
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H-Bonding-mediated binding and charge reorganization of proteins on gold nanoparticles†
Brahmaiah
Meesaragandla
ab,
Isabel
García
 c,
Doreen
Biedenweg
b,
Jhoan
Toro-Mendoza
c,
Ivan
Coluzza
cd,
Luis M.
Liz-Marzán
cd and
Mihaela
Delcea
*abe
c,
Doreen
Biedenweg
b,
Jhoan
Toro-Mendoza
c,
Ivan
Coluzza
cd,
Luis M.
Liz-Marzán
cd and
Mihaela
Delcea
*abe
aInstitute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17489 Greifswald, Germany. E-mail: delceam@uni-greifswald.de
bZIK HIKE – Center for Innovation Competence “Humoral Immune Reactions in Cardiovascular Diseases”, University of Greifswald, Fleischmannstr. 42-44, 17489 Greifswald, Germany
cCIC biomaGUNE and CIBER de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Passeo de Miramón 182, 20014 Donostia-San Sebastián, Spain
dIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
eDZHK (Deutsches Zentrum für Herz-Kreislauf-Forschung), Partner site Greifswald, Germany
First published on 31st January 2020
Abstract
Once introduced into the human body, nanoparticles often interact with blood proteins, which in turn undergo structural changes upon adsorption. Although protein corona formation is a widely studied phenomenon, the structure of proteins adsorbed on nanoparticles is far less understood. We propose a model to describe the interaction between human serum albumin (HSA) and nanoparticles (NPs) with arbitrary coatings. Our model takes into account the competition between protonated and unprotonated polymer ends and the curvature of the NPs. To this end, we explored the effects of surface ligands (citrate, PEG-OMe, PEG-NH2, PEG-COOH, and glycan) on gold nanoparticles (AuNPs) and the pH of the medium on structural changes in the most abundant protein in blood plasma (HSA), as well as the impact of such changes on cytotoxicity and cellular uptake. We observed a counterintuitive effect on the ζ-potential upon binding of negatively charged HSA, while circular dichroism spectroscopy at various pH values showed an unexpected pattern in the reduction of α-helix content, as a function of surface chemistry and curvature. Our model qualitatively reproduces the decrease in α-helix content, thereby offering a rationale based on particle curvature. The simulations quantitatively reproduce the charge inversion measured experimentally through the ζ-potential of the AuNPs in the presence of HSA. Finally, we found that AuNPs with adsorbed HSA display lower toxicity and slower cell uptake rates, compared to functionalized systems in the absence of protein. Our study allows examining and explaining the conformational dynamics of blood proteins triggered by NPs and corona formation, thereby opening new avenues toward designing safer NPs for drug delivery and nanomedical applications.
Introduction
The use of gold nanoparticles (AuNPs) in biomedical applications such as drug delivery, cellular targeting, imaging, photodynamic therapy, and tissue engineering1–5 results from the unique combination of their distinctive physicochemical and optical properties, i.e. low toxicity, biodegradability and localized surface plasmon resonances.6–8 AuNPs can readily form stable conjugates with proteins through either covalent bonds or physical interactions. Upon entering the human body, NPs may interact with blood proteins, forming a so-called “protein corona”,9 which governs the ultimate fate of the biological activity of the NPs.10–14 The formation of a corona can, however, lead to changes in the structures of the adsorbed proteins, thereby affecting their physiological functions and potentially inducing unexpected biological reactions such as immunostimulation or immunosuppression.15,16In particular, various properties of AuNPs such as size, shape and surface chemistry have been reported to influence the binding of proteins to nanoparticles, in terms of protein structure and flexibility, which, in turn, influence how nanoparticles interact with cells and/or tissues.17,18 The interplay between all such properties has been investigated by numerical simulations19,20 and, experimentally, by the application of techniques such as circular dichroism (CD) spectroscopy and sum frequency generation vibrational spectroscopy.21,22 The analysis pursues the effects of the curvature, morphology and chemical nature of the NPs on the conformational changes of proteins under given pH and temperature conditions. For example, Douglas et al. reported the interaction of common blood proteins (e.g., albumin) with citrate coated AuNPs of various sizes.23 They demonstrated that smaller NPs seem to retain a native-like protein structure when compared with larger NPs. In another study, Cañaveras et al.24 studied the role of nanoparticle functionalization using both citrate- and 6-mercaptopurine-capped AuNPs interacting with HSA. These authors showed that the formation of AuNP–HSA bioconjugates mainly stems from electrostatic interactions, which induce partial unfolding of the protein when such bioconjugates are formed in the presence of low protein concentration. It has additionally been demonstrated that the strength of the interaction between albumin and tannic acid-coated AuNPs depends on the density of tannic acid molecules on the NPs’ surfaces, such that larger NPs with a dense tannic acid coating react more strongly with the protein, as compared to smaller ones containing a low concentration of tannic acid.25
In this respect, electrostatic charges have been proposed to be the driving force behind the initial attachment upon reorientation, and possible subsequent reconfiguration.26 This idea has been exploited to propose a three stage model for attachment, involving the following steps:27 (i) reversible association; (ii) rearrangement or reorientation of the protein; and (iii) strong, irreversible binding. Unfortunately, the link between the reconfiguration, the stability of the binding and the overall electrodynamic properties of the system is still unclear.28 We, therefore, studied the effects of varying the surface chemistry of AuNPs and the medium pH on the structure of human serum albumin (HSA), forming a protein corona, and aimed to correlate conformational changes with NP toxicity and cellular uptake. We propose a combined approach toward establishing the relationship between conformational changes and electrodynamic properties obtained from experiments, and the specific redistribution of charges within the protein upon adsorption, obtained from coarse-grained simulations. Our main hypothesis is that the rearrangement of charges should provide a qualitative explanation for some of the results, while the creation of H-bonds is required to fully understand the overall behavior. To that aim, we used HSA, the most abundant protein in blood plasma, which is involved in the transport of many endogenous small ligands,29 as a model protein to investigate the formation of AuNP–HSA bioconjugates, using AuNPs with various surface modifications (namely citrate, thiol terminated PEG-OMe, PEG-NH2, PEG-COOH, and glycans) and the results were analyzed by means of coarse-grained simulations at constant pH. A recent report suggests that constant charge calculations can be used to predict the induction of large pKa shifts in the amino acids of proteins, in the presence of charged surfaces even at physiological pH, instead of more demanding calculations at constant pH.30
Another important aspect connected to the surface chemistry of NPs is their compatibility with the immune system, so that an appropriate surface modification can reduce their immunotoxicity. The most common chemical approach to introduce AuNPs into biological environments involves surface coating with poly(ethyleneglycol) (PEG) chains.31 A promising alternative to PEG is based on the use of glycan ligands, which have been shown to be biocompatible and to inhibit protein adsorption with no loss of colloidal stability.32 It is well-established that misfolded proteins often aggregate and interact with other cellular components, thereby affecting cell viability and eventually leading to cell death. Therefore, assessing the toxicity of NPs after contact with proteins is a critical step in manipulating the immune response and the corresponding cytotoxic effects. It has also been shown that NPs carrying biomolecules at their surfaces induce differences in adhesion and cell internalization, as compared to bare NPs. For example, Lesniak et al. reported that silica NPs with adsorbed serum proteins preferentially accumulate at lysosomes, whereas the same NPs without serum proteins showed a higher adhesion to the cell membrane and greater internalization.33 In another study, the same group reported that carboxylated polystyrene NPs exhibit preferential adhesion to the cell membrane without serum, as compared to the same NPs with serum.34 To further develop the use of PEGylated or other coatings, it is mandatory to evaluate in detail the conformational changes of proteins, induced by the coating and subsequent corona formation. Our study provides a unified picture of the roles of NP size and surface chemistry in protein adsorption and protein corona formation, combined with NPs’ toxicity in cell culture.
