
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular dynamics simulations reveal distinct differences in conformational dynamics and thermodynamics between the unliganded and CD4-bound states of HIV-1 gp120†
Yi
Li
 ab,
Lei
Deng
a,
Jing
Liang
a,
Guang-Heng
Dong
a,
Yuan-Ling
Xia
a,
Yun-Xin
Fu
*c and
Shu-Qun
Liu
*a
ab,
Lei
Deng
a,
Jing
Liang
a,
Guang-Heng
Dong
a,
Yuan-Ling
Xia
a,
Yun-Xin
Fu
*c and
Shu-Qun
Liu
*a
aState Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan & School of Life Sciences, Yunnan University, Kunming 650091, China. E-mail: shuqunliu@ynu.edu.cn
bCollege of Mathematics and Computer Science, Dali University, Dali 671003, China
cHuman Genetics Center and Division of Biostatistics, School of Public Health, The University of Texas Health Science Center, Houston, USA. E-mail: Yunxin.Fu@uth.tmc.edu
First published on 2nd March 2020
Abstract
The entry of human immunodeficiency virus type I (HIV-1) into host cells is initiated by binding to the cell-surface receptor CD4, which induces a conformational transition of the envelope (Env) glycoprotein gp120 from the closed, unliganded state to the open, CD4-bound state. Despite many available structures in these two states, detailed aspects on the dynamics and thermodynamics of gp120 remain elusive. Here, we performed microsecond-scale (μs-scale) multiple-replica molecular dynamics (MD) simulations to explore the differences in the conformational dynamics, protein motions, and thermodynamics between the unliganded and CD4-bound/complexed forms of gp120. Comparative analyses of MD trajectories reveal that CD4 binding promotes the structural deviations/changes and conformational flexibility, loosens the structural packing, and complicates the molecular motions of gp120. Comparison of the constructed free energy landscapes (FELs) reveals that the CD4-complexed gp120 has more conformational substates, larger conformational entropy, and lower thermostability than the unliganded form. Therefore, the unliganded conformation represents a structurally and energetically stable “ground state” for the full-length gp120. The observed great increase in the mobility of V1/V2 and V3 along with their more versatile movement directions in the CD4-bound gp120 compared to the unliganded form suggests that their orientations with respect to each other and to the structural core determine the differences in the conformational dynamics and thermodynamics between the two gp120 forms. The results presented here provide a basis by which to better understand the functional and immunological properties of gp120 and, furthermore, to deploy appropriate strategies for the development of anti-HIV-1 drugs or vaccines.
Introduction
The HIV-1 envelope glycoprotein Env trimer, which is composed of three gp120/gp41 heterodimers (also called protomers), is the membrane fusion machine that mediates viral entry into host cells.1–3 In particular, because the gp120 subunit is exposed on the surface of Env trimer and responsible for interactions with the cell-surface receptors, it becomes the principal target for anti-viral neutralizing antibodies.4,5 As a result, Knowledge of the structure–dynamics–function relationship of gp120 is essential for understanding the mechanisms of HIV-1 infection and immune evasion and, further, for developing anti-HIV drugs and vaccines.6In contrast to the standard type I membrane fusion mechanism of the enveloped viruses,7 HIV-1 has evolved a two-step mechanism to enter the target cell via sequential binding of gp120 to two distinct receptors, the receptor CD4 and the coreceptor C–C chemokine receptor type 5 (CCR5) or C–X–C chemokine receptor type 4 (CXCR4).8–10 Structural studies have revealed that before CD4 binding, Env trimer adopts a closed unliganded conformation,11–14 in which the inter-protomer noncovalent contacts among V1/V2 and V3 of gp120 subunits lock the trimer crown and the V1/V2 region packs against the V3 loop and buries the coreceptor-binding site. Binding of the initial receptor, CD4, induces substantial conformational changes in gp120, including dissociation of contacts among V1/V2 and V3 at the apex of the trimer crown, reorientation of V1/V2 away from V3 and toward CD4, and coalescence of separate elements of coreceptor-binding site, ultimately leading to the opening of the trimer crown and the formation/exposure of the coreceptor-binding site.15–19 Subsequent coreceptor binding is considered to promote additional conformational changes in gp41, leading to the formation of a energetically stable six-helix bundle necessary for the fusion of viral and cellular membranes.20,21 To avoid semantic confusion, in this paper we refer to the gp120 conformations before and after CD4 binding as the unliganded (or ligand-free) and the CD4-bound (or CD4-complexed) states, respectively.
Although X-ray crystallographic and cryo-electron microscopy studies have provided detailed structural information about various conformational states of Env/gp120,11–14,16,19,20,22,23 these static snapshots cannot fully reflect the dynamic/thermodynamic nature of gp120, including the conformational flexibility, molecular motions, distributions of and transitions between different conformational states, and how these are influenced by receptor binding, which are important for a complete understanding of the gp120 biological functions.
Myszka et al.24 determined the thermodynamic parameters for gp120-CD4 binding reaction using isothermal titration calorimetry (ITC); the results revealed the unexpected large changes in enthalpy, entropy, and heat capacity, thus implying considerable conformational flexibility and extensive structural rearrangements of gp120 upon CD4 binding. Using hydrogen–deuterium exchange (HDX) to measure the rates of deuterium incorporation into backbone amides in solution, Guttman et al.15 probed the changes in conformational dynamics of the soluble Env trimers induced by CD4 binding; their results showed that the major structural reorganizations occurred in V1/V2 and V3, bridging-sheet elements, and much of the inner domain of gp120 as well as in gp41 fusion subunit. However, the data from both ITC and HDX cannot provide a clear atomic-level picture of the protein dynamics and, hence, the differences in the conformational flexibility/dynamics between the unliganded and CD-bound gp120s still remain elusive. Munro et al.25 investigated the thermodynamic distributions of the populations of gp120 states in the context of the native Env trimers on the viral surface by using single-molecule fluorescence resonance energy transfer (smFRET). Although their observations revealed that the ligand-free Env/gp120 sampled three distinctly different states for which the relative populations were remodeled by receptor and antibody binding, the representation of the free energy landscape (FEL) capable of characterizing the conformational diversity and detailed populations of different states/substates of gp120 near the native conformations is still lacking.
Thanks to GPU-accelerated algorithms, molecular dynamics (MD) simulations can now sample the protein conformational space more efficiently than ever before, thus providing reliable atomic-level information on the conformational flexibility and molecular motions of a protein and, furthermore, allowing the construction of the FEL near the protein native state.26–28 In this paper, we have performed μs-scale multiple-replica MD simulations on two structural models, the near full-length unliganded monomeric gp120 and the gp120-CD4 complex (hereafter, the gp120 in the complex is referred to as the CD4-complexed gp120), to explore the differences in the conformational dynamics, molecular motions, and FEL between the unliganded and CD4-complexed forms of gp120. Our results reveal that CD4 binding increases the conformational dynamics/entropy/diversity, complicates the protein motions, and lowers the thermostability of gp120 through promoting the mobility of V1/V2 and V3, thus explaining why the activated CD4-bound state is susceptible to antibody neutralization and is prone to spontaneous inactivation. The implications of the distinctly different dynamic and thermodynamic behaviors between the two states were also discussed in terms of the functional and immunological properties of gp120 and the development of anti-HIV drugs and vaccines.
