
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transient IR spectroscopy identifies key interactions and unravels new intermediates in the photocycle of a bacterial phytochrome†
Joachim
Kübel‡
 a,
Manoop
Chenchiliyan‡
a,
Saik Ann
Ooi
a,
Emil
Gustavsson
a,
Linnéa
Isaksson
a,
Valentyna
Kuznetsova
b,
Janne A.
Ihalainen
b,
Sebastian
Westenhoff
*a and
Michał
Maj
*a
a,
Manoop
Chenchiliyan‡
a,
Saik Ann
Ooi
a,
Emil
Gustavsson
a,
Linnéa
Isaksson
a,
Valentyna
Kuznetsova
b,
Janne A.
Ihalainen
b,
Sebastian
Westenhoff
*a and
Michał
Maj
*a
aDepartment of Chemistry and Molecular Biology, University of Gothenburg, Gothenburg 40530, Sweden. E-mail: michal.maj@gu.se; westenho@chem.gu.se
bNanoscience Center, Department of Biological and Environmental Science, University of Jyväskylä, Jyväskylä 40014, Finland
First published on 3rd March 2020
Abstract
Phytochromes are photosensory proteins in plants, fungi, and bacteria, which detect red- and far-red light. They undergo a transition between the resting (Pr) and photoactivated (Pfr) states. In bacterial phytochromes, the Pr-to-Pfr transition is facilitated by two intermediate states, called Lumi-R and Meta-R. The molecular structures of the protein in these states are not known and the molecular mechanism of photoconversion is not understood. Here, we apply transient infrared absorption spectroscopy to study the photocycle of the wild-type and Y263F mutant of the phytochrome from Deinococcus radiodurans (DrBphP) from nano- to milliseconds. We identify two sequentially forming Lumi-R states which differ in the local structure surrounding the carbonyl group of the biliverdin D-ring. We also find that the tyrosine at position 263 alters local structure and dynamics around the D-ring and causes an increased rate of Pfr formation. The results shed new light on the mechanism of light-signalling in phytochrome proteins.
1 Introduction
Phytochromes are photoreceptor proteins that regulate various physiological processes in plants, bacteria, and fungi.1–5 Their photosensory module (PSM) comprises a Per/Arnt/Sim (PAS), a cGMP phosphodiesterase, adenylyl cyclase, FhlA (GAF), and a phytochrome-specific GAF-related (PHY) domain. Bacterial phytochromes carry a biliverdin chromophore, which contains four rings (A–D) embedded in the chromophore binding domain (CBD), formed by the PAS and GAF domains. The connection between PHY and GAF domains includes a structurally unique hairpin loop, often referred to as the tongue.6 Phytochromes typically photoconvert between two stable conformers, termed as Pr and Pfr based on their respective absorption maxima in the red and far-red spectral region. Generally, the Pr is the resting state, and the Pfr is the biologically active state, but in some bacterial phytochromes, Pfr is the dark-adapted resting state.7,8 The photoconversion is initiated by the light absorption of the chromophore that induces photoisomerization of the C15![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C16 methine double bond between the C and D pyrrole ring, which triggers the reversible photochromic switch between the Pr and Pfr states.9,10 Thus, by cycling between Pr and Pfr states, phytochromes act as a light-activated molecular switch that coordinates key biological processes, essential for the growth and sustainability of many organisms on Earth.
C16 methine double bond between the C and D pyrrole ring, which triggers the reversible photochromic switch between the Pr and Pfr states.9,10 Thus, by cycling between Pr and Pfr states, phytochromes act as a light-activated molecular switch that coordinates key biological processes, essential for the growth and sustainability of many organisms on Earth.
Upon photoexcitation, the D-ring in the chromophore undergoes Z to E isomerization that triggers a series of subsequent changes in the protein structure.11 Previous studies show that, in bacterial phytochromes, the photocycle involves three intermediate states named Lumi-R, Meta-Ra and Meta-Rc.12 The first intermediate, Lumi-R, forms from a short-lived excited state within hundreds of picoseconds.13,14 In tens of microseconds, the Lumi-R thermally decays to the Meta-Ra state. The subsequent thermal relaxation to the Meta-Rc, which involves transient deprotonation of the chromophore, occurs within a few milliseconds. Finally, in hundreds of milliseconds, the Pfr photoproduct is formed. A re-protonation of the chromophore occurs from the external medium in this last step.15
It is known that several amino acid residues surrounding the chromophore in the binding pocket (Fig. 1) and at the GAF/PHY-tongue interface are highly conserved in phytochromes.17–19 The collective interactions between these conserved residues as well as water molecules around the chromophore are critical for the phytochrome photochemistry, photoisomerization, and photoconversion.14,20–22 Among various conserved residues, the Y263 resides close to the chromophore at the GAF/PHY-tongue interface in bacterial phytochromes. Y263 has been proposed to play a significant role in Pr to Pfr photointerconversion.23–26 It is one of the integral residues that form a hydrophobic cavity around the pyrrole D-ring.25 Through the interaction with the D207–R466 salt bridge, the Y263 residue stabilizes the interaction between the tongue and the GAF domain that facilitates the phytochrome to maintain its Pr conformation in the dark.16 In a recent study, we replaced the tyrosine at position 263 with phenylalanine (Y263F) in the phytochrome from the bacterium Deinococcus radiodurans and solved the crystal structure of the PSM.27 The overall fold of the phytochrome was retained by the mutation. However, the mutation resulted in the tongue adopting an α-helical conformation regardless of the chromophore state, which suggested an uncoupling between the chromophore conformation and the protein structure. By combining steady-state FTIR and fluorescence spectroscopy with the X-ray scattering results, we further proposed the possible existence of an alternative signaling routes to the tongue to accomplish the photocycle. In another recent report, we documented the photocycle pathways of the wild type (WT) DrBphP phytochrome using step-scan FTIR spectroscopy.28