Experimental
Materials
Human serum albumin (HSA), tetrachloroauric acid (HAuCl4), trisodium citrate and O-[2-(3-mercaptopropionylamino)ethyl]-O′-methylpoly(ethylene glycol) 5 kDa (PEG), PEG-COOH, PEG-OMe and PEG-NH2 were purchased from Sigma-Aldrich. The neoglycoconjugate of lactose (Lac), (β-D-galactopyranosyl-(1–4)-D-glucose) was synthesized as described previously.35 Dialysis membranes with a molecular weight cut-off (MWCO) of 100 kDa (cellulose ester) and ultrafiltration membranes (regenerated cellulose) were purchased from Millipore (Billerica, MA, USA). Phosphate-buffered saline (PBS; Dulbecco w/o Ca2+/Mg2+) was purchased from Biochrom GmbH, (Berlin, Germany). EtOH, HCl, and NaOH were supplied by Roth (Karlsruhe, Germany). A stock solution of HSA was prepared in PBS, at pH 7.4. PBS solutions with various pH values were prepared by adding concentrated HCl or NaOH to PBS at pH 7.4. The concentration of HSA was measured spectrophotometrically, considering a molar absorptivity of 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mol−1 cm−1 at 280 nm. All chemicals were used as received. Water was purified through an ultrapure water system (Millipore system, Sartorius Stedim Biotech, Göttingen, Germany).
000 mol−1 cm−1 at 280 nm. All chemicals were used as received. Water was purified through an ultrapure water system (Millipore system, Sartorius Stedim Biotech, Göttingen, Germany).
Fourier-transform infrared (FTIR) spectroscopy
FTIR spectra of the functionalized nanoparticles, in a powder form of KBr pellets, were recorded using a PerkinElmer Frontier FTIR spectrometer by placing the solid on the surface of a diamond attenuated total reflectance crystal. The spectra were obtained at regular time intervals in the region of 4000–600 cm−1 at a resolution of 4 cm−1 (128 scans).:1 v/v) for 24 h, diluted with 2% HNO3 for two days and then subjected to ICP-OES analysis.
MD simulations
We performed coarse-grained molecular dynamics simulations of HSA in the presence of charged spherical nanoparticles using the package LAMMPS.37 In order to characterize the protein structural changes induced by the adsorption on the NPs, we carried out computational studies which enabled an efficient description of conformational changes near the native state. To this end, we represented HSA using a GO-type protein model developed by Baumketner et al.38 In such model types, the folded state is imposed to be the global free-energy minimum. The rationale behind such an approach is that the folding landscape follows the minimum-frustration principle introduced by Wolynes and Onuchic.39 Hence, the configurational space close to the native state is modelled accurately.40 All attractive interactions (residue–residue and opposite charged residue–NP) are represented with a sigmoidal-type potential UATT used in coarse-grained protein models to represent residue–residue interactions.40The solvent is implicit. On the other hand, all excluded volume interactions are represented with a Weeks–Chandler–Andersen potential that is a Lennard-Jones potential shifted to be zero from the distance corresponding to the minimum to infinite.41
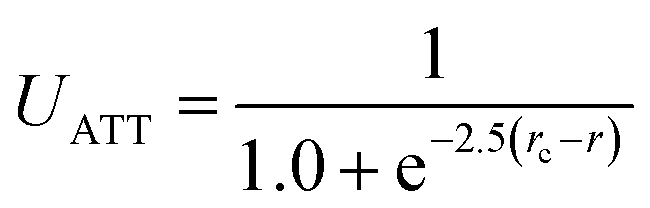
Although the representation of the electrostatic interaction is not physically accurate, it still captures the short-ranged attractions that hold the protein on to the surfaces of the NPs and cause protein conformational changes, all at a fraction of the computational cost. Moreover, we did not see the point in coupling GO-potentials to accurate electrostatic interactions. Our approach left us a free parameter, namely the relative ratio between the residue–residue and the residue–NP interactions. Thanks to the efficiency of the simulation, we could scan for a wide range of values [1–300] and identify a window compatible with the experimental observations. As a first approximation to model the coated NP, we assumed a spherical rigid object interacting with the complete HSA structure. The high grafting density of the polymer brushes justifies our approach to model all the NPs as rigid spheres with size given by the experimentally determined hydrodynamic radius.
We outline next the operative steps in performing the calculations. We downloaded the crystal structure of HSA in a PDB file obtained from the Protein Database.42 We then determined the protonated/deprotonated residues of the protein, and we tested the structure stability of the GO-model by performing an unfolding–refolding simulation. Then, we introduced the nanoparticle modelled as a hard-sphere with a homogeneously charged surface. HSA is represented as 579 beads interconnected according to the specific bonding, angular and dihedral parameters. The simulation box (large enough to allow the protein to fully unfold) contains the NP (no angular and dihedral parameters specified) and HSA. The particle–HSA interaction parameters are set to represent the attraction of specific residues (beads) located on the surface of the protein with the different NP coatings.
We performed all the calculations at a constant temperature and in implicit solvent, in an NVT ensemble using a Langevin thermostat for 1 M step single runs. We then analyzed the results in terms of the root mean square displacement of HSA from its native structure, and the radial distribution function of the surface charged residues. The values of the parameters in LJ units were fixed at temperature = 1, mass = 1, epsilon = 1, timestep = 0.002, and sigma = 4. We placed the particles in the center of a cubic box large enough to avoid self-interactions of HSA with its periodic images.
Results and discussion
Ligand exchange reactions were carried out on 14 nm diameter, citrate-coated AuNPs in water, using 100 molecules per nm2. AuNP spheres were incubated with either PEGylated ligands terminated with carboxylic (COOH), amino (NH2) or methoxy (OMe) groups, or with lactose ligands, showing in all cases excellent stability in water. Citrate-capped AuNPs showed a localized surface plasmon resonance (LSPR) band at 520 nm, which was preserved after all different surface modifications (Fig. S1, ESI†). A comparison of the corresponding UV-Vis spectra shows a spectral red-shift (3–6 nm) in the LSPR band, typically related to minor changes in the dielectric function near the metal nanoparticle surface, upon functionalization. The absence of significant shifts in the LSPR band indicates that surface-modified AuNPs do not aggregate during the ligand exchange process. Successful ligand exchange was also proven by FTIR analysis (Fig. S2, ESI†). After modification with PEGylated ligands, we identified the characteristic absorption bands centered at 1112 cm−1 arising from the C–O–C stretching vibration in PEG. Functionalized AuNPs also showed strong absorption bands at 1880 cm−1, which can be assigned to C–H stretching. A new relevant band at 1733 cm−1, which is related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, appears for carboxylic group-terminated PEG ligands. Finally, the hydroxyl peak (around 3400 cm−1) is enhanced by the presence of lactose moieties in the case of glycan-modified AuNPs.