Results
Structural models
The structural models of the unliganded gp120 and gp120-CD4 complex are shown in Fig. 1A and B, respectively. To clearly demonstrate the CD4-bound/complexed state, CD4 was removed from the complex and only the model of gp120 is shown in Fig. 1C. Both the unliganded and CD4-complexed models are composed of a relatively conserved structural core, a bridging-sheet minidomain, and five peripheral variable regions V1–V5. According to the X-ray structures (PDB IDs: 1GC1, 1G9M, and 1G9N) of the gp120 core determined by Kwong et al.,29,30 the core can be divided into the inner and outer domains. The inner domain comprises the N-, C-termini, a seven-stranded β-sandwich, and three topological layers emanating from the β-sandwich, with each layer being associated with a helix, i.e., layers 1, 2, and 3 with α0, α1, and α5, respectively.31 The outer domain mainly consists of two end-to-end stacked β-barrels and a long α-helix α2. It is clear from Fig. 1A and C that many of the secondary structural elements within the structural core are conserved between the unliganded and CD4-bound states, although slight differences in the orientation and packing can be observed. In particular, a large hydrophobic cavity, which is located under the bridging sheet and between the inner and outer domains, can be observed in both gp120 forms; this indicates that the unliganded state does not hinder CD4 binding.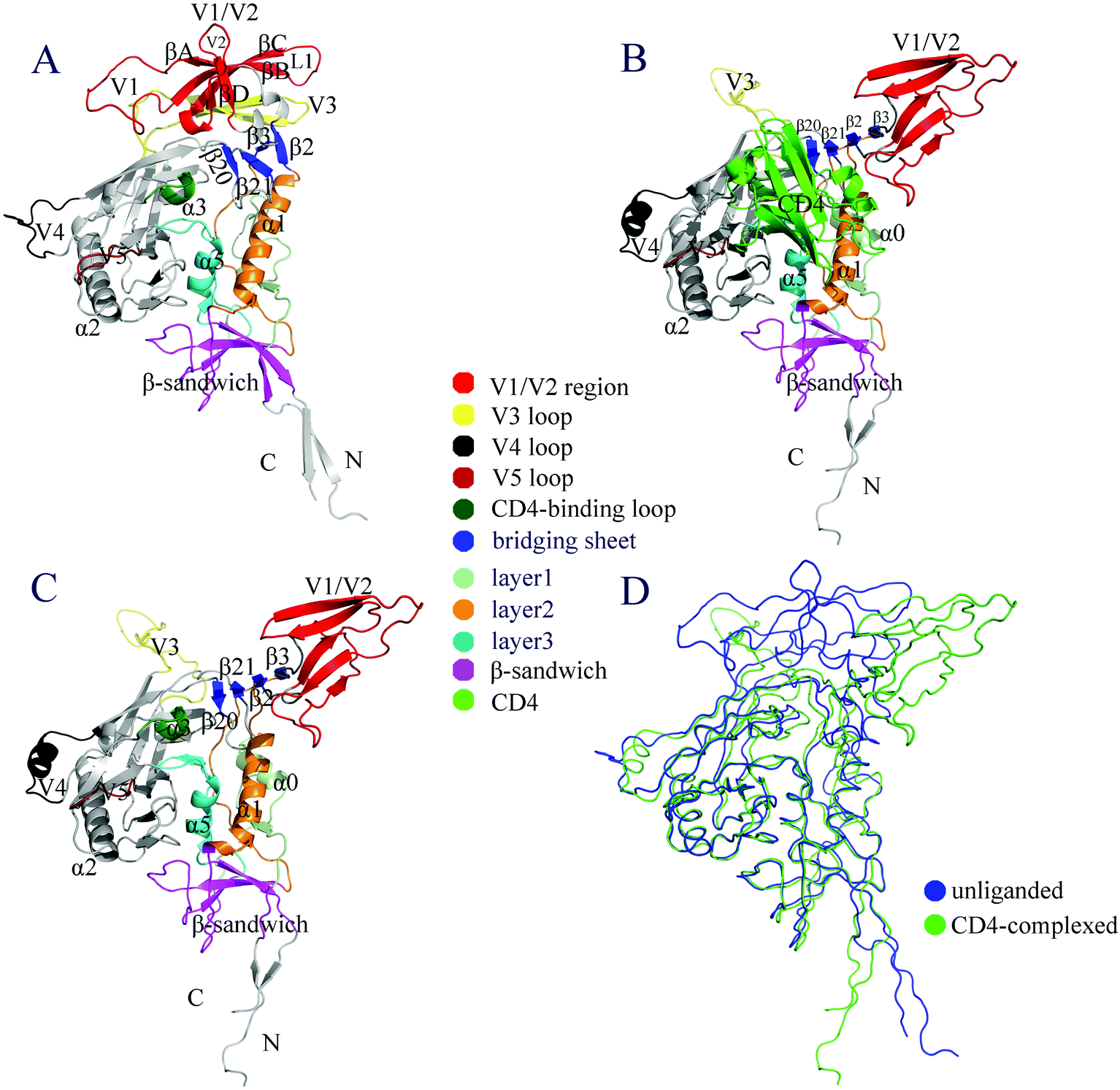 | ||
| Fig. 1 Structural models. (A) Monomeric unliganded gp120. (B) gp120-CD4 complex. (C) CD4-complexed/bound gp120 model without presence of CD4. (D) Backbone superposition of the unliganded (blue) and CD4-bound (green) models. In (A) to (C), various structural regions/elements are labeled and differently colored; note that in (A), the four β-strands in the “Greek key”-like V1/V2 region are labeled βA to βD, and in (B), the CD4 D1 domain is colored light green. | ||
The most pronounced differences between the two models were observed in the relative orientations of V1/V2 and V3 with respect to each other and to the gp120 core. In the unliganded model, the V1/V2 region, which emanates from the inner domain and adopts a “Greek key”-like topology,4 packs against the V3 loop, which in turn packs against the distal end of the gp120 core, with V3's tip making direct contacts with the bridging sheet. In the CD4-complexed model, the V1/V2 region opens up and moves away from V3, which in turn protrudes away from the gp120 core, making the overall orientation between V1/V2 and V3 look like a letter “Y”. One other key difference lies in the ordering and positioning of the bridging-sheet elements. In the unliganded gp120, two two-stranded antiparallel β-sheets, β2–β3 and β20–β21, are positioned alongside one another with a parallel β-sheet formed between β3 and β21; therefore, the bridging-sheet elements are in the order β2–β3–β21–β20 (Fig. 1A). However, in the CD4-bound model, β2–β3 flips over to form a β-sheet between β2 and β21, leading to the order β3–β2–β21–β20, in which the three β-sheets, β3–β2, β2–β21, and β21–β20, are all antiparallel (Fig. 1B and C). In this paper we refer to β3–β2–β21–β20 in the CD4-bound state and β2–β3–β21–β20 in the unliganded state as the mature and premature bridging sheets, respectively. Since β2–β3 forms the stem of the V1/V2 region, its flip-over not only directs V1/V2 away from the gp120 core but also leads to the formation and exposure of the mature bridging sheet, which, together with the fully protruded V3 loop, forms the coreceptor binding site as observed in the CD4-bound/complexed model (Fig. 1C). As shown in Fig. 1D, the superimposed backbones of the two models clearly demonstrate the structurally conserved core and the distinctly different orientations of V1/V2 and V3. Quantitatively, the Cα root mean square deviation (RMSD) values after least square superposition of the structural cores of these two models are 0.89, 2.84, 6.60, and 15.42 Å for the structural core, the whole structure, V3, and V1/V2, respectively.
Structural stability during MD simulations
To evaluate the overall structural stability of the unliganded and CD4-complexed gp120s, time evolutions of the backbone RMSD values relative to their respective starting structures were calculated. For the unliganded monomeric gp120, all 10 replicas require less than 5 ns to reach relatively stable RMSD values (Fig. 2A), whereas for the CD4-complexed gp120, some replicas require a longer duration (about 10 ns) to reach a RMSD plateau (Fig. 2B). Moreover, it is clear that the equilibrated portions of RMSD curves exhibit a narrower range for the unliganded gp120 than for the CD4-complexed gp120, with the former ranging between 0.3 and 0.6 nm while the latter between 0.4 and 1.1 nm. The other obvious difference is in the fluctuation amplitudes of RMSD curves, i.e., most of the 10 CD4-complexed curves exhibit much larger amplitudes than the unliganded curves. Taken together, the above results reveal that during MD simulations, the CD4-complexed gp120 experienced larger structural deviations from the starting conformation and more dramatic conformational changes than the unliganded gp120, indicating that the former has a lower structural stability and a stronger capability to alter conformation than the latter.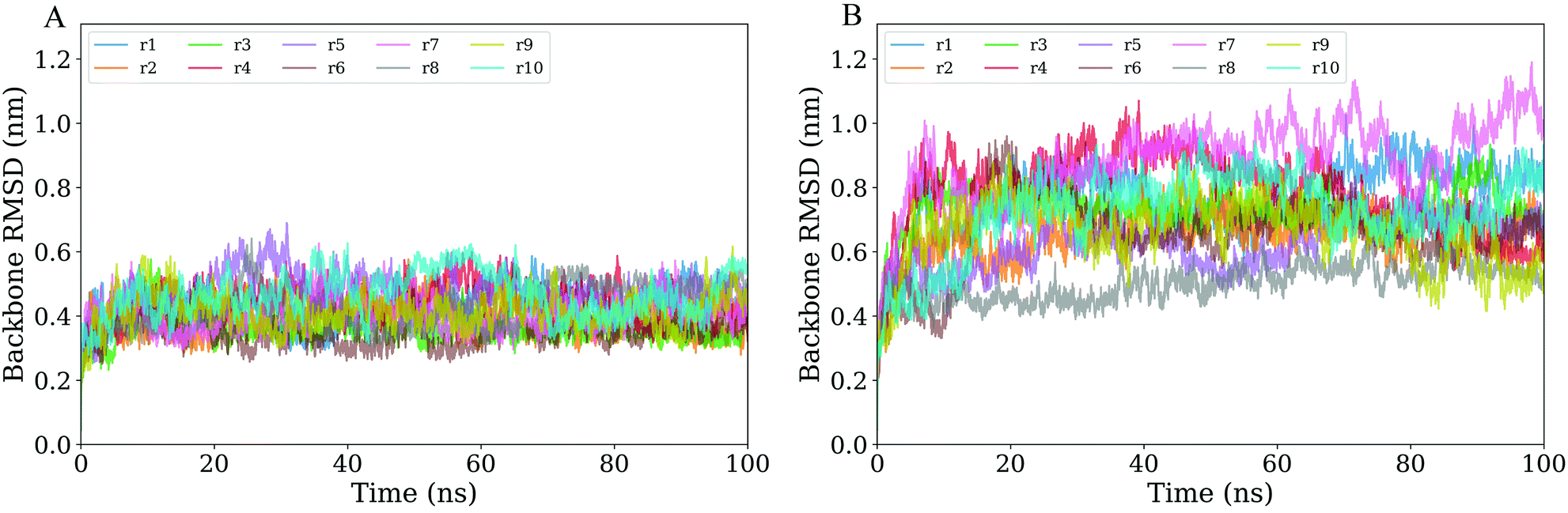 | ||
| Fig. 2 Time evolutions of the backbone RMSD values of the two gp120 forms with respect to their respective starting structures calculated from the 10 MD simulation replicas (r1 to r10). (A) Unliganded gp120. (B) CD4-complexed gp120. | ||
For each simulation system, the first 10 ns trajectories of the 10 replicas were discarded, and the remaining equilibrated portions (10–100 ns) were concatenated into a single joined trajectory (900 ns). Comparison of the cosine contents of the first three eigenvectors derived from essential dynamics (ED) analyses of the joined equilibrium trajectories and the replicas 1–10 reveals that, for both systems, the cosine contents are effectively lowered by the concatenation of the individual replicas (Table S1, ESI†), and this indicates that the joined trajectories have improved the system ergodicity and achieved a higher degree of sampling convergence compared to a single replica. Therefore, all the subsequent analyses were performed based on the joined equilibrium trajectories.