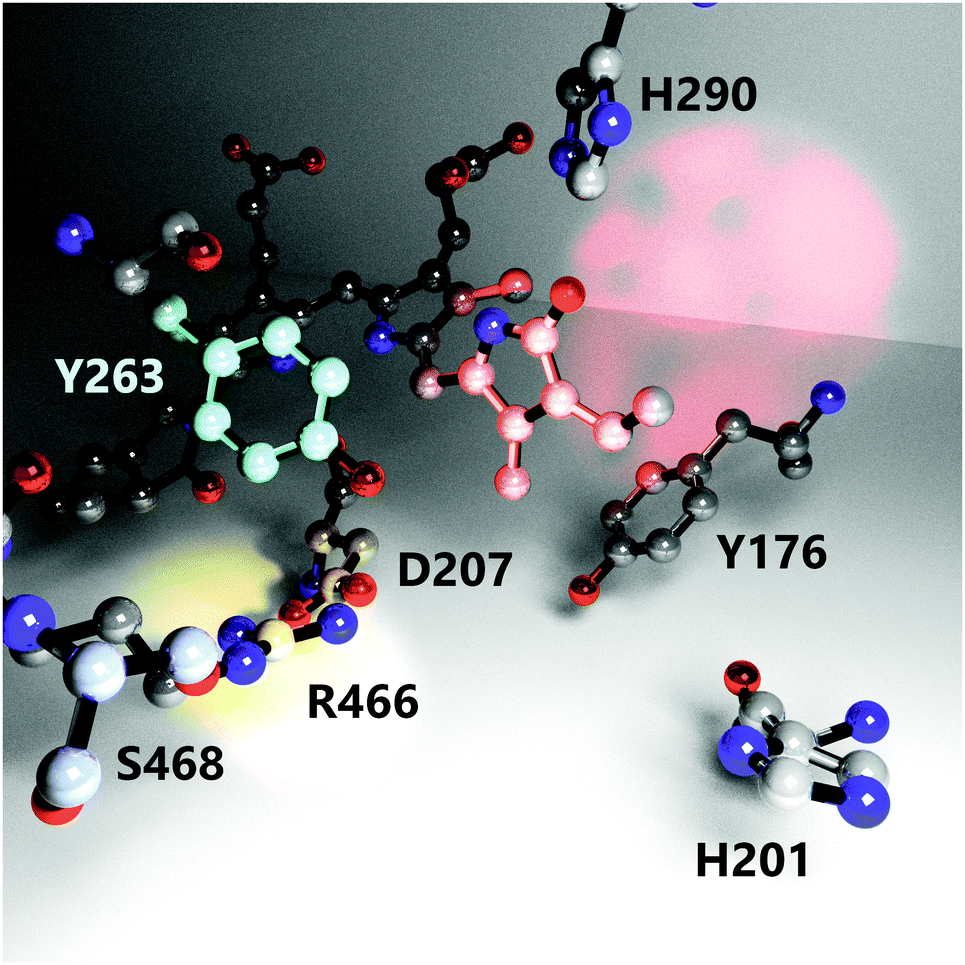 | ||
| Fig. 1 The nearest environment of the biliverdin chromophore D-ring showing positioning and orientation of relevant amino acids involved in the photoconversion process. Red, cyan and yellow light spots highlight the D-ring, residue Y263 and the D207–R466 salt bridge, respectively. The structure is reproduced from PDB ID: 4o0p.16 Hydrogens, protein backbone, and water molecules are omitted for clarity. | ||
To unravel the exact photocycle routes of the DrBphP and its Y263F mutant, we employed ns Vis pump – fs mid-IR probe spectroscopy that follows the structural changes on the nanosecond to millisecond timescales. There are many studies on phytochromes in which transient IR techniques are applied; however, to the best of our knowledge, none of these achieved to follow the complete photocycle routes from Pr to Pfr states.14,29–32 The majority of the previous spectroscopic studies on phytochromes were carried out using either femtosecond (fs) laser spectroscopy,13,14,30,33 (resonance) Raman,10,11,34,35 or step-scan FTIR methods.28 These techniques each have a different set of advantages, but the common problem is their rather limited experimental time window. In fs experiments, the delay between the visible and infra-red pulses is usually controlled by a mechanical translation stage, which allows reaching the relative delay of several nanoseconds, at most. Implementations with two synchronized amplifiers extend the maximum delay by orders of magnitude, but are rather sophisticated and costly.33 On the other hand, the usual time window of step-scan FTIR measurements lies between tens of nanoseconds and several milliseconds, but recording data over several orders of magnitude is experimentally demanding and time-consuming.36,37 Consequently, the photochemical events that occur after tens to hundreds of ns after the excitation are often not resolved. The experimental setup used in our study allows us to cover a much broader range of timescales. Furthermore, the present pump–probe spectra have a spectral resolution of about 5.5 cm−1 which is higher than the spectra reported in the recent step-scan study. The higher resolution in our study allows us resolve the spectral features of each intermediate in the photocycle.28 All the spectra were recorded at room temperature, which is fundamentally important for understanding the biologically significant dynamics within the intermediate states of the photocycle.35,38
The experimental data identify a new state, which forms before the previously identified Lumi-R state in the photocycle. We name the new state early Lumi-R. The formation of this intermediate is sequential and, thus, the state must be an inherent part of the photocycle. From time-resolved infrared spectra we identify that the biggest structural changes between the early and late Lumi-R states are located in close proximity to the chromophore D-ring.
Our spectroscopic investigation of the Y263F mutant shows that the Y263 modifies the local structure and dynamics around the D-ring and has influence on the rate of Pfr formation.
2 Materials and methods
2.1 Expression of the WT and mutant phytochromes
The expression plasmids and the protocols for expression and purification of CBD-PHY have been published in detail before.23,27 In brief, the WT and Y263F mutant were expressed in Escherichia coli strain BL21 (DE3). The biliverdin chromophore was incorporated in all protein lysates overnight on ice. The uniformly isotope-labeled protein was expressed in a minimal medium supplemented with 13C-enriched glucose and 15NH4Cl. The proteins were purified using affinity chromatography (HisTrap HP 5 ml, GE Healthcare), followed by size exclusion chromotography (Superdex 200, GE Healthcare) using Äkta pure protein purification system (GE Healthcare). The purified proteins were eluted in 30 mM Tris–HCl (pH 8), concentrated to 10 mg ml−1, and flash frozen. All purifications were performed in the dark.2.2 Experimental setup
The steady-state visible absorption spectra were measured using a commercial UV/Vis spectrometer (Hitachi U-2910).The transient IR spectra were measured using a nanosecond Vis pump – femtosecond mid-IR probe spectroscopy setup. The samples were excited at 660 nm by a Nd:YAG Q-switched nanosecond laser (Ekspla NT342B, 20 Hz, (3–5) ns). The fs mid-IR probe laser pulses at 5 kHz were generated using noncollinear difference frequency generation (NDFG) of the signal and idler beams produced by a commercial optical parametric amplifier (Light Conversion TOPAS-Twins, 120 fs) pumped by the output of a regenerative amplifier (Spitfire Ace, Spectra Physics). The central wavelength of the mid-IR probe was either 5950 nm, or 6350 nm and the overlapping spectral window were stitched during the data processing to have complete IR spectra presented here. The mid-IR probe and a corresponding reference beam are obtained using a BaF2 wedge. Probe and pump beams, focused to ca. 120 μm and 250 μm, respectively, are overlapped at the sample position, with the reference beam spatially offset. The probe and reference beams, after passing through the sample, were detected by a commercial spectrograph (Horiba i320) equipped with a dual row MCT array with 64 pixels each (Infrared Systems). The pump fluence was set to 4.5 mJ cm−2. In order to recover the protein to the Pr state, we illuminated an area of approximately 3 × 8 mm around the pumped spot with a CW solid state laser at 780 nm for ca. 40 ms prior to each excitation pulse. The fluence was similar or higher than the one of the pump laser and we verified that the molecules returned to the Pr state by minimizing the offset at negative times. To minimize protein degradation the (x,y)-sample stages were moved synchronized with the acquisition. Due to purely electronic signal contributions measured on the detector that depend on the electronic delay (and with intensities similar to those of the light-induced signal), for each 40 pump shots on the sample with the shutter open (with pump light going to the sample), 40 pump shots with the shutter closed (no pump light on the sample) were recorded on the same sample spot.