O stretching, appears for carboxylic group-terminated PEG ligands. Finally, the hydroxyl peak (around 3400 cm−1) is enhanced by the presence of lactose moieties in the case of glycan-modified AuNPs.
Surface-modified AuNPs were also characterized by transmission electron microscopy (TEM) and dynamic light scattering (DLS). Fig. S3 (ESI†) shows representative TEM images of AuNPs with an average size of 13.44 ± 2.19 nm. No morphological changes were observed after surface modification, as indicated by the absence of aggregation. The average hydrodynamic diameter of citrate-capped AuNPs measured by DLS was 19.31 ± 0.1 nm, but it was found to increase after surface modification (Fig. S4 and Table S1, ESI†). The size increase after surface modification is due to the high molecular weight and chain length of the polymers used in the ligand exchange. All the prepared AuNPs were stable in water, in agreement with the recorded ζ-potential values (Table S1, ESI†). The steric hindrance of the polymer molecules at the AuNPs’ surfaces is also likely to stabilize the NPs in water, as well as in phosphate buffer saline (PBS).
Interactions of HSA with different surface-modified AuNPs at various pH values
HSA is a globular protein, which is known to undergo pH-dependent conformational changes.43–46 At a physiological pH of 7.4, HSA has a normal heart-shaped structure, which changes into an ellipsoid at pH values greater than 9, whereas at acidic pH below 4.7, the protein is assumed to exist in a highly charged, rapidly migrating form.The interaction of HSA with functionalized AuNPs was monitored using UV-Vis, DLS, ζ-potential and circular dichroism (CD) techniques. Scheme 1 presents the preparation of AuNP–HSA bioconjugates, the interaction between AuNPs and HSA, and the molecular structures of the ligands.
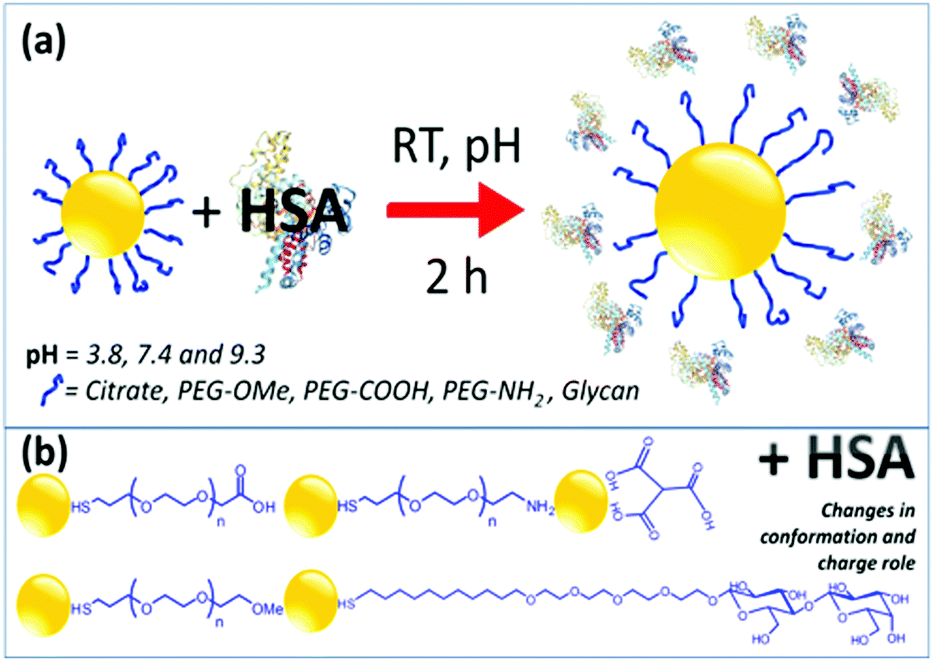 | ||
| Scheme 1 Schematic representations of the formation of AuNP–HSA bioconjugates and of the various functional groups used to functionalize AuNPs. | ||
The formation of a protein corona in Au–HSA bioconjugates was monitored by DLS. Fig. S5 (ESI†) shows the DLS data for Au–HSA bioconjugates with different surface-modified AuNPs, at selected pH values (3.8, 7.4 and 9.3). In the case of citrate-capped AuNPs, the hydrodynamic diameter of the bioconjugates increased from 20 up to 45 nm, which indicates protein corona formation. However, for PEG-OMe, PEG-COOH, PEG-NH2, and glycan functionalized AuNPs, the hydrodynamic diameter of the bioconjugates did not increase significantly (Fig. S5, ESI†), likely due to the shielding effect of both PEG and glycans, or to an insufficient amount (2.4 × 10−6 M) of HSA to form a protein corona. Therefore, we further increased the concentration of HSA (7.5 × 10−3 M) prior to the formation of AuNP–HSA bioconjugates. Fig. 1a shows DLS data for AuNPs and AuNP–HSA bioconjugates with different surface modifications, at pH 7.4.
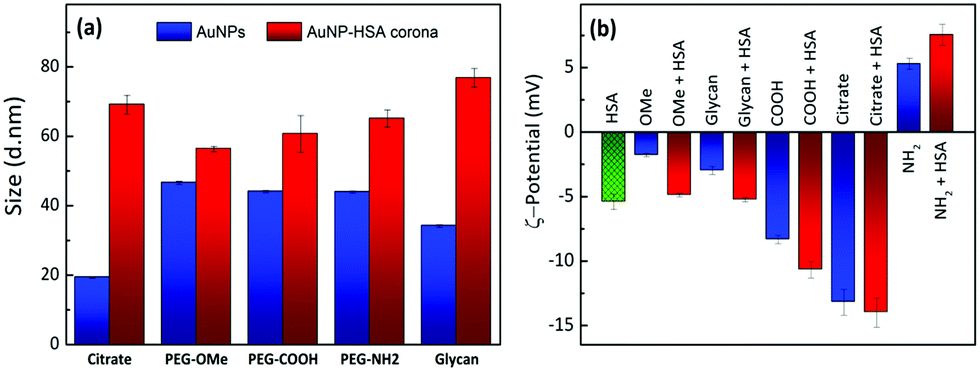 | ||
| Fig. 1 (a) Hydrodynamic diameters measured by DLS for AuNPs and AuNP–HSA bioconjugates with different surface-modifications, at pH 7.4. (b) ζ-Potentials of HSA, surface-modified AuNPs, and their corresponding AuNP–HSA bioconjugates, at pH 7.4. | ||
An increase in the hydrodynamic diameter was found for all the bioconjugates at higher HSA concentration, which suggests that an excess amount of HSA is required to generate a well-defined corona. The difference in hydrodynamic diameter for the various bioconjugates was also found to vary with the type of ligand at the AuNPs’ surfaces. For example, bioconjugates formed by both citrate- and glycan-functionalized AuNPs showed a large increment in the hydrodynamic diameter, as compared to their counterparts lacking the protein. In a previous report,32 it was shown that glycans prevent the adsorption of proteins on coated NPs, compared to higher molecular weight PEG-coated systems. In that case, the study was performed in a richer medium, in the presence of at least 10 different proteins in competition for the surface. In contrast, in our case, we can detect the specific effect of the surface chemistry on HSA adsorption, and no further treatment was performed, such as centrifugation and washing. Glycan-functionalized AuNPs present –OH groups at the surface and thus allow strong hydrogen bonding, resulting in an increase in diameter from 35 nm to 79 nm. PEG-functionalized AuNPs showed smaller changes in the hydrodynamic diameter upon interaction with HSA, indicating weak interaction of HSA with PEG-AuNPs or the formation of a soft protein corona. These results suggest that, in contrast to PEG-functionalized AuNPs which did not allow much HSA adsorption, both citrate- and glycan-AuNPs lead to the multilayer adsorption of HSA on the NPs’ surfaces. It is obvious that proteins adsorb on the AuNPs, forming multilayers, when the concentration of protein exceeds the monolayer formation.47 For example, in the case of citrate-AuNPs, HSA directly binds to the gold core through various chemical groups, especially free thiol groups due to the fact that gold has high affinity for the sulfur containing groups. Once HSA is bound to the AuNP's surface, the total surface will have a negative charge which repels other HSA molecules (isoelectric point pI = 4.7) by creating an electrostatic barrier. In the presence of an excess of HSA molecules, the electrostatic barrier is overcome with increased kinetic energy and multilayer formation is observed on the NPs’ surfaces, which results in an increase in the size of the protein corona. A similar trend was also observed in the case of glycan-AuNPs where the availability of a large number of –OH groups at their surfaces allows hydrogen bonding interactions with HSA molecules to further undergo multiple adsorption of HSA on the NPs’ surfaces.