Structural properties
To quantitatively compare the differences in structural properties between the unliganded and CD4-complexed gp120s, the average values of several structural/geometrical parameters were computed from the joined equilibrium trajectories. As listed in Table 1, the differences in the calculated parameters are not large between these two forms of gp120. However, the relatively small differences can still reflect the variations in average structural features of gp120 upon CD4 binding. The unliganded model differs from the CD4-complexed form in higher average values of the number of hydrogen bonds (NHB) and the number of close interatomic contacts (NCIC). The more interatomic interactions in the unliganded gp120 imply an increase in the overall structural stability compared to the CD4-complexed gp120, in agreement with the above RMSD analysis. In addition, the smaller average values of solvent accessible surface area (SASA) and radius of gyration (Rg) in the unliganded gp120 indicate that it has a more compact packing and smaller size than the CD4-complexed gp120. These results can be expected, since CD4 binding induces considerable conformational changes in gp120; for instance, upon binding, the dissociations between V1/V2 and V3 and between V3 and the gp120 core contribute to decreasing the number of interatomic interactions; the protruding orientations of V1/V2 and V3, together with the full exposure of the mature bridging sheet, contribute to increasing Rg and SASA of gp120.| NHBa | NCICb | SASAc (Å) | R g (Å) | SSCe | ||||
|---|---|---|---|---|---|---|---|---|
| α-Helix | β-Strand | Turn | Coil | |||||
| a Number of hydrogen bonds. A hydrogen bond is defined as being present if the donor–acceptor distance is less 3.5 Å and the donor–hydrogen–acceptor angle is greater than 120°. b Number of close interatomic contacts. A close contact is defined as being made if the distance between two atoms is less than 6 Å. c Solvent accessible surface area. SASA is defined as the total area of the protein surface that is accessible to a sphere solvent probe with radius of 1.4 Å. d Radius of gyration. e Secondary structure content. SSC is defined as the number of residues in a given secondary structural type. | ||||||||
| Unliganded | 323.7 (10.9) | 493941 (3159) | 24031 (491) | 25.8 (0.03) | 43.2 (4) | 167 (8.4) | 46 (5.7) | 130 (7.2) |
| CD4-complexed | 316.9 (10.1) | 482019 (2654) | 26177 (448) | 27.8 (0.06) | 45.2 (4) | 154 (9.7) | 37 (5.8) | 148 (8.3) |
Compared to the CD4-complexed gp120, the unliganded gp120 has a slightly lower α-helical content. This is likely due to the retention of α-helices in V4 and layer 1 of the CD4-complexed gp120 (Fig. 1C) during simulations, which are absent in the starting model of the unliganded gp120 (Fig. 1A). Nevertheless, the unliganded gp120 has obviously more contents of β-strands and turns while less coil content than the CD4-complexed gp120, indicating that CD4 binding deforms some regular β-strands and turns into irregular coils. A typical example is the two-stranded β-sheet observed in the buried V3 of the unliganded gp120 (Fig. 1A), which is destructured into a protruding coil loop upon CD4 binding (Fig. 1B and C).
Conformational flexibility
Per-residue Cα root mean square fluctuation (RMSF), an often-used indicator for evaluating protein conformational flexibility based on MD simulations, were computed from the joined equilibrium trajectories. Fig. 3 shows the Cα RMSF values of the unliganded and CD4-complexed gp120s as a function of residue number. Clearly, the V1/V2 region and V3 loop have significantly higher RMSF values in the CD4-complexed gp120; this is not surprising, as CD4 binding disrupts their association with respect to each other and, furthermore, makes them fully expose to solvent. With the exception of V1/V2 and V3, the other regions exhibit similar RMSF profiles for the two gp120 forms, with the regular secondary structural elements in the core featuring low values while the N-, C-termini and core-peripheral loops characterized by relatively high values. However, close inspection reveals that most of the core and core-peripheral regions still have higher RMSF values in the CD4-complexed gp120. | ||
| Fig. 3 Per-residue Cα RMSF profiles of the unliganded (blue line) and CD4-complexed (green line) gp120s. The average RMSF values are shown as dotted lines and some structural regions are labelled (for details, see sequence alignment shown in Fig. S1, ESI†). | ||
For the unliganded gp120, the average RMSF values of the regular secondary structure elements and connecting loops within the core are 0.21 ± 0.15 and 0.28 ± 0.21 nm, respectively, and the value of the entire structure is 0.26 ± 0.18 nm; in the case of the CD4-complexed gp120, the corresponding values are 0.26 ± 0.11, 0.33 ± 0.19, and 0.46 ± 0.42 nm, respectively. These results indicate that CD4 binding not only facilitates the fluctuations of the core but also enhances the overall conformational flexibility of gp120.
There are only three regions that exhibit higher RMSF values in the unliganded gp120 than in the CD4-complexed form, i.e., residues 58–63 (a portion of layer 1), 426–432 (the tip of β20–β21 hairpin), and 494–500 (a portion of the C-terminus). Residues 58–63 is a loop located between β![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) and α0 (note that α0 does not exists in the unliganded model). This loop suffers fewer structural restrictions in the unliganded gp120 due to its more extended conformation than in the CD4-complexed gp120. The tip of β20–β21 is located at the top of the large hydrophobic cavity between the inner and outer domains and hence can fluctuate relatively freely in the cavity not occupied by CD4; upon CD4 binding, Trp427 on this tip interacts directly with CD4 Phe43 and, furthermore, CD4 covers much of the tip and hence restricts its fluctuations.
and α0 (note that α0 does not exists in the unliganded model). This loop suffers fewer structural restrictions in the unliganded gp120 due to its more extended conformation than in the CD4-complexed gp120. The tip of β20–β21 is located at the top of the large hydrophobic cavity between the inner and outer domains and hence can fluctuate relatively freely in the cavity not occupied by CD4; upon CD4 binding, Trp427 on this tip interacts directly with CD4 Phe43 and, furthermore, CD4 covers much of the tip and hence restricts its fluctuations.
Collective motions
In addition to traditional comparative analyses in terms of structural/geometrical properties and conformational flexibility, ED analyses were employed to investigate the differences in the collective motions between the two forms of gp120. For the unliganded and CD4-complexed gp120s, the values of total mean square fluctuations (TMSF) obtained by diagonalization of the Cα covariance matrices are 39.0 and 158.3 nm2, respectively, indicating a significant increase in the fluctuation amplitude of gp120 upon CD4 binding, in agreement with the above flexibility analysis.Fig. 4 shows the eigenvalues of the first 30 eigenvectors (main plot) and the cumulative contribution of eigenvectors to TMSF (inset plot). For both gp120 forms, the first eigenvector has the largest eigenvalue, while as the eigenvector index increases, eigenvalues decrease until reach a plateau slightly above 0 nm2. Notably, the first few eigenvectors of the CD4-complexed gp120 have significantly larger eigenvalues than those of the unliganded gp120, indicating that CD4 binding greatly facilitates the global large-scale collective motions in gp120. Moreover, for the unliganded gp120, the cumulative TMSF contribution rates of the first 4 and 10 eigenvectors are 53.8% and 76.9%, respectively, and for the CD4-complexed gp120, the corresponding values are 73.1% and 89.5% (Fig. 4, inset plot), respectively. Since the conformational space of gp120 is highly multi-dimensional (i.e., spanned by 462 × 3 eigenvectors), it can be considered that the first 10 eigenvectors, in particular the first 4 eigenvectors, make a substantial contribution to TMSF in the space. Therefore, the first 4 eigenvectors constitute an essential subspace within which the largest-amplitude protein motions take place.