The spectral resolution is ca. 5.5 cm−1 when using a grating with 75 grooves per mm. The samples at (0.5–1) mm in D2O buffer are placed in a custom sample cell between CaF2 windows separated by a 25 μm Teflon spacer. The absorption bands of water vapour were used for spectral calibration. All measurements were performed at ambient temperature, (295 ± 2) K.
The electronic delay τi between pump and probe lasers is controlled between −50 ns and 150 μs. Longer delays are obtained by calculating signals from the 20 consecutive probe shots after the ns pump pulse with individual delays of
| Δti,n = τi + (n − 1) 200 μs. |
All laser shots are recorded and afterwards processed using home-written Matlab scripts. During data processing the noise reduction algorithm introduced by Ge is applied.39,40
3 Results
3.1 Steady-state UV/Vis absorption of WT CBD-PHY and its Y263F mutant
The visible absorption spectra of the DrBphP WT–PSM and Y263F–PSM phytochromes are presented in Fig. 2. The absorption maximum of the WT protein in the Pr form lays at 700 nm, and becomes red-shifted to 750 nm upon photoconversion. The absorption spectra of the Y263F–PSM mutant are similar to those of the WT protein. The dark-adapted Pr state of the Y263F mutant becomes red-shifted by about 2 nm, whereas the light-induced Pfr spectrum is blue-shifted by ∼10 nm with respect to the WT spectrum.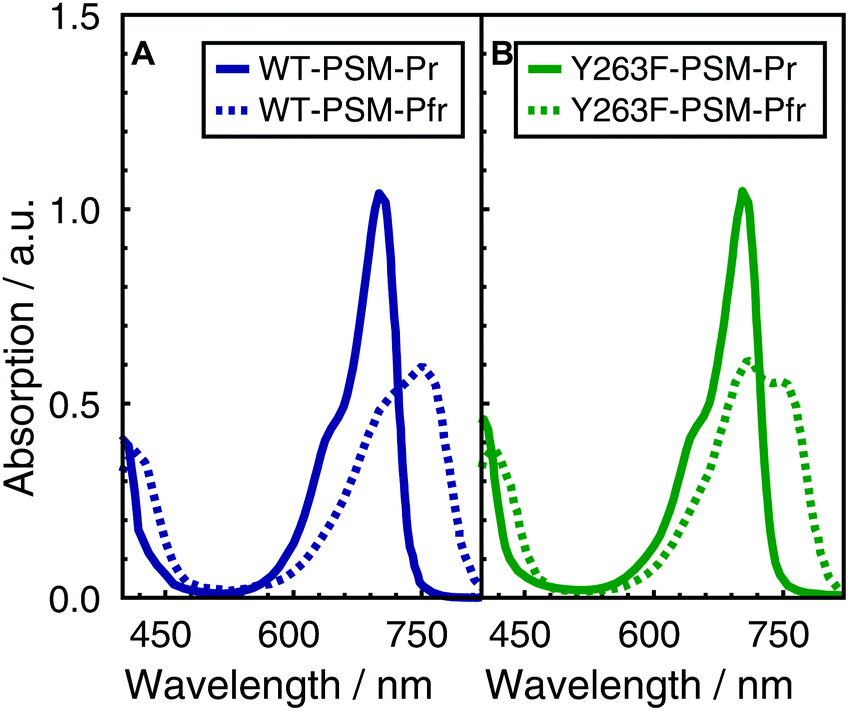 | ||
| Fig. 2 Normalized absorption spectra of WT–PSM (A) and Y263F–PSM (B). The dark-adapted Pr and the light induced Pfr spectra are respectively represented by solid and dotted lines. | ||
3.2 The photocycle routes in WT and Y263F CBD-PHY revealed by ns Vis pump – fs mid-IR probe spectroscopy
To probe the photocycle pathways of the WT and Y263F mutant proteins, transient infrared absorption spectra (Fig. 3) were measured on nano- to millisecond timescales, which covers the complete photocycle from Pr to Pfr. To ensure that the Pfr state is reached within the experimental time window, we directly compare our transient IR data with the Pfr–Pr difference FTIR spectra (Fig. S3, ESI†). The spectra were recorded in the [1470–1740] cm−1 range, which allowed us to simultaneously follow the chromophore-specific structural changes with the conformational transitions in the protein backbone.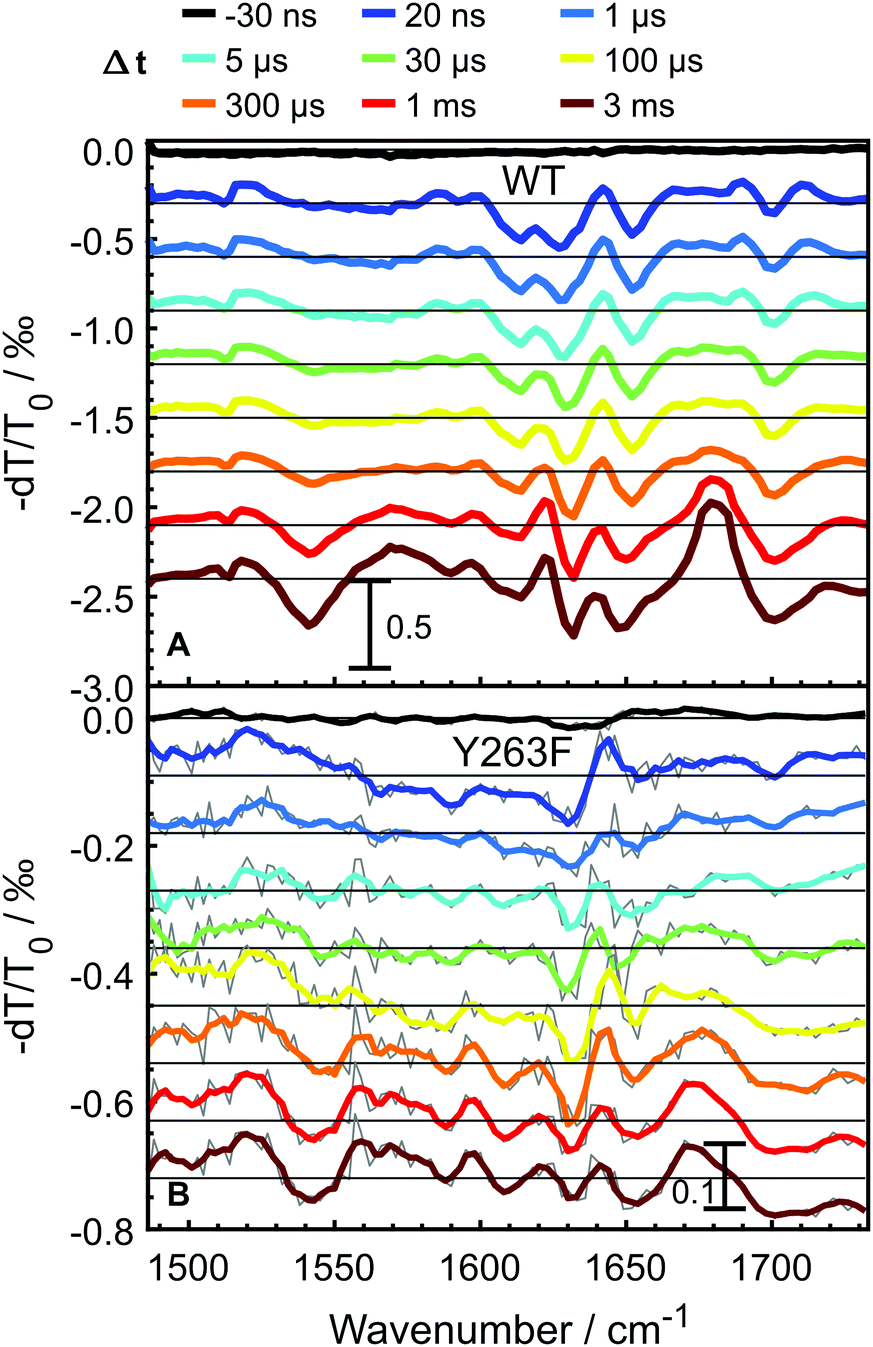 | ||
| Fig. 3 Transient infrared absorption spectra for WT–PSM (A) and Y263F–PSM (B) at selected delay times. The spectra are shifted by offsets of 3 × 10−4 and 9 × 10−5, in panels A and B, respectively. The gray curves in panel B are raw data, while the colored curves are smoothed using a Savitzky–Golay filter (2-order, eight points). | ||
Transient spectra recorded at early delay times, contain mainly contributions from the excited-state of the photoexcited biliverdin chromophore and are fairly similar in shape and intensity for both WT and Y263F mutant. With the decay of the excited state, the signal intensity drops considerably for the wild-type protein; the drop is even more severe in the case of the mutant.