To further understand the multilayer adsorption of HSA at different pH values, we again carried out protein corona formation using a high concentration of HSA at acidic and basic pH (see Fig. S6, ESI†). We found that NPs coated with PEG-OMe-/COOH- show a similar increment in size under all pH conditions, whereas PEG-NH2-AuNPs show a change in the hydrodynamic diameter with the pH of the medium. A slight increment in the hydrodynamic diameter was observed for PEG-NH2-HSA composites at acidic and basic pH compared to neutral pH, suggesting the multilayer formation of HSA. This might be due to the slight changes in the electrostatic behavior between PEG-NH2-AuNPs and HSA molecules under various pH conditions. Unlike PEG-NH2-, glycan-AuNPs show the opposite trend in the hydrodynamic diameter where the size is increased at physiological pH compared to acidic and basic pH. This could be due to hydrogen bonding interactions which may reduce the multilayer adsorption of HSA (compared to neutral pH) due to the availability of H+ and OH− ions at acidic and basic pH. However, citrate-AuNPs showed multilayer adsorption of HSA under all pH conditions.
Fig. 1b provides a comparison between the ζ-potential values for HSA, various surface-modified AuNPs and their corresponding bioconjugates, recorded at pH 7.4. HSA has a negative ζ-potential at physiological pH, but we found that in the bioconjugates the overall net charge changes in a non-trivial manner. The change in the surface charge of AuNPs upon incubation with HSA again indicates protein corona formation. ζ-Potential measurements in PBS, in the absence of HSA, showed that, except for PEG-NH2-AuNPs, all the other surface-modified AuNPs have negative ζ-potential values. The positive value for the amine-terminated ligand is counterintuitive because addition of HSA leads to an increase in the overall positive potential. Considering the negative charge of HSA, we would expect a reduction of the total positive charge. Such an unusual behavior offers the ground for an interesting interpretation of the interplay between electrostatic interactions and hydrogen bonding, which we discuss in more detail below. All the negatively charged systems showed increased negative ζ-potentials upon protein conjugation. Notably, the systems with higher ζ-potential values exhibit a larger size upon contact with HSA. This observation strongly supports the idea that corona formation is driven – at least at the initial stage – by electrostatic interactions. A three-step model for protein adsorption has been proposed: (i) reversible association; (ii) rearrangement or reorientation of the protein; and (iii) strong, irreversible binding.27
Optical spectra also support the formation of a protein corona on AuNPs observed by DLS, since plasmon resonances in AuNPs reveal information about the local environment, including interparticle distance and eventual aggregation. UV-vis spectra for AuNP–HSA bioconjugates from different surface modified AuNPs, at pH 3.8, pH 7.4 and pH 9.3, are shown in Fig. S7 (ESI†). With the exception of citrate-AuNPs, all the other surface-modified AuNPs show small differences in both the maximum absorbance wavelength and the bandwidth (FWHM), i.e. no obvious changes in the LSPR band (at around 524 nm) after conjugation with HSA at different pH values. Moreover, no significant changes in the line shape of the band were observed, indicating that the bioconjugates are stable in PBS, even at different pH values. In the case of citrate-AuNPs, the LSPR band became wider and was red-shifted to 530 nm, which may be related to a more direct interaction of the gold cores with free –SH, –COOH or –NH2 groups in HSA. In the case of surface-modified AuNPs, direct interaction between gold cores and HSA may be hindered due to shielding provided by thiol terminated PEG and glycan molecules on the gold cores. In general, all protein–NP bioconjugates obtained through this simple mixing process remained stable for at least 48 h, allowing further experimental studies.
Modeling surface charge inversion
