
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for lactone formation during infrared multiple photon dissociation spectroscopy of bromoalkanoate doped salt clusters†
Nina K.
Bersenkowitsch
,
Milan
Ončák
 *,
Jakob
Heller
,
Tobias F.
Pascher
,
Christian
van der Linde
and
Martin K.
Beyer
*
*,
Jakob
Heller
,
Tobias F.
Pascher
,
Christian
van der Linde
and
Martin K.
Beyer
*
Institut für Ionenphysik und Angewandte Physik, Universität Innsbruck, Technikerstraße 25, 6020 Innsbruck, Austria. E-mail: milan.oncak@uibk.ac.at; martin.beyer@uibk.ac.at
First published on 18th May 2020
Abstract
Reaction mechanisms of organic molecules in a salt environment are of fundamental interest and are potentially relevant for atmospheric chemistry, in particular sea-salt aerosols. Here, we found evidence for lactone formation upon infrared multiple photon dissociation (IRMPD) of non-covalent bromoalkanoate complexes as well as bromoalkanoate embedded in sodium iodide clusters. The mechanism of lactone formation from bromoalkanoates of different chain lengths is studied in the gas phase with and without salt environment by a combination of IRMPD and quantum chemical calculations. IRMPD spectra are recorded in the 833–3846 cm−1 range by irradiating the clusters with tunable laser systems while they are stored in the cell of a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. The measurements of the binary complex Br(CH2)mCOOH·Br(CH2)mCOO− for m = 4 indicate valerolactone formation without salt environment while lactone formation is hindered for longer chain lengths. When embedded in sodium iodide clusters, butyrolactone formation from 4-bromobutyrate seems to take place already during formation of the doped clusters in the electrospray process, evidenced by the infrared (IR) signature of the lactone. In contrast, IRMPD spectra of sodium iodide clusters containing 5-bromovalerate contain signatures for both valerate as well as valerolactone. In both cases, however, a neutral fragment corresponding to the mass of valerolactone is eliminated, indicating that ring formation can be activated by IR light in the salt cluster. Quantum chemical calculations show that already complexation with one sodium ion significantly increases the barrier for lactone formation for all chain lengths. IRMPD of sodium iodide clusters doped with neutral bromoalkanoic acid molecules proceeds by elimination of HI or desorption of the intact acid molecule from the cluster.
Introduction
When it comes to modelling the climate, the investigation of aerosols plays an important role.1–5 Tropospheric aerosols contribute to processes such as cloud formation.6 As the ocean covers more than 70% of the Earths’ surface, marine aerosols7 constitute a significant fraction of the tropospheric aerosol inventory. They are produced via the mechanical disruption of the sea surface.8 Marine aerosols are composed of a complex mixture of species that include not only salt, but also a large amount of organic matter.9–12 The exposure to intense solar radiation induces complex photoprocessing of organic matter in sea-salt aerosols.13,14 It is known that carbonaceous particles with acidic character appear with a quite high concentration in marine aerosols.15,16 Volatile organic compounds17 (VOCs) from anthropogenic and biogenic sources undergo rapid transformation via photolysis.18Halogens are very reactive species, and brominated alkanes are reported to be a source of bromine in the stratosphere19 where bromine participates in the catalytic destruction of ozone.20 Several studies reported the presence of n-alkanes and n-alkanoic acids in aerosols.21–24 Ground water pollution25–29 is a highly relevant topic as it affects directly human health27,30,31 and environment.32 Inorganic33,34 and organic35–38 pollutants as some (di-)bromoalkanoic pesticides39–41 and volatile halogenated bromoalkanes42 have already been identified. 3-Bromopyruvic acid is a potential anti-cancer drug and, if applied to patients, will make its way via wastewater to the oceans.43 Vogel et al.44 studied the reactivity of some mono- and di-bromoalkanes in aqueous buffers. A large variety of halogenated organic molecules are formed as disinfection byproducts in the reaction of disinfectants with natural organic matter and bromide or iodide.45 These pollutants can easily get into the ocean, and can be therefore present in sea salt aerosols.
Visible or near-infrared one photon vibrational overtone excitation has the potential to trigger atmospheric reactions.46 In laboratory experiments, however, infrared multiple photon dissociation (IRMPD) is usually performed at longer wavelengths to obtain action spectra of clusters and ions.47–50 It is well established that resonant infrared irradiation may activate chemical reactions in clusters, used e.g. to characterize ion–molecule reaction products.51,52 A detailed IRMPD study of N2O decomposition on rhodium clusters by Mackenzie and co-workers revealed the influence of co-adsorbed oxygen.53 However, even black-body infrared radiation is able to trigger N2O decomposition on small rhodium clusters, showing that no mode-specificity is involved.54 We have recently investigated the IR induced decomposition of copper formate,55,56 and measured IRMPD spectra of CO2˙−(H2O)n and Co(CO2)(H2O)n−.57,58 Of particular interest to the present work, however, are our recent studies on salt clusters doped with organic molecules, where we used IRMPD to elucidate structural properties59,60 and UV excitation to analyze the influence of the salt environment on glyoxylate photochemistry.61
To get a better understanding of the chemistry of brominated organic molecules in a salt environment, we performed IRMPD on anionic complexes of n-bromoalkanoic acid (n = 5, 8) and the corresponding n-bromoalkanoate, i.e. Br(CH2)n−1COOH·Br(CH2)n−1COO−. We then repeated the experiments with sodium iodide clusters in which one I− ion was replaced by a bromoalkanoate ion, Na6I4Br(CH2)n−1COO+, as well as sodium iodide cluster with one molecule of bromoalkanoic acid adsorbed, Na6I5Br(CH2)n−1COOH+. Elimination of neutral (CH2)n−1OCO, which indicates lactone formation, is exclusively observed for bromoalkanoate species. Complexation with salt reduces the propensity for (CH2)n−1OCO elimination. Quantum chemical calculations provide a molecular level understanding of the observed IRMPD pathways.
Experimental and computational details
The detailed setup of the experiment was already described elsewhere.62 Briefly, the main setup consists of a Bruker APEX Qe 9.4 T Fourier-Transform Ion Cyclotron Resonance (FT-ICR) Mass Spectrometer, equipped with an ESI/MALDI Dual Source II and a Nanobay Console. Doped sodium iodide clusters are generated by electrospray ionization (ESI) of a 5 mM NaI solution in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeOH:H2O, containing the respective bromoalkanoic acid at a concentration of 1–5 mM. All chemicals were purchased from Sigma-Aldrich, with a purity of at least 98%. Mass selected cluster ions are irradiated with light from two tunable laser systems, emitting IR light at 3846–2234 cm−1 (EKSPLA NT277, 1000 Hz repetition rate, typical power 25–100 mW) and at 2234–833 cm−1 (EKSPLA NT273-XIR, 1000 Hz repetition rate, typical power 2–30 mW). A plot of the laser power as a function of wavelength is available as ESI,† Fig. S1. IR absorption cross sections are derived by assuming sequential photon absorption following first-order kinetics, as described in detail before,59 not considering radiative cooling. The contribution of ambient black-body infrared radiative dissociation (BIRD)63 was taken into account as much as possible. For single photon cross sections, the rate of unimolecular decomposition due to BIRD59 was measured by storing the ions for different time intervals without laser irradiation. For multiphoton cross sections, the fragment intensity due to BIRD was subtracted before further analysis. IRMPD spectra are recorded by scanning the laser frequency and monitoring the intensity of the dissociation products by mass spectrometry. The influence of the laser pulse energy is corrected within the calculation of the displayed cross sections and IRMPD yield. Overview spectra were obtained in the region where absorptions could be expected by scanning the laser typically in steps of 20–30 nm. The absorption bands identified in this way were re-measured with smaller steps and are shown here. Overview spectra are available in the ESI† (Fig. S2 and S3).
:1 mixture of MeOH:H2O, containing the respective bromoalkanoic acid at a concentration of 1–5 mM. All chemicals were purchased from Sigma-Aldrich, with a purity of at least 98%. Mass selected cluster ions are irradiated with light from two tunable laser systems, emitting IR light at 3846–2234 cm−1 (EKSPLA NT277, 1000 Hz repetition rate, typical power 25–100 mW) and at 2234–833 cm−1 (EKSPLA NT273-XIR, 1000 Hz repetition rate, typical power 2–30 mW). A plot of the laser power as a function of wavelength is available as ESI,† Fig. S1. IR absorption cross sections are derived by assuming sequential photon absorption following first-order kinetics, as described in detail before,59 not considering radiative cooling. The contribution of ambient black-body infrared radiative dissociation (BIRD)63 was taken into account as much as possible. For single photon cross sections, the rate of unimolecular decomposition due to BIRD59 was measured by storing the ions for different time intervals without laser irradiation. For multiphoton cross sections, the fragment intensity due to BIRD was subtracted before further analysis. IRMPD spectra are recorded by scanning the laser frequency and monitoring the intensity of the dissociation products by mass spectrometry. The influence of the laser pulse energy is corrected within the calculation of the displayed cross sections and IRMPD yield. Overview spectra were obtained in the region where absorptions could be expected by scanning the laser typically in steps of 20–30 nm. The absorption bands identified in this way were re-measured with smaller steps and are shown here. Overview spectra are available in the ESI† (Fig. S2 and S3).