The data were fitted globally using a five-component and four-component sequential scheme for the WT and Y263F mutant, respectively. The resulting species associated differential spectra (SADS) from the fit of the WT–PSM are presented in Fig. 4, and compared with the SADS of the Y263–PSM in Fig. 5. The initial components decaying within the time-resolution of our experiment, that correspond to the excited state, are not shown. The characteristic lifetimes are listed in Table 1. The five component approximation applied for the WT data assumes the existence of four states in the photocycle (excluding the excited state), contrary to the three states reported in the previous studies. The quality of the fit is significantly improved using the four state model (see Fig. S2, ESI†). A new short lived state (of ca. 0.4 μs, black curve in Fig. 4A) is obtained from our analysis, which was not resolved in former studies on DrBphP. The lifetimes of the remaining SADS components, corresponding to the subsequent states in the photocycle, are in good agreement with the earlier reports.6,16,28 We confirm that these three SADS represent the transitions between Lumi-R, Meta-R and Pfr states, respectively.
 | ||
| Fig. 4 Time-resolved infrared absorption spectra on DrBphP WT–PSM. The transient spectra were analyzed globally and the resulting SADS are presented in A along with their characteristic time constants in microseconds. The SADS in black, red, green and blue respectively correspond to the early Lumi-R, late Lumi-R, Meta-R, and Pfr photo-conversion states. The inset shows the evolution of the transient spectra between 1695 cm−1 and 1725 cm−1 to emphasize the distinction of early and late Lumi-R state. The evolution of the normalized populations of each species in the sequential reaction scheme employed in the global analysis is presented in B. | ||
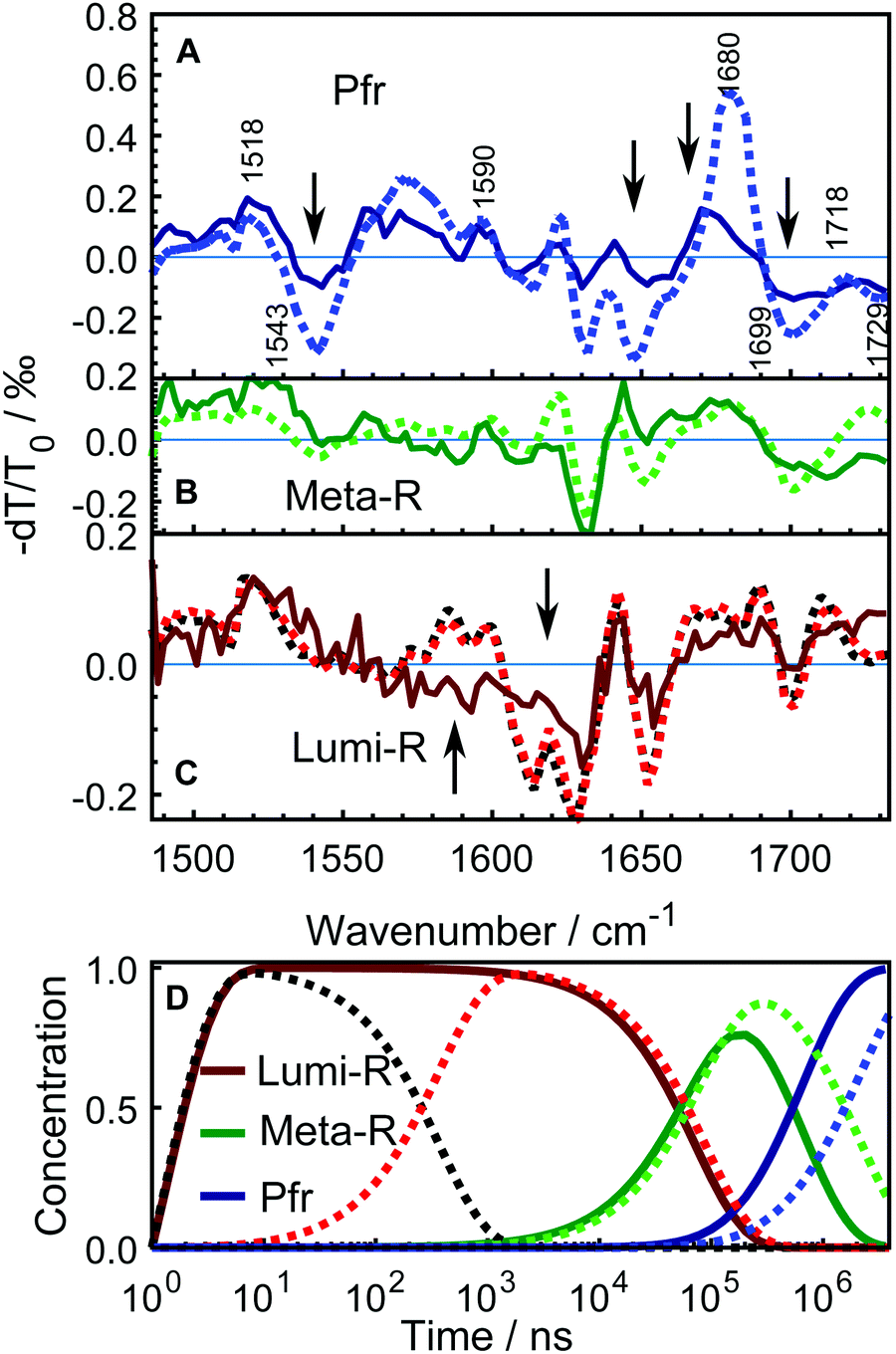 | ||
| Fig. 5 Comparison of the SADS between WT–PSM (dotted curves) and Y263F–PSM (solid curves). The spectra from WT protein that correspond to different photo-conversion states are similarly colored as in Fig. 4. The SADS components presented in panel A and B correspond to the Pfr, Meta-R states. The lone Lumi-R state of the Y263–PSM is compared with the early (black dotted curve) and the late (red dotted curve) Lumi-R states of the WT–PSM in panel C. The major vibrational bands are indicated in A according to the previous assignments.27,28,41–43 The Y263F mutant has similar spectral features in every state as the WT sample except at the spectral regions highlighted by arrows. The mutant signal is scaled a factor of 2.5. Panel D shows the evolution of the normalized populations of each SADS species in the sequential reaction scheme employed in the global analysis for WT–PSM (dotted lines) and Y263F–PSM (solid lines). | ||
The newly resolved extra state has spectral features similar to those of the Lumi-R state except the characteristic chromophore absorption above 1700 cm−1. In view of the similarity between the additional state and the Lumi-R state (red curve), we infer that this extra state is a Lumi-R sub state, and hereafter we name these two state as early (black curve) and late (red curve) Lumi-R states. The presence of the early Lumi-R state in a phytochrome photocycle will be further addressed in the discussion section. The existence of the early and late Lumi-R state is fully reproducible across the data sets and is also confirmed by the studies on the isotope-labeled protein (see Fig. S1, ESI†). The population changes as a function of time are provided in Fig. 4B. No Meta-R sub-states, such as Meta-Ra and Meta-Rc observed in other species of phytochromes, were discerned in the transient IR spectra presented in this report.