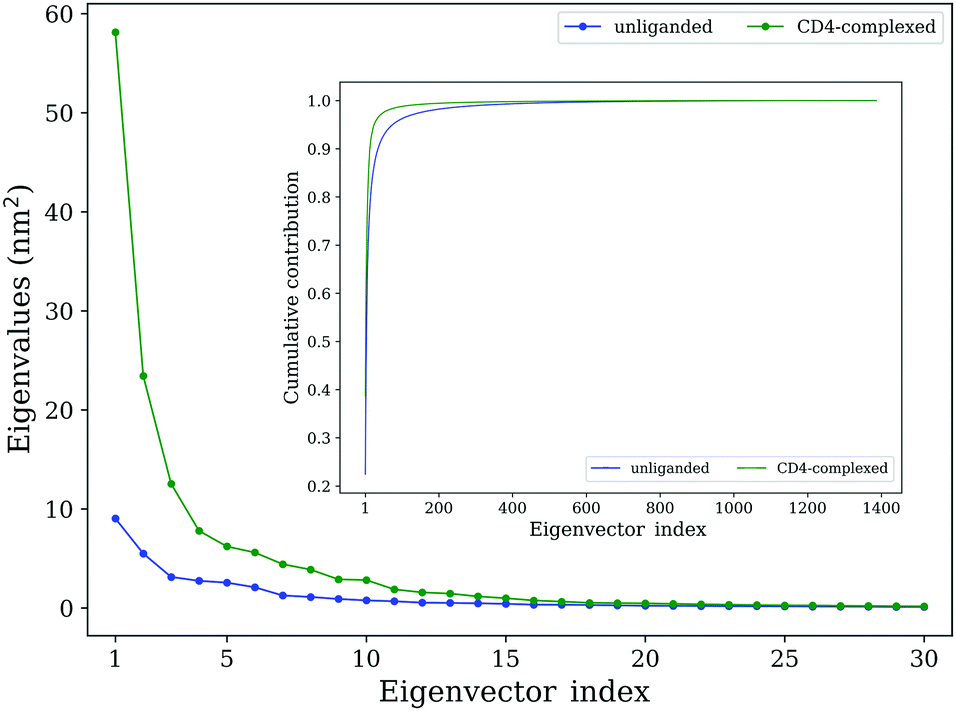 | ||
| Fig. 4 Eigenvalues of eigenvectors derived from ED analyses of the joined equilibrium trajectories of the unliganded (blue line) and CD4-complexed (green line) gp120 models. The main plot shows eigenvalues of the first 30 eigenvectors; the inset plot shows the cumulative contribution of all eigenvectors (1386) to TMSF. | ||
Fig. 5 shows the porcupine plots representing the collective motions (or motion modes) of the two gp120 forms along the first four eigenvectors. In these plots, a cone was drawn on each Cα atom, with its pointing direction and length representing the moving direction and fluctuation amplitude of the atom, respectively. In all the four modes of the unliganded gp120 (Fig. 5A–D), the regions exhibiting a large fluctuation amplitude mainly involve the N-, C-termini, core-peripheral loops (V4 and V5), and the three connecting loops within the V1/V2 region (i.e., the V1 loop connecting between βA and βB, L1 between βB and BC, and V2 between βC and βD), whereas much of the structural core exhibits a moderate or relatively small fluctuation amplitude. Nevertheless, with the exception of the N-, C-termini, most of the peripheral regions (V4, V5, and loops in the V1/V2 region) move in concert (i.e., in the same or similar directions) with their adjacent core regions, making the collective motions of the entire unliganded structure look simple and integrated. In the case of the CD4-complexed gp120, the isolated V1/V2 region exhibits the largest amplitude in all the four modes (Fig. 5E–H), while the N-, C-termini, V3 loop, and core-peripheral loops exhibit, in different modes, fluctuation amplitudes that are either larger than, or similar to that of the structural core. These observations, together with the versatile fluctuation directions of the peripheral regions relative to the structural core, indicate that CD4 binding diversifies and complicates the motion modes of gp120.
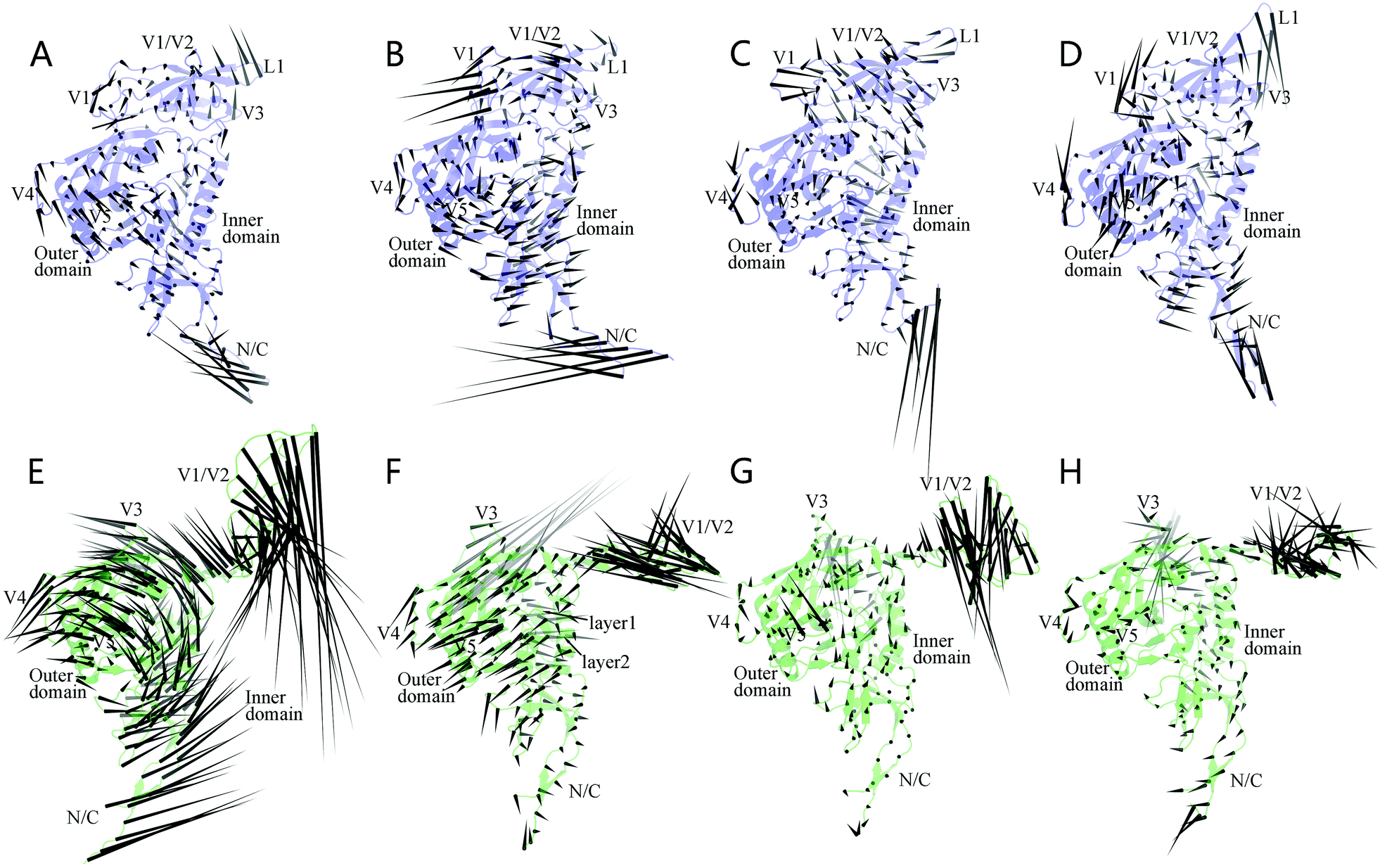 | ||
| Fig. 5 Porcupine plots of the first four motion modes derived from ED analyses. (A–D) Collective motions of the unliganded gp120 along eigenvectors 1–4, respectively. (E–H) Collective motions of the CD4-complexed gp120 along eigenvectors 1–4, respectively. | ||
In the first ranked mode of the unliganded gp120 (Fig. 5A), the outer and inner domains appear to move toward each other, and much of the V1/V2 region (with the exception of L1 tip and its neighboring partial β-strands) moves toward the structural core, thus resulting in the overall contraction of gp120 structure and the shrink of the hydrophobic cavity. The second mode of the unliganded gp120 (Fig. 5B) is characterized by a common anticlockwise rotation of the inner domain, the outer domain, and the V1/V2 region around an axis running through the center of the hydrophobic cavity; such a vortex-like mode results in the twisting of gp120 structure. The mode along the third eigenvector (Fig. 5C) seems to elongate gp120 structure to some extent since much of the V1/V2 region and the upper half of the inner domain move collectively in the left-up direction while the lower halves of the inner and outer domains move in the right-down direction. The fourth mode (Fig. 5D) is slightly more complicated: V1/V2 and its neighboring core regions rotate collectively to form a small clockwise vortex, thus twisting the upper part of the hydrophobic cavity; the lower halves of the inner and outer domains exhibit a trend to move apart from each other, thereby widening to some extent the lower part of the structural core.
For the CD4-complexed gp120, the first ranked motion mode (Fig. 5E) is characterized by a large vortex formed by common rotations of the structural core and its peripheral loops around an axis parallel to the inner and outer domains; while the distant V1/V2 region and N-, C-termini move with the largest amplitudes in the downward and right-up directions, respectively. In the second mode (Fig. 5F), much of the structural core shifts in the leftward direction with the exception of the layers 1 and 2, which move in the rightward and right-down directions, respectively, thus widening the hydrophobic cavity to some extent; of note is that the V3 loop does not moves in concert with the core but shifts with the largest amplitude in the opposite direction relative to that of the core. In the third (Fig. 5G) and fourth modes (Fig. 5H), the gp120 core exhibits reduced fluctuation amplitudes when compared to those in the first two modes, whereas V1/V2 and V3 still have the largest amplitudes. Nevertheless, close inspection reveals that the collective motions along eigenvector 3 slightly widen the hydrophobic cavity, while those along eigenvector 4 lead to a slight twisting of the core.