The clusters were investigated using methods of computational chemistry at the B3LYP/def2TZVP level of theory, considering several isomers for each ion. For the bromoalkanoic acid/alkanoate complex, different conformations of the COOH hydrogen bond were considered, with and without lactone formation. For salt complexes, we employed different Na6I5+ clusters59 and replaced one I− ion by the respective alkanoate. Calculated IR spectra were scaled by a factor of 0.97 and displayed with Gaussian broadening with a full width at half maximum (FWHM) of 30 cm−1. All structures represent local minima on the potential energy surface. Relative energies are reported with zero-point energy correction, but without thermal corrections. All calculations were performed in the Gaussian program.64
Results and discussion
Evidence for lactone formation in Br(CH2)mCOOH·Br(CH2)mCOO− (m = 4, 7)
First, we focus on the anionic complexes Br(CH2)mCOOH·Br(CH2)mCOO− to identify potential reaction pathways of bromoalkanoic acids and bromoalkanoates in the gas phase, in the absence of a salt environment. Fig. 1a and d shows the IRMPD yield spectra normalized to the most intense absorption, and Fig. 1b and e illustrates the experimental multiphoton cross sections of these species for m = 4, 7. Calculated structures of selected low-lying isomers are shown in Fig. 2 with the corresponding spectra in Fig. 1c and f. Two structure types are considered, with and without pre-formed lactone. For m = 4, the more stable isomers Ia–c contain lactone while isomers Id–f feature the intact alkanoate Br(CH2)4COO−. For m = 7, the structures containing lactone, isomers IId–e, lie higher in energy than the alkanoate. Table 1 summarizes the reactions, energetics and branching ratios of the fragments, where the latter were calculated via the integrated experimental curves. The notations IR1, IR2 and IR3 indicate different infrared regions, the respective wavenumber range is noted in the table. Note that for m = 3, the Br(CH2)3COOH·Br(CH2)3COO− precursor could not be obtained in the experiment; the spectrum for m = 10 is shown in the ESI,† Fig. S4 and S5. The kinetics for m = 4, 7, 10 are shown in Fig. S6–S8 (ESI†).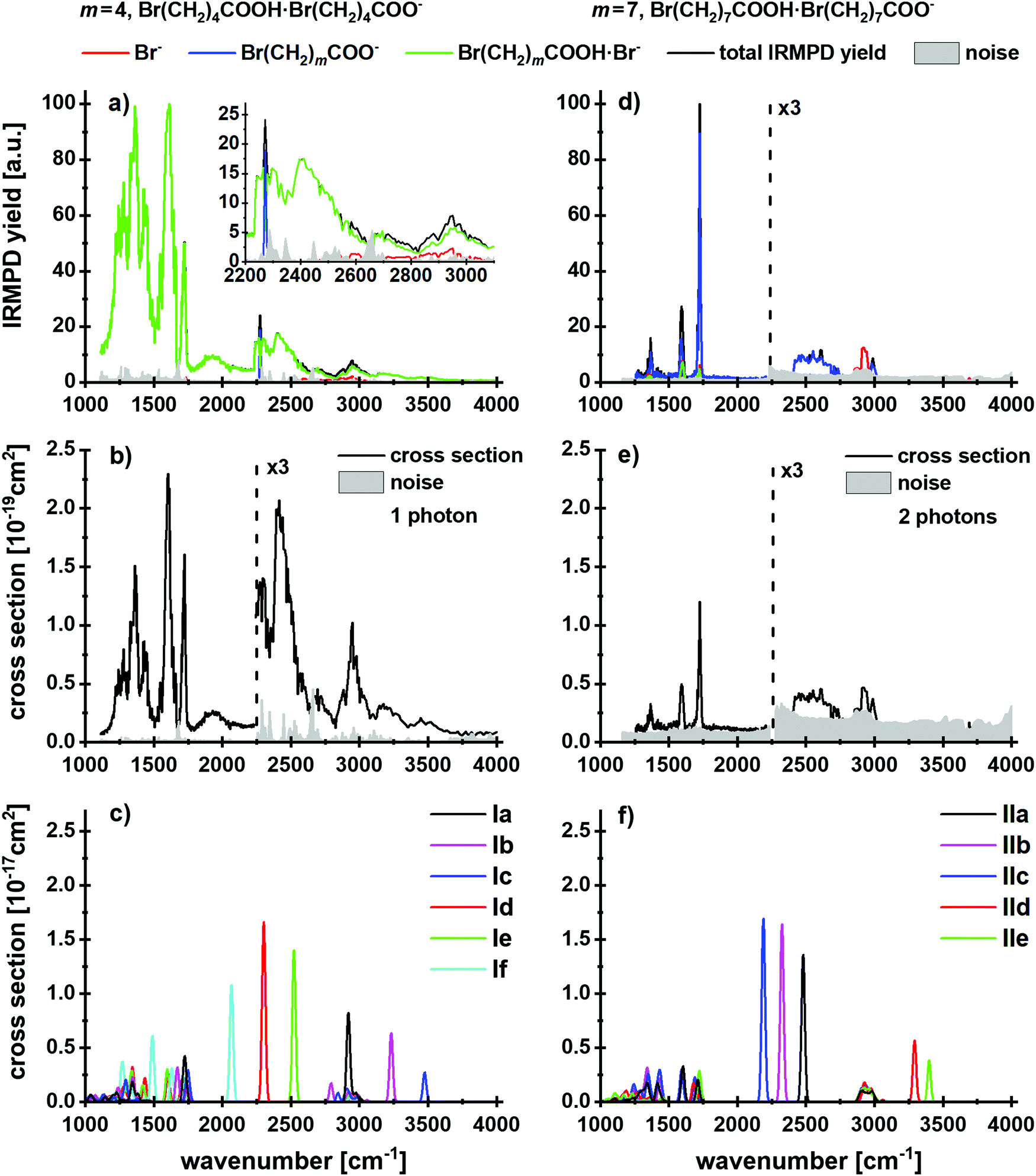 | ||
| Fig. 1 (a and d) IRMPD yield spectra with the total yield (black line) and the corresponding fragment contributions (red, blue and green lines), (b and e) experimental and (c and f) theoretical cross sections of the cluster Br(CH2)mCOO·Br(CH2)mCOOH− with m = 4 and m = 7. In (d), the Br− and Br(CH2)7COOH·Br− fragments are superimposed at 1500–1750 cm−1. Calculated at the B3LYP/def2TZVP level of theory. | ||
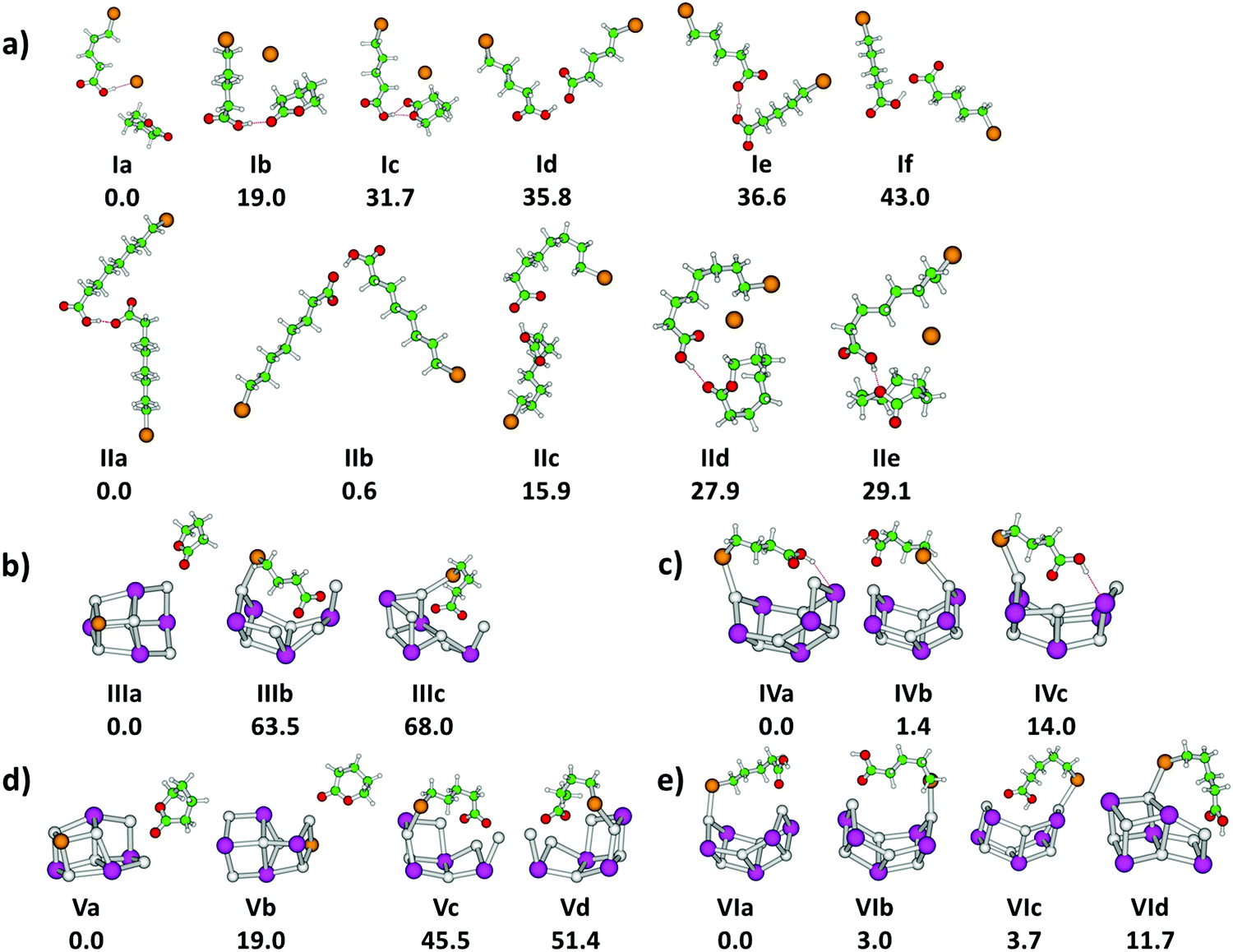 | ||
| Fig. 2 (a) n-Bromoalkanoic acid–alkanoate pairs; (b) 4-bromobutyrate/Na6I4; (c) 4-bromobutyric acid/Na6I5; (d) 5-bromovalerate/Na6I4; (e) 5-bromovaleric acid/Na6I5. Calculated at the B3LYP/def2TZVP level of theory, relative energy is given in kJ mol−1. Color code: Na grey, I violet, Br brown, C green, O red, H white. | ||
| Parent ion | Products | ΔE [kJ mol−1] | IR1a [%] | IR2b [%] | IR3c [%] |
|---|---|---|---|---|---|
| a 833–1500 cm−1 for salt environment. b 1000–2200 cm−1 for anionic complexes; 1600–1800 cm−1 for salt environment. c 2200–4000 cm−1 for anionic complexes; 2600–4000 cm−1 for salt environment. d Reaction energies for a cluster with/without lactone formation. | |||||
| Br(CH2)4COOH·Br(CH2)4COO− | Br− + (CH2)4COO·Br(CH2)4COOH | 114 | — | 1.1 | 5.2 |
| Br(CH2)4COOH·Br− + (CH2)4OCO | 42 | — | 98.5 | 94.8 | |
| Br(CH2)4COO− + Br(CH2)4COOH | 90 | — | 0.4 | — | |
| Br(CH2)7COOH·Br(CH2)7COO− | Br− + (CH2)7COO·Br(CH2)7COOH | 61 | — | 11.6 | 28.6 |
| Br(CH2)7COOH·Br− + (CH2)7OCO | 34 | — | 7.5 | — | |
| Br(CH2)7COO− + Br(CH2)7COOH | 76 | — | 80.9 | 71.4 | |
| Na6I4(Br(CH2)3COO)+ | Na6I4Br+ + (CH2)3OCO | 99 | 88.2 | 100 | 94.5 |
| Na2Br+ + (NaI)4(CH2)3COO | 174 | 4.4 | — | — | |
| Na2I+ + Na4I3(Br(CH2)3COO) | 178 | 4.9 | — | 3.0 | |