To further understand the behavior of the ζ-potential at a physiological pH of 7.4, and to model the charge inversion observed for PEG-NH2-AuNPs, we performed two independent numerical analyses. In the first modeling step, we estimated the number of charged groups on the surfaces of PEG-NH2-AuNPs and PEG-COOH-AuNPs, and on the surface of an HSA molecule. We obtained the structure of HSA from the protein database listed under the code 4K2C (inset of Fig. 2a). We identified the balance of total exposed charged amino acids as 42 positives, 55 negatives, and 48 neutrals, by using the software PDB2PQR.48 Among the 48 neutrals, 22 residues could eventually act as either donors or receptors. Shown in Fig. 2a are the two outer positive and negative iso-surface electrostatic potentials, obtained by solving the Poisson–Boltzmann model via the application of APBS49 and the respective VMD50 visualization software. The negative contribution of electric charges is stronger than that coming from positive charges. This result indicates that the contribution to the surface potential from positive charges (+1.6 mV) is overcompensated for by the negative potential of −4.6 mV, in agreement with the net negative charge exhibited by HSA. To quantify the total surface charge at the ζ-potential plane σζ, we can use the expression obtained by Oshima:51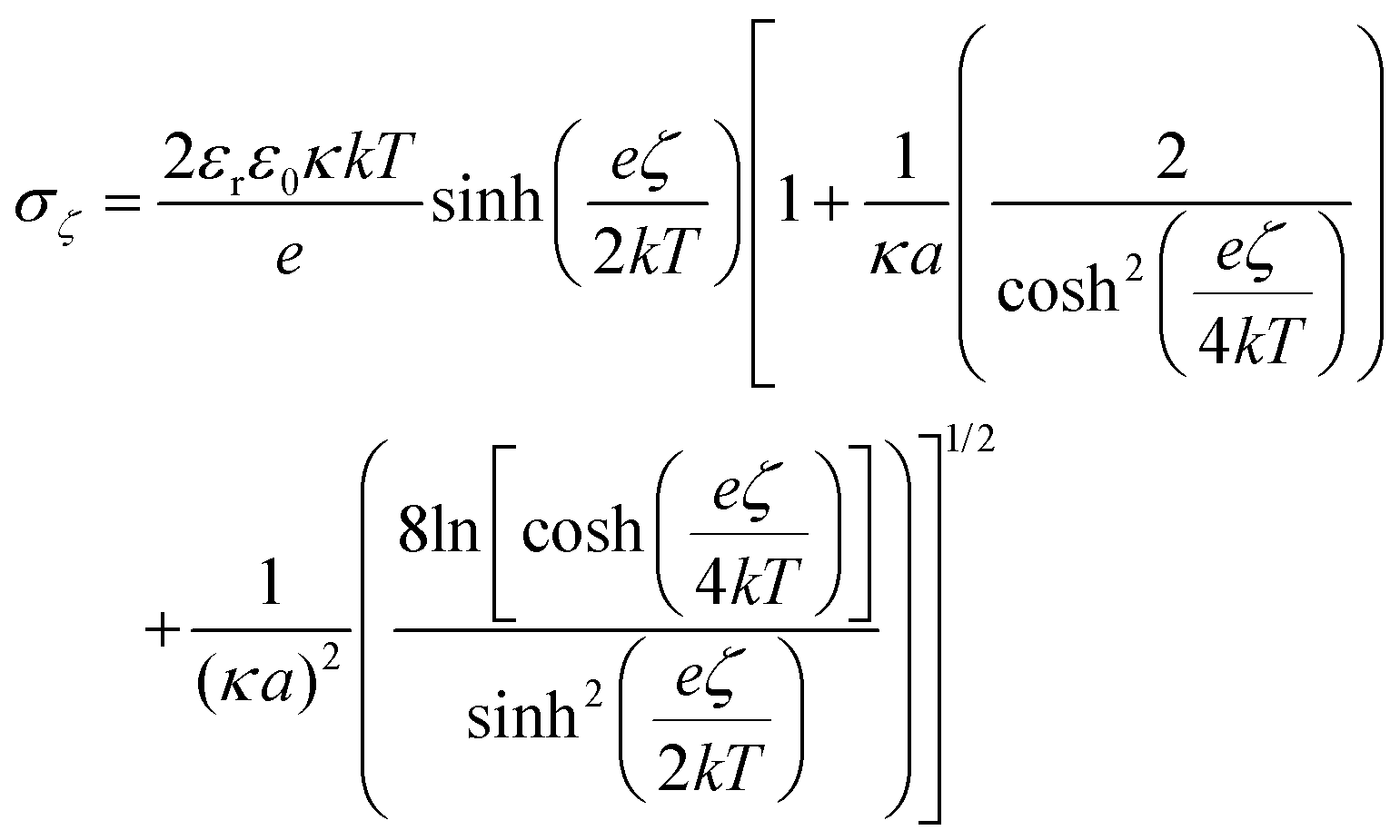 | (1) |
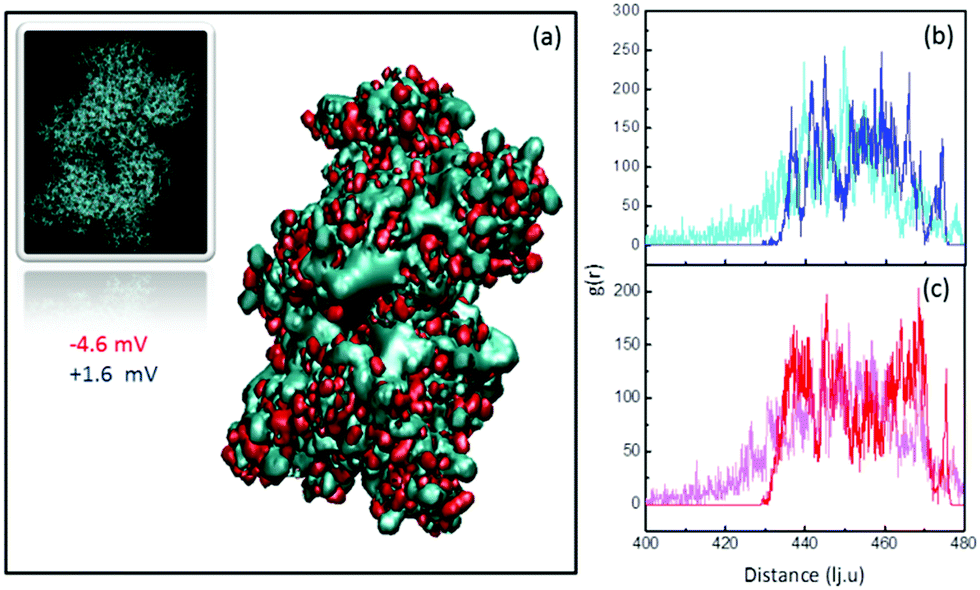 | ||
| Fig. 2 (a) Surface electric potential of HSA and (b) evolution of the radial distribution function of positive surface charges of HSA during adsorption compared to the (c) negative surface charges. In (b) and (c) the lighter colors represent the initial configurations and the darker colors represent the final configurations of the positive (b) and negative (c) surface residues of HSA in the presence of NP-NH2, respectively. The grey zone corresponds to the outer face of HSA. | ||
To model in more detail the charge inversion process, we included additional features in the analysis and obtained information on the surface exposure of the charged residues. Therefore, we performed molecular dynamics simulations of HSA in the presence of charged spherical nanoparticles (see numerical details in the Experimental section). In particular, we introduced attractive/repulsive forces to model the electrostatic interactions between protonated/deprotonated residues of the protein and the charged surface. As a first approximation to model the coated NP, we assumed a spherical rigid object interacting with each residue in HSA.
As stated above, the charge present in all of the used coatings suggests that adsorption is affected by electrostatic interactions. Specifically, the charges present at the polymer terminal groups should bind onto sites with an opposite charge at the HSA surface. Concerning negatively charged NPs, the observed increase in ζ-potential in all cases points toward electrostatic binding between positive protein residues and negative ligands on the NP surface. Interestingly, the case of PEG-NH2 in the presence of HSA indicates that electrostatic interactions are not enough to satisfactorily explain the observed increase in positive ζ-potential. Fig. 2b and c show the radial distribution function of the surface charges of HSA while attaching to PEG-NH2 coated NPs. Positive charges (Fig. 2b) slightly redistribute towards the outer face of the protein, while negative ones (Fig. 2c) redistribute towards the face in contact with the positively charged surface. A comparison between the areas below the curves for the initial and final configurations shows that the opposite-to-NP face of HSA displays an increased positive charge density by ∼46%. Notwithstanding, the density of negative charges also increases by ∼23% in the outer face, as a response to the overall relocation. The total amount of exposed charges results in a smaller gain of positive charges by only 5–6 per protein. The final charge increase is hence 37–44%, which is very close to the increment observed in the ζ-potential value of ∼42%. Hence, we can explain the increase in the positive ζ-potential of PEG-NH2 coated NPs by weighing the relative contributions of electrostatic and hydrogen bonding interactions. Obviously, our analysis would also work for the negative charge case, but this is unnecessary because HSA presents excess negative charge. Hence, it is not surprising that the ζ-potential would become more negative.