Free energy landscape
Fig. 6 shows the FELs of the unliganded and CD4-complexed gp120s using the projection of the joined equilibrium trajectory onto the first two eigenvectors as the reaction coordinates. Note that for the unliganded and CD4-complexed gp120s, the first two eigenvectors cumulatively contribute 40.5% and 51.8% to TMSF, respectively. Our previous work28,32,33 and other studies26,34 have shown that the first few eigenvector projections are good reaction coordinates capable of distinguishing among different conformational states/substates sampled by MD simulations; furthermore, the evaluation of conformational sampling with cosine contents of the first few eigenvectors indicates that our μs-scale multiple-replica MD simulations have achieved a sufficient convergence of the conformational sampling. Therefore, our constructed FELs are accurate enough to provide reliable information on the thermodynamics and kinetics of the two forms of gp120 near their starting states. | ||
| Fig. 6 Free energy landscapes (FELs) of the two gp120 forms as a function of the projection of the joined equilibrium trajectory onto the two-dimensional essential subspace spanned by eigenvectors 1 and 2. (A) FEL of the unliganded gp120. (B) FEL of the CD4-complexed gp120. The color bar denotes the relative free-energy level in kJ mol−1. | ||
It is clear from Fig. 6 that the FEL of the CD4-complexed gp120 (Fig. 6B) exhibits a significantly larger free-energy surface than that of the unliganded gp120 (Fig. 6A), e.g., the former spans from −18 to 18 nm and from −12 to 13 nm along eigenvectors 1 and 2, respectively, while the corresponding spanning ranges of the latter are between −6 and 11 nm and between −7 and 5 nm, respectively. As a result, the CD4-complexed gp120 sampled a larger range in the essential subspace during MD simulations, thus implying an increase in gp120 conformational entropy upon CD4 binding. Furthermore, the CD4-complexed FEL features a more rough/rugged surface due to its more free-energy minima/basins compared to the unliganded FEL. There are about seven and three discernible minima with a free-energy level lower than −12 kJ mol−1 in FELs of the CD4-complexed and unliganded gp120s, respectively, indicating that CD4 binding greatly increases the conformational diversity and kinetic complexity of gp120. If those with a free-energy level lower than −14 kJ mol−1 are considered, only one single basin can be observed in the unliganded FEL, while there are five minima in the CD4-complexed FEL. Interestingly, the single large basin has a larger size and a lower minimum free-energy value (−16 kJ mol−1) than each of the five minima, indicating that the main (single) substate of the unliganded gp120 has a larger population and is more thermally stable than the main substates of the CD4-complexed gp120.
Discussion
In this paper, two structural models, the near full-length monomeric gp120 and the gp120-CD4 complex were subjected to multiple-replica MD simulations to investigate the differences in the conformational dynamics, molecular motions, and thermodynamics between the unliganded and CD4-bound forms of gp120. Although it can be expected that the type and size of glycans would influence the fluctuations of the protein regions neighboring the N-linked glycosylation sites due to steric hindrance and weight contribution from glycans, previous MD simulation studies on gp120s with the glycosylated and non-glycosylated V3 loop showed no significant differences in the Cα-atom fluctuations between these two forms of gp120, especially between V3 loops.35,36 Therefore, comparative MD simulations of the unliganded gp120 and the gp120-CD4 complex, despite containing no glycan, could most likely reflect the true differences in the conformational dynamics and thermodynamics between the unliganded and CD4-bound forms of gp120.Comparative analyses of the joined equilibrium trajectories in terms of RMSD, structural/geometrical parameters, RMSF, and FEL point to a common conclusion: the CD4-complexed gp120 is more structurally unstable and conformationally flexible/mobile than the unliganded gp120. This, together with the smFRET data evidencing that the unliganded, closed state is most prevalent among the three states (i.e., the unliganded and CD4-bound states and the transition intermediate between them) of the ligand-free Env/gp120,25 suggests that the unliganded state is the stable “ground state” of gp120. This is contradictory to previous experimental8 and MD studies,37 which argue that the CD4-bound conformation is the ground state since (i) the unliganded state is only maintained by quaternary restraints in the context of Env trimer, whereas the monomeric gp120 tends to spontaneously assume the CD4-bound state; and (ii) the CD4-bound state adopts a more compact and stable conformation than the unliganded state, and gp120 can transition spontaneously from the unliganded to CD4-bound states due to a lower free-energy level of the CD4-bound state. The reason for this contradictory is that whether the truncated or full-length gp120 was used in specific studies. In ref. 8, all the four X-ray crystallographically determined structures (referred to as the extended core or coree) lack the V1/V2 region while containing the V3 stem, which forms hydrogen bonds with neighboring residues on the β20–β21 stem and hence stabilizes the mature bridging sheet (β3–β2–β21–β20) and confines the coree conformation in the CD4-bound state. In,37 the two homology models used for MD simulations, although including intact V3, also do not contain V1/V2; moreover, the unliganded model of the HIV-1 gp120 core was built using the X-ray structure of the ligand-free/unliganded simian immunodeficiency virus (SIV) gp120 core38 as the template, which has a low sequence identity with the target (35%) and differs distinctly in orientation and packing of certain secondary structural elements from the CD4-bound model of the HIV-1 gp120 core. For example, the two β-hairpins, β2–β3 and β20–β21, are separated by a long distance of ∼23 Å in the unliganded SIV gp120 core, while in the CD4-bound HIV-1 gp120 core are arranged alongside one another to form the mature bridging sheet. As a result, it is not surprising that the loosely packed structural model of the unliganded state was more unstable than the compactly packed model of the CD4-bound one during MD simulations.
In this work, the near full-length gp120 models contain both the V3 and V1/V2 regions. Although the unliganded model was generated from a template extracted from the Env trimer crystallographic structure, small-angle X-ray scattering data39 and MD simulations40 suggest that the monomeric, full-length, ligand-free gp120 structure closely resembles the conformation of the gp120 subunit in the unliganded Env trimer. Actually, for both the present unliganded and CD4-bound gp120 models, their structural cores are highly similar to each other (with Cα RMSD value of 0.89 Å) and to the experimentally determined unliganded gp120 coree structures (Cα RMSDs < 1.4 Å),8 whereas the most pronounced differences lie in the relative orientations of V1/V2 and V3 with respect to each other and to the structural core and, in the ordering and positioning of the bridging-sheet elements (Fig. 1). These on the one hand explain why the X-ray structures of the unliganded (or ligand-free) gp120 coree assume the so-called CD4-bound conformation (which is very similar to the unliganded conformation regarding merely the structural core, i.e., the conformations of the structural core are very similar in the unliganded and CD4-bound forms of gp120); on the other hand, indicate that the mutual orientation between V1/V2 and V3 is a crucial determinant for discriminating between the unliganded and CD4-bound states of the full-length gp120.
As can be seen from Fig. 3, the regions V3 and V1/V2 have significantly higher flexibility in the CD4-complexed gp120 than in the unliganded form. A significant increase in the mobility of V1/V2 and V3 upon CD4 binding was also observed in a recent MD study.41 These results provide an explanation for why the structure of the V1/V2/V3-containing gp120 monomer has resisted crystallization in the CD4-bound state.8,29 Moreover, despite the complete departure of these two regions from the core of the CD4-complexed gp120, the V3 loop connects between β12 and β13 that are located at the center of the distal end β-barrel in the outer domain, and the V1/V2 region is tied to the inner domain via the bridging-sheet element β2–β3. It is highly possible that the strong mobility (or high kinetic energy) of V1/V2 and V3 could be transmitted via the neighborhood effect to these β-strands, for which the increased mobility could be further propagated to the other regions of the structural core via either the network of intramolecular interactions or the hinge-bending mechanism,33,42,43 ultimately enhancing the global flexibility of the CD4-complexed gp120. As a result, when compared to the unliganded gp120, the observed increased conformational dynamics of the CD4-complexed gp120 could be attributed to the greatly increased mobility/flexibility of V1/V2 and V3 arising from the dissociation and full exposure of them. Interestingly, it has been shown that specific amino acid substitutions in V1/V2 and V3 capable of weakening the attractive interactions among these loops in the unliganded state can lead to increases in the viral infectivity and neutralization sensitivity to anti-V3 antibodies, indicating that the enhanced dynamics of V1/V2 and V3 caused by their easier dissociation can mechanically modulate the biological phenotypes of HIV-1.40 This is supported by another study using MD simulations combined with the Markov state model construction, which showed that the destabilization of the hydrophobic core flanked by V1/V2 and V3 could promote transition of gp120 from the closed unliganded state to the open CD4-bound state.41