| Na6I5(Br(CH2)3COOH)+ | Na6I4(Br(CH2)3COO)+ + HI | 14/77d | 38.5 | 29.5 | 26.5 |
| Na6I5+ + Br(CH2)3COOH | 99 | 60.3 | 70.5 | 73.5 | |
| Na6I4(Br(CH2)4COO)+ | Na6I4Br+ + (CH2)4OCO | 104 | 71.0 | 90.7 | 69.0 |
| Na2I+ + Na4I3(Br(CH2)4COO) | 222 | 29.1 | — | — | |
| Na4I2(Br(CH2)4COO)+ + (NaI)2 | 183 | — | 9.3 | 28.6 | |
| Na6I5(Br(CH2)4COOH)+ | Na6I4(Br(CH2)4COO)+ + HI | 27/72d | 85.0 | 89.9 | 75.0 |
| Na6I4Br+ + HI + (CH2)4OCO | 130 | 6.5 | — | 1.4 | |
| Na6I5+ + Br(CH2)4COOH | 105 | 8.5 | 8.3 | 23.0 | |
Three different fragments are observed: Br(CH2)mCOO− is obviously formed by loss of neutral Br(CH2)mCOOH, while Br− and Br(CH2)mCOOH·Br− suggest lactone formation upon IRMPD, see Table 1. For m = 4, the by far dominant IRMPD fragment is Br(CH2)4COOH·Br−, the eliminated (CH2)4OCO is most likely valerolactone. Scheme 1 depicts the mechanism of the SN2 reaction that transforms Br(CH2)4COO− to (CH2)4OCO + Br−. Although the alternative reaction, decarboxylation and (CH2)4 cyclobutane formation is thermochemically slightly favored with a reaction energy of ΔE = 34 kJ mol−1, calculations indicate a high barrier of 168 kJ mol−1 in Br(CH2)4COO− due to an energetically demanding charge transfer. Valerolactone is therefore the most plausible product. The bromide fragment appears selectively around 2600–3000 cm−1 where high photon energies and high laser power provide more energy to trigger secondary reactions, in this case lactone formation followed by dissociation of the non-covalent Br(CH2)4COOH·Br− complex. In line with experiment, the calculated reaction energies in Table 1 predict formation of Br(CH2)4COOH·Br− to be the most probable channel; evaporation of the pre-formed lactone requires only 42 kJ mol−1. Other channels lie considerably higher in energy.
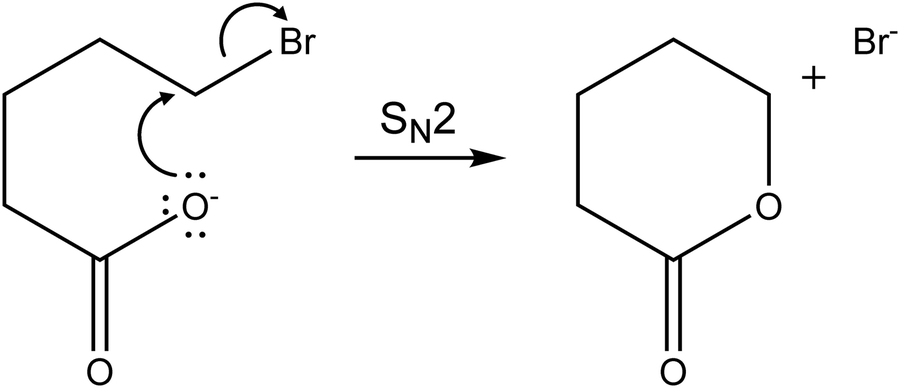 | ||
| Scheme 1 Formation of valerolactone via an intramolecular SN2 reaction from Br(CH2)4COO−. | ||
The main fragment observed for m = 7 is Br(CH2)7COO− with evaporation of Br(CH2)7COOH, while Br− and Br(CH2)7COOH·Br− are present only in minor amounts. Br− is observed again selectively around 2900 cm−1, while both Br− and Br(CH2)7COOH·Br− appear almost with the same yield at 1500–1750 cm−1. The calculated energies, Table 1, indicate that lactone is not present in major amounts on the cluster, as it would be eliminated preferentially, requiring 34 kJ mol−1, followed by Br− production as the second most important channel. Instead, Br(CH2)7COO− is formed from Br(CH2)7COOH·Br(CH2)7COO− in the experiment, with a dissociation energy of 76 kJ mol−1.
Analysis of the IR spectra supports the conclusions drawn on a thermochemical basis. The calculated isomers can be discriminated based on the position of the O–H stretch vibration. For m = 4, the broad absorption at 2200–2600 cm−1 is assigned to the O–H stretching mode shifted by the interaction with the COO− group of the alkanoate in isomers Id–f. The tail of the spectrum at 3100–3600 cm−1, however, can be assigned to the interaction of the O–H group hydrogen bonded in the lactone complexes Ia–c and C–H stretching vibrations. The peak positions in the fingerprint region of 1100–1700 cm−1 support the presence of both Ia–c and Id–f isomer classes. However, no specific isomers can be assigned here. The peak centered at 1606 cm−1 is likely due to isomers Id–f, see also Fig. S5 (ESI†) for a zoom in the fingerprint region. The observed fragmentation can be induced by only one photon. The calculated and experimental cross sections, Fig. 1b and c, differ by one to two orders of magnitude, which we attribute in part to the high structural flexibility of the alkanoic acid/alkanoate-system, in particular the extreme broadening of the O–H stretching mode upon hydrogen bonding to the carboxylate group, which goes along with efficient energy redistribution. Radiative cooling may also contribute to the low experimental IRMPD yield.
In the IR spectrum for m = 7, the noise in the high-energy region above 3000 cm−1 is too high to identify or rule out the presence of octalactone based on the O–H vibrations. Besides the dominant evaporation of the intact acid, the fragments indicating lactone elimination, Br− and Br(CH2)7COOH·Br−, are observed. All features in the spectrum can be assigned to isomer IIa without lactone. While there is an overlap with isomers containing lactone at the absorptions around 3000 cm−1 and 1700 cm−1, the calculated intense O-H stretch features of isomers IId,e at 3300–3400 cm−1 are missing in the experimental spectrum. Thus, the lactone containing isomers IId,e are at most a minor fraction of the experimental mixture. According to the calculated thermochemistry, at least two photons are required to induce the observed fragmentation; the experimental and calculated absorption cross sections differ again by two orders of magnitude.
IRMPD of n-bromoalkanoic acids and n-bromoalkanoates in salt environment (n = 4, 5, 8, 11)
To investigate the influence of a salt environment on the potential lactone formation, sodium iodide clusters were prepared by ESI, where one I− ion was replaced by a bromoalkanoate ion. Sodium iodide clusters doped with a neutral molecule of bromoalkanoic acid were studied for comparison. Table 1 shows the reaction energies and branching ratios of the fragments with an intensity above 4%, the detailed breakdown of all observed fragments is given in Table S1 (ESI†). Fig. 3 shows the absorption spectrum of Na6I4(Br(CH2)3COO)+ and Na6I5(Br(CH2)3COOH)+ in the 833–3846 cm−1 region. The spectra for Na6I4(Br(CH2)3COO)+ were measured with 15 s (region IR1), 7 s (region IR2) and 5 s (region IR3) irradiation time, while the ones for Na6I5(Br(CH2)3COOH)+ were measured with 15 s (region IR1), 6 s (region IR2) and 3 s (region IR3). The calculated structures of the most stable isomers are illustrated in Fig. 2b–e. For the Na6I4(Br(CH2)3COO)+ ion, the most intense fragmentation channel over the whole spectral range is Na6I4Br+, which indicates butyrolactone elimination from the cluster. In line with this interpretation, the most stable structure of Na6I4(Br(CH2)3COO)+ is Na6I4Br+ with (CH2)3OCO non-covalently attached, isomer IIIa in Fig. 2b. Since structures with a bromobutyrate moiety lie significantly higher in energy, isomer IIIa is also the most likely structure to appear in the experiment. The kinetics suggests that lactone elimination is a primary process (Fig. S10, ESI†).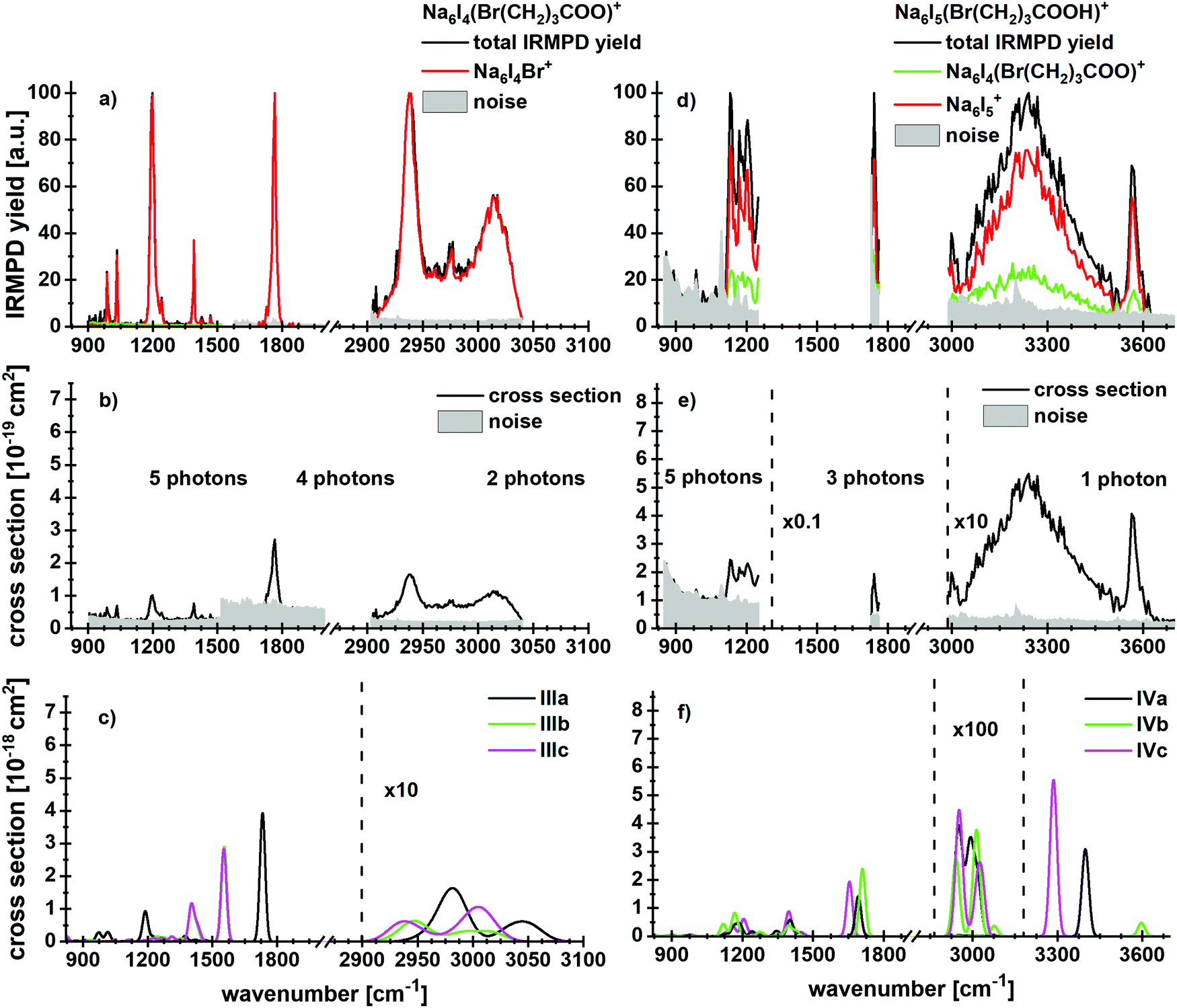 | ||
| Fig. 3 Experimental IRMPD yield spectra of (a) Na6I4(Br(CH2)3COO)+ and (d) Na6I5(Br(CH2)3COOH)+ with contribution of the most intense fragmentation products. (b and e) Multiphoton photodissociation cross sections derived from (a and d). (c and f) Calculated spectra in harmonic approximation for Na6I4(Br(CH2)3COO)+ and Na6I5(Br(CH2)3COOH)+, respectively, for 833–3846 cm−1. Here, the regions measured in detail are shown. The overview spectra are provided in Fig. S2 (ESI†). The theoretical spectra are scaled with a factor of 0.97. | ||
The peaks at 2938 cm−1 and 3013 cm−1 do not allow for an unambiguous assignment of isomers, since C–H absorptions are only moderately influenced by the cluster structure. The observed features are consistent with any of the three calculated isomers, or a mixture of them. However, the sharp C–O stretch peak at 1761 cm−1, the missing absorption at 1540 cm−1 and the absorptions at 1195, 1032 and 987 cm−1 are consistent with the dominant presence of isomer IIIa. This indicates that butyrolactone is formed in the electrospray process, before the clusters are stored in the ICR cell and heated by IR irradiation.
The dissociation into Na6I4Br+, observed as the main reaction channel in the experiment, requires 99 kJ mol−1, Table 1. This implies that about two photons are needed in the 2903–3040 cm−1 region and four to five photons in the fingerprint region. The absolute values of the experimental and theoretical cross section derived using this assumption (Fig. 3b and c) match well in the C–H stretch region, while the deviation is about an order of magnitude in the fingerprint region. Due to the low laser power in this region and the large number of degrees of freedom, irradiation times are long, which favors radiative cooling. This effect is not accounted for in our multiple photon analysis.
When neutral bromobutyric acid is attached to the sodium iodide cluster, resulting in Na6I5(Br(CH2)3COOH)+, the most intense fragmentation channel corresponds to evaporation of the intact 4-bromobutyric acid molecule from the cluster, evidenced by the detection of Na6I5+ (see Fig. 3d–f for the spectra, Table 1 for the reaction energies and Fig. S11 (ESI†) for the kinetics). The second most intense channel is HI elimination. Evaporation of 4-bromobutyric acid or HI requires 99 kJ mol−1 or 77 kJ mol−1, respectively. Following HI elimination, the remaining bromobutyrate affords lactone formation, which reduces the overall reaction energy to 14 kJ mol−1. However, both HI elimination and lactone formation are hindered by barriers (see also below). We note that HI release was already observed in a previous study where cesium iodide clusters were doped with small peptides.60 In both cases, HI elimination probably follows a similar mechanism as the release of HCl from a bulk sea-salt surface following uptake of HNO3, which was reported by Haan and Finlayson-Pitts.65
The IR spectrum shows that the cluster is present in several conformations. For example, in the O–H stretching region, the peak at 3569 cm−1 results from a free O–H vibration (e.g. in isomer IVb), whereas the broad absorption centered at 3241 cm−1 is induced by the OH⋯I− interaction (isomers IVa and IVc). In the 2800–3000 cm−1 region, C–H vibrations contribute to the experimental spectral intensity. For the fragmentation in this wavelength region, at least one photon is needed. The peak at 1739 cm−1 points towards isomer IVb or similar conformations, with at least three photons needed for the dissociation. The experimental and theoretical cross section values are comparable in this region. Since the signal and the absorption around 1200 cm−1 were very weak, no assignment to a specific isomer is possible.
Fig. 4 shows experimental and calculated IR spectra of Na6I4(Br(CH2)4COO)+ and Na6I5(Br(CH2)4COOH)+. The spectra for Na6I4(Br(CH2)4COO)+ were measured with 20 s (region IR1), 10 s (region IR2) and 3 s (region IR3) irradiation time, the irradiation times for Na6I5(Br(CH2)4COOH)+ were 10 s (region IR1), 10 s (region IR2) and 3 s (region IR3). For 5-bromovalerate complexed with salt, several different fragmentation patterns are observed (Table 1), where the dominant fragment Na6I4Br+ is consistent with valerolactone elimination. In lower yields, Na2I+ and Na4I2(Br(CH2)4COO)+ are observed. Na4I2Br+ appears with less intensity and may be created during secondary reactions. The most stable structure in Fig. 2d, isomer Va, shows that lactone formation is again energetically preferred.
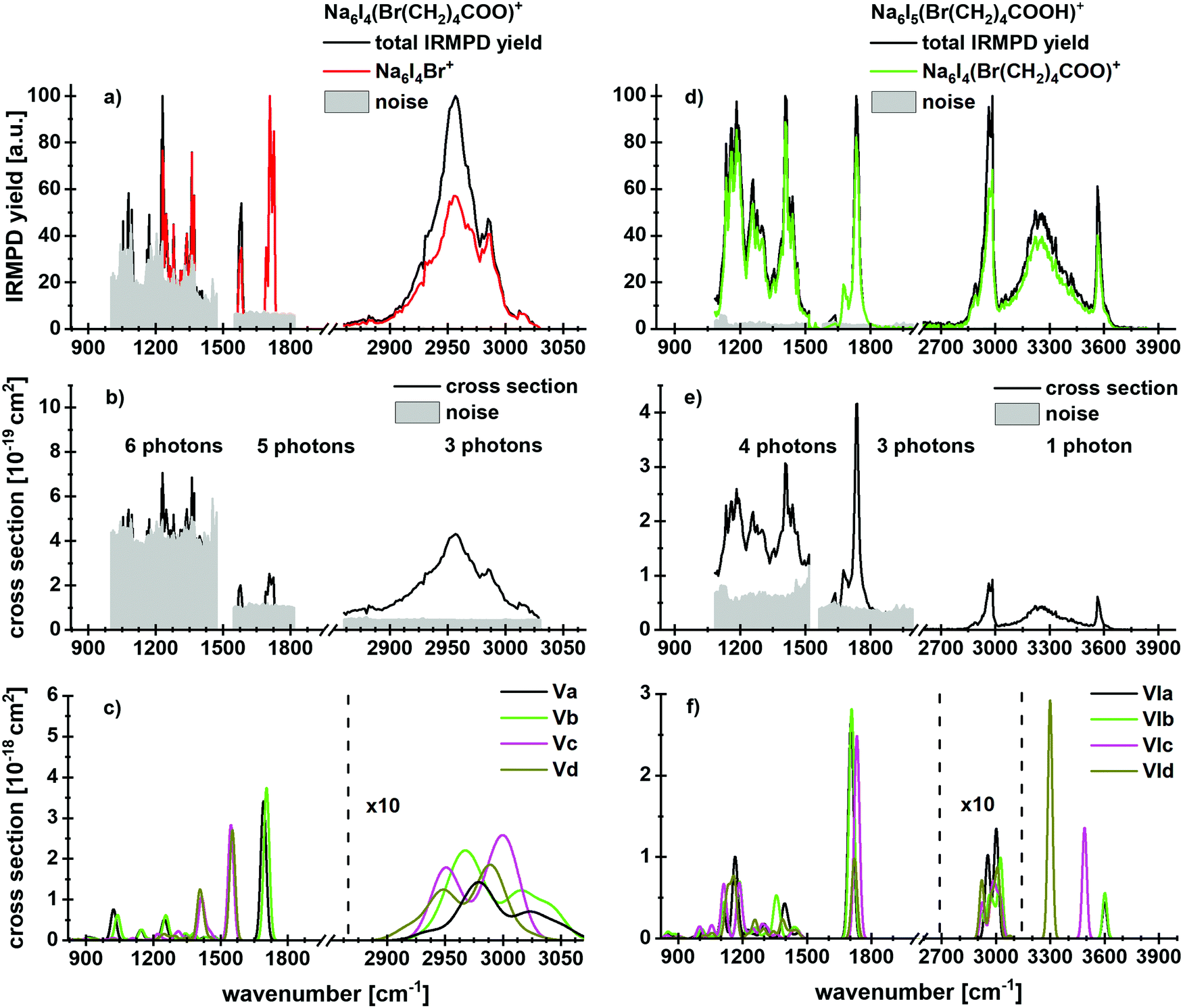 | ||
| Fig. 4 Experimental IRMPD yield spectra of (a) Na6I4(Br(CH2)4COO)+ and (d) Na6I5(Br(CH2)4COOH)+ with contribution of the most intense fragmentation products. (b and e) Multiphoton photodissociation cross sections derived from (a and d). (c and f) Calculated spectra in harmonic approximation for Na6I4(Br(CH2)4COO)+ and Na6I5(Br(CH2)4COOH)+, respectively, for 833–3846 cm−1. Here, the regions measured in detail are shown. The overview spectra are provided in Fig. S3 (ESI†). The theoretical spectra are scaled with a factor of 0.97. | ||
In contrast to 4-bromobutyrate, both the valerolactone and 5-bromovalerate seem to be present in the experimental ion population, evidenced by the peaks in the C–O stretch region. The lactone structure (isomers Va,b) is identified by the peak at 1712 cm−1. The presence of 5-bromovalerate (isomers Vc,d) is responsible for the absorption at 1575 cm−1. Since the Na6I4Br+ fragment appears also following this excitation, we conclude that lactone is formed from 5-bromovalerate activated by IR light. The kinetics (Fig. S12, ESI†) shows that lactone elimination is again a primary process. The kinetic fit requires absorption of multiple photons. The spectral features in the C–H stretch region around 3000 cm−1 and the fingerprint region of 850–1480 cm−1 cannot be assigned to specific isomers. The theoretical and experimental cross sections are comparable in the C–H and fingerprint region, while a difference of more than an order of magnitude is observed for the C–O modes.
The spectrum and fragmentation behavior changes for 5-bromovaleric acid, see Fig. 4d–f. As also evident from the kinetics, Fig. S13 (ESI†), the most intense fragmentation channel is the evaporation of HI from the cluster, with a low reaction energy of 72 kJ mol−1, or 27 kJ mol−1 if lactone is formed in parallel. Thus, at least one photon is needed for decomposition at 2540–3840 cm−1. The Na6I5+ fragment appears as the second most abundant fragment, with a calculated reaction energy of 105 kJ mol−1. Interestingly, also Na6I4Br+ is observed, which according to the fit is a secondary product. It is thus seamlessly explained as HI elimination followed by lactone formation and loss.
The absorption centered at 3564 cm−1 originates from a free O–H vibration of the acid, the broad absorption at 3250 cm−1 again from the OH⋯I− interaction. The doublet around 2970 cm−1 and a weak transition at 2886 cm−1 can be assigned to C–H vibrations, based on the comparison with the spectrum in Fig. 4a and the calculations.
For the C–O absorption observed at 1720 cm−1, Na6I4(Br(CH2)4COO)+ with HI elimination is the most intense fragmentation channel while Na6I5+ appears with lower intensity. In the 1100–1500 cm−1 region, HI elimination is again the favored dissociation pathway. The absorption centered at 1400 cm−1 results from a CH2 bending mode and indicates the presence of isomer VIa or VIb, showing a local absorption in this region. Two further peaks with similar intensities, induced by coupled C–C and C–O vibrations, again point out that more than one isomer is needed to interpret the IR spectrum.
The measurements were repeated with bromoalkanoates and bromoalkanoic acids with longer aliphatic chains. These experiments did not yield clear evidence for lactone formation. For 8-bromooctanoate, loss of [NaI]2 is the preferred fragmentation channel (Fig. S14, ESI†), while no fragment that would correspond to lactone formation is present in the investigated IR region. Upon doping the salt cluster with intact 8-bromooctanoic acid, the dominant fragment channels are loss of HI competing with loss of the complete molecule.
In the case of 11-bromoundecanoate (Fig. S15 and S16, ESI†), loss of [NaI]2 again is the dominant reaction channel. Interestingly, the C–Br bond becomes activated in these smaller salt clusters, evidenced by Na4I2(I(CH2)10COO)+ and Na2(I(CH2)10COO)+, the dominant secondary fragments. Obviously, I− replaces Br− in an SN2 reaction.66 This is the reverse of the standard SN2 reaction, most likely mediated by the interaction with the salt. A fragment that could point to lactone formation, Na2Br+, appears late with low intensity. However, it is more likely formed by loss of Na2I(I(CH2)10COO) from Na4I2(Br(CH2)10COO)+, which has already rearranged to Na4IBr(I(CH2)10COO)+.
Reaction paths for lactone formation
The experimental observation of Br− ions, either bare, complexed to bromoalkanoic acid, or embedded in a [NaI]x cluster, is evidence for lactone formation. To get a better idea how and when lactone formation is feasible in the experiment, we calculated the reaction path for m = 3, 4 and 7 in three model environments as shown in Fig. 5: bare bromoalkanoate, bromoalkanoate complexed with Na+, and a simplified bromoalkanoate–bromoalkanoic acid complex. For bare bromoalkanoate, the barrier increases monotonically along the m = 3, 4, 7 series from 18 kJ mol−1 to 58 kJ mol−1. To get closer to the relevant barrier of lactone formation in the experiment, we repeated this calculation with a simplified model of the studied complex, the complex Br(CH2)COOH·Br(CH2)mCOO− in which the aliphatic chain of the neutral molecule was shortened to reduce both the conformational flexibility and the number of electrons. Here, one can expect a barrier of about 61 and 119 kJ mol−1 for lactone formation with m = 4 and 7, respectively. Due to the high barrier for the longer chain, lactone formation in the isolated complex can be ruled out, since direct dissociation is both energetically preferred, Table 1, and with a loose transition state also mechanistically favored. For m = 7, fragments involving lactone elimination therefore most likely result from isomers IId,e formed in the ESI process. The kinetics for m = 7, 10 in Fig. S6 and S7 (ESI†) also reveal that the potential lactone elimination takes place much less efficiently for longer alkanoate chains than for the complex with m = 4, Fig. S8 (ESI†), further hinting that the ring has to be present before irradiation. Lactone elimination is also induced by ambient black-body infrared radiation for m = 4 with a very small rate of 0.006 s−1, see Fig. S9 (ESI†) for the BIRD kinetics.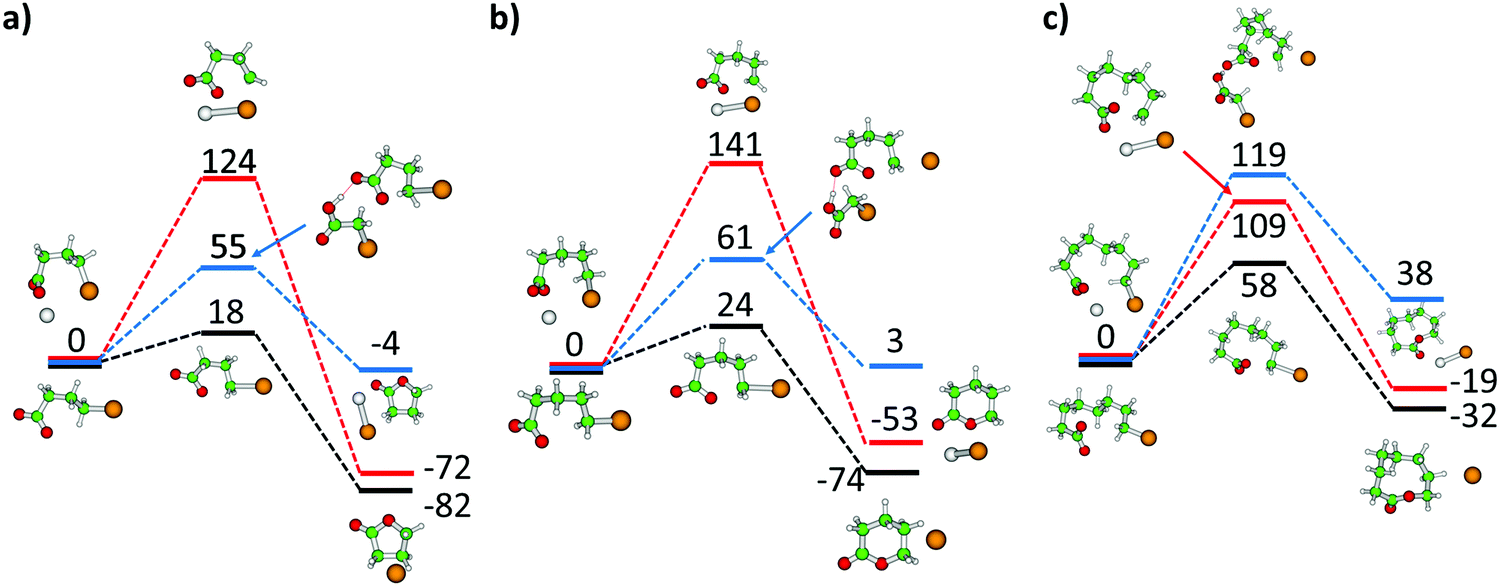 | ||
| Fig. 5 Reaction path to form lactones from bromoalkanoate Br(CH2)mCOO− in the gas phase (black curve), in the presence of a sodium ion NaBr(CH2)mCOO (red curve) and in the Br(CH2)COOH·Br(CH2)mCOO− model complex (blue curve) for (a) m = 3, (b) m = 4, (c) m = 7. Calculated at the B3LYP/def2TZVP level, energies are given in kJ mol−1. Color code: Na grey, Br brown, C green, O red, H white. | ||
The lactone formation path for bromobutyrate and the bromobutyrate–sodium complex NaBr(CH2)3COO is provided in Fig. 5a. Compared to butyrolactone formation in the gas phase with a small barrier of 18 kJ mol−1 and exothermicity of 82 kJ mol−1, the presence of the Na+ counterion increases the barrier to 124 kJ mol−1, since the interaction between the CO2− group, Br and Na+ stabilizes the reactant compared to the transition state structure. The reaction energy stays almost unaffected by the presence of Na+. This suggests that lactone formation takes place during the electrospray process, since complexation of bromobutyrate with the salt cluster increases the barrier and slows down the reaction.
For 5-bromovalerate, lactone formation faces a higher barrier and is less exothermic than for 4-bromobutyrate (Fig. 5a and b). Compared to the barrier in the gas phase of 24 kJ mol−1, the barrier increases to 141 kJ mol−1 when the ion complexes with Na+. This can explain the presence of both isomers in the experimental mixture. Lactones may be formed in the ESI source, with a slightly higher reaction barrier compared to 4-bromovalerate. The reaction is then hindered as soon as the organic ion is complexed with salt.
We found transition states for lactone formation for 8-bromooctanoate and 11-bromoundecanoate in the gas phase, with barriers of 58 and 44 kJ mol−1, respectively, about twice the value of 5-bromovalerate. We consider it highly unlikely to happen in the salt environment during IRMPD, as the long bromoalkyl chain has to be in the correct orientation to form the lactone ring, which is exceedingly improbable.
Conclusion
IRMPD of negatively charged complexes with n-bromoalkanoic acids (n = 5, 8) clustered with the corresponding n-bromoalkanoates, as well as positively charged sodium iodide clusters doped with the intact acids and alkanoates (n = 4, 5) was investigated experimentally and theoretically. The anionic complexes showed that lactone formation can be initiated by IR light for n = 5, and some fragments indicating formation of rings were also observed for n = 8. However, the formation via IR light is not expected for the latter case due to steric effects and a concomitant higher barrier, implying that a small fraction of clusters already contained lactone before irradiation. The failure to generate these complexes for n = 4 by ESI suggests that lactone formation is even more facile for this small system, making the complex unstable under the experimental conditions.The fragmentation behavior of the cationic salt clusters including an attached intact bromoalkanoic acid molecule shows the loss of HI, [NaI]x and the intact acid. The situation changes for bromoalkanoates embedded in salt environment. Here, lactone evaporation was observed with 4-bromobutyrate, and the spectra suggest that butyrolactone is already present before irradiation of the cluster with laser light, formed during the ESI process. 5-Bromovalerate is slightly more stable, the valerolactone is formed both before and during laser irradiation. Experiment and calculations show that the salt environment tends to stabilize bromoalkanoates against lactone formation by increasing the barrier with respect to the free bromoalkanoate in the gas phase for 4-bromobutyrate and 5-bromovalerate.
In a real sea-salt aerosol, similar reaction mechanisms may be operative, albeit not driven by multiple infrared photons. Due to their larger size and the high-pressure environment in the atmosphere, they are efficiently thermalized. Reactions in the aerosol will be either thermally activated, or photochemically by visible or ultraviolet light. Moreover, sea-salt aerosols consist mainly of NaCl, which will behave differently from the NaI clusters studied here. Nevertheless, our work shows that the barriers for thermally activated lactone formation from bromoalkanoates via an intramolecular SN2 reaction depend sensitively on the local environment of the carboxylate group and the bromine atom involved in the reaction. We suggest that this applies also to SN2 reactions in real sea-salt aerosols.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Austrian Science Fund FWF within the DK-ALM: W1259-N27 and stand-alone project P28896. The computational results presented have been achieved using the HPC infrastructure LEO of the University of Innsbruck. The tunable OPO systems are part of the Innsbruck Laser Core Facility, financed by the Austrian Federal Ministry of Education, Science and Research.References
- B. J. Finlayson-Pitts, Reactions at surfaces in the atmosphere: integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols, Phys. Chem. Chem. Phys., 2009, 11, 7760–7779 RSC.
- M. O. Andreae, Atmospheric aerosols: biogeochemical sources and role in atmospheric chemistry, Science, 1997, 276, 1052–1058 CrossRef CAS.
- U. Pöschl, Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem., 2005, 44, 7520–7540 CrossRef PubMed.
- S. J. Ghan and S. E. Schwartz, Aerosol properties and processes: a path from field and laboratory measurements to global climate models, Bull. Am. Meteorol. Soc., 2007, 88, 1059–1084 CrossRef.
- J. Haywood and O. Boucher, Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: a review, Rev. Geophys., 2000, 38, 513–543 CrossRef CAS.
- M. O. Andreae and D. Rosenfeld, Aerosol–cloud–precipitation interactions. Part 1. The nature and sources of cloud-active aerosols, Earth-Sci. Rev., 2008, 89, 13–41 CrossRef.
- C. D. O'Dowd and G. de Leeuw, Marine aerosol production: a review of the current knowledge, Philos. Trans. R. Soc., A, 2007, 365, 1753–1774 CrossRef PubMed.
- C. D. O'Dowd, M. H. Smith, I. E. Consterdine and J. A. Lowe, Marine aerosol, sea-salt, and the marine sulphur cycle: a short review, Atmos. Environ., 1997, 31, 73–80 CrossRef.
- C. D. O'Dowd, M. C. Facchini, F. Cavalli, D. Ceburnis, M. Mircea, S. Decesari, S. Fuzzi, Y. J. Yoon and J.-P. Putaud, Biogenically driven organic contribution to marine aerosol, Nature, 2004, 431, 676–680 CrossRef PubMed.
- A. I. Gogou, M. Apostolaki and E. G. Stephanou, Determination of organic molecular markers in marine aerosols and sediments: one-step flash chromatography compound class fractionation and capillary gas chromatographic analysis, J. Chromatogr. A, 1998, 799, 215–231 CrossRef CAS.
- D. Hansell, C. Carlson, D. Repeta and R. Schlitzer, Dissolved organic matter in the ocean: a controversy stimulates new insights, Oceanography, 2009, 22, 202–211 CrossRef.
- A. Virkkula, K. Teinilä, R. Hillamo, V.-M. Kerminen, S. Saarikoski, M. Aurela, I. K. Koponen and M. Kulmala, Chemical size distributions of boundary layer aerosol over the atlantic ocean and at an antarctic site, J. Geophys. Res., 2006, 111, 21695 CrossRef.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of atmospheric brown carbon, Chem. Rev., 2015, 115, 4335–4382 CrossRef CAS PubMed.
- T. Lou and H. Xie, Photochemical alteration of the molecular weight of dissolved organic matter, Chemosphere, 2006, 65, 2333–2342 CrossRef CAS PubMed.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. Gouw, M. de, Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef CAS.
- R. Fisseha, J. Dommen, M. Sax, D. Paulsen, M. Kalberer, R. Maurer, F. Höfler, E. Weingartner and U. Baltensperger, Identification of organic acids in secondary organic aerosol and the corresponding gas phase from chamber experiments, Anal. Chem., 2004, 76, 6535–6540 CrossRef CAS PubMed.
- R. Atkinson, Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 2000, 34, 2063–2101 CrossRef CAS.
- I.-S. Kang, J. Xi and H.-Y. Hu, Photolysis and photooxidation of typical gaseous VOCs by UV irradiation: removal performance and mechanisms, Front. Environ. Sci. Eng., 2018, 12, 73 Search PubMed.
- S. N. Kozlov, V. L. Orkin, R. E. Huie and M. J. Kurylo, OH reactivity and UV spectra of propane, n-propyl bromide, and isopropyl bromide, J. Phys. Chem. A, 2003, 107, 1333–1338 CrossRef CAS.
- Y. L. Yung, J. P. Pinto, R. T. Watson and S. P. Sander, Atmospheric bromine and ozone perturbations in the lower stratosphere, J. Atmos. Sci., 1980, 37, 339–353 CrossRef CAS.
- M.-A. Sicre, J.-C. Marty and A. Saliot, n-Alkanes, fatty acid esters, and fatty acid salts in size fractionated aerosols collected over the mediterranean sea, J. Geophys. Res., 1990, 95, 3649 CrossRef CAS.
- P. Fu, K. Kawamura and K. Miura, Molecular characterization of marine organic aerosols collected during a round-the-world cruise, J. Geophys. Res., 2011, 116, 482603 Search PubMed.
- B. R. T. Simoneit, Compound-specific carbon isotope analyses of individual long-chain alkanes and alkanoic acids in Harmattan aerosols, Atmos. Environ., 1997, 31, 2225–2233 CrossRef CAS.
- N. Yassaa, B. Youcef Meklati, A. Cecinato and F. Marino, Particulate n-alkanes, n-alkanoic acids and polycyclic aromatic hydrocarbons in the atmosphere of Algiers city area, Atmos. Environ., 2001, 35, 1843–1851 CrossRef CAS.
- S. Kurwadkar, Groundwater pollution and vulnerability assessment, Water Environ. Res., 2017, 89, 1561–1579 CrossRef CAS PubMed.
- A. D. McEachran, D. Shea, W. Bodnar and E. G. Nichols, Pharmaceutical occurrence in groundwater and surface waters in forests land-applied with municipal wastewater, Environ. Toxicol. Chem., 2016, 35, 898–905 CrossRef CAS PubMed.
- R. P. Schwarzenbach, T. Egli, T. B. Hofstetter, U. Gunten and B. von, Wehrli, Global water pollution and human health, Annu. Rev. Env. Resour., 2010, 35, 109–136 CrossRef.
- P. J. Squillace, J. C. Scott, M. J. Moran, B. T. Nolan and D. W. Kolpin, VOCs, pesticides, nitrate, and their mixtures in groundwater used for drinking water in the United States, Environ. Sci. Technol., 2002, 36, 1923–1930 CrossRef CAS PubMed.
- Q. Sui, X. Cao, S. Lu, W. Zhao, Z. Qiu and G. Yu, Occurrence, sources and fate of pharmaceuticals and personal care products in the groundwater: a review, Emerging Contam., 2015, 1, 14–24 CrossRef.
- R. A. Mandour, Human health impacts of drinking water (surface and ground) pollution Dakahlyia Governorate, Egypt, Appl. Water Sci., 2012, 2, 157–163 CrossRef CAS.
- M. Haseena, M. Faheem Malik, A. Javed, S. Arshad, N. Asif, S. Zulfiqar and J. Hanif, Water pollution and human health, Environ. Risk Assess. Rem., 2017, 01, 16–19 Search PubMed.
- Water challenges of an urbanizing world, ed. M. Glavan, InTech, 2018 Search PubMed.
- J. A. Camargo and A. Alonso, Ecological and toxicological effects of inorganic nitrogen pollution in aquatic ecosystems: a global assessment, Environ. Int., 2006, 32, 831–849 CrossRef CAS PubMed.
- K. Mohankumar, V. Hariharan and N. P. Rao, Heavy metal contamination in groundwater around industrial estate vs. residential areas in Coimbatore, India, J. Clin. Diagn. Res., 2016, 10, BC05–BC07 CAS.
- D. J. Lapworth, N. Baran, M. E. Stuart and R. S. Ward, Emerging organic contaminants in groundwater: a review of sources, fate and occurrence, Environ. Pollut., 2012, 163, 287–303 CrossRef CAS PubMed.
- L. Lamastra, M. Balderacchi and M. Trevisan, Inclusion of emerging organic contaminants in groundwater monitoring plans, MethodsX, 2016, 3, 459–476 CrossRef PubMed.
- C. Rosik-Dulewska, T. Ciesielczuk and M. Krysiński, Organic pollutants in groundwater in the former airbase, Arch. Environ. Prot., 2012, 38, 305 Search PubMed.
- B. C. J. Zoeteman, K. Harmsen, J. B. H. J. Linders, C. F. H. Morra and W. Slooff, Persistent organic pollutants in river water and ground water of the Netherlands, Chemosphere, 1980, 9, 231–249 CrossRef CAS.
- S. J. Nelson, M. Iskander, M. Volz, S. Khalifa and R. Haberman, Studies of DBCP in subsoils, Sci. Total Environ, 1981, 21, 35–40 CrossRef CAS.
- P. J. Isaacson, L. Hankin and C. R. Frink, Boiling drinking water removes ethylene dibromide, Science, 1984, 225, 672 CrossRef CAS PubMed.
- R. A. Weintraub, G. W. Jex and H. A. Moye, Chemical and microbial degradation of 1,2-dibromoethane (EDB) in Florida ground water, soil, and sludge, in Evaluation of pesticides in ground water, ed. W. Y. Garner, R. C. Honeycutt and H. N. Nigg, American Chemical Society, Washington, DC, 1986, vol. 315, pp. 294–310 Search PubMed.
- R. P. Schwarzenbach, W. Giger, C. Schaffner and O. Wanner, Groundwater contamination by volatile halogenated alkanes: abiotic formation of volatile sulfur compounds under anaerobic conditions, Environ. Sci. Technol., 1985, 19, 322–327 CrossRef CAS PubMed.
- F. Ferreira da Silva, N Varella do, M. T. Jones, N. C. Vrønning Hoffmann, S. Denifl, S. Bald and I. Kopyra, J. Electron-induced reactions in 3-bromopyruvic acid, Chem. – Eur. J., 2019, 25, 5498–5506 CrossRef CAS PubMed.
- T. M. Vogel and M. Reinhard, Reaction products and rates of disappearance of simple bromoalkanes, 1,2-dibromopropane, and 1,2-dibromoethane in water, Environ. Sci. Technol., 1986, 20, 992–997 CrossRef CAS PubMed.
- S. D. Richardson, Environmental mass spectrometry: emerging contaminants and current issues, Anal. Chem., 2010, 82, 4742–4774 CrossRef CAS PubMed.
- D. J. Donaldson, A. F. Tuck and V. Vaida, Atmospheric photochemistry via vibrational overtone absorption, Chem. Rev., 2003, 103, 4717–4730 CrossRef CAS PubMed.
- J. Roithová, Characterization of reaction intermediates by ion spectroscopy, Chem. Soc. Rev., 2012, 41, 547–559 RSC.
- N. Heine, T. I. Yacovitch, F. Schubert, C. Brieger, D. M. Neumark and K. R. Asmis, Infrared photodissociation spectroscopy of microhydrated nitrate-nitric acid clusters NO3−(HNO3)m(H2O)n, J. Phys. Chem. A, 2014, 118, 7613–7622 CrossRef CAS PubMed.
- M. C. Thompson, J. Ramsay and J. M. Weber, Solvent-driven reductive activation of CO2 by bismuth: switching from metalloformate complexes to oxalate products, Angew. Chem., Int. Ed., 2016, 55, 15171–15174 CrossRef CAS PubMed.
- J. Oomens, D. T. Moore, G. Meijer and G. von Helden, Infrared multiple photon dynamics and spectroscopy of cationic PABA and its dehydroxylated fragment ion, Phys. Chem. Chem. Phys., 2004, 6, 710–718 RSC.
- V. J. F. Lapoutre, B. Redlich, A. F. G. van der Meer, J. Oomens, J. M. Bakker, A. Sweeney, A. Mookherjee and P. B. Armentrout, Structures of the dehydrogenation products of methane activation by 5d transition metal cations, J. Phys. Chem. A, 2013, 117, 4115–4126 CrossRef CAS PubMed.
- O. W. Wheeler, M. Salem, A. Gao, J. M. Bakker and P. B. Armentrout, Sequential activation of methane by Ir+: an IRMPD and theoretical investigation, Int. J. Mass Spectrom., 2019, 435, 78–92 CrossRef CAS.
- A. C. Hermes, S. M. Hamilton, W. S. Hopkins, D. J. Harding, C. Kerpal, G. Meijer, A. Fielicke and S. R. Mackenzie, Effects of coadsorbed oxygen on the infrared driven decomposition of N2O on isolated Rh5+ clusters, J. Phys. Chem. Lett., 2011, 2, 3053–3057 CrossRef CAS.
- I. S. Parry, A. Kartouzian, S. M. Hamilton, O. P. Balaj, M. K. Beyer and S. R. Mackenzie, Chemical reactivity on gas-phase metal clusters driven by blackbody infrared radiation, Angew. Chem., Int. Ed., 2015, 54, 1357–1360 CrossRef CAS PubMed.
- T. F. Pascher, M. Ončák, C. van der Linde and M. K. Beyer, Release of formic acid from copper formate: hydride, proton-coupled electron and hydrogen atom transfer all play their role, ChemPhysChem, 2019, 20, 1420–1424 CAS.
- T. F. Pascher, M. Ončák, C. van der Linde and M. K. Beyer, Decomposition of copper formate clusters: insight into elementary steps of calcination and carbon dioxide activation, ChemistryOpen, 2019, 8, 1453–1459 CrossRef CAS PubMed.
- A. Herburger, M. Ončák, C.-K. Siu, E. G. Demissie, J. Heller, W. K. Tang and M. K. Beyer, Infrared spectroscopy of size-selected hydrated carbon dioxide radical anions CO2˙− (H2O)n (n = 2–61) in the C–O stretch region, Chem. – Eur. J., 2019, 25, 10165–10171 CrossRef CAS PubMed.
- E. Barwa, M. Ončák, T. F. Pascher, A. Herburger, C. van der Linde and M. K. Beyer, Infrared multiple photon dissociation spectroscopy of hydrated cobalt anions doped with carbon dioxide CoCO2(H2O)n−, n = 1–10, in the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch region, Chem. – Eur. J., 2020, 26, 1074–1081 CrossRef CAS PubMed.
O stretch region, Chem. – Eur. J., 2020, 26, 1074–1081 CrossRef CAS PubMed. - N. K. Bersenkowitsch, M. Ončák, J. Heller, C. van der Linde and M. K. Beyer, Photodissociation of sodium iodide clusters doped with small hydrocarbons, Chem. – Eur. J., 2018, 24, 12433–12443 CrossRef CAS PubMed.
- J. Heller, M. Ončák, N. K. Bersenkowitsch, C. van der Linde and M. K. Beyer, Infrared multiple photon dissociation of cesium iodide clusters doped with mono-, di- and triglycine, Eur. J. Mass Spectrom., 2019, 25, 122–132 CrossRef CAS PubMed.
- N. K. Bersenkowitsch, M. Ončák, C. van der Linde, A. Herburger and M. K. Beyer, Photochemistry of glyoxylate embedded in sodium chloride clusters, a laboratory model for tropospheric sea-salt aerosols, Phys. Chem. Chem. Phys., 2018, 20, 8143–8151 RSC.
- A. Herburger, C. van der Linde and M. K. Beyer, Photodissociation spectroscopy of protonated leucine enkephalin, Phys. Chem. Chem. Phys., 2017, 19, 10786–10795 RSC.
- R. C. Dunbar, BIRD (blackbody infrared radiative dissociation): evolution, principles, and applications, Mass Spectrom. Rev., 2004, 23, 127–158 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, 2016 Search PubMed.
- D. O. Haan and B. J. de Finlayson-Pitts, Knudsen cell studies of the reaction of gaseous nitric acid with synthetic sea salt at 298 K, J. Phys. Chem. A, 1997, 101, 9993–9999 CrossRef.
- J. Xie, R. Otto, J. Mikosch, J. Zhang, R. Wester and W. L. Hase, Identification of atomic-level mechanisms for gas-phase X− + CH3Y SN2 reactions by combined experiments and simulations, Acc. Chem. Res., 2014, 47, 2960–2969 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00272k |
| This journal is © the Owner Societies 2020 |