Conformational changes of HSA structure in AuNP–HSA bioconjugates at different pH values
We further monitored the influence of AuNP functionalization on the structure of HSA molecules using circular dichroism (CD) spectroscopy. Far-UV CD spectra of native HSA at pH 3.8 (black), 7.4 (red), and 9.3 (blue) are shown in Fig. S8 (ESI†). The CD spectra of HSA displayed a characteristic band with two negative minima at 208 and 222 nm, corresponding to the α-helical structure of the protein. The difference in the ellipticity of HSA at various pH values is due to the different conformational states of the albumin.44 CD spectra of functionalized AuNPs were also recorded in the absence of HSA, showing no CD signal (Fig. S9, ESI†). In the case of AuNP–HSA bioconjugates (Fig. S10, ESI†), the ellipticity values vary with both the type of ligand at the NPs’ surfaces and the pH of the medium.Fig. 3a displays the CD spectra and ellipticity values of native HSA and of HSA in conjugates with different surface-modified AuNPs, at pH 7.4. Upon addition of different surface modified AuNPs, the CD signal of HSA showed a decrease in ellipticity at both 208 and 222 nm. At a physiological pH of 7.4, only citrate- (black) and PEG-NH2 (brown) AuNPs have an effect on the secondary structure of HSA. Because the pH is maintained, we speculate that the secondary structure is affected by the interaction between protein residues and AuNP ligands. Both citrate and PEG-NH2 are susceptible to electrostatic and H-bond type interactions. We can thus assume that the interaction between positively charged PEG-NH2-AuNPs and negatively charged HSA molecules (pI = 4.7) would lead to strong electrostatic attraction. Conversely, the negatively charged citrate-AuNPs could interact with the positively charged lysine residues in HSA. Negatively charged PEG-COOH-AuNPs and neutral ligands on AuNPs (PEG-OMe and glycan) did not show any changes in the secondary structure of HSA at pH 7.4.
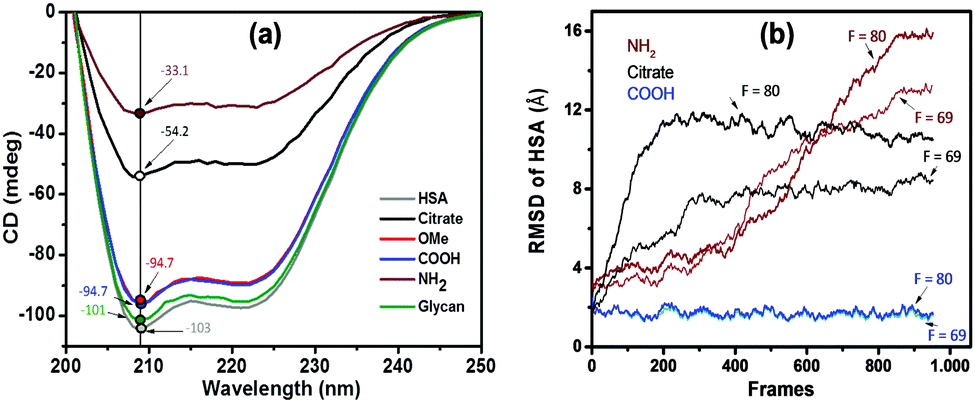 | ||
| Fig. 3 (a) CD spectra of native HSA (2.4 × 10−6 M) and the same concentration in Au–HSA bioconjugates with different surface modified AuNPs, at pH 7.4. Ellipticity values (n = 3) at 208 nm for HSA and AuNP–HSA bioconjugates at pH 7.4 are highlighted and indicated with arrows. (b) Root mean square displacements representing the structural modification of HSA in the presence of three differently coated NPs, estimated from MD simulations at two different magnitudes of the attractive forces in simulation reduced units (numbers highlighted therein). The value of F = 69 is the first one where an appreciable modification induced by NH2 was observed. The same trend is valid up to F = 80, above which we see a jump to F = 100, where citrate also becomes deformed. In order to see changes produced by COOH, it is necessary to reach F = 200 (data not shown). | ||
At an acidic pH of 3.8 (Fig. S10a, ESI†), we observed smaller changes in the ellipticity values for PEG-OMe, PEG-NH2 and citrate-AuNPs, whereas glycan- and PEG-COOH-AuNPs showed a strong decrease in the ellipticity values, most likely due to strong hydrogen bonding or electrostatic interactions between the –OH groups of the ligands and HSA molecules. A strong electrostatic interaction between negatively charged PEG-COOH functionalized AuNPs and positively charged residues in HSA would induce a large decrease in ellipticity, whereas hydrogen bonding would be dominant in the case of glycan-AuNPs (between the –OH groups of AuNPs and the –COOH groups of HSA molecules). Neutral PEG-OMe molecules most probably lead to weak interactions between AuNPs and HSA. Positively charged PEG-NH2-AuNPs allow electrostatic repulsions between AuNPs and the HSA protein, which will not induce significant conformational changes in the bioconjugates. Similarly, at basic pH (Fig. S10b, ESI†), both glycan- and PEG-COOH-AuNPs induce larger changes in the secondary structure of the protein, compared to the other types of AuNPs. A moderate change in the secondary structure was observed for PEG-NH2-AuNPs, whereas no such changes were observed for both citrate- and PEG-OMe-AuNPs.
At both acidic and basic pH conditions, glycan-AuNPs induce larger changes in the HSA structure due to the strong hydrogen bonding interaction. It is known that HSA molecules become unfolded at acidic and basic pH values, where –COOH and –NH2 groups will be at the surface and may strongly interact with –OH groups of the glycan molecules inducing a change in the conformational structure. All these results show that both electrostatic and hydrogen bonding interactions have an influence on the protein secondary structure which is further influenced by the pH of the medium. Secondary structural elements were further quantified using the online algorithms BeStSel (http://bestsel.elte.hu/index.php).54 Table S2 (ESI†) shows the percentages of secondary structures of HSA and the same in the presence of different surface-modified AuNPs at various pH values. At physiological pH, both citrate- and PEG-NH2-AuNPs showed a decrease in the percentage of α-helix content (from 48.8% to 38.4 and 35.2%, respectively) and an increase in the percentage of β-sheet content (from 11.3 to 15.4 and 18.6%, respectively). At acidic pH, both PEG-COOH- and glycan-AuNPs showed a decrease in the percentage of α-helix content (from 42.4% to 31.3 and 33.8%, respectively) and an increase in the percentage of β-sheet content (from 13.5 to 19.7 and 20.5%, respectively). Similarly, at basic pH, both PEG-COOH- and glycan-AuNPs showed a decrease in the percentage of α-helix and an increase in the percentage of β-sheet content.
Overall, we observed that neutral ligands on AuNPs both induce insignificant conformational changes in HSA and retain the protein secondary structure even after pH change in the medium (PEG-OMe-AuNPs), or they induce larger changes in the protein at both acidic and basic pH than at physiological pH (glycan-AuNPs). The investigated positively charged ligand (PEG-NH2-AuNPs) induces conformational changes in HSA under all pH conditions. Negatively charged ligands (e.g. citrate-AuNPs) induce changes in the secondary structure at acidic and physiological pH, whereas PEG-COOH-AuNPs induce changes in the protein structure at acidic and basic pH. PEG-OMe-AuNPs can be considered safe for drug delivery applications because they do not induce any changes in protein structure. The ellipticity values at 208 nm for HSA and HSA within bioconjugates at physiological pH 7.4 are highlighted in Fig. 3a. It can be seen that both citrate- and PEG-NH2-AuNPs show a predominant decrease in the ellipticity values at 208 nm, suggesting that strong electrostatic interactions induce changes in the secondary structure of HSA upon interaction with these AuNPs. Neutral PEG-OMe- and glycan-AuNPs, as well as negatively charged PEG-COOH-AuNPs, show no major decrease in ellipticity values at 208 nm. Our results indicate that the type of interaction between AuNP ligands and HSA plays a crucial role in the conformational behavior of HSA within the bioconjugates.