What are the functional consequences of the observed differences in the conformational dynamics and thermodynamics between the unliganded and CD4-complexed gp120s? Before CD4 binding, the gp120 conformation is well maintained near the closed unliganded state by stable intramolecular interactions and compact packing, with a small probability to sample the other states/substates (Fig. 6A).25,32 In such a stable ground state, the vulnerable epitopes recognized by the highly potent neutralizing antibodies are buried, whereas the immunodominant epitopes recognized by the non-neutralizing or less potent antibodies are exposed.16,44,45 Therefore, before viral infection, the stable maintenance of gp120 close to the unliganded state provides a thermodynamic basis for HIV-1 neutralization resistance. However, the unliganded gp120 still retains the capacity to bind to CD4 or transition to the CD4-bound state with or without induction by CD4. This is because (i) the large hydrophobic cavity occupied by CD4 in the CD4-complexed gp120 also presents in the unliganded gp120, although the differences in the cavity architecture do exist; (ii) the tip of the bridging-sheet β-hairpin β20–β21 exhibits higher flexibility in the unliganded gp120 (Fig. 3), thus increasing the probabilities of disrupting the premature bridging sheet and forming the CD4-Phe43-binding pocket,46,47 which allow the repositioning of the bridging-sheet elements and selective binding of CD4;48 (iii) in the largest-amplitude motion modes of the unliganded gp120, although the entire V1/V2 region fluctuates in concert with the structural core, some of its structural elements still exhibit the tendency to detach from the core, e.g., in the modes along eigenvectors 1 to 3 (Fig. 5A-C), the L1 loop and its neighboring partial β-strands move with a large amplitude in the directions that may help to disrupt the adhesion to the V3 loop, and in the mode along eigenvector 4 (Fig. 5D), the twisting of V1/V2 will result in the departure of the loop V1 from while the approach of L1 to the core; and (iv) the contraction, twisting, elongation, or widening of the entire structure/hydrophobic cavity arising from the collective motions of the unliganded gp120 may either contribute to maintaining the unliganded state or be conducive to the initial association with CD4 or even initiating the spontaneous transition to the CD4-bound state.32
Upon CD4 binding, the gp120 structure becomes unstable, exhibiting increased flexibility, more diversified motion modes, and more complicated thermodynamic and kinetic behaviors compared to that before CD4 binding. This seems to be a double-edged sword regarding the gp120 functions. The high mobility of the protruding V3 has been considered crucial in sensing and trapping the cell-surface coreceptor (CCR5 or CXCR4),16,41,49 whose binding was thought to promote additional conformational changes in gp41 to establish an energetically stable six-helix bundle for virus-cell membrane fusion.20,21,31 In the context of the Env trimer, one gp41 subunit associates non-covalently with one gp120 subunit to form a heterodimer/protomer50,51 and, furthermore, a “priming network”, which spans from the CD4-binding cavity via layers 1 to 3 of the gp120 inner domain to the HR1 of gp41, has been proposed to be responsible for the inter-subunit communication and priming the gp41 fusogenic properties.15,31 Therefore, it is possible that the observed increased mobility of the network-forming regions in gp120 upon CD4 binding signifies the activation of this network, which in turn will facilitate the formation of the gp41 prehairpin intermediate.21
In the CD4-bound state, the mature bridging sheet, which forms not only a part of the coreceptor-binding site but is also the major target of the CD4-induced antibodies, has been formed and fully exposed. Therefore, the CD4-bound state harbors the risk of being neutralized if the coreceptor is not timely trapped; nevertheless, since the bridging sheet is located at the base of the “Y”-orientated V1/V2 and V3 (Fig. 1C), the high mobility of these two regions increases, to some extent, the probability of shielding the bridging sheet, thus lowering the risk of being neutralized. Indeed, in our previous MD study,28 we found that the increased conformational diversity of the CD4-bound state arose mainly from the variable orientation between V1/V2 and V3, with closely adjacent orientations that occlude the entrance to the bridging sheet being observed in some of the sampled substates. The MD simulations of the CD4-bound gp120 conducted by Da and Lin also revealed that the V3 loop was able to explore multiple orientations relative to V1/V2 and the structural core, with its closed orientation covering the mature bridging sheet.41 Furthermore, it is also possible that the enhanced mobility of the other core-peripheral regions could impart increased lability to certain antibody-binding epitopes, thus disturbing the recognition and binding of relevant antibodies to gp120 in the CD4-bound state.
Notably, the enhanced dynamics upon CD4 binding also bring about disadvantageous consequences to gp120. Although the widening and twisting of the CD4-binding cavity could be conducive to the orientation adjustment of CD4, the increased fluctuations of the cavity-forming residues increase the likelihood of the cavity deformation and hence facilitate the dissociation of CD4 from gp120. Once this occurs, the neutralization-sensitive epitopes such as CD4-induced and CD4-binding-site ones become fully exposed, thus increasing the probability of antibody-mediated neutralization. Nevertheless, a remediation measure still exists, as the smFRET data25 and our constructed FELs28 indicate that CD4 removal could shift the gp120 conformational equilibrium from the activated CD4-bound state toward the inactive unliganded state (or ground state). Moreover, the CD4-induced inactivation has been proposed to be primarily a consequence of gp120 shedding from the Env trimer,52–54i.e., upon CD4 binding and achieving the CD4-bound state, the gp120 subunit will shed from the trimer with a high probability if coreceptor binding is impeded or delayed. Although the weak noncovalent association between gp120 and gp41 is necessary for gp120 shedding, we highlight that the greatly enhanced conformational flexibility/dynamics of the CD4-complexed gp120 in comparison to the unliganded gp120 is a sufficient condition for its release when activated.
Materials and methods
Structural models
The 3D structure of the gp120-CD4 complex was extracted from an atomistic model of HIV Env trimer obtained from Protein Data Bank (PDB) (http://www.rcsb.org) with PDB ID 3J70,18 where the gp120 subunits contain all variable loops and are in complex with CD4 and antibody 17b. It should be noted that although 3J70 is a computationally constructed structural model, the segments (core, V1/V2, V3, V4, and N- and C-termini) used for the assembly of the complete gp120 structure have relatively high resolutions ranging from 1.89 to 3.51 Å, and the low-resolution cryo-electron microscopy map (20 Å) was only used for fitting of the gp120 model, CD4, and 17b to form the trimeric complex, for which the quality assessment revealed a high stereochemical quality corresponding to high-resolution (<2 Å) structures.18The unliganded gp120 structural model was constructed using the homology modeling procedure implemented in MODELLER v9.17.55 The gp160 sequence of the HIV-1 HXBc2 isolate (clade B) was obtained from UniProtKB database (http://www.uniprot.org) with accession number P04578. Residues of the signal peptide, partial gp120 N-terminus, and gp41 were removed, and the finally obtained gp120 target sequence comprises 462 amino acid residues. The gp120 atomic coordinate extracted from a closed unliganded Env trimer crystal structure (PDB ID: 5FYJ (chain G); 3.11 Å resolution56) from HIV-1 clade G was used as the template. Despite the different clades of gp120, the high sequence identity (67%) and few amino acid insertions and deletions between the target and template ensure the reliability of homology modeling. After aligning the target sequence to the template sequence (Fig. S1, ESI†), 20 structural models were generated, followed by a refinement using molecular dynamics simulated annealing. Finally, only the model with the lowest molecular probability density function score was selected as the “best” unliganded model, for which the stereochemistry was checked using PROCHECK,57 with the result (Fig. S2, ESI†) demonstrating a good stereochemical quality.
MD simulations
All MD simulations were performed using GROMACS 5.1.458 with the AMBER99SB-ILDN force field.59 Before simulations, the structural models of the unliganded gp120 and gp120-CD4 complex were separately solvated in a periodic dodecahedron box of TIP3P water model60 with a minimum solute-wall distance of 1.18 and 0.85 nm, respectively. Cl− (134 and 155 for the systems of the unliganded gp120 and gp120-CD4 complex, respectively) and Na+ (138 and 155 for these two systems, respectively) ions were also introduced to achieve the electroneutral system at a salt concentration of 150 mM. Each system was subjected to a steepest descent energy minimization until no significant energy change could be detected, followed by four consecutive 200 ps position-restrained MD simulations with protein heavy atoms restrained by decreasing harmonic potential force constants (Kposres = 1000, 100, 10, and 0 kJ mol−1 nm−2) to effectively “soak” the solute into the solvent.61To improve the conformational sampling efficiency, each system was subjected to the multi-replica production MD simulations,34 where 10 independent 100 ns MD runs (with an accumulative simulation time of 1 μs) were initialized with different atomic initial velocities assigned from a Maxwell distribution at 300 K. The protocols used in the production runs were as follows: integration time step was 2 fs with the LINCS algorithm62 to constrain all bond lengths; the long-range electrostatic interactions were handled using the particle-mesh Ewald (PME) method,63 with a real-space cut-off of 1.0 nm, Fourier grid spacing of 0.12 nm, and interpolation order of 4; the van der Waals (vdW) interactions were treated using twin-range cut-offs of 1.0 nm for the short-range neighbor list and 1.4 nm for the vdW cut-off, respectively; the solute and solvent were separately coupled to the velocity-rescaling thermostat64 with a reference temperature of 300 K and a time constant of 0.1 ps; the Parrinello–Rahman barostat65 was used to maintain system pressure at 1 atm with a time constant of 0.5 ps; and the system coordinates were saved every 2 ps.