Fig. 5 compares the SADS obtained for the Y263F–PSM with the WT counterpart and the characteristic lifetimes of the states from the Y263F mutant are listed in Table 1. The SADS corresponding to the Pfr state (Fig. 5A) matches closely with the steady-state FTIR difference spectrum (Pfr minus Pr,28 and Fig. S3, ESI†), which indicates that the complete photocycle has been resolved in the time window of the experiment. The major vibrational bands in the Pfr state are indicated in Fig. 5A. The spectral bands around 1518 cm−1 (+) and 1543 cm−1 (−) arise due to the amide II vibrations. The bands at 1573 cm−1 (+) and 1590 cm−1 (−) result from the chromophore B–C methine stretches. The peaks at 1680 cm−1 (+)/1699 cm−1(−) and 1718 cm−1 (+)/1729 cm−1 (−) originate from the carbonyl vibrations of the chromophore D and A ring, respectively.
The SADS obtained from the Y263F–PSM mutant exhibit similar spectral features as the WT–PSM in each intermediate state. However, in contrast to the WT protein, we detect only one Lumi-R state.
In the Pfr state, shown in Fig. 5A, four spectral regions, with the most substantial mismatch between the WT and mutant samples are highlighted. A negative signal at 1543 cm−1 is weaker in Y263F–PSM and may correspond to changes of the Amide II.
The peak at 1622 cm−1, which presumably comes from the Amide I vibration of β-sheets, and negative peaks at (1630 and 1650) cm−1, are considerably weaker in Y263F–PSM. The positive peak associated with the chromophore D-ring carbonyl vibrations at 1679 cm−1 in WT–PSM is red-shifted by 6 cm−1 in Y263F–PSM. We also observe some minor differences in the spectra corresponding to the chromophore A ring carbonyl vibrations (≥1700 cm−1).
The spectra of the Meta-R states of WT and Y263F mutant largely agree (Fig. 5B). The features in the range of (1600–1650) cm−1 likely correspond to structural changes in the protein backbone and are not significantly influenced by the mutation. A deviation is the lack of the positive band at 1620 cm−1. The characteristic signal of the D-ring carbonyl in Meta-R at 1730 cm−1 for the WT is not found for the mutant. Instead, a broad positive band is found just below 1680 cm−1. This could indicate that the D-ring carbonyl remains H-bonded in the meta-R state in the Y263F mutant.
A comparison of the spectra between the wild type and Y263F mutant in the first photocycle intermediate, Lumi-R, is presented in Fig. 5C. To simplify the figure, the subtle distinction between the early and late Lumi-R states in WT–PSM is excluded. The amplitude of the spectral band corresponding to the Amide I vibrations of β-sheets, around (1610–1630) cm−1, is considerably weaker in Y263F–PSM compared to the WT–PSM.
The photoinduced positive peak of the WT–PSM chromophore D-ring carbonyl vibrations is broad and at the same position as the bleach of the D-ring carbonyl.28 It can be seen as two peaks at 1690 cm−1 and 1710 cm−1, which mark the wings of the peak. While the D-ring carbonyl bleach signature is clearly visible in the Lumi-R spectrum of the mutant, the broad peaks around that band suggest a likewise dynamic H-bond environment in the Lumi-R state as was suggested for the WT.28
In contrast to the generally matching spectra of the states involved in the photocycle, the dynamics of WT–PSM and Y263F–PSM are characterized by different Meta-R → Pfr rates. The time-dependent concentration changes of each species in the WT and mutant protein are presented in Fig. 5D. The total population of the species at all time points are normalized to unity. In the Y263F mutant, the Meta-R is depopulated two to three times faster than in the WT–PSM.
4 Discussion
4.1 The revisited photocycle of the wild-type CBD-PHY
The photocycle of the DrBphP has long been assumed to involve three intermediate states, i.e. the Lumi-R, Meta-Ra, and Meta-Rc. However, it is also known that the photocycle exhibits quite substantial variations across species. For instance, the meta-Ra and Meta-Rc states, which are distinguishable in cyanobacterial (Cph) phytochrome, are yet to be discovered in DrBphP.The presence of two Lumi-R states is not entirely unexpected as the inherently difficult timescale of tens to hundreds of ns is often not accessible in common experimental setups especially for time-resolved infrared spectroscopy. Our setup uses electronic synchronization of fs and ns lasers of very different repetition rates, enabling us to determine the dynamics of the early and late Lumi-R states and beyond in a single experiment. Furthermore, we have shown that the spectral differences between early and late Lumi-R states are rather small and mostly involve the nearest environment surrounding the D-ring carbonyl stretch vibration of the chromophore. We also observe an additional minor disparity between the early and late Lumi-R around 1625 cm−1, which could indicate a possible rearrangement of a secondary structural element in the protein (Fig. 4A). The shifts cannot be assigned to the excited state, because they occur on the hundreds of nanosecond time scale and the excited state has a lifetime of several hundred picoseconds.14 For SaBphP1 (from organism Stigmatella aurantiaca) a 100 ns-process was assigned as deactivation pathway.33 In contrast, the similarity of early and late Lumi-R spectra observed here suggests a successive occurrence of the two states.
Based on our experimental findings, we propose a refined model of the DrBphP photocycle. The schematic of the photochemical transitions in the photocycle is depicted in Fig. 6a. The red light illumination at 660 nm initiates the photocycle. Within hundreds of ps after the excitation, the first Lumi-R intermediate is formed. The early Lumi-R state then thermally relaxes to the late Lumi-R state with a lifetime of about 0.4 μs. Within 88 μs the late Lumi-R thermally relaxes to the Meta-R state, consistent with earlier data.28 We do not show distinct sub-Meta-R states as they have not been detected in our experiment. Eventually, with a time constant of ca. 2 ms, the Pfr state is reached,13,44 with the nearest environment around the chromophore adapted to the new protein conformation. The Pfr state converts back to the Pr state either in the dark or after illumination with 780 nm. The reverse photocycle is not presented, but it has been shown that it proceeds through Lumi-F and Meta-F intermediates.45 It is yet to be determined if distinct Lumi states form in the reverse photocycle.
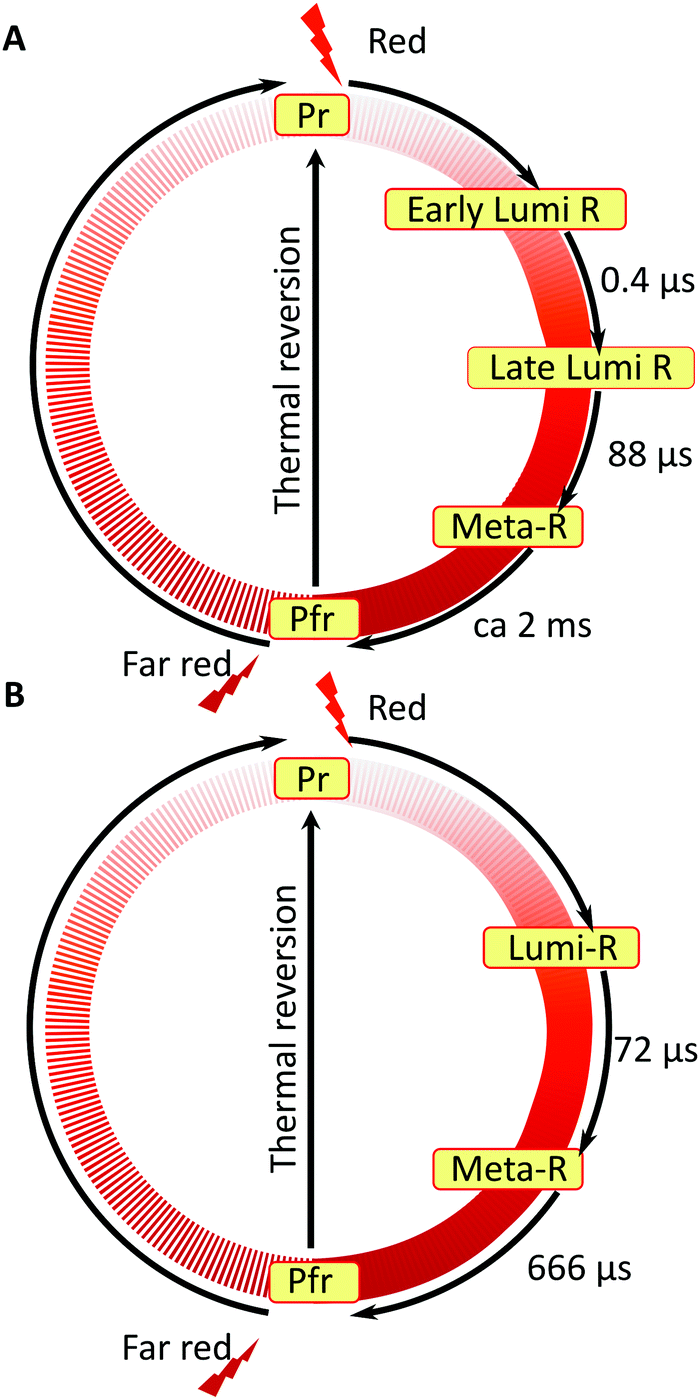 | ||
| Fig. 6 Comparison of the photocycle pathways between WT–PSM (A) and Y263F–PSM (B). The states involved in the reverse photocycle from Pfr to Pr are not studied in this report. | ||
As observed by us previously,28 the positive band corresponding to the D-ring CO group, which is observed either in the early or late Lumi-R spectra, appears to be significantly broader compare to the cryo-trapped Lumi-R spectrum.41,46 The broad peak indicates that the carbonyl vibrations of the D ring experiences a dynamic chemical environment in both Lumi-R substates:28 molecular modelling was used to show that a highly dynamic H-bonding patterns exists around the D-ring CO, which is bound to one or two NH groups of R466 and to a single water molecule. The water molecule was also in a close proximity to the conserved Y263 residue. Based on the new Lumi-R state found in the present paper, we suggest that the structural redistribution around the D-ring carbonyl occurs sequentially and is an intrinsic part of the photocycle.
Although it is the first time to resolve two distinct Lumi-R states in a bacterial phytochrome, more complex photocycles have been reported for plant phytochromes. Most importantly, the existence of two sequential Lumi-R states has been previously proposed by Zhang and co-workers for the oat phytochrome.47 The dynamics of those two Lumi-R states was elucidated by laser photolysis spectroscopy and the lifetimes measured at 283 K were found to be 7.4 μs and 89.5 μs for the early and late Lumi-R states, respectively. Despite the fact that the two Lumi-R states could not be resolved at room temperature in their study, the lifetime of the late Lumi-R at 283 K is in reasonable agreement with our experiments. We are currently investigating if the early structural changes found in the oat phytochrome correspond to the same mechanism as in the case of DrBphP studied here.
4.2 Alternative photocycle routes in Y263F–PSM
The conserved residues around the chromophore in phytochromes have long been regarded as essential residues for the photoconversion and the stability of the resting and biologically active states. The functional importance of many of these residues has been investigated using site-specific mutations.25,48–50 The Y263 is one of the hydrophobic residues, along with Y176 and F203, in DrBphP that provides an adjustable hydrophobic surrounding to assist the amphipathic chromophore D-ring in both Pr and Pfr states.25 In a recent report on Y263F, the possibility of an alternative signaling route in this mutant has been suggested; however, the complete photocycle has not been experimentally assessed.27Analysis of the transient infrared absorption spectra of the Y263F mutant suggests faster dynamics for the Pr → Pfr transition. This may be rationalized based on earlier reports (such as ref. 27) showing the decoupling of the tongue structure from the chromophore in the Y263F mutant. The presence of Pfr-like secondary structure in the Pr state of the mutant reduces the degree of structural reorganization needed for the photoconversion. As suggested for the Y263F mutant of DrBphP,27 and previously for the Y263F mutant of Cph1,51 we observe a reduced quantum yield for the Lumi-R formation. Noteworthy, the scaling factor of 2.5, which was applied to the mutant spectra shown in Fig. 5, is consistent with the relative quantum yield determined in the case of the related Cph1 mutant.51
Nonetheless, our data show that similar dynamics occur up to the meta-R state. This can be interpreted in terms of redundant signalling components in DrBphP: despite the decoupling of a specific signaling route, such as tongue refolding, the protein remains photo-switchable. We find that the consecutive nature of protein dynamics initiated by light excitation persists in the Y263F mutant. The photocycle pathway of the Y263F mutant is illustrated in Fig. 6b.
It has been reported that the protein backbone experiences conformational changes in the Lumi-R and Pfr but not in Meta-R state.28 This indicates that the backbone structure adapted during the Lumi-R state carries on to the Meta-R state, although with minor changes. The simulated Lumi-R structure of the WT DrBphP shows that the Y263 residue is within a hydrogen bonding distance to a water molecule.28 According to the crystal structure of the Y263F mutant, such hydrogen bond can no longer form.27 As a consequence, the local environment around the chromophore D-ring may be significantly altered. The discrepancies noticed in the Lumi-R and Pfr spectra of the WT protein and the mutant support this hypothesis. Most notably, the changes at (1580–1630) cm−1 between WT an Y263F mutant are consistent with different structure of the protein backbone in the Lumi-R state of the Y263F mutant. Actual structural information of sub-states during photoconversion is sparse; notably, for a bathy phytochrome (an Agp2 variant from Agrobacterium fabrum) the crystal structure of the meta-F state (considering the Pfr–Pr transition) could be solved.34
It has been suggested earlier that the dynamic chemical environment present in the WT Lumi-R is critical for the further evolution toward the Meta-R by sampling multiple pathways.28
Earlier reports suggest that weaker interactions with the Y263 residue destabilize the tongue region and, thus, the phytochrome is more likely to adopt a Pfr-like structure.27,52 This could also have implications for the reduced light responsiveness of the Y263F mutant, which is characterized by a lower yield of formation of the Lumi-R state.27
The spectrum of the Pfr state of Y263F indicates that structural changes also occur around the chromophore in the final photoproduct. The solved Pfr crystal structures of the WT DrBphP show that the CO group of the D-ring has a stable hydrogen bond to H201. Moreover, the carboxylate group of D207 forms a hydrogen bond with S468, which is located in the tongue region, as well as with the Y263 in the GAF domain. The Y263 moves closer to the tongue as the D-ring flips and slides in the Pfr state.48 The stable hydrogen bond between the chromophore D-ring CO and H201 in the Pfr state features a narrow IR peak pair at 1679 cm−1 (+)/1699 cm−1 (−) in the WT protein.28 However, this band pair becomes broader in Y263F and the position of the positive band is red-shifted by 6 cm−1, indicating a modified local environment for the chromophore D-ring in the Pfr state of Y263F. This is well in line with a computational evidence of OH–π interactions between Y263 and the chromophore D-ring.53 Furthermore, the observed spectral mismatch between WT and Y263F above 1700 cm−1 possibly suggest that the structural arrangement around the chromophore A-ring in the Pfr state is also affected by the mutation. Despite the mentioned differences in the IR spectra, the structure of the Pfr state most likely closely resembles that of the WT protein, as evidenced by the recent crystallographic data.
5 Conclusions
In this report, we employed transient absorption spectroscopy in the IR to study the complete photocycle pathways of CBD-PHY from DrBphP and its Y263F mutant. The broad experimental time window combined with high spectral and temporal resolution of the applied technique allowed us to deliver new structural insights about the light-induced photoswitching process in bacterial phytochromes. For the first time, we have provided the experimental evidence for the existence of two sequentially forming Lumi-R states, which differ by the local environment around the carbonyl group of the chromophore D-ring. The early Lumi-R state, which forms directly from the excited state, decays with a lifetime of ca. 400 ns, whereas the late Lumi-R state is characterized by a much longer lifetime of 88 μs.The experiments on the Y263F mutant confirmed that Y263 is a key residue that controls the structural evolution around the D-ring carbonyl throughout the photocycle.
The mutation does not completely inhibit the phytochrome from reaching the biologically active state, although the structural evolution is altered. This observation points to the earlier suggestion of the redundancy of components involved in the signaling pathway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MC acknowledges the grants UPD2018-0271 and UPD2019-0229 from the Wenner-Gren Foundations. SW thanks the Knut and Alice Wallenberg Foundation for an Academy Fellowship. JAI acknowledges the Academy of Finland (296135), Jane and Aatos Erkko foundation, and the Magnus Ehrnrooth foundation.Notes and references
- N. C. Rockwell, Y.-S. Su and J. C. Lagarias, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS PubMed.
- A. Blumenstein, K. Vienken, R. Tasler, J. Purschwitz, D. Veith, N. Frankenberg-Dinkel and R. Fischer, Curr. Biol., 2005, 15, 1833–1838 CrossRef CAS PubMed.
- E. Giraud, S. Zappa, L. Vuillet, J.-M. Adriano, L. Hannibal, J. Fardoux, C. Berthomieu, P. Bouyer, D. Pignol and A. Vermglio, J. Biol. Chem., 2005, 280, 32389–32397 CrossRef CAS PubMed.
- H. A. Borthwick, S. B. Hendricks, M. W. Parker, E. H. Toole and V. K. Toole, Proc. Natl. Acad. Sci. U. S. A., 1952, 38, 662–666 CrossRef CAS PubMed.
- S. J. Davis, A. V. Vener and R. D. Vierstra, Science, 1999, 286, 2517–2520 CrossRef CAS PubMed.
- A. Björling, O. Berntsson, H. Lehtivuori, H. Takala, A. J. Hughes, M. Panman, M. Hoernke, S. Niebling, L. Henry, R. Henning, I. Kosheleva, V. Chukharev, N. V. Tkachenko, A. Menzel, G. Newby, D. Khakhulin, M. Wulff, J. A. Ihalainen and S. Westenhoff, Sci. Adv., 2016, 2, e1600920 CrossRef PubMed.
- X. Yang, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14715–14720 CrossRef CAS PubMed.
- T. Lamparter, M. Carrascal, N. Michael, E. Martinez, G. Rottwinkel and J. Abian, Biochemistry, 2004, 43, 3659–3669 CrossRef CAS PubMed.
- W. Rüdiger, F. Thümmler, E. Cmiel and S. Schneider, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 6244–6248 CrossRef PubMed.
- F. Andel, J. C. Lagarias and R. A. Mathies, Biochemistry, 1996, 35, 15997–16008 CrossRef CAS PubMed.
- M. A. Mroginski, D. H. Murgida and P. Hildebrandt, Acc. Chem. Res., 2007, 40, 258–266 CrossRef CAS PubMed.
- B. Borucki, D. von Stetten, S. Seibeck, T. Lamparter, N. Michael, M. A. Mroginski, H. Otto, D. H. Murgida, M. P. Heyn and P. Hildebrandt, J. Biol. Chem., 2005, 280, 34358–34364 CrossRef CAS PubMed.
- J. Dasgupta, R. R. Frontiera, K. C. Taylor, J. C. Lagarias and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1784–1789 CrossRef CAS PubMed.
- N. Lenngren, P. Edlund, H. Takala, B. Stucki-Buchli, J. Rumfeldt, I. Peshev, H. Hkknen, S. Westenhoff and J. A. Ihalainen, Phys. Chem. Chem. Phys., 2018, 20, 18216–18225 RSC.
- J. J. van Thor, B. Borucki, W. Crielaard, H. Otto, T. Lamparter, J. Hughes, K. J. Hellingwerf and M. P. Heyn, Biochemistry, 2001, 40, 11460–11471 CrossRef CAS PubMed.
- H. Takala, A. Björling, O. Berntsson, H. Lehtivuori, S. Niebling, M. Hoernke, I. Kosheleva, R. Henning, A. Menzel, J. A. Ihalainen and S. Westenhoff, Nature, 2014, 509, 245–248 CrossRef CAS PubMed.
- B. Borucki, Photochem. Photobiol. Sci., 2006, 5, 553–566 RSC.
- B. Karniol, J. R. Wagner, J. M. Walker and R. D. Vierstra, Biochem. J., 2005, 392, 103–116 CrossRef CAS PubMed.
- T. Rohmer, C. Lang, W. Grtner, J. Hughes and J. Matysik, Photochem. Photobiol., 2010, 86, 856–861 CrossRef CAS PubMed.
- X. Yang, E. A. Stojkovic, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12571–12576 CrossRef CAS PubMed.
- E. S. Burgie, T. Wang, A. N. Bussell, J. M. Walker, H. Li and R. D. Vierstra, J. Biol. Chem., 2014, 289, 24573–24587 CrossRef CAS PubMed.
- L. Hennig and E. Schfer, J. Biol. Chem., 2001, 276, 7913–7918 CrossRef CAS PubMed.
- J. R. Wagner, J. S. Brunzelle, K. T. Forest and R. D. Vierstra, Nature, 2005, 438, 325–331 CrossRef CAS PubMed.
- L.-O. Essen, J. Mailliet and J. Hughes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14709–14714 CrossRef CAS PubMed.
- J. R. Wagner, J. Zhang, D. von Stetten, M. Gnther, D. H. Murgida, M. A. Mroginski, J. M. Walker, K. T. Forest, P. Hildebrandt and R. D. Vierstra, J. Biol. Chem., 2008, 283, 12212–12226 CrossRef CAS PubMed.
- A. T. Ulijasz, G. Cornilescu, C. C. Cornilescu, J. Zhang, M. Rivera, J. L. Markley and R. D. Vierstra, Nature, 2010, 463, 250–254 CrossRef CAS PubMed.
- H. Takala, H. K. Lehtivuori, O. Berntsson, A. Hughes, R. Nanekar, S. Niebling, M. Panman, L. Henry, A. Menzel, S. Westenhoff and J. A. Ihalainen, J. Biol. Chem., 2018, 293, 8161–8172 CrossRef CAS PubMed.
- J. A. Ihalainen, E. Gustavsson, L. Schroeder, S. Donnini, H. Lehtivuori, L. Isaksson, C. Thing, V. Modi, O. Berntsson, B. Stucki-Buchli, A. Liukkonen, H. Hkknen, E. Kalenius, S. Westenhoff and T. Kottke, J. Am. Chem. Soc., 2018, 140, 12396–12404 CrossRef CAS PubMed.
- F. Velazquez Escobar, D. von Stetten, M. Gnther-Lkens, A. Keidel, N. Michael, T. Lamparter, L.-O. Essen, J. Hughes, W. Grtner, Y. Yang, K. Heyne, M. A. Mroginski and P. Hildebrandt, Front. Mol. Biosci., 2015, 2, 37 Search PubMed.
- J. J. van Thor, K. L. Ronayne and M. Towrie, J. Am. Chem. Soc., 2007, 129, 126–132 CrossRef CAS PubMed.
- C. Schumann, R. Gro, N. Michael, T. Lamparter and R. Diller, ChemPhysChem, 2007, 8, 1657–1663 CrossRef CAS PubMed.
- K. Heyne, O. F. Mohammed, A. Usman, J. Dreyer, E. T. J. Nibbering and M. A. Cusanovich, J. Am. Chem. Soc., 2005, 127, 18100–18106 CrossRef CAS PubMed.
- T. Mathes, J. Ravensbergen, M. Kloz, T. Gleichmann, K. D. Gallagher, N. C. Woitowich, R. Peter, S. E. Kovaleva, E. A. Stojković and J. T. M. Kennis, J. Phys. Chem. Lett., 2015, 6, 239–243 CrossRef CAS PubMed.
- A. Schmidt, L. Sauthof, M. Szczepek, M. F. Lopez, F. V. Escobar, B. M. Qureshi, N. Michael, D. Buhrke, T. Stevens, D. Kwiatkowski, D. von Stetten, M. A. Mroginski, N. Krau, T. Lamparter, P. Hildebrandt and P. Scheerer, Nat. Commun., 2018, 9, 4912 CrossRef PubMed.
- C. Kneip, P. Hildebrandt, W. Schlamann, S. E. Braslavsky, F. Mark and K. Schaffner, Biochemistry, 1999, 38, 15185–15192 CrossRef CAS PubMed.
- C. J. Manning, R. A. Palmer and J. L. Chao, Rev. Sci. Instrum., 1991, 62, 1219–1229 CrossRef CAS.
- T. Kottke, V. A. Lrenz-Fonfra and J. Heberle, J. Phys. Chem. B, 2017, 121, 335–350 CrossRef CAS PubMed.
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- Y. Feng, I. Vinogradov and N.-H. Ge, Opt. Express, 2017, 25, 26262–26279 CrossRef CAS PubMed.
- Y. Feng, I. Vinogradov and N.-H. Ge, Opt. Express, 2019, 27, 20323–20346 CrossRef CAS PubMed.
- H. Foerstendorf, C. Benda, W. Grtner, M. Storf, H. Scheer and F. Siebert, Biochemistry, 2001, 40, 14952–14959 CrossRef CAS PubMed.
- P. Schwint, H. Foerstendorf, Z. Hussain, W. Grtner, M.-A. Mroginski, P. Hildebrandt and F. Siebert, Biophys. J., 2008, 95, 1256–1267 CrossRef PubMed.
- E. A. Stojkovi ć, K. C. Toh, M. T. A. Alexandre, M. Baclayon, K. Moffat and J. T. M. Kennis, J. Phys. Chem. Lett., 2014, 5, 2512–2515 CrossRef PubMed.
- Y. Yang, M. Linke, T. von Haimberger, J. Hahn, R. Matute, L. Gonzlez, P. Schmieder and K. Heyne, J. Am. Chem. Soc., 2012, 134, 1408–1411 CrossRef CAS PubMed.
- E. Chen, V. N. Lapko, P.-S. Song and D. S. Kliger, Biochemistry, 1997, 36, 4903–4908 CrossRef CAS PubMed.
- H. Foerstendorf, E. Mummert, E. Schfer, H. Scheer and F. Siebert, Biochemistry, 1996, 35, 10793–10799 CrossRef CAS PubMed.
- C. F. Zhang, D. L. Farrens, S. C. Bjorling, P. S. Song and D. S. Kliger, J. Am. Chem. Soc., 1992, 114, 4569–4580 CrossRef CAS.
- E. Burgie, J. Zhang and R. Vierstra, Structure, 2016, 24, 448–457 CrossRef CAS PubMed.
- X. Yang, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15639–15644 CrossRef CAS PubMed.
- A. J. Fischer and J. C. Lagarias, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17334–17339 CrossRef CAS PubMed.
- V. Sineshchekov, J. Mailliet, G. Psakis, K. Feilke, J. Kopycki, M. Zeidler, L.-O. Essen and J. Hughes, Photochem. Photobiol., 2014, 90, 786–795 CAS.
- P. H. Quail, Nat. Rev. Mol. Cell Biol., 2002, 3, 85–93 CrossRef CAS PubMed.
- R. Gonzlez and M. A. Mroginski, J. Phys. Chem. B, 2019, 123, 9819–9830 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp06995j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2020 |