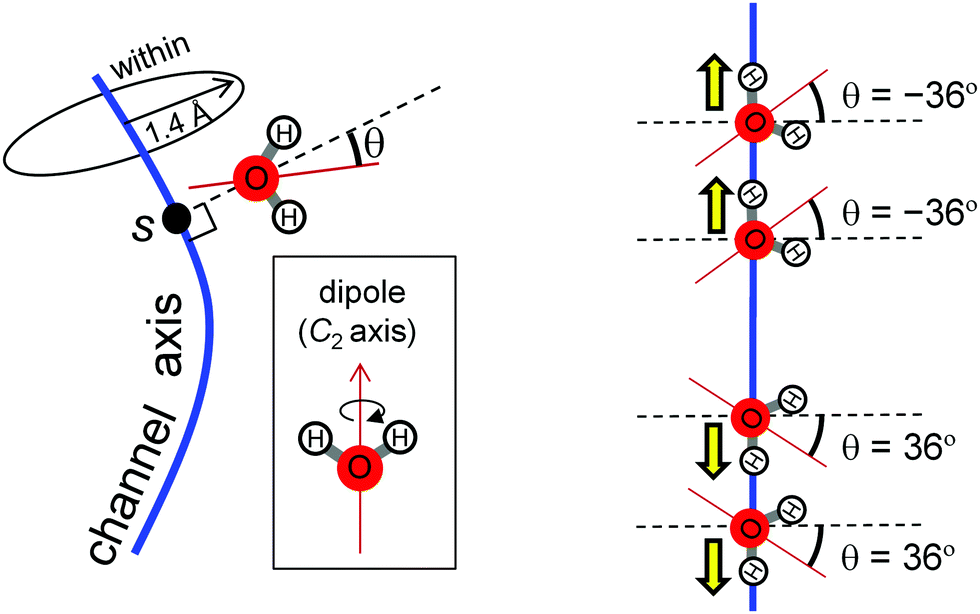 | ||
| Fig. 5 Two variables for statistical analysis based on MD simulations. The coordinate, s, on the channel axis and the angle, θ, between the normal vector of the channel axis and the dipole vector (the C2 rotational axis) of the water molecule. We accounted for water molecules with oxygen atoms within 1.4 Å (corresponding to the van der Waals radius of a water molecule) of the channel axis. When the direction of water-molecule H-bonds is oriented along the channel axis, θ is ±36°. | ||
Results and discussion
Water-binding sites
Using MD simulations, we identified the water biding sites (P1–P9 for PSII and W0–W8 for AQP) (Fig. 4 and 6a–d). These sites correspond exactly to the positions of water molecules observed in the original crystal structures (Fig. 4),12,33 except for the W7 site in AQP and the P6 site in PSII. In AQP, the space of W7 was occupied by a glycerol molecule. In PSII, P6 corresponded to an additional water molecule, which entered the channel during the simulation and was not contained in the initial structure (the 1.9 Å crystal structure12 of PSII) used in the simulation (Fig. 4a–c). Note that this additional water molecule, P6, was observed at the same position in the X-ray free electron laser structure (Fig. S1, ESI†).44 The appearance of the water-binding sites indicates that most of the water molecules maintained the same positions as those in the crystal structure, even when dynamics were considered.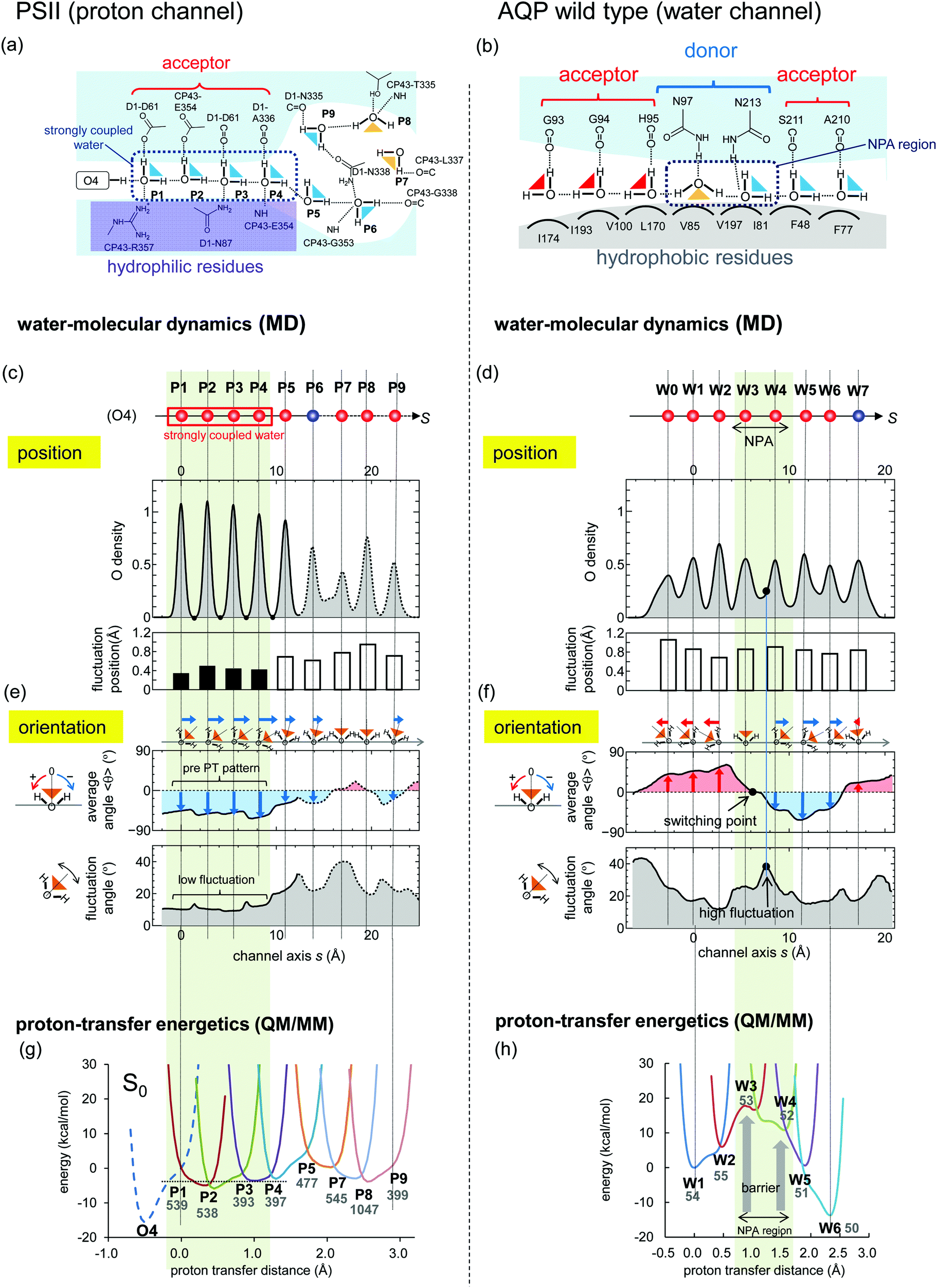 | ||
Fig. 6 Comparison of water-molecular dynamics and proton-transfer energetics between PSII (left: a, c, e and g) and AQP (right: b, d, f and h). (a and b) H-bond patterns of water molecules in the protein environments. (c–f) Statistical analysis of the MD simulation of PSII and AQP. (c and d) Distribution of water-molecule oxygen atoms (O density) on the coordinate, s, of the channel axis (upper) and position fluctuation of the oxygen atoms of water molecules (lower). Red spheres corresponds to the crystal water molecule, whereas blue spheres have no corresponding water molecule (see Fig. 4). The function, f(s), of the O density was normalized to satisfy  , where 〈N〉 is the average number of water molecules analyzed. The origin of the channel axis was defined as the peak of P1 and W1. (e and f) The average angle, 〈θ〉, of the water orientation (upper) and angle fluctuation, 〈(θ − 〈θ〉)2〉1/2 (lower). The angle, θ, was defined as the angle between the normal vector of the channel axis and the dipole vector (the C2 axis) of the water molecule (see Fig. 5). Schematic geometries of water molecules are shown at the top. (g and h) Proton-transfer potential along the channel obtained from QM/MM calculations based on the crystal structures. , where 〈N〉 is the average number of water molecules analyzed. The origin of the channel axis was defined as the peak of P1 and W1. (e and f) The average angle, 〈θ〉, of the water orientation (upper) and angle fluctuation, 〈(θ − 〈θ〉)2〉1/2 (lower). The angle, θ, was defined as the angle between the normal vector of the channel axis and the dipole vector (the C2 axis) of the water molecule (see Fig. 5). Schematic geometries of water molecules are shown at the top. (g and h) Proton-transfer potential along the channel obtained from QM/MM calculations based on the crystal structures. | ||
Next, we defined a channel axis as an interpolating curve smoothly connecting the water-binding sites (Fig. 4c and f). Corresponding to the water-binding sites, we observed maximum points of oxygen density along the channel axes (Fig. 6c and d). The peaks of the maximum points of P1–P4 in PSII are very sharp as compared with the others, and there is zero oxygen density at the intermediate points of these peaks. This reflects that these water molecules (P1–P4) exhibit less exchange of their binding sites. Indeed, the positional fluctuation of the oxygen atoms of P1–P4 is sufficiently small as compared with that of both the other water molecules (P5–P9) in PSII and all water molecules (W0–W9) in AQP (Fig. 6c and d).
In PSII, the four water molecules from the O4 side (P1–P4) clearly exhibit different properties from the other water molecules (P5–P9). Although the water molecules (P1–P4) are arranged in single file along the long and thin pores, the P5–P9 water molecules aggregate in the larger pore (Fig. 4c and 6a), which should be regarded as a pool of water rather than a water chain. Below, we mainly focus on the P1–P4 region of the channel.
H-Bond direction
We statistically analyzed the orientations of water molecules along the channel axis (Fig. 5). The results show that the O–H covalent bonds of P1–P4 in PSII are directed forward (O–H⋯O–H⋯O–H⋯) along the channel axis forming the pre-PT H-bond pattern (Fig. 6e). These well-ordered water molecules facilitate immediate proton transfer to the inner area of the channel upon entry. In contrast, the direction of the O–H covalent bonds of W0–W9 in AQP switches at W3 from the backward (H–O⋯H–O⋯) to the forward (O–H⋯O–H⋯) orientation (Fig. 6f) (i.e., they are directed toward the outer face in both of the two ends of the channel). These water orientations inhibit proton entrance into the channel from both ends and proton transfer via the Grotthuss-like mechanism, as reported previously.19,20The difference in the orientation of water molecules between PSII and AQP originates from the differences in H-bond partners with the water molecules. In PSII, all of the water molecules have H-bond acceptors [D1-Asp61, -Ala336 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and CP43-Glu354] located on the same side of the channel (Fig. 6a), which facilitates the ordering of water-molecule orientation in the same direction. However, in AQP, these partners are not all H-bond acceptors [W3 and W4 located at the NPA region have exceptionally H-bond donors (Asn97 and Asn213)] (Fig. 6b). The exceptional H-bond donors, Asn97 and Asn213, disturb the unidirectional arrangement of the water molecules. In the N97D/N213D mutant, all of the water molecules are ordered in the same direction (see Appendix A).
O), and CP43-Glu354] located on the same side of the channel (Fig. 6a), which facilitates the ordering of water-molecule orientation in the same direction. However, in AQP, these partners are not all H-bond acceptors [W3 and W4 located at the NPA region have exceptionally H-bond donors (Asn97 and Asn213)] (Fig. 6b). The exceptional H-bond donors, Asn97 and Asn213, disturb the unidirectional arrangement of the water molecules. In the N97D/N213D mutant, all of the water molecules are ordered in the same direction (see Appendix A).
Orientational fluctuations of water molecules
We then statistically analyzed fluctuations in water molecule orientations. The results shows that the orientational fluctuation observed in the region of P1–P4 in PSII is significantly smaller than that observed in both the P5–P9 region and the entire region (W0–W7) in AQP (Fig. 6e and f). These small orientational fluctuations are consistent with the small positional fluctuations (Fig. 6c), which indicates that P1–P4 are rigidly fixed to their sites and do not change their H-bond direction, thus forming the pre-PT pattern. Note that the small fluctuations observed in P1–P4 are consistent with their corresponding water molecules in crystal structures exhibiting small B-factor values (Table S1, ESI†). In contrast, fluctuations in AQP are large, especially in the region between W3 and W4 located in proximity to the NPA motif (Fig. 6f). Large fluctuations in water-molecule orientation indicate the high degree of water-molecule mobility allowing water-transport ability in AQP.The difference in water-molecule fluctuations between PSII and AQP also originates from differences in hydrophobicity of the protein environments, including water-molecule H-bond partners. In the PSII proton channel, water molecules (especially P1–P4) are surrounded by hydrophilic residues (e.g., D1-Asp61 and -Asn87; CP43-Arg357 and -Glu354) in all directions (Fig. 1b and 6a) that work as H-bond partners to fix the position of the water molecules. However, in the AQP water channel, there exist hydrophilic residues (H-bond partners; e.g., Asn213, Asn97, and the backbones of Ser211 and His95) only on one side of the channel, with the other side consisting of hydrophobic residues (no H-bond partners; e.g., Val85, Leu170, and Ile197) (Fig. 2b and 6b). This structural difference results in a notable variation in water fluctuation. Because of the presence of hydrophilic residues (rich in H-bond partners), especially around P1–P4 in the proton channel, these water molecules are rigidly fixed to their sites and strongly coupled to one another. In contrast, hydrophobic residues in the water channel enable water-molecule mobility.
Barriers in proton-transfer potential
We compared the proton-transfer potential profile along the channel axis, using a QM/MM approach based on crystal structures. The proton-transfer potential profile of AQP shows that an energy barrier located in proximity to the NPA motif prevents proton transfer (Fig. 6h), which is consistent with previous theoretical studies.17,19–31,45–47 In contrast, the PSII proton-transfer potential5 shows a barrier-less profile in the region of the P1–P4 water molecules, even in the S0 state (Fig. 6g). This finding indicates that proton transfer is possible, even in the S0 state, although protons will not be transferred due to the absence of the driving force of the deprotonation from the O4 on the Mn4CaO5 cluster. When the S0 state transits to the S1 state, the proton transfer becomes downhill,5 and the Mn4CaO5 cluster can release a proton (Fig. S3a and b, ESI†). During this transition, the P1–P4 region shows large energy differences (∼25 kcal mol−1) as the driving force increases (Fig. S3c, ESI†). The concerted proton transfer on the P1–P4 water molecules correlates with the observed unidirectional H-bonds and the low oriental/positional fluctuations in the P1–P4 water molecules.The proton-transfer potential profile of AQP shows a barrier around W3 and W4 in the NPA region (Fig. 6h), whereas that of PSII is barrier-less (Fig. 6g). The reason for this difference can be explained in terms of electrostatic interactions. In AQP, water molecules (W3 and W4) located in the NPA region accept donations of H-bonds from the NH2 group of Asn97 and Asn213 in the NPA motif (Fig. 6b). The partial positive charge (δ+) of the H atom from the NH2 group destabilizes the proton on W3 and W4 [i.e., decreases the pKa (H3O+/H2O) of W3 and W4], thereby elevating the potential energy of H3O+ around W3 and W4 (Fig. 6h). On the other hand, in PSII, all of the water molecules (P1–P4) donate H-bonds to the acceptors of carbonyl (backbones of D1-Asp61 and -Ala336) or carboxyl groups (side chains of D1-Asp61 and CP43-Glu354) containing oxygen atoms (Fig. 6a). The partial negative charge (δ–) of the oxygen atoms in these groups stabilizes the proton [i.e., increases the pKa (H3O+/H2O)], thereby lowering the potential energy (Fig. 6g). Therefore, the differences in the proton-transfer potential profile between PSII and AQP are due to the differences in H-bond partnering with water molecules. In the N97D/N213D AQP, proton transfer occurs (see Appendix A), whereas in the CP43-E354Q PSII, proton transfer does not occur (see Appendix B). In summary, the proton donor and acceptor have opposite electrostatics, which directly affect the height of the energy barrier for proton transfer. They also provide different orientations for H-bonds along the channel. Thus, the orientation of the H-bonds along the channel and the height of the energy barrier are equivalent in terms of electrostatics.
Proton channel versus water channel
For proton transfer via the Grotthuss-like mechanism, because water molecules do not move themselves, but rather mediate proton orientation, water molecules need to be rigidly fixed (Fig. 7a). The fluctuation of water molecules at the entrance region (P1–P4) in the proton channel of PSII is very small (Fig. 6c and e), and proton transfer is barrier-less (Fig. 6g). | ||
| Fig. 7 Factors that differentiate the functions between the (a) proton-transport channel in PSII and the (b) water-transport channel in AQP. They are (i) the types of water-molecule H-bond partners, (ii) the hydrophobicity of the residues surrounding the water molecules, (iii) water-molecule fluctuation, (iv) the direction of the H-bonds among water molecules, and (v) the barrier associated with the proton-transfer potential. | ||
However, in the AQP water channel, because water molecules need to move, they also need to be flexible (Fig. 7b). In the AQP water channel, water-molecule fluctuation is elevated across the entire region (Fig. 6d and f) as compared with that observed at the entrance region (P1–P4) of the PSII proton channel (Fig. 6c and e). This flexibility also causes another effect on proton transfer as a byproduct: the proton is unlikely to transfer via the Grotthuss-like mechanism, because the direction of the H-bonds is disordered by the fluctuations. This effect is attributed to the proton-transfer energetics, with the barrier observed in the proton-transfer potential profile at the W3 and W4 region in the AQP water channel (Fig. 7b).
The principle described here also applies to other proton and water channels. Gramicidin has a polar channel, and the fast Grotthuss-like proton transfer were reported.48 Water permeates the hydrophobic water channels in carbon nanotubes.49
Classical molecular dynamics simulations versus static quantum calculations
To theoretically investigate proton transfer via water chains, we needed to calculate the free energy profile. Previous studies of AQP investigated the free energy profile associated with the Grotthuss-like proton transfer in detail using the potential meaning force (PMF) method.25–27,50 On the other hand, proton transfer from the O4 site in PSII was originally reported using a QM/MM approach based on the crystal structure.5 The energy profile based on the static crystal structure differs from the free energy profile, because it includes only enthalpic effects while lacking the entopic effects. Because the region of the O4 water chain is in the inner core of the membrane protein and the Mn4CaO5 cluster is stabilized by a number of ligand residues from the protein subunits, the enthalpic energy profile based on the crystal structure is likely to be consistent with the free energy profile, as demonstrated in the present study. Entopic effects on free energy can be estimated by configuration sampling of protein structures through molecular dynamics (MD) simulations.Remarkably, the present results shows that the characteristic behaviors of P1–P4 in PSII and W3 and W4 in the NPA region of AQP were clearly observed in both the dynamics (from MD; small fluctuation of P1–P4 and large fluctuation of W3 and W4) and the energetics (from QM/MM; the absence and presence of the energy barrier in PSII and AQP, respectively), indicating that the dynamics of water fluctuations are strongly correlated with the energetics of proton transfer. This is because these behaviors originate from H-bond partners in protein environments. Because both the dynamics of water molecules and the enthalpic potential profile depend on H-bond partners, the enthalpic potential profile should qualitatively reflect the free energy profile. The barrier on the AQP potential profile was also observed in the free energy profile of proton transfer in previous studies.17,19–31,45–47 Based on these observations, it seems plausible that the enthalpic potential profile can substantially provide the same results and conclusions for proton transfer along water channels as estimation of the real free energy.
Conclusion
In the O4-water chain in PSII, hydrophobic residues (e.g., D1-Asp61, -Asn87, and CP43-Glu354) serving as H-bond acceptors rigidly fix the water molecules in H-bond pattern, which can facilitate proton transfer via the Grotthuss-like mechanism, thereby helping to form the barrier-less proton-conducting wire (Fig. 7a). In contrast, in the water channel in AQP, two H-bond donors (Asn97 and Asn213) located at the central NPA motif and other hydrophobic residues (e.g., Val85, Leu170, and Ile197) destabilize the positive charge of the proton, helping to prevent proton passage by a high energy barrier. These residues also enhance water-molecule fluctuation, resulting in a disordered orientation preventing proton transfer via the Grotthuss-like mechanism and promoting water-molecule permeation (Fig. 7b).The conclusions obtained from the enthalpic potential profile and those obtained from water-molecule dynamics obtained by classical MD simulation without estimating the real free energy are the same, because the H-bonded partners in the water chain and the protein simultaneously determine both the energetics of proton transfer and the dynamics of water-molecule fluctuation, thereby determining the transport target of the water chain. When the water chain is surrounded by H-bond acceptors (e.g., Asp, Glu, and backbone carbonyl groups), it is capable of proton transport. In contrast, when the water chain is surrounded by hydrophobic sidechains or H-bond donors (e.g., the NH2 groups of Asn and Gln), it is capable of transporting only water.
Conflicts of interest
There are no conflicts to declare.Appendix
A Proton conducting N97D/N213D AQP mutant
Recent studies proposed that the NPA motif, which has Asn97 and Asn213, was not prerequisite for inhibition of proton transfer in AQP.29–31,51 We performed MD simulations on the N97D/N213D AQP mutant. MD simulations show that the N97D/N213D AQP mutant is stable only when the two aspartates are protonated. When the two aspartates are protonated, Asp213 serves as either an H-bond donor (Fig. 8b and Fig. S4c, ESI†). or acceptor (Fig. 8a and Fig. S4b, ESI†). Notably, when Asp213 serves acceptor, the Grotthuss-like H-bond pattern forms (Fig. 8c). QM/MM calculations show no energy barrier along the channel, which allows proton transfer (Fig. 8e). These results suggest that the presence of the H-bond donor in the protein environment interferes formation of the Grotthuss-like H-bond pattern, induces the energy barrier, and prevents proton transfer.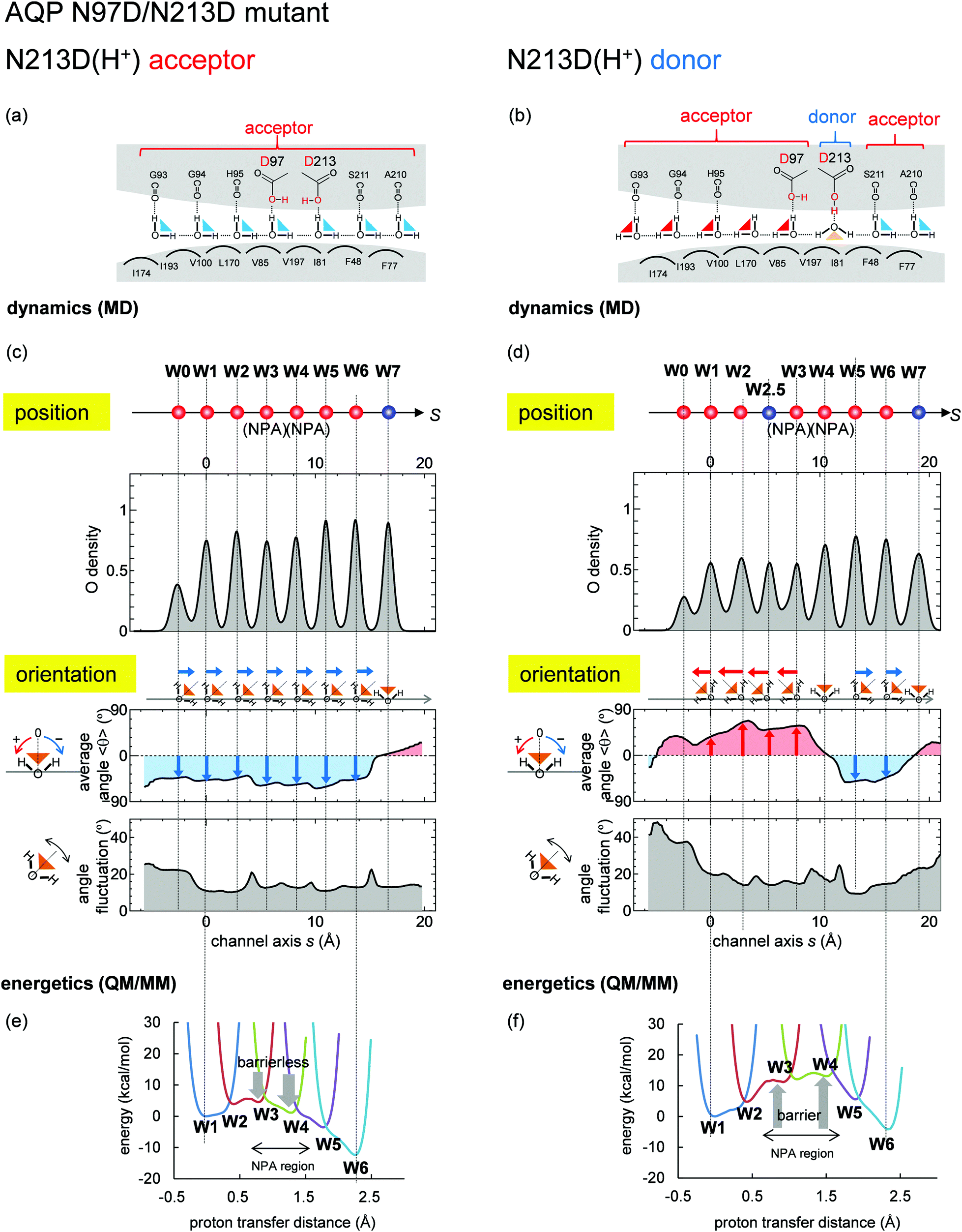 | ||
| Fig. 8 Water-molecular dynamics and proton-transfer energetics for the N97D/N213D AQP mutant. Graphs are essentially the same as those shown in Fig. 6. Both aspartates were protonated, and the QM/MM proton-transfer potentials were calculated based on the mutant structure modeled from the crystal structure of the wild type. (left: a, c and e) Results when N213D serves as H-bond acceptors (corresponding to Fig. S4b, ESI†). (right: b, d and f) Results when N213D serves as the H-bond donor (corresponding to Fig. S4c, ESI†), where an additional water molecule (W2.5) entered the channel (see Fig. S5, ESI†). | ||
B Absence of proton transfer in the CP43-E354Q PSII mutant
It was reported that a majority fraction of the CP43-E354Q PSII mutant is unable to advance beyond the S2 or S3 states52 probably because CP43-E354 is a ligand of Mn3 of the Mn4CaO5 cluster.12 MD simulations show that CP43-E354Q serves as an H-bond donor (Fig. 9a) in contrast to CP43-Glu354 serving as an H-bond acceptor in wild type, which leads to reorientation of the H-bond at P2 (Fig. 9c). QM/MM calculations show the presence of the energy barrier at P2 (Fig. 9d), which prevents proton transfer. It seems likely that CP43-Glu354 not only serves as a ligand of the Mn4CaO5 cluster but also maintains the appropriate H-bond pattern, facilitating proton transfer. | ||
| Fig. 9 Water-molecular dynamics and proton-transfer energetics for the CP43-E354Q PSII mutant. Graphs are essentially the same as those shown in Fig. 6. The QM/MM proton-transfer potentials were calculated based on the mutant structure modeled from the crystal structure of the wild type. | ||
Acknowledgements
This research was supported by JST CREST (JPMJCR1656), JSPS KAKENHI (JP18H01186 to K. S., JP16H06560 to K. S. and H. I., and JP26105012 and JP18H05155 to H. I.), Japan Agency for Medical Research and Development (AMED; 18ak0101084h0002), and Interdisciplinary Computational Science Program in CCS, University of Tsukuba.References
- H. Dau, I. Zaharieva and M. Haumann, Curr. Opin. Chem. Biol., 2012, 16, 3–10 CrossRef CAS PubMed.
- J. R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- E. Schlodder and H. T. Witt, J. Biol. Chem., 1999, 274, 30387–30392 CrossRef CAS PubMed.
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2005, 44, 1708–1718 CrossRef CAS PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- T. Takaoka, N. Sakashita, K. Saito and H. Ishikita, J. Phys. Chem. Lett., 2016, 7, 1925–1932 CrossRef CAS PubMed.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L. J. Yu, T. Sakamoto, T. Motomura, J. H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J. R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- T. Shimizu, M. Sugiura and T. Noguchi, J. Phys. Chem. B, 2018, 122, 9460–9470 CrossRef CAS PubMed.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273–295 CrossRef CAS.
- M. Eigen, Angew. Chem., Int. Ed. Engl., 1964, 3, 1–19 CrossRef.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- K. Ogata, T. Yuki, M. Hatakeyama, W. Uchida and S. Nakamura, J. Am. Chem. Soc., 2013, 135, 15670–15673 CrossRef CAS PubMed.
- A. G. Gabdulkhakov, V. G. Kljashtorny and M. V. Dontsova, Crystallogr. Rep., 2015, 60, 83–89 CrossRef CAS.
- N. Sakashita, H. C. Watanabe, T. Ikeda, K. Saito and H. Ishikita, Biochemistry, 2017, 56, 3057–3094 CrossRef PubMed.
- A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1813–1814 CrossRef CAS PubMed.
- A. Burykin and A. Warshel, FEBS Lett., 2004, 570, 41–46 CrossRef CAS PubMed.
- K. Murata, K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel and Y. Fujiyoshi, Nature, 2000, 407, 599–605 CrossRef CAS PubMed.
- E. Tajkhorshid, P. Nollert, M. O. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud and K. Schulten, Science, 2002, 296, 525–530 CrossRef CAS PubMed.
- B. L. de Groot and H. Grubmuller, Science, 2001, 294, 2353–2357 CrossRef CAS PubMed.
- A. Yarnell, Chem. Eng. News, 2004, 82, 42–44 CrossRef.
- M. O. Jensen, E. Tajkhorshid and K. Schulten, Biophys. J., 2003, 85, 2884–2899 CrossRef CAS PubMed.
- G. V. Miloshevsky and P. C. Jordan, Biophys. J., 2004, 87, 3690–3702 CrossRef CAS PubMed.
- B. Ilan, E. Tajkhorshid, K. Schulten and G. A. Voth, Proteins: Struct., Funct., Bioinf., 2004, 55, 223–228 CrossRef CAS PubMed.
- H. N. Chen, Y. J. Wu and G. A. Voth, Biophys. J., 2006, 90, L73–L75 CrossRef CAS PubMed.
- H. N. Chen, B. Ilan, Y. J. Wu, F. Q. Zhu, K. Schulten and G. A. Voth, Biophys. J., 2007, 92, 46–60 CrossRef CAS PubMed.
- H. Li, H. N. Chen, C. Steinbronn, B. H. Wu, E. Beitz, T. Zeuthen and G. A. Voth, J. Mol. Biol., 2011, 407, 607–620 CrossRef CAS PubMed.
- M. L. Groot, N. P. Pawlowicz, L. J. G. W. van Wilderen, J. Breton, I. H. M. van Stokkum and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13087–13092 CrossRef CAS PubMed.
- N. Chakrabarti, B. Roux and R. Pomes, J. Mol. Biol., 2004, 343, 493–510 CrossRef CAS PubMed.
- M. Kato, A. V. Pisliakov and A. Warshel, Proteins: Struct., Funct., Bioinf., 2006, 64, 829–844 CrossRef CAS PubMed.
- A. Burykin and A. Warshel, Biophys. J., 2003, 85, 3696–3706 CrossRef CAS PubMed.
- E. Beitz, B. H. Wu, L. M. Holm, J. E. Schultz and T. Zeuthen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 269–274 CrossRef CAS PubMed.
- J. D. Ho, R. Yeh, A. Sandstrom, I. Chorny, W. E. Harries, R. A. Robbins, L. J. Miercke and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7437–7442 CrossRef CAS PubMed.
- X. Chen, H. Kalbacher and S. Grunder, J. Gen. Physiol., 2006, 127, 267–276 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, R. S. LeGrand, T. LuchkoLuo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- N. Sakashita, H. C. Watanabe, T. Ikeda and H. Ishikita, Photosyn. Res., 2017, 1–11 Search PubMed.
- K. Saito and H. Ishikita, Biochim. Biophys. Acta, 2014, 1837, 159–166 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- A. D. MacKerell, Jr., D. Bashford, R. L. Bellott, R. L. Dunbrack, Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- QSite, Schrödinger LLC, New York, NY, 2012.
- K. Saito, T. Ishida, M. Sugiura, K. Kawakami, Y. Umena, N. Kamiya, J.-R. Shen and H. Ishikita, J. Am. Chem. Soc., 2011, 133, 14379–14388 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- B. L. de Groot, A. Engel and H. Grubmuller, J. Mol. Biol., 2003, 325, 485–493 CrossRef CAS PubMed.
- B. L. de Groot, T. Frigato, V. Helms and H. Grubmuller, J. Mol. Biol., 2003, 333, 279–293 CrossRef CAS PubMed.
- N. Chakrabarti, E. Tajkhorshid, B. Roux and R. Pomes, Structure, 2004, 12, 65–74 CrossRef CAS PubMed.
- R. Pomes and B. Roux, Biophys. J., 1996, 71, 19–39 CrossRef CAS PubMed.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef CAS PubMed.
- B. Ilan, F. Q. Zhu, E. Tajkhorshid, K. Schulten and G. A. Voth, Biophys. J., 2005, 88, 111a Search PubMed.
- A. Burykin and A. Warshel, FEBS Lett., 2004, 570, 41–46 CrossRef CAS PubMed.
- M. A. Strickler, H. J. Hwang, R. L. Burnap, J. Yano, L. M. Walker, R. J. Service, R. D. Britt, W. Hillier and R. J. Debus, Philos. Trans. R. Soc. Lond. B. Biol. Sci., 2008, 363, 1179–1187 CrossRef CAS PubMed ; discussion 1187–1178.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00295j |
| This journal is © the Owner Societies 2020 |