Analysis methods
The degree of sampling convergence was assessed by calculating the cosine contents of the first few eigenvectors derived from ED analyses of MD trajectories. When the cosine contents of the first few eigenvectors are close to 1, the largest-amplitude protein motions during a simulation resemble random diffusion, representative of insufficient sampling;26,33,66 whereas the cosine contents close to 0 represent an adequate convergence of sampling in the MD simulation.RMSD, RMSF, and the structural/geometrical parameters including NHB, NCIC, SASA, Rg, and SSC, were calculated using a variety of trajectory analysis tools implemented in GROMACS. The ED method, which is based on the diagonalization of the covariance matrix built from atomic fluctuations in a MD trajectory, was used to study the collective motions of gp120 structural models; the obtained eigenvectors and corresponding eigenvalues represent the motion modes in the sampled conformational space and the atomic fluctuation amplitudes along the corresponding modes, respectively. The GROMACS tools “gmx covar” and “gmx anaeig” were employed to build/diagonalize the Cα covariance matrix and to project the trajectory onto the eigenvectors, respectively. The porcupine plots, which were used to visualize the largest-amplitude motion modes along the first few eigenvectors, were obtained using the modevectors.py script (available from: https://pymolwiki.org/index.php/Modevectors) with the two extremes along an eigenvector projection as the input. The two-dimensional FELs using the first two eigenvectors as reaction coordinates were reconstructed by the equation F(s) = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(Ni/Nmax), where kB is the Boltzmann's constant, T is the simulation temperature in Kelvin, Ni is an estimate of the probability density function defining the probability of finding the system in a particular state i, and Nmax are the probability of the most probable state. The rationale of the above equation is that the probability of finding the system in a particular state characterized by the reaction coordinate is proportional to e−F(s)/kBT, where F(s) is the free energy of the state.
ln(Ni/Nmax), where kB is the Boltzmann's constant, T is the simulation temperature in Kelvin, Ni is an estimate of the probability density function defining the probability of finding the system in a particular state i, and Nmax are the probability of the most probable state. The rationale of the above equation is that the probability of finding the system in a particular state characterized by the reaction coordinate is proportional to e−F(s)/kBT, where F(s) is the free energy of the state.
Conclusions
Through performing μs-scale multiple-replica MD simulations on structural models of the near full-length unliganded gp120 and the gp120-CD4 complex, we found that CD4 binding promotes the global structural deviations/changes, loosens the structural packing, enhances the overall conformational flexibility, complicates the molecular motions, and increases the conformational entropy/diversity while lowering the thermostability of gp120. Our results, in conjunction with the previous smFRET study evidencing that the unliganded conformation was the most populated among the three observed states of the ligand-free Env/gp120,25 lead us to conclude that the unliganded and CD4-bound conformations should be the stable “ground state” and unstable “excited state” of gp120, respectively. The relative orientations of V1/V2 and V3 with respect to each other and to the gp120 core are not only the major hallmark for distinguishing between the unliganded and CD4-bound states, but also determine the differences in the conformational dynamics and thermodynamics between these two states. Although the results presented in this study were obtained by MD simulations of the structural models of gp120 from the CD4-dependent but neutralization-sensitive HXBc2 isolate, they extend in detail and in generality a better understanding of the functional and immunological properties of HIV-1 gp120: (i) both the stable maintenance of the unliganded state and the increased conformational dynamics/flexibility/diversity of the CD4-bound state provide thermodynamic advantages for immune evasion, although gp120 from the neutralization-sensitive isolate is more structurally unstable/flexible and prone to spontaneous transition to the CD4-bound state than that from the neutralization-resistant isolate;32 (ii) for the unliganded gp120, the observed high mobility of the β20–β21 hairpin tip and the outward-moving trend of V1/V2 endow gp120 with the capacity to transition to the CD4-bound state with or without induction by CD4; (iii) for the CD4-complexed/bound gp120, its enhanced conformational dynamics would on the one hand be advantageous for the gp120-coreceptor association and gp120–gp41 communication necessary for triggering the gp41-entry machinery and, on the other hand, bring about the side effect of spontaneous inactivation via facilitating gp120 shedding from the Env trimer. Last but not least, we propose several strategies for the development of anti-HIV-1 drugs or vaccines: (i) engineering the conformationally fixed Env trimers as vaccine antigens that could elicit the production of Env-directed antibodies with potent neutralization activity against the gp120 unliganded state; (ii) discovering or designing small molecules as potential inhibitors that could effectively locks gp120 into the unliganded state via either fixing the association between V1/V2 and V3 or suppressing fluctuations of the β20–β21 hairpin, thereby blocking the transition of the Env trimer to the fusion-active state; (iii) developing CD4-mimetic compounds capable of competitively binding to gp120 with a higher affinity than the cell-surface CD4 so as to inactivate HIV-1 via prematurely triggering the unstable CD4-bound state of gp120 (and hence its shedding) before attaching to the cell surface.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been funded by the National Natural Sciences Foundation of China (No. 31370715) and Program for Donglu Scholar in the Yunnan University. We acknowledge the computational support from the High Performance Computing Center, Yunnan University.Notes and references
- A. B. Ward and I. A. Wilson, Immunol. Rev., 2017, 275, 21–32 CrossRef CAS PubMed.
- A. B. Ward and I. A. Wilson, Trends Biochem. Sci., 2015, 40, 101–107 CrossRef CAS PubMed.
- R. W. Doms and J. P. Moore, J. Cell Biol., 2000, 151, F9–F14 CrossRef CAS PubMed.
- J. S. McLellan, M. Pancera, C. Carrico, J. Gorman, J. P. Julien, R. Khayat, R. Louder, R. Pejchal, M. Sastry, K. Dai, S. O'Dell, N. Patel, S. Shahzad-ul-Hussan, Y. Yang, B. Zhang, T. Zhou, J. Zhu, J. C. Boyington, G. Y. Chuang, D. Diwanji, I. Georgiev, Y. D. Kwon, D. Lee, M. K. Louder, S. Moquin, S. D. Schmidt, Z. Y. Yang, M. Bonsignori, J. A. Crump, S. H. Kapiga, N. E. Sam, B. F. Haynes, D. R. Burton, W. C. Koff, L. M. Walker, S. Phogat, R. Wyatt, J. Orwenyo, L. X. Wang, J. Arthos, C. A. Bewley, J. R. Mascola, G. J. Nabel, W. R. Schief, A. B. Ward, I. A. Wilson and P. D. Kwong, Nature, 2011, 480, 336–343 CrossRef CAS PubMed.
- R. Pantophlet and D. R. Burton, Annu. Rev. Immunol., 2006, 24, 739–769 CrossRef CAS PubMed.
- P. Zhan, C. Pannecouque, E. De Clercq and X. Liu, J. Med. Chem., 2016, 59, 2849–2878 CrossRef CAS PubMed.
- D. M. Eckert and P. S. Kim, Annu. Rev. Biochem., 2001, 70, 777–810 CrossRef CAS PubMed.
- Y. D. Kwon, A. Finzi, X. Wu, C. Dogo-Isonagie, L. K. Lee, L. R. Moore, S. D. Schmidt, J. Stuckey, Y. Yang, T. Zhou, J. Zhu, D. A. Vicic, A. K. Debnath, L. Shapiro, C. A. Bewley, J. R. Mascola, J. G. Sodroski and P. D. Kwong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5663–5668 CrossRef CAS PubMed.
- Q. J. Sattentau, Structure, 1998, 6, 945–949 CrossRef CAS PubMed.
- E. A. Berger, P. M. Murphy and J. M. Farber, Annu. Rev. Immunol., 1999, 17, 657–700 CrossRef CAS PubMed.
- M. Pancera, T. Zhou, A. Druz, I. S. Georgiev, C. Soto, J. Gorman, J. Huang, P. Acharya, G. Y. Chuang, G. Ofek, G. B. Stewart-Jones, J. Stuckey, R. T. Bailer, M. G. Joyce, M. K. Louder, N. Tumba, Y. Yang, B. Zhang, M. S. Cohen, B. F. Haynes, J. R. Mascola, L. Morris, J. B. Munro, S. C. Blanchard, W. Mothes, M. Connors and P. D. Kwong, Nature, 2014, 514, 455–461 CrossRef CAS PubMed.
- L. Rutten, Y. T. Lai, S. Blokland, D. Truan, I. J. M. Bisschop, N. M. Strokappe, A. Koornneef, D. van Manen, G. Y. Chuang, S. K. Farney, H. Schuitemaker, P. D. Kwong and J. P. M. Langedijk, Cell Rep., 2018, 23, 584–595 CrossRef CAS PubMed.
- J. P. Julien, A. Cupo, D. Sok, R. L. Stanfield, D. Lyumkis, M. C. Deller, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, A. B. Ward and I. A. Wilson, Science, 2013, 342, 1477–1483 CrossRef CAS PubMed.
- D. Lyumkis, J. P. Julien, N. de Val, A. Cupo, C. S. Potter, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, B. Carragher, I. A. Wilson and A. B. Ward, Science, 2013, 342, 1484–1490 CrossRef CAS PubMed.
- M. Guttman, N. K. Garcia, A. Cupo, T. Matsui, J. P. Julien, R. W. Sanders, I. A. Wilson, J. P. Moore and K. K. Lee, Structure, 2014, 22, 974–984 CrossRef CAS PubMed.
- C. C. Huang, M. Tang, M. Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski, I. A. Wilson, R. Wyatt and P. D. Kwong, Science, 2005, 310, 1025–1028 CrossRef CAS PubMed.
- L. Wu, N. P. Gerard, R. Wyatt, H. Choe, C. Parolin, N. Ruffing, A. Borsetti, A. A. Cardoso, E. Desjardin, W. Newman, C. Gerard and J. Sodroski, Nature, 1996, 384, 384179 CrossRef PubMed.
- M. Rasheed, R. Bettadapura and C. Bajaj, Structure, 2015, 23, 1138–1149 CrossRef CAS PubMed.
- H. Wang, A. A. Cohen, R. P. Galimidi, H. B. Gristick, G. J. Jensen and P. J. Bjorkman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7151–E7158 CrossRef CAS PubMed.
- G. Ozorowski, J. Pallesen, N. de Val, D. Lyumkis, C. A. Cottrell, J. L. Torres, J. Copps, R. L. Stanfield, A. Cupo, P. Pugach, J. P. Moore, I. A. Wilson and A. B. Ward, Nature, 2017, 547, 360–363 CrossRef CAS PubMed.
- R. Wyatt and J. Sodroski, Science, 1998, 280, 1884–1888 CrossRef CAS PubMed.
- L. Scharf, H. Wang, H. Gao, S. Chen, A. W. McDowall and P. J. Bjorkman, Cell, 2015, 162, 1379–1390 CrossRef CAS PubMed.
- A. Bartesaghi, A. Merk, M. J. Borgnia, J. L. Milne and S. Subramaniam, Nat. Struct. Mol. Biol., 2013, 20, 1352–1357 CrossRef CAS PubMed.
- D. G. Myszka, R. W. Sweet, P. Hensley, M. Brigham-Burke, P. D. Kwong, W. A. Hendrickson, R. Wyatt, J. Sodroski and M. L. Doyle, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9026–9031 CrossRef CAS PubMed.
- J. B. Munro, J. Gorman, X. Ma, Z. Zhou, J. Arthos, D. R. Burton, W. C. Koff, J. R. Courter, A. B. Smith, 3rd, P. D. Kwong, S. C. Blanchard and W. Mothes, Science, 2014, 346, 759–763 CrossRef CAS PubMed.
- E. Papaleo, P. Mereghetti, P. Fantucci, R. Grandori and L. De Gioia, J. Mol. Graphics Modell., 2009, 27, 889–899 CrossRef CAS PubMed.
- I. Tavernelli, S. Cotesta and E. E. Di Iorio, Biophys. J., 2003, 85, 2641–2649 CrossRef CAS PubMed.
- Y. Li, L. Deng, L. Q. Yang, P. Sang and S. Q. Liu, Int. J. Mol. Sci., 2019, 20, 260 CrossRef PubMed.
- P. D. Kwong, R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski and W. A. Hendrickson, Nature, 1998, 393, 648–659 CrossRef CAS PubMed.
- P. D. Kwong, R. Wyatt, S. Majeed, J. Robinson, R. W. Sweet, J. Sodroski and W. A. Hendrickson, Structure, 2000, 8, 1329–1339 CrossRef CAS PubMed.
- M. Pancera, S. Majeed, Y. E. A. Ban, L. Chen, C. C. Huang, L. Kong, Y. D. Kwon, J. Stuckey, T. Zhou, J. E. Robinson, W. R. Schief, J. Sodroski, R. Wyatt and P. D. Kwong, Proc. Natl. Acad. Sci. U. S. A., 2009, 107, 1166–1171 CrossRef PubMed.
- Y. Li, L. Deng, S. M. Ai, P. Sang, J. Yang, Y. L. Xia, Z. B. Zhang, Y. X. Fu and S. Q. Liu, RSC Adv., 2018, 8, 14355–14368 RSC.
- P. Sang, Q. Yang, X. Du, N. Yang, L. Q. Yang, X. L. Ji, Y. X. Fu, Z. H. Meng and S. Q. Liu, Int. J. Mol. Sci., 2016, 17, 254 CrossRef PubMed.
- L. S. Caves, J. D. Evanseck and M. Karplus, Protein Sci., 1998, 7, 649–666 CrossRef CAS PubMed.
- N. T. Wood, E. Fadda, R. Davis, O. C. Grant, J. C. Martin, R. J. Woods and S. A. Travers, PLoS One, 2013, 8, e80301 CrossRef PubMed.
- M. Yokoyama, S. Naganawa, K. Yoshimura, S. Matsushita and H. Sato, PLoS One, 2012, 7, e37530 CrossRef CAS PubMed.
- P. Sang, L. Q. Yang, X. L. Ji, Y. X. Fu and S. Q. Liu, PLoS One, 2014, 9, e104714 CrossRef PubMed.
- B. Chen, E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley and S. C. Harrison, Nature, 2005, 433, 834–841 CrossRef CAS PubMed.
- M. Guttman, M. Kahn, N. K. Garcia, S. L. Hu and K. K. Lee, J. Virol., 2012, 86, 8750–8764 CrossRef CAS PubMed.
- M. Yokoyama, M. Nomaguchi, N. Doi, T. Kanda, A. Adachi and H. Sato, Front. Microbiol., 2016, 7, 110 Search PubMed.
- L. T. Da and M. Lin, Phys. Chem. Chem. Phys., 2019, 21, 26003–26016 RSC.
- Y. Tao, Z. H. Rao and S. Q. Liu, J. Biomol. Struct. Dyn., 2010, 28, 143–158 CrossRef CAS PubMed.
- L. Q. Yang, P. Sang, Y. Tao, Y. X. Fu, K. Q. Zhang, Y. H. Xie and S. Q. Liu, J. Biomol. Struct. Dyn., 2014, 32, 372–393 CrossRef CAS PubMed.
- T. R. Fouts, J. M. Binley, A. Trkola, J. E. Robinson and J. P. Moore, J. Virol., 1997, 71, 2779–2785 CrossRef CAS PubMed.
- Y. D. Kwon, M. Pancera, P. Acharya, I. S. Georgiev, E. T. Crooks, J. Gorman, M. G. Joyce, M. Guttman, X. Ma, S. Narpala, C. Soto, D. S. Terry, Y. Yang, T. Zhou, G. Ahlsen, R. T. Bailer, M. Chambers, G. Y. Chuang, N. A. Doria-Rose, A. Druz, M. A. Hallen, A. Harned, T. Kirys, M. K. Louder, S. O'Dell, G. Ofek, K. Osawa, M. Prabhakaran, M. Sastry, G. B. Stewart-Jones, J. Stuckey, P. V. Thomas, T. Tittley, C. Williams, B. Zhang, H. Zhao, Z. Zhou, B. R. Donald, L. K. Lee, S. Zolla-Pazner, U. Baxa, A. Schon, E. Freire, L. Shapiro, K. K. Lee, J. Arthos, J. B. Munro, S. C. Blanchard, W. Mothes, J. M. Binley, A. B. McDermott, J. R. Mascola and P. D. Kwong, Nat. Struct. Mol. Biol., 2015, 22, 522–531 CrossRef PubMed.
- D. R. Langley, S. R. Kimura, P. Sivaprakasam, N. Zhou, I. Dicker, B. Mcauliffe, T. Wang, J. F. Kadow, N. A. Meanwell and M. Krystal, Proteins, 2015, 83, 331–350 CrossRef CAS PubMed.
- I. H. Shrivastava, K. Wendel and J. M. LaLonde, Biochemistry, 2012, 51, 7783–7793 CrossRef CAS PubMed.
- A. Herschhorn, C. Gu, F. Moraca, X. Ma, M. Farrell, A. B. Smith, M. Pancera, P. D. Kwong, A. Schön, E. Freire, C. Abrams, S. C. Blanchard, W. Mothes and J. G. Sodroski, Nat. Commun., 2017, 8, 1049 CrossRef PubMed.
- S. Liu, S. Fan and Z. Sun, J. Mol. Model., 2003, 9, 329–336 CrossRef CAS PubMed.
- P. Zhu, H. Winkler, E. Chertova, K. A. Taylor and K. H. Rouxl, PLoS Pathog., 2008, 4, e1000203 CrossRef PubMed.
- P. L. Earl, R. W. Doms and B. Moss, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 648–652 CrossRef CAS PubMed.
- H. Haim, Z. Si, N. Madani, L. Wang, J. R. Courter, A. Princiotto, A. Kassa, M. DeGrace, K. McGee-Estrada, M. Mefford, D. Gabuzda, A. B. Smith, 3rd and J. Sodroski, PLoS Pathog., 2009, 5, e1000360 CrossRef PubMed.
- T. K. Hart, R. Kirsh, H. Ellens, R. W. Sweet, D. M. Lambert, S. R. J. Petteway, J. Leary and P. J. Bugelski, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 2189–2193 CrossRef CAS PubMed.
- S. P. Layne, M. J. Merges, M. Dembo, J. L. Spouge, S. R. Conley, J. P. Moore, J. L. Raina, H. Renz, H. R. Gelderblom and P. L. Nara, Virology, 1992, 189, 695–714 CrossRef CAS PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 Search PubMed.
- G. B. Stewart-Jones, C. Soto, T. Lemmin, G. Y. Chuang, A. Druz, R. Kong, P. V. Thomas, K. Wagh, T. Zhou, A. J. Behrens, T. Bylund, C. W. Choi, J. R. Davison, I. S. Georgiev, M. G. Joyce, Y. D. Kwon, M. Pancera, J. Taft, Y. Yang, B. Zhang, S. S. Shivatare, V. S. Shivatare, C. C. Lee, C. Y. Wu, C. A. Bewley, D. R. Burton, W. C. Koff, M. Connors, M. Crispin, U. Baxa, B. T. Korber, C. H. Wong, J. R. Mascola and P. D. Kwong, Cell, 2016, 165, 813–826 CrossRef CAS PubMed.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Proteins, 2014, 82, 195–215 CrossRef CAS PubMed.
- D. J. Price and C. L. Brooks III, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed.
- Y. L. Xia, J. H. Sun, S. M. Ai, Y. Li, X. Du, P. Sang, L. Q. Yang, Y. X. Fu and S. Q. Liu, RSC Adv., 2018, 8, 29698–29713 RSC.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 65, 031910 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp06706j |
| This journal is © the Owner Societies 2020 |