Modelling conformational changes in HSA upon corona formation
From the analysis of surface charges within HSA, we estimated that the protein undergoes a conformational change associated with the redistribution of charged residues. Naively, it would appear reasonable to assume that the only relevant parameter is the interaction between the protein and the ligand coating on the nanoparticles. However, this simplification would imply that negatively charged particles would all behave similarly, while the experimental data in Fig. 3a indicate that this assumption is qualitatively incorrect. Indeed, COOH and citrate coatings have very different effects on the protein structure.In order to quantify the modification of the protein, we measured the root mean square displacements (RMSD) in the presence of coated NPs (COOH, NH2, and citrate). Since we do not know the exact interaction strength between the amino acid and the functional surfaces, we tested a range of different magnitudes of the attractive interaction force F (given in reduced simulation units). As can be expected for large forces, all scenarios lead to a deformation of the protein upon adsorption. Conversely, weak forces leave the protein unaltered (see Fig. S11, ESI†). At intermediate values of F ∼ [60–80], we could qualitatively reproduce the behaviour observed in the CD spectra (see Fig. 3). We observe that HSA in the presence of COOH-NPs exhibits a small deformation (RMSD < 2 Å), regardless of the whole range of applied forces. Instead, in the presence of NH2, the protein quickly deforms (large RSMD values >10 Å), leading to the charge redistribution presented above. It is important to stress that the hydrodynamic radius for citrate coated AuNPs is much smaller than that for COOH. However, both COOH and citrate systems display the same protein–colloid interaction. Contrary to the COOH system, we now observe an intermediate structural change of the protein, meaning that the different radius is critical to producing the significant structural change observed for citrate-AuNPs.
Overall, we qualitatively reproduce the trend observed in the CD spectra (compare Fig. 3a with Fig. 3b). We conclude that the structural stability depends on the curvature of the surface (the higher the curvature, the smaller the deformation) and on the specific locations of HSA sites interacting with the NP. As a reminder, negatively charged COOH and citrate surfaces show attractive interactions with positively charged surface residues, whereas the opposite effect occurs with NH2.
Cytotoxicity of HSA–AuNP bioconjugates
Finally, we investigated whether the conformational changes of HSA in bioconjugates would affect NP uptake and consequently cytotoxicity at physiological pH, using human breast cancer cells (MDA-MB-231). Fig. 4a shows the uptake of AuNPs and their corresponding HSA–AuNP bioconjugates in MDA-MB-231 cells. Samples were incubated for 4 h at 37 °C with 5% CO2, and FBS-free DMEM medium was used to avoid any unspecific interactions of proteins with AuNPs. We found that the cell uptake rates do vary for different types of ligands at the AuNPs’ surfaces. For example, the uptake of positively charged PEG-NH2-AuNPs was significantly faster than those of negatively charged PEG-COOH/citrate-AuNPs and neutral PEG-OMe-AuNPs. After HSA conjugation, the uptake rate of AuNPs was found to slow down, regardless of their surface charge and ligand chemistry. We propose that this effect arises from HSA molecules, lowering the AuNP surface energy and thereby reducing unspecific interactions of AuNP–HSA complexes with the cell membrane, as compared to the other surface-modified AuNPs.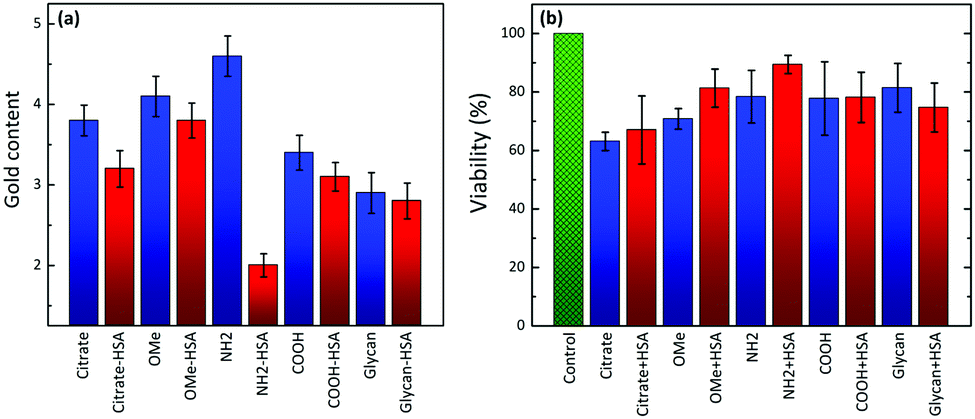 | ||
| Fig. 4 (a) Gold contents for all of the employed AuNPs and the corresponding HSA–AuNP bioconjugates, in MDA-MB-231 cells at physiological pH (AuNP concentration = 1.1 × 1010 particles per mL). (b) Cytotoxicity assay for different NPs and the corresponding HSA–AuNP bioconjugates, in MDA-MB-231 cells at physiological pH (AuNP concentration = 2 × 1010 particles per mL). | ||
Negatively charged PEG-COOH-AuNPs and neutral PEG-OMe-AuNP conjugates, which did not induce conformational changes in the HSA structure, showed a decreased cellular uptake compared to non-coated PEG-COOH-AuNPs and PEG-OMe-AuNPs. However, larger cellular uptake differences were observed in the case of PEG-NH2-AuNP–HSA bioconjugates, which showed higher ellipticity changes with respect to the free protein. The complex made of HSA and PEG-NH2-AuNPs led to the lowest cell uptake among all the AuNPs investigated in this study. It appears that the “unfolded” protein reduces the internalization efficiency compared to non-coated AuNPs or those coated with less unfolded HSA, likely because they do not trigger any receptor-mediated phagocytosis process.
Fig. 4b shows the cytotoxicity results for functionalized AuNPs and their bioconjugates with HSA. Compared to citrate-AuNPs, all the other types of AuNPs and their bioconjugates present a lower cytotoxicity. The bioconjugation of AuNPs with HSA maintains (for PEG-COOH-AuNPs) or slightly reduces (for glycan-AuNPs) or slightly increases (for citrate-, PEG-OMe- and PEG-NH2-AuNPs) the percentage of cellular viability for the different NP types. The difference in the viability for the bioconjugates is closely related not only to the cellular uptake percentage, but also to the subcellular distribution and elimination of NPs (possible adverse effects on organelles). It is also evident that the cellular toxicities of the nanoparticles depend on the various properties such as composition, charge density/polarity, surface chemical groups, and medium. Because both HSA and PEG-COOH molecules are negatively charged at physiological pH, it is assumed that both have similar effects on cytotoxicity. We also believe that multilayer protein coating also has an impact on the AuNP toxicity in cell culture. In the case of glycan-coated AuNPs, a decrease in the viability was observed after HSA adsorption compared to glycan-coated NPs alone. On the one hand, this could be attributed to the increase in the size by multilayer adsorption of HSA, which slightly lowers (almost no effect) the uptake rates than glycan-coated NPs alone, and thus decreases the viability. On the other hand, a very slight increase in the viability for citrate-AuNPs after HSA adsorption was observed, which could also be due to multilayer adsorption.
Consequently, PEG-NH2-AuNPs, which induced significant conformational changes in HSA upon bioconjugation and displayed the lowest uptake values, were shown to be the least toxic ones upon bioconjugation. Therefore, the capacity of NPs to induce protein structural changes does have an effect on cellular uptake and therefore may also be able to modulate cytotoxicity.
Conclusions
We studied the effect of AuNP surface functionalization and the pH of the medium on the structure of HSA upon conjugation with AuNPs carrying various ligands. Changes in the secondary structure of HSA in AuNP–HSA bioconjugates were found to vary with both the type of ligand on the nanoparticle surface and the pH of the medium. The counterintuitive results found in the increased positive charge upon binding of negatively charged HSA and the effects of the complex interplay between surface chemistry and NP size on protein conformational changes were successfully explained. In particular, we provided the answer to these questions from coarse-grained modelling: (1) charge inversion can be explained by taking into account the fraction of uncharged NH2 ends and the redistribution of protein charges upon adsorption; and (2) we demonstrated the separate roles of the NP surface charge and size in the conformational change experienced by the protein. The specific distribution of negative charges on the surface of HSA makes it more susceptible to a positively charged NP (highest change). More generally, numerical and theoretical analyses indicate that electrostatic interactions and hydrogen bonding play an important role in NP-protein binding. Neutral PEG-OMe-AuNPs do not induce changes in the protein structure at all the investigated pHs and we propose that they may be considered safe for drug delivery applications, whereas positively charged PEG-NH2-AuNPs do induce conformational changes in HSA under all pH conditions. Moreover, PEG-NH2-AuNPs in conjugation with HSA resulted in relatively low cellular uptake, indicating that they may be expected to fail in reaching a target, upon interaction with blood proteins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ina Buchholz for help with CD analysis and Martin Kulke for critical reading of the manuscript. This work was financially supported by the German Federal Ministry of Education and Research (BMBF) within the project NanoImmun (FKZ03Z22C51) to M. D. Support from the Ministerio de Economía y Competitividad (MINECO) (FIS2017-89471-R to I. C.; MAT2017-86659-R to L. M. L.-M.) is acknowledged. This work was partly carried out in the framework of the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency (Grant MDM-2017-0720).References
- A. B. Serrano-Montes, J. Langer, M. Henriksen-Lacey, D. Jimenez de Aberasturi, D. M. Solís, J. M. Taboada, F. Obelleiro, K. Sentosun, S. Bals, A. Bekdemir, F. Stellacci and L. M. Liz-Marzán, J. Phys. Chem. C, 2016, 120, 20860–20868 CrossRef CAS.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- L. A. Austin, B. Kang and M. A. El-Sayed, Nano Today, 2015, 10, 542–558 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- F. Charbgoo, M. Nejabat, K. Abnous, F. Soltani, S. M. Taghdisi, M. Alibolandi, W. T. Shier, T. W. J. Steele and M. Ramezani, J. Controlled Release, 2018, 272, 39–53 CrossRef CAS PubMed.
- E. Y. Kim, D. Kumar, G. Khang and D.-K. Lim, J. Mater. Chem. B, 2015, 3, 8433–8444 RSC.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- F. Ding, S. Radic, R. Chen, P. Chen, N. K. Geitner, J. M. Brown and P. C. Ke, Nanoscale, 2013, 5, 9162–9169 RSC.
- A. Assali, S. Razzazan, O. Akhavan, F. Mottaghitalab, M. Adeli and F. Atyabi, Colloids Surf., B, 2019, 173, 891–898 CrossRef CAS PubMed.
- J. Lazarovits, S. Sindhwani, A. J. Tavares, Y. Zhang, F. Song, J. Audet, J. R. Krieger, A. M. Syed, B. Stordy and W. C. W. Chan, ACS Nano, 2019, 13, 8023–8034 CrossRef CAS PubMed.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469–478 CrossRef CAS PubMed.
- Q. Jiao, L. Li, Q. Mu and Q. Zhang, BioMed Res. Int., 2014, 426028 Search PubMed.
- M. Henry, C. Dupont-Gillain and P. Bertrand, Langmuir, 2003, 19, 6271–6276 CrossRef CAS.
- F. Höök, M. Rodahl, B. Kasemo and P. Brzezinski, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12271–12276 CrossRef PubMed.
- H. Arkin and W. Janke, Phys. Rev. E, 2017, 96, 062504 CrossRef PubMed.
- S. Wei, L. S. Ahlstrom and C. L. Brooks III, Small, 2017, 13, 1603748 CrossRef PubMed.
- X. Jiang, J. Jiang, Y. Jin, E. Wang and S. Dong, Biomacromolecules, 2005, 6, 46–53 CrossRef CAS PubMed.
- C. Zhang, J. N. Myers and Z. Chen, Soft Matter, 2013, 9, 4738–4761 RSC.
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- F. Cañaveras, R. Madueño, J. M. Sevilla, M. Blázquez and T. Pineda, J. Phys. Chem. C, 2012, 116, 10430–10437 CrossRef.
- S. Sekowski, E. Tomaszewska, K. Soliwoda, G. Celichowski and J. Grobelny, Eur. Biophys. J., 2017, 46, 49–57 CrossRef CAS PubMed.
- W. Norde, Cells Mater., 1995, 5, 9 Search PubMed.
- A. Wang, K. Vangala, T. Vo, D. Zhang and N. C. Fitzkee, J. Phys. Chem. C, 2014, 118, 8134–8142 CrossRef CAS.
- S. Goy-López, J. Juárez, M. Alatorre-Meda, E. Casals, V. F. Puntes, P. Taboada and V. Mosquera, Langmuir, 2012, 28, 9113–9126 CrossRef PubMed.
- T. Peters, Jr., All about albumin: biochemistry, genetics, and medical applications, Academic Press, 1995 Search PubMed.
- F. M. Boubeta, G. Soler-Illia and M. Tagliazucchi, Langmuir, 2018, 34, 15727–15738 CrossRef CAS PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- I. García, A. Sánchez-Iglesias, M. Henriksen-Lacey, M. Grzelczak, S. Penadés and L. M. Liz-Marzán, J. Am. Chem. Soc., 2015, 137, 3686–3692 CrossRef.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- A. G. Barrientos, J. M. de la Fuente, T. C. Rojas, A. Fernández and S. Penadés, Chem. – Eur. J., 2003, 9, 1909–1921 CrossRef CAS PubMed.
- B. V. Enustun and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 3317–3328 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- A. Baumketner, A. Jewett and J. E. Shea, J. Mol. Biol., 2003, 332, 701–713 CrossRef CAS.
- J. N. Onuchic and P. G. Wolynes, Curr. Opin. Struct. Biol., 2004, 14, 70–75 CrossRef CAS.
- I. Coluzza, Mol. Phys., 2015, 113, 2905–2912 CrossRef CAS.
- J. D. Weeks, D. Chandler and H. C. Andersen, J. Chem. Phys., 1971, 54, 5237–5247 CrossRef CAS.
- Y. Wang, H. Yu, X. Shi, Z. Luo, D. Lin and M. Huang, J. Biol. Chem., 2013, 288, 15980–15987 CrossRef CAS PubMed.
- J. Wilting, M. M. Weideman, A. C. J. Roomer and J. H. Perrin, Biochim. Biophys. Acta, 1979, 579, 469–473 CrossRef CAS.
- A. K. Shaw and S. K. Pal, J. Photochem. Photobiol., B, 2008, 90, 69–77 CrossRef CAS PubMed.
- F. S. