
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent dynamics play a decisive role in the complex formation of biologically relevant redox proteins†
Ellen M.
Adams
a,
Oliver
Lampret
b,
Benedikt
König
a,
Thomas
Happe
 b and
Martina
Havenith
*a
b and
Martina
Havenith
*a
aLehrstuhl für Physkalische Chemie II, Ruhr Universität Bochum, 44801 Bochum, Germany. E-mail: martina.havenith@rub.de
bAG Photobiotechnologie, Ruhr Universität Bochum, 44801 Bochum, Germany
First published on 26th March 2020
Abstract
Electron transfer processes between proteins are vital in many biological systems. Yet, the role of the solvent in influencing these redox reactions remains largely unknown. In this study, terahertz-time domain spectroscopy (THz-TDS) is used to probe the collective hydration dynamics of flavoenzyme ferredoxin-NADP+-reductase (FNR), electron transfer protein ferredoxin-1 (PetF), and the transient complex that results from their interaction. Results reveal changes in the sub-picosecond hydration dynamics that are dependent upon the surface electrostatic properties of the individual proteins and the transient complex. Retarded solvent dynamics of 8–9 ps are observed for FNR, PetF, and the FNR:PetF transient complex. Binding of the FNR:PetF complex to the substrate NADP+ results in bulk-like solvent dynamics of 7 ps, showing that formation of the ternary complex is entropically favored. Our THz measurements reveal that the electrostatic interaction of the protein surface with water results in charge sensitive changes in the solvent dynamics. Complex formation between the positively charged FNR:NADP+ pre-complex and the negatively charged PetF is not only entropically favored, but in addition the solvent reorganization into more bulk-like water assists the molecular recognition process. The change in hydration dynamics observed here suggests that the interaction with the solvent plays a significant role in mediating electron transfer processes between proteins.
Introduction
In the center of photosynthetic metabolic redox-reactions of plants and other photosynthetic microorganisms, ferredoxins (FDXs) serve as universal electron shuttles by distributing electrons originating from photosynthetic water oxidation and fermentation to specific FDX-dependent enzymes in the chloroplast.1–4 Plant-type FDXs are small metalloproteins that usually have low redox potentials (−300 to −460 mV) and harbor [2Fe–2S]-clusters.1–8In the photosynthetic electron transport chain of the well characterized unicellular green algae Chlamydomonas reinhardtii, FDX1 (also known as PetF) transfers electrons from photosystem I (PSI) to the flavoenzyme ferredoxin-NADP+-reductase (FNR) to generate nicotinamide adenine dinucleotide phosphate (NADPH), an important energy carrier in most biological systems. Regardless of whether occurring in vivo under physiological conditions or in vitro, electron transfer reactions between two or more proteins require the formation of transient protein complexes.9 A reversible two-electron transfer between PetF and FNR initiates the hydride (H−)-transfer to generate NADPH;10,11 protein crystallography revealed that this process takes place as a ternary complex between FNR, NADP+ and PetF, in which FNR and NADP+ form a pre-complex.12–14 The general interaction pattern of this redox-couple has already been well characterized by protein crystallography, biochemical methods and nuclear magnetic resonance (NMR) experiments. These studies revealed that the intermolecular recognition and subsequent stabilization of this complex is mainly driven by electrostatic charge–charge interactions between basic residues of FNR and acidic residues of PetF (Fig. 1a and Fig. S1 in the ESI†).13,15–17 This transient complex is further stabilized by several amino acids from both proteins that form a hydrophobic environment near the two redox centers which were reported to be in close proximity to each other (∼6 Å) to ensure fast electron transfer (Fig. S2, ESI†).15
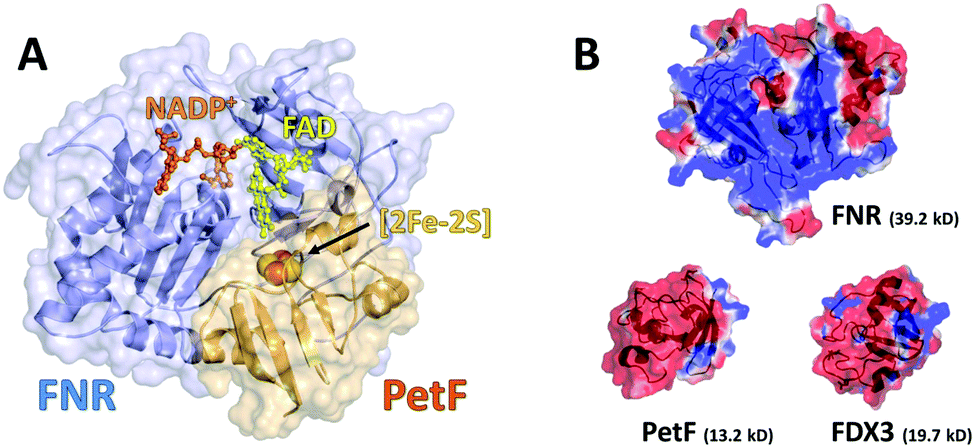 | ||
| Fig. 1 (a) Structural model of the transient FNR–NADP+–PetF complex from Chlamydomonas reinhardtii as a cartoon and surface model (based on PDB ID 1GAQ). The redox-centers ([2Fe–2S]-cluster, FAD and NADP+) are displayed as sticks and spheres. (b) Visualization of the surface electrostatic potential of FNR (upper panel, structural model, based on PDB ID 1EWY), PetF (left, PDB ID: 2MH7) and FDX3 (right, structural model, based on PDB ID 2MH7). Surface charge distribution has been calculated using APBS. Red represents regions of negative charges and blue represents regions of positive charge. | ||
Interestingly, despite the presence of at least 13 FDX-isoforms in C. reinhardtii, which are similar in their sequence and share structural homology (∼40% sequence similarity), only a few of these isoforms (including PetF, FDX2, and FDX7) show an interaction with FNR.6,8 Most importantly, the relevant acidic amino acids (including D58 of PetF) are strictly conserved in FDX-isoforms 1–3. However, the fact that FDX3 is not able to interact with FNR indicates that the processes of molecular recognition and complex stabilization cannot be solely attributed to the function of individual amino acids as these patterns are mainly conserved in FDX isoforms.
In recent years the importance of the aqueous solvent in influencing protein structure, function, and dynamics has been recognized.18,19 Coupling of water molecules to the protein surface results in an interfacial region in which water molecules within this region have distinctly different properties than bulk water. However, the structure and dynamics within this interfacial region are still not easy to access experimentally. Studies of protein solutions by surface sensitive techniques such as sum frequency generation (SFG) spectroscopy have found that organization of water in the interfacial region largely depends on the structure and net charge of the protein.20–22 Furthermore, probing of water near specific molecular groups relevant to proteins such as carboxylic acids, alcohols, and amines have found that the hydrogen-bonding network is largely influenced by the local electric field (i.e. charge), hydrophobicity, and the ability of the molecular group to form hydrogen bonds.23–25 This technique, however, is restricted to water near air/liquid or liquid/solid interfaces.
Terahertz (THz) spectroscopy has been shown to be a powerful tool to investigate solvent dynamics in bulk solutions. Radiation in the THz regime is directly sensitive to the low frequency collective intermolecular hydrogen-bonding vibrations of water (0.3–6 THz or 10–200 cm−1), and thus to any changes in the hydrogen-bonding network.18 Changes in these sub-picosecond collective motions, such as protein–water interactions, can be probed by changes in the measured THz absorption. Individual hydration shells of proteins have been shown to contribute largely to structure–function relationships and ultimately modulate the binding properties of proteins.26–30 However, solvation dynamics are still poorly characterized in protein–protein complexes in spite of the fact that they may play a key role in unraveling the molecular recognition of redox-protein couples. In this study, the transient protein complex between PetF and FNR was studied by THz-time domain spectroscopy (THz-TDS). By comparing the native redox couple PetF:FNR with the non-functional couple FDX3:FNR at different protein concentrations, compelling evidence is provided that solvation water plays a crucial role for the molecular recognition and electron transfer processes of redox proteins. Investigation of the ternary complex PetF:FNR:NADP+ reveals a change in solvent dynamics upon substrate binding which could promote release of the reduced substrate after the electron transfer process.
Experimental
Protein expression and purification
FNR, PetF, and FDX3 proteins were expressed and purified according to previously established protocols (see ESI† for details).16 All protein samples were stored at −80 °C until further use. Specific concentrations were prepared from dilution of the stock solution. All mixtures were freshly prepared on the day of measurement prior to injection into the sample cell.Terahertz-time domain spectroscopy
THz-TDS measurements were carried out with a custom-built spectrometer setup similar to that previously described.31,32 Briefly, the 800 nm output of a Ti:sapphire oscillator (Mai Tai EHP, Spectra Physics, United States) with a repetition rate of 80 MHz and pulse duration of 70 fs is split into pump and probe beams. The pump beam propagates to a photoconductive antenna (Tera-SED 3, Gigaoptics, Germany) that is biased at 20 V and modulated at 92 kHz to produce THz radiation. The generated THz radiation is collimated and focused by two off-axis parabolic mirrors to the sample cell. The THz beam transmitted through the cell is collected and focused by two off-axis parabolic mirrors to a 0.5 mm ZnTe(110) crystal for electro-optical sampling (EOS). A hole in the last parabolic mirror allows a collinear sampling geometry with the probe beam. Based on the principles of EOS, the electric field of the THz pulse generates a birefringence in the ZnTe crystal which induces a rotation in the polarization state of the probe beam that is directly proportional to the electric field of the THz pulse. The probe beam is split into orthogonal polarization components with a Wollaston prism and directed to a balanced photodetector (Nirvana Auto-Balanced Optical Receiver, Model 2007, New Focus, United States), where the intensity difference of the two components is measured. The signal of the photodetector is processed by a lock-in amplifier (HF2-LI, Zurich Instruments, Switzerland) with a 6 dB filter and 5 μs time constant, and the phase of the lock-in amplifier is set such that the signal in the X-component is maximized. A time window of approximately 20 ps is probed through the use of a rapid delay stage (ScanDelay, Model 150, APE, Germany) in the pump beam path which operates at 2 Hz. All measurements were conducted at 30 °C, where the temperature was held constant by an external chiller (±0.1 °C). Humidity within the sampling chamber was fixed at ≤5%.The sample cell was a commercially available demountable cell (Bruker, Germany), in which two z-cut quartz windows were used. Sample thickness of 100 μm was achieved with a Teflon spacer inserted between the windows. The front window had two holes bored in it, allowing for the sample to be injected. An overall solution volume of 200 μL was needed to fill the cell. The frequency dependent refractive index n(ν) and absorption coefficient α(ν) were calculated with eqn (1) and (2)
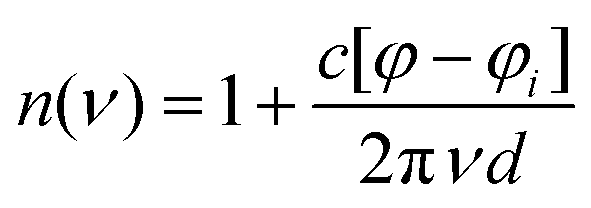 | (1) |
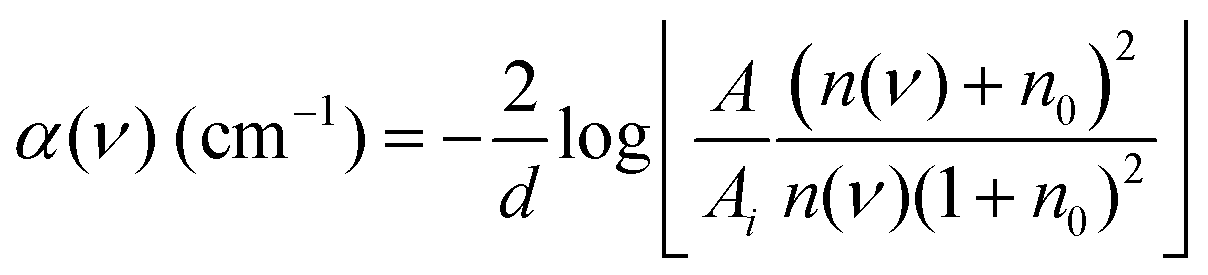 | (2) |
Results and discussion
Fig. 2 shows the THz-TDS absorption spectra of solutions containing FNR and PetF with concentrations ranging from 0 to 2 mM. The absorption coefficient increases nearly linearly over the measured frequency range for all spectra. With increasing protein concentration a decrease in the absorption coefficient occurs, consistent with a decrease in absorption due to a decrease in bulk water concentration as similarly observed in previous measurements of other proteins.34–37 If the decrease in absorption is occurring solely due to a lowered water concentration, a linear decrease at every measured frequency as a function of protein concentration would be observed. In the case of PetF, it can be clearly seen that changes in the absorption coefficient are frequency dependent, where the absorption coefficient is largely independent of concentration below 1 THz, but decreases significantly at higher frequencies. Based on this observation, the average absorption coefficient (αavg) for frequency ranges of 0.3–1, 1–1.75, and 1.75–2.5 THz were determined, and provides the same information as an individual frequency within these ranges. Results shown here will focus on the range 1.75–2.5 THz (the other two regions are shown in Fig. S3, ESI†). The larger absorption coefficient at higher frequencies means that changes in the αavg due to water exclusion will be more pronounced in this regime. Any deviations from the expected water exclusion value report on the hydrodynamic solvation shell of the protein. However, similar effects can be observed at other frequency ranges and will be discussed where pertinent. Fig. 3 shows αavg of FNR and PetF solutions from 1.75–2.5 THz for protein concentrations ranging from 0 to 2 mm. The black line in Fig. 3 represents the expected decrease in αavg of the buffer solution by excluding a volume of bulk water equal to the volume of the protein in which spherical volume of the protein was assumed to be RFNR = 29 Å and RPetF = 17 Å from the radii measured with dynamic light scattering in previous literature.38 The αavg of both individual FNR and PetF solutions deviates from this black line which takes into account solely bulk water exclusion. At all volume fractions FNR has an increased absorption relative to the black line while PetF features a decreased absorption. The nonlinear behavior in αavg observed here for both proteins indicates that the water molecules directly interacting with the protein surface, i.e. the hydration shell, have a distinct absorption which differs from bulk water, consistent with previous studies of proteins in solution.18,36,37,39 However, the manner in which water interacts with the two proteins is different, evidenced by the opposite behavior in αavg. Coupling of water to the protein depends upon its surface properties, in which the local topography influences interactions with water. For instance, water molecules within hydrophobic pockets or grooves form strong hydrogen-bonds mostly due to sterical constraints,40 whereas coordination of water molecules to hydrophilic sites appears to be correlated with charge.41 The resulting dynamical solvation shell emerges from the cooperative effect of water solvating the heterogeneous surface of the protein.42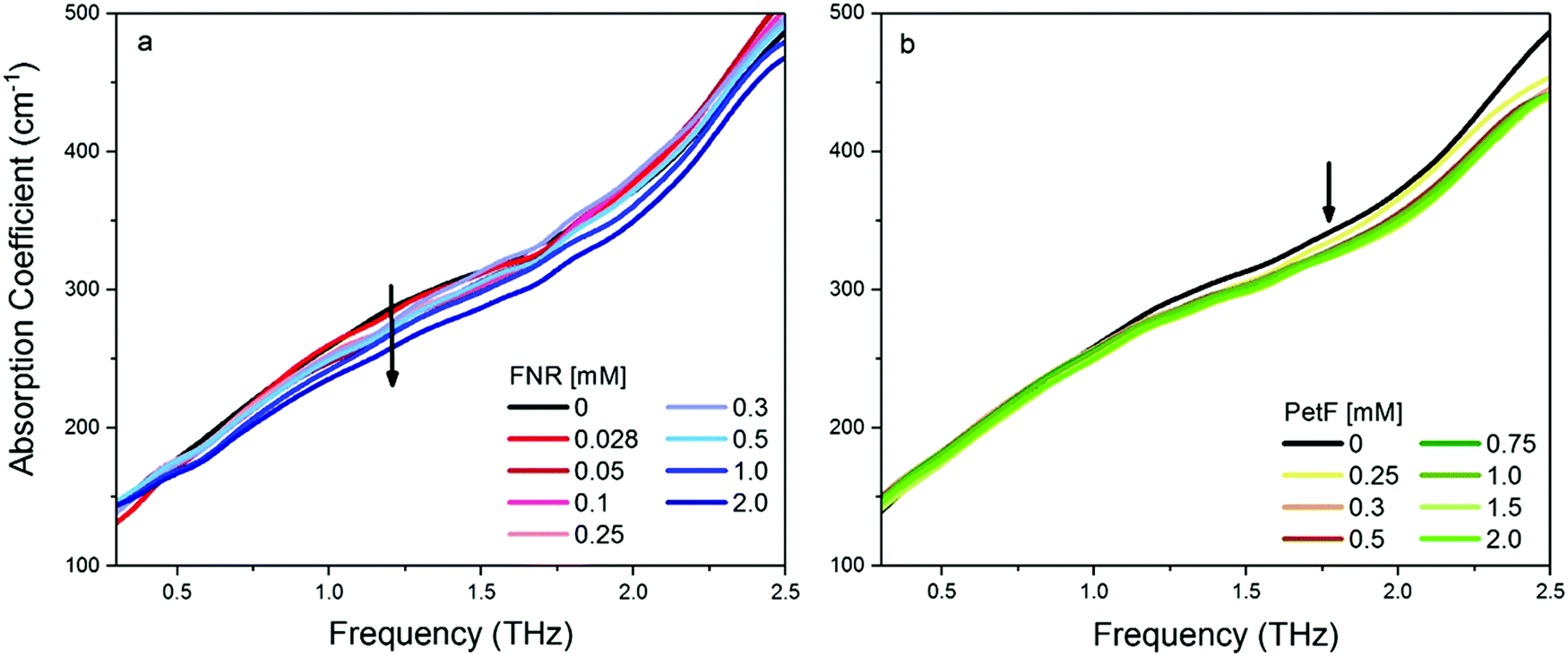 | ||
| Fig. 2 THz-TDS absorption spectra of aqueous solutions at pH 8 of (a) FNR and (b) PetF. The error of each measurement is on average 0.6 cm−1. The absorption coefficient decreases with increasing protein concentration, consistent with a decreasing water content. | ||
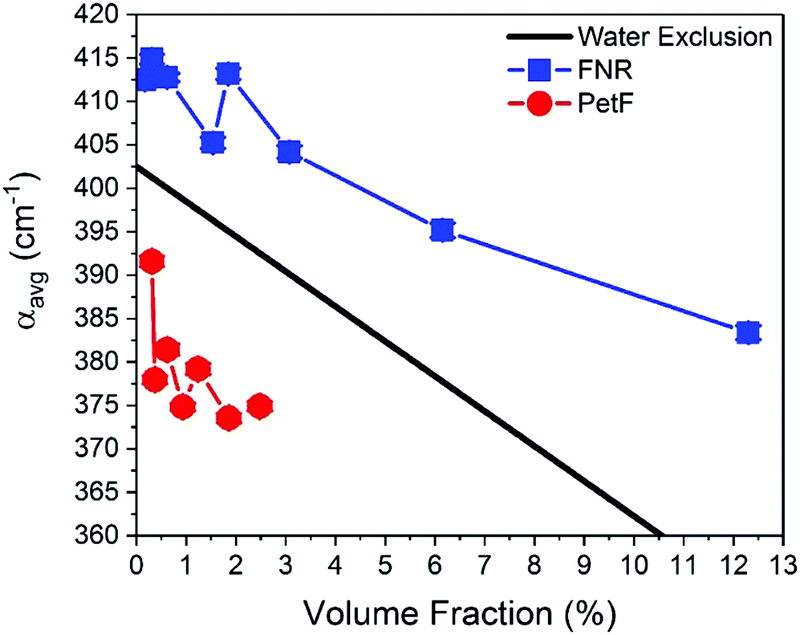 | ||
| Fig. 3 The average absorption coefficient (αavg) in the 1.75–2.5 THz range of aqueous solutions containing either FNR (blue) or PetF (red) as a function of volume fraction of protein in solution. The black line shows the expected decrease in αavg based on exclusion of water equal to the volume of protein in solution. Error bars are on average 0.6 cm−1. | ||
Mapping of the surface electrostatic potential of FNR and PetF at pH 8 (Fig. 1b, based on available PDB structures) shows that the surface of FNR has two distinct regions of negative and positive charges while the surface of PetF is largely negatively charged.43,44 The opposite surface electrostatic potentials of these proteins influence water to different extents, and is reflected in their corresponding THz spectra. Ab initio MD (AIMD) simulations and THz spectra of glycine and valine have shown that positively charged groups, such as aminos (NH3+), result in an increased THz intensity in the low frequency regime (below 100 cm−1, 3 THz). This increased intensity was attributed to cage rattling modes of the solvating water molecules at 65 and 70 cm−1 (1.8 and 2.0 THz).45–47 While the surface of FNR has both positively and negatively charged regions, the positively charged region covers a larger surface area, and the results obtained here show that the contribution of the positively charged groups dominates the THz response. In contrast, PetF has a negatively charged surface, which is mostly comprised of aspartic acid and glutamic acid residues. MD and AIMD simulations on glycine have shown that water molecules solvating carboxylate groups (COO−) are well ordered and have a cage librational mode, i.e. a correlated solute–water mode, in the THz regime centered at 2.4 THz (80 cm−1). The THz intensity below 150 cm−1 is predicted to be suppressed for the negative form of the carboxylic acid group compared to the neutral form.45,47,48 Measurements of glycine confirm this, where it was observed that the negatively charged molecule has a larger intensity than the zwitterionic molecule,49 and is consistent with results shown in Fig. 2 for PetF. This observation demonstrates how solvent dynamics are sensitive to differences in the surface properties of individual proteins, and can be used to report on changes in the electrostatics of protein surfaces.
THz absorption spectra for solutions of the native redox couple FNR:PetF, the non-functional couple FNR:FDX3, and the ternary complex FNR:PetF:NADP+ (NADP+ purchased from Roth GmbH, >97% purity) are shown in Fig. 4. All solutions were prepared with equimolar amounts of each component. Fig. 5 shows the corresponding αavg determined for each solution in the 1.75–2.5 THz range (see Fig. S4, ESI† for other frequency regions). The αavg follows the expected water exclusion for FNR:PetF solutions (assuming R = 30 Å for the transient complex),38 while increased values are observed for FNR:FDX3 solutions (assuming R = 17 Å for FDX3). FDX3 alone has decreased αavg similar to PetF as shown in Fig. S5 and S6 (ESI†). This result demonstrates differences in the collective water dynamics in the hydration shell of proteins that remain unbound in solution (FNR and FDX3) compared to those that form a complex (FNR:PetF). Formation of site-specific salt bridges is imperative for the complexation of FNR and PetF.14,43,50,51 Positively charged lysine and arginine residues on the surface of FNR couple to negatively charged glutamic acid and aspartic acid residues on PetF (Fig. S1, ESI†).16,17,44 Neutralization of these surface charges upon complexation results in a change in the surface electrostatic potential such that the solute is overall neutral. Our results demonstrate that the THz responses of FNR, PetF, and the FNR:PetF complex are clearly distinguishable.
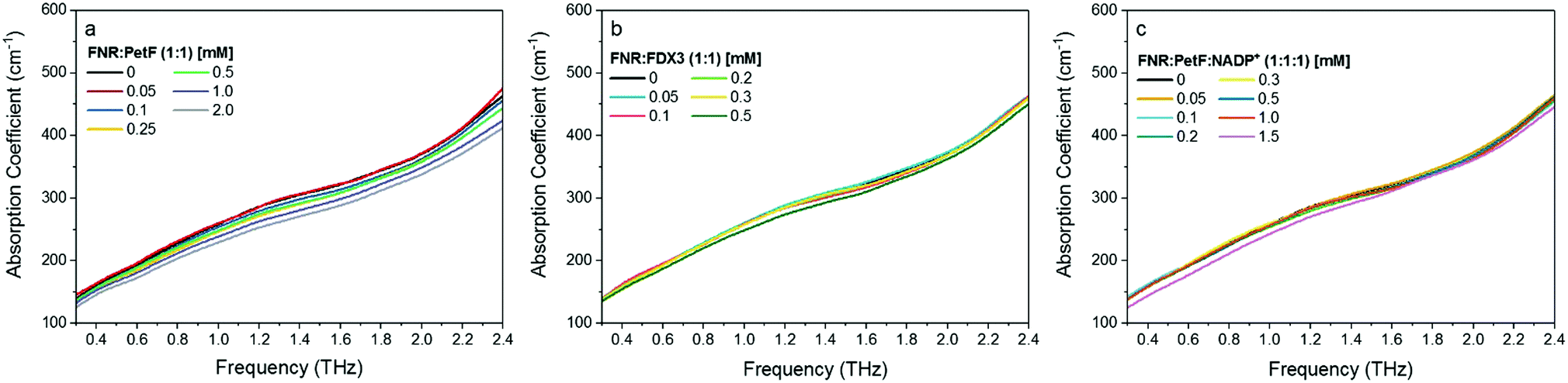 | ||
| Fig. 4 THz-TDS absorption spectra of aqueous solutions at pH 8 of equimolar mixtures of (a) FNR:PetF (b) FNR:FDX3 and (c) FNR:PetF:NADP+. The error of each measurement is on average 0.6 cm−1. The absorption coefficient decreases with increasing protein concentration, consistent with decreasing water content. | ||
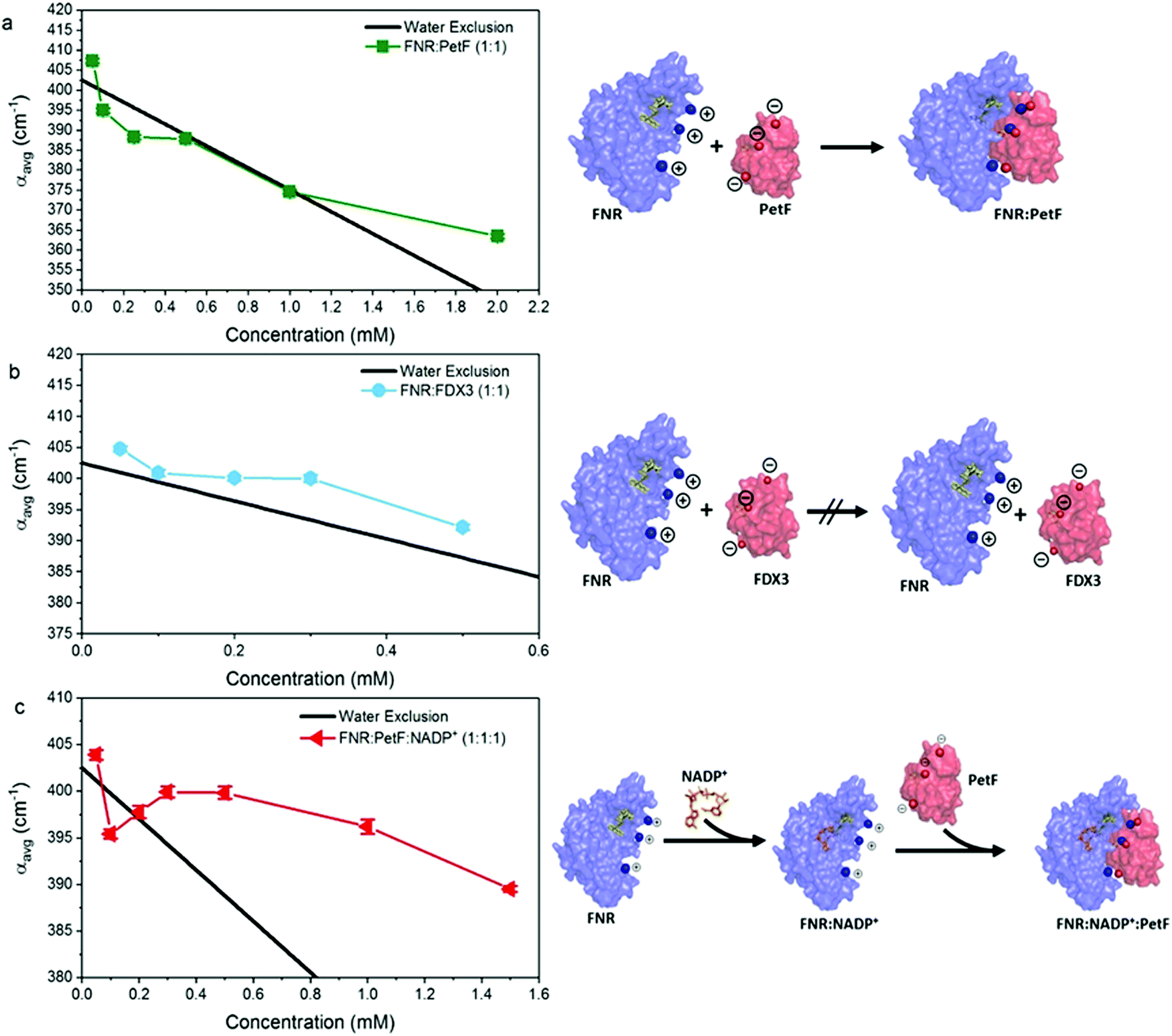 | ||
| Fig. 5 The average absorption coefficient (αavg) in the 1.75–2.5 THz range of aqueous solutions of (a) FNR:PetF (b) FNR:FDX3 and (c) FNR:PetF:NADP+ as a function of concentration. See Fig. S4 in ESI† for other frequency ranges. The black line in each panel shows the expected decrease in αavg based on exclusion of water equal to the volume of protein in solution. Error bars are on average 0.6 cm−1. The complexation state of proteins in solution is illustrated on the right hand side. | ||
Fig. 5c shows that the αavg of the FNR:PetF:NADP+ ternary complex in aqueous solutions deviates significantly from the expected water exclusion, where an increased αavg is observed for concentrations greater than 0.3 mM. This result is surprising as it is well known that no major changes in surface morphology of the complex occur upon NADP+ binding (Fig. S2, ESI†), thus yielding valuable insight. NADP+ binds within the interior of FNR prior to complexation with PetF (Fig. 5c and Fig. S2, ESI†) and is consequentially solvent inaccessible. Measurements of NADP+ show that it has no influence on the measured THz absorption (Fig. S7 and S8, ESI†), and therefore cannot explain the observed increase in absorption. Thus, we propose binding of the NADP+ alters the charge distribution at the surface of the complex, which consequently influences hydration water.
Evaluation of the complex dielectric response of the protein solutions (see ESI† for details) reports on the water solvent dynamics, specifically the time scale of fluctuations in the hydrogen-bonding network. Here, the complex dielectric response was modeled with a double Debye function,52 which reveals a slow relaxation process, τ1, associated to the cooperative rearrangement of the hydrogen bonded network, and a fast relaxation process, τ2, assigned to the rotational modes of individual polar water molecules.53,54Fig. 6 shows values of τ1 determined for all protein solutions (see Fig. S9, ESI† for τ2). For solutions of FNR and PetF (Fig. 6a), τ1 is observed to increase from the initial 7.0 ps value at 0 mM up to 9 ps and 8 ps, respectively, with increasing protein concentration. This trend is consistent with MD simulations that found water molecules in the hydration shell of proteins have significantly slower reorientation times than pure, bulk water.26,40,42,52 The slower relaxation times observed for FNR relative to PetF are indicative of its predominantly hydrophobic nature, in which water molecules near the hydrophobic surface will form stronger hydrogen-bonds with neighboring water molecules to minimize interaction with the hydrophobic surface which ultimately results in retarded dynamics.
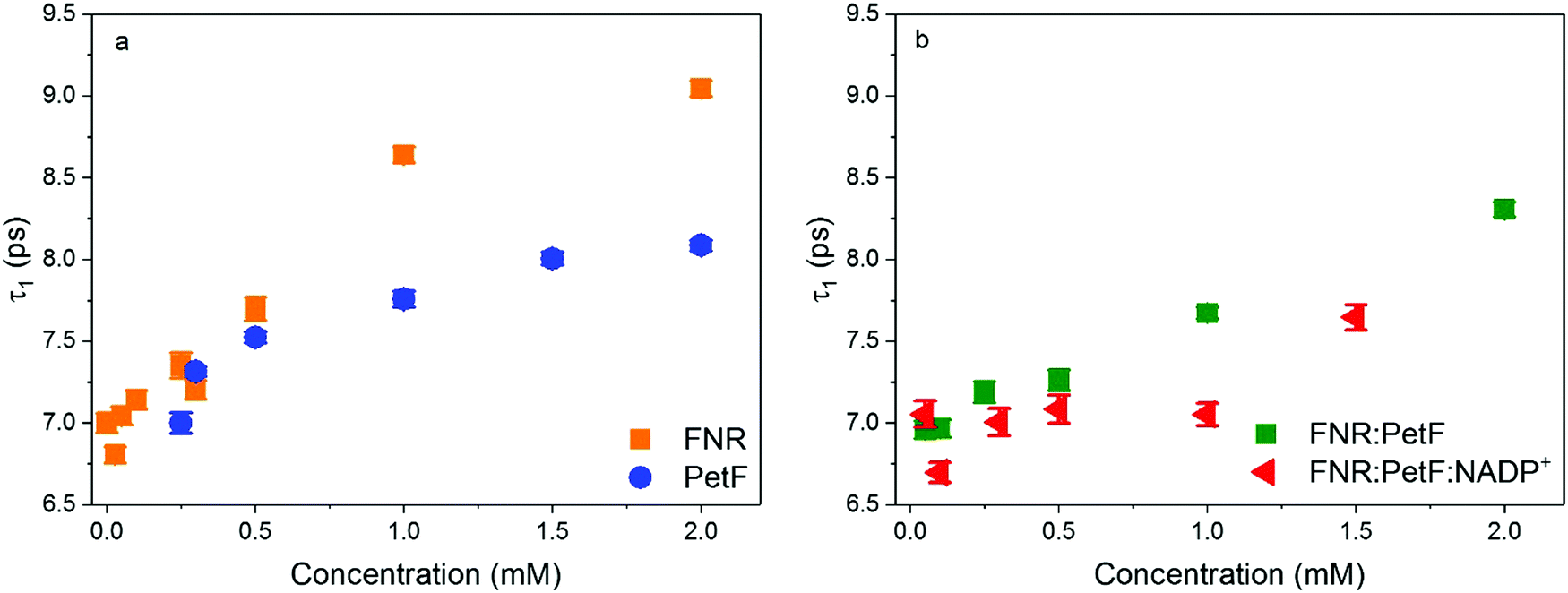 | ||
| Fig. 6 Relaxation time constants τ1 of aqueous solutions of (a) FNR and PetF (b) FNR:PetF and FNR:PetF:NADP+. | ||
Similarly, an increase in τ1 with increasing protein concentration is observed for the transient FNR:PetF complex (Fig. 6b). In contrast, the presence of the substrate NADP+ does not initially affect the relaxation dynamics (up to 0.5 mM). For the FNR:PetF:NADP+ complex the values of τ1 remain relatively constant around 7.0 ps until a concentration of 1.5 mM, where a sudden increase to 7.6 ps occurs. The relaxation dynamics for the ternary complex are similar to pure bulk water while the binary complex has retarded dynamics.
Changes in the solvent dynamics upon complex formation indicate the solvent contributes to changes in the free energy of the system (ΔG = ΔH − TΔS). Water molecules tightly bound to the protein surfaces have the potential to act as an entropic reservoir to offset the enthalpic and entropic costs of forming the complex. FNR and PetF solutions show retarded water dynamics stemming from water molecules tightly bound to charged surface residues. As illustrated in Fig. 7, these tightly bound water molecules (shown in blue) must be displaced and released into the bulk phase in order to favor binding of PetF to the pre-formed FNR:NADP+ complex. The bulk-like solvent dynamics observed for the ternary complex indicate that water molecules within its hydration shell have a low frequency response which is similar to that of pure water (i.e. bulk-like water), meaning that binding of the complex in the presence of the NADP+ is entropically favorable. The increased THz absorption upon embedding of NADP+ into its binding pocket is on the other hand an indication of a change of the surface charge distribution due to binding. For a binding reaction the overall change of ΔG = ΔH − TΔS is decisive. Whereas most of the time there is a focus on ΔH, the magnitude of TΔS is mostly of the same size. Here, we show indications for a change in solvent dynamics which favors ternary complex formation. Our results suggest that the solvent is a vital participant in the electron transfer process, where changed solvent dynamics upon binding of the substrate NADP+ will most likely serve as a prompt for the following step in the redox reaction. In electron transfer processes, dissociation of the transient complex after electron transfer to the substrate is usually the rate-limiting step.55–57 The bulk-like solvent dynamics observed here upon NADP+ binding is proposed to help facilitate dissociation of the complex so that the formed NADPH molecule can be released after the electron transfer.
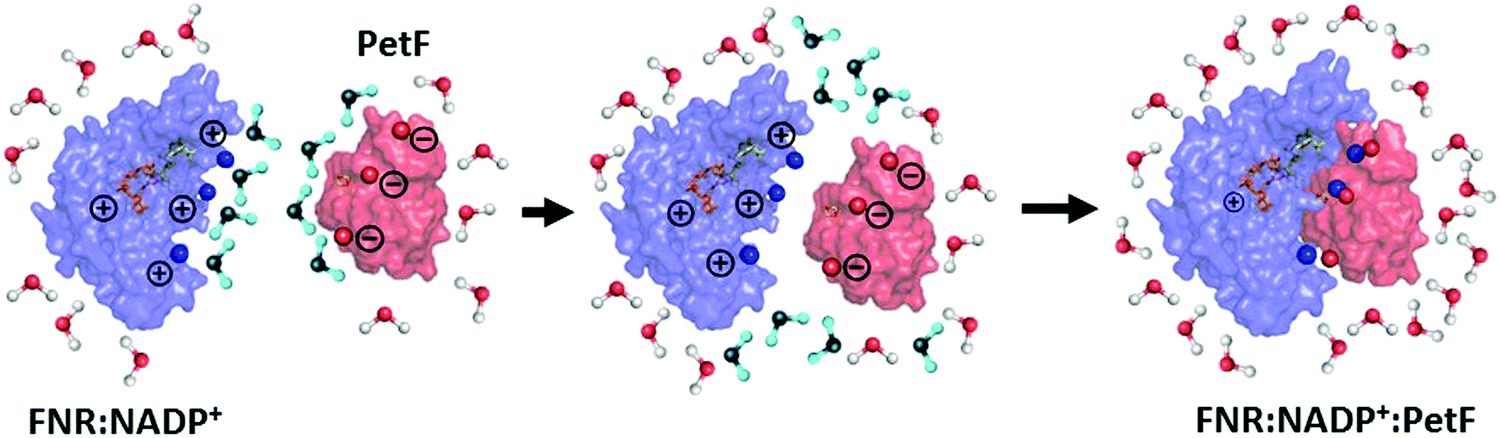 | ||
| Fig. 7 Formation of the FNR:NADP+:PetF ternary complex is entropically favorable. Tightly bound, retarded water molecules (shown in blue) near the charged surface residues on the pre-formed FNR:NADP+ complex and PetF protein are released as the charged surfaces approach each other, resulting in a hydration shell that is more bulk-like in nature. | ||
Conclusions
These results demonstrate that THz-spectroscopy is a powerful technique to study the role of water in redox protein couples in solution that are of biological and biotechnological relevance. While the transient FNR:PetF complex has already been characterized extensively by several biochemical and spectroscopic methods, results here shed light on the role of the solvent. We show the THz response of the solvent is sensitive to surface properties of the individual proteins, with a change in the THz response observed upon protein complexation. Our results further reveal that the interaction of proteins does not solely account to individual amino acids on the protein surface as PetF and FDX3 share a conserved amino acid pattern, which were shown to be relevant for a mutual interaction with FNR. Furthermore, we find that formation of the ternary complex FNR:NADP+:PetF, in which the substrate NADP+ binds first to FNR, results in bulk-like hydration dynamics. The change in solvent dynamics from a more retarded to a bulk-like nature is entropically favorable and facilitates complex formation to trigger electron transfer. In general, solvation dynamics play a decisive factor for the molecular recognition and interaction of redox proteins.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. MH acknowledges ERC Advanced Grant 695437 THz calorimetry.References
- P. Gou, G. T. Hanke, Y. Kimata-Ariga, D. M. Standley, A. Kubo, I. Taniguchi, H. Nakamura and T. Hase, Higher Order Structure Contributes to Specific Differences in Redox Potential and Electron Transfer Efficiency of Root and Leaf Ferredoxins †, Biochemistry, 2006, 45, 14389–14396 CrossRef CAS PubMed.
- M. Winkler, A. Hemschemeier, J. Jacobs, S. Stripp and T. Happe, Multiple ferredoxin isoforms in Chlamydomonas reinhardtii – Their role under stress conditions and biotechnological implications, Eur. J. Cell Biol., 2010, 89, 998–1004 CrossRef CAS PubMed.
- H. Kameda, K. Hirabayashi, K. Wada and K. Fukuyama, Mapping of Protein-Protein Interaction Sites in the Plant-Type [2Fe-2S] Ferredoxin, PLoS One, 2011, 6, e21947 CrossRef CAS PubMed.
- A. Hemschemeier and T. Happe, Alternative photosynthetic electron transport pathways during anaerobiosis in the green alga Chlamydomonas reinhardtii, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 919–926 CrossRef CAS PubMed.
- K. Fukuyama, Structure and Function of Plant-Type Ferredoxins, Photosynth. Res., 2004, 81, 289–301 CrossRef CAS PubMed.
- E. A. Peden, M. Boehm, D. W. Mulder, R. Davis, W. M. Old, P. W. King, M. L. Ghirardi and A. Dubini, Identification of Global Ferredoxin Interaction Networks in Chlamydomonas reinhardtii, J. Biol. Chem., 2013, 288, 35192–35209 CrossRef CAS PubMed.
- J. Noth, D. Krawietz, A. Hemschemeier and T. Happe, Pyruvate:Ferredoxin Oxidoreductase Is Coupled to Light-independent Hydrogen Production in Chlamydomonas reinhardtii, J. Biol. Chem., 2013, 288, 4368–4377 CrossRef CAS PubMed.
- A. Sawyer and M. Winkler, Evolution of Chlamydomonas reinhardtii ferredoxins and their interactions with [FeFe]-hydrogenases, Photosynth. Res., 2017, 134, 307–316 CrossRef CAS PubMed.
- S. Jayashree, P. Murugavel, R. Sowdhamini and N. Srinivasan, Interface residues of transient protein-protein complexes have extensive intra-protein interactions apart from inter-protein interactions, Biol. Direct, 2019, 14, 1 CrossRef CAS PubMed.
- N. Carrillo and E. A. Ceccarelli, Open questions in ferredoxin-NADP+ reductase catalytic mechanism, Eur. J. Biochem., 2003, 270, 1900–1915 CrossRef CAS PubMed.
- J. R. Peregrina, B. Herguedas, J. A. Hermoso, M. Martínez-Júlvez and M. Medina, Protein Motifs Involved in Coenzyme Interaction and Enzymatic Efficiency in Anabaena Ferredoxin-NADP+ Reductase, Biochemistry, 2009, 48, 3109–3119 CrossRef CAS PubMed.
- C. J. Batie and H. Kamin, Electron Transfer by Ferredoxin:NADP+ Reductase, J. Biol. Chem., 1984, 259, 11976–11985 CAS.
- J. A. Hermoso, T. Mayoral, M. Faro, C. Gómez-Moreno, J. Sanz-Aparicio and M. Medina, Mechanism of Coenzyme Recognition and Binding Revealed by Crystal Structure Analysis of Ferredoxin–NADP+ Reductase Complexed with NADP+, J. Mol. Biol., 2002, 319, 1133–1142 CrossRef CAS PubMed.
- P. N. Palma, B. Lagoutte, L. Krippahl, J. J. G. Moura and F. Guerlesquin, Synechocystis ferredoxin/ferredoxin-NADP+-reductase/NADP+ complex: Structural model obtained by NMR-restrained docking, FEBS Lett., 2005, 579, 4585–4590 CrossRef CAS PubMed.
- G. Kurisu, M. Kusunoki, E. Katoh, T. Yamazaki, K. Teshima, Y. Onda, Y. Kimata-Ariga and T. Hase, Structure of the electron transfer complex between ferredoxin and ferredoxin-NADP+ reductase, Nat. Struct. Biol., 2001, 8, 5 CrossRef PubMed.
- K. Wiegand, M. Winkler, S. Rumpel, D. Kannchen, S. Rexroth, T. Hase, C. Farès, T. Happe, W. Lubitz and M. Rögner, Rational redesign of the ferredoxin-NADP+-oxido-reductase/ferredoxin-interaction for photosynthesis-dependent H2-production, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 253–262 CrossRef CAS PubMed.
- S. Rumpel, J. F. Siebel, C. Farès, J. Duan, E. Reijerse, T. Happe, W. Lubitz and M. Winkler, Enhancing hydrogen production of microalgae by redirecting electrons from photosystem I to hydrogenase, Energy Environ. Sci., 2014, 7, 3296–3301 RSC.
- S. Ebbinghaus, S. J. Kim, M. Heyden, X. Yu, U. Heugen, M. Gruebele, D. M. Leitner and M. Havenith, An extended dynamical hydration shell around proteins, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20749–20752 CrossRef CAS PubMed.
- P. Ball, Water as an Active Constituent in Cell Biology, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- J. Wang, S. M. Buck and Z. Chen, Sum Frequency Generation Vibrational Spectroscopy Studies on Protein Adsorption, J. Phys. Chem. B, 2002, 106, 11666–11672 CrossRef CAS.
- K. Engelhardt, W. Peukert and B. Braunschweig, Vibrational sum-frequency generation at protein modified air–water interfaces: Effects of molecular structure and surface charging, Curr. Opin. Colloid Interface Sci., 2014, 19, 207–215 CrossRef CAS.
- K. Meister, C. J. Moll, S. Chakraborty, B. Jana, A. L. DeVries, H. Ramløv and H. J. Bakker, Molecular structure of a hyperactive antifreeze protein adsorbed to ice, J. Chem. Phys., 2019, 150, 131101 CrossRef CAS PubMed.
- G. R. Bell, C. D. Bain and R. N. Ward, Sum-frequency vibrational spectroscopy of soluble surfactants at the air/water interface, Faraday Trans., 1996, 92, 515 RSC.
- D. E. Gragson, B. M. McCarty and G. L. Richmond, Surfactant/Water Interactions at the Air/Water Interface Probed by Vibrational Sum Frequency Generation, J. Phys. Chem., 1996, 100, 14272–14275 CrossRef CAS.
- P. B. Miranda, Q. Du and Y. R. Shen, Interaction of water with a fatty acid Langmuir film, Chem. Phys. Lett., 1998, 286, 1–8 CrossRef CAS.
- M. Heyden and M. Havenith, Combining THz spectroscopy and MD simulations to study protein-hydration coupling, Methods, 2010, 52, 74–83 CrossRef CAS PubMed.
- S. Ebbinghaus, K. Meister, B. Born, A. L. DeVries, M. Gruebele and M. Havenith, Antifreeze Glycoprotein Activity Correlates with Long-Range Protein−Water Dynamics, J. Am. Chem. Soc., 2010, 132, 12210–12211 CrossRef CAS PubMed.
- M. Grossman, B. Born, M. Heyden, D. Tworowski, G. B. Fields, I. Sagi and M. Havenith, Correlated structural kinetics and retarded solvent dynamics at the metalloprotease active site, Nat. Struct. Mol. Biol., 2011, 18, 1102–1108 CrossRef CAS PubMed.
- V. Conti Nibali and M. Havenith, New Insights into the Role of Water in Biological Function: Studying Solvated Biomolecules Using Terahertz Absorption Spectroscopy in Conjunction with Molecular Dynamics Simulations, J. Am. Chem. Soc., 2014, 136, 12800–12807 CrossRef CAS PubMed.
- Y. Xu, A. Bäumer, K. Meister, C. G. Bischak, A. L. DeVries, D. M. Leitner and M. Havenith, Protein–water dynamics in antifreeze protein III activity, Chem. Phys. Lett., 2016, 647, 1–6 CrossRef CAS.
- H. Wirtz, S. Schäfer, C. Hoberg and M. Havenith, Differences in Hydration Structure Around Hydrophobic and Hydrophilic Model Peptides Probed by THz Spectroscopy, J. Infrared, Millimeter, Terahertz Waves, 2018, 39, 816–827 CrossRef CAS.
- C. Hoberg, P. Balzerowski and M. Havenith, Integration of a rapid scanning technique into THz time-domain spectrometers for nonlinear THz spectroscopy measurements, AIP Adv., 2019, 9, 035348 CrossRef CAS PubMed.
- K. Aoki, K. Shiraki and T. Hattori, Observation of salt effects on hydration water of lysozyme in aqueous solution using terahertz time-domain spectroscopy, Appl. Phys. Lett., 2013, 103, 173704 CrossRef.
- J. Xu, K. W. Plaxco and S. J. Allen, Collective Dynamics of Lysozyme in Water: Terahertz Absorption Spectroscopy and Comparison with Theory, J. Phys. Chem. B, 2006, 110, 24255–24259 CrossRef CAS PubMed.
- N. Q. Vinh, S. J. Allen and K. W. Plaxco, Dielectric Spectroscopy of Proteins as a Quantitative Experimental Test of Computational Models of Their Low-Frequency Harmonic Motions, J. Am. Chem. Soc., 2011, 133, 8942–8947 CrossRef CAS PubMed.
- J. W. Bye, S. Meliga, D. Ferachou, G. Cinque, J. A. Zeitler and R. J. Falconer, Analysis of the Hydration Water around Bovine Serum Albumin Using Terahertz Coherent Synchrotron Radiation, J. Phys. Chem. A, 2014, 118, 83–88 CrossRef CAS PubMed.
- H. Vondracek, J. Dielmann-Gessner, W. Lubitz, M. Knipp and M. Havenith, THz absorption spectroscopy of solvated β-lactoglobulin, J. Chem. Phys., 2014, 141, 22D534 CrossRef PubMed.
- Y. Kimata-Ariga, H. Kubota-Kawai, Y.-H. Lee, N. Muraki, T. Ikegami, G. Kurisu and T. Hase, Concentration-dependent oligomerization of cross-linked complexes between ferredoxin and ferredoxin–NADP+ reductase, Biochem. Biophys. Res. Commun., 2013, 434, 867–872 CrossRef CAS PubMed.
- F. Novelli, S. Ostovar Pour, J. Tollerud, A. Roozbeh, D. R. T. Appadoo, E. W. Blanch and J. A. Davis, Time-Domain THz Spectroscopy Reveals Coupled Protein–Hydration Dielectric Response in Solutions of Native and Fibrils of Human Lysozyme, J. Phys. Chem. B, 2017, 121, 4810–4816 CrossRef CAS PubMed.
- E. Duboué-Dijon and D. Laage, Comparative study of hydration shell dynamics around a hyperactive antifreeze protein and around ubiquitin, J. Chem. Phys., 2014, 141, 22D529 CrossRef PubMed.
- D. Laage, G. Stirnemann and J. T. Hynes, Why Water Reorientation Slows without Iceberg Formation around Hydrophobic Solutes, J. Phys. Chem. B, 2009, 113, 2428–2435 CrossRef CAS PubMed.
- D. Laage, T. Elsaesser and J. T. Hynes, Water Dynamics in the Hydration Shells of Biomolecules, Chem. Rev., 2017, 117, 10694–10725 CrossRef CAS PubMed.
- I. B. Kovalenko, A. N. Diakonova, A. M. Abaturova, G. Y. Riznichenko and A. B. Rubin, Direct computer simulation of ferredoxin and FNR complex formation in solution, Phys. Biol., 2010, 7, 026001 CrossRef CAS PubMed.
- A. N. Diakonova, S. S. Khrushchev, I. B. Kovalenko, G. Y. Riznichenko and A. B. Rubin, Influence of pH and ionic strength on electrostatic properties of ferredoxin, FNR, and hydrogenase and the rate constants of their interaction, Phys. Biol., 2016, 13, 056004 CrossRef CAS PubMed.
- J. Sun, G. Niehues, H. Forbert, D. Decka, G. Schwaab, D. Marx and M. Havenith, Understanding THz Spectra of Aqueous Solutions: Glycine in Light and Heavy Water, J. Am. Chem. Soc., 2014, 136, 5031–5038 CrossRef CAS PubMed.
- A. Baumer, Terahertz-Spektroscopie solvatisierter Biomoleküle, Ruhr Universitat Bochum, 2017 Search PubMed.
- A. Esser, H. Forbert, F. Sebastiani, G. Schwaab, M. Havenith and D. Marx, Hydrophilic Solvation Dominates the Terahertz Fingerprint of Amino Acids in Water, J. Phys. Chem. B, 2018, 122, 1453–1459 CrossRef CAS PubMed.
- M. G. Campo, Molecular dynamics simulation of glycine zwitterion in aqueous solution, J. Chem. Phys., 2006, 125, 114511 CrossRef PubMed.
- A. Bäumer, Terahertz-Spektroscopie solvatisierter Biomoleküle, Ruhr Universität Bochum, 2017 Search PubMed.
- T. Mayoral, M. Martínez-Júlvez, I. Pérez-Dorado, J. Sanz-Aparicio, C. Gómez-Moreno, M. Medina and J. A. Hermoso, Structural analysis of interactions for complex formation between Ferredoxin-NADP+ reductase and its protein partners, Proteins: Struct., Funct., Bioinf., 2005, 59, 592–602 CrossRef CAS PubMed.
- J. K. Hurley, G. Tollin, M. Medina and C. Gómez-Moreno, in Photosystem I, ed. J. H. Golbeck, Springer Netherlands, Dordrecht, 2006, vol. 24, pp. 455–476 Search PubMed.
- N. Q. Vinh, M. S. Sherwin, S. J. Allen, D. K. George, A. J. Rahmani and K. W. Plaxco, High-precision gigahertz-to-terahertz spectroscopy of aqueous salt solutions as a probe of the femtosecond-to-picosecond dynamics of liquid water, J. Chem. Phys., 2015, 142, 164502 CrossRef CAS PubMed.
- J. T. Kindt and C. A. Schmuttenmaer, Far-Infrared Dielectric Properties of Polar Liquids Probed by Femtosecond Terahertz Pulse Spectroscopy †, J. Phys. Chem., 1996, 100, 10373–10379 CrossRef CAS.
- T. Sato and R. Buchner, Dielectric Relaxation Processes in Ethanol/Water Mixtures, J. Phys. Chem. A, 2004, 108, 5007–5015 CrossRef CAS.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Climbing Nitrogenase: Toward a Mechanism of Enzymatic Nitrogen Fixation, Acc. Chem. Res., 2009, 42, 609–619 CrossRef CAS PubMed.
- Q. Bashir, S. Scanu and M. Ubbink, Dynamics in electron transfer protein complexes: Dynamics in electron transfer protein complexes, FEBS J., 2011, 278, 1391–1400 CrossRef CAS PubMed.
- T.-Y. Lin, T. Werther, J.-H. Jeoung and H. Dobbek, Suppression of Electron Transfer to Dioxygen by Charge Transfer and Electron Transfer Complexes in the FAD-dependent Reductase Component of Toluene Dioxygenase, J. Biol. Chem., 2012, 287, 38338–38346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00267d |
| This journal is © the Owner Societies 2020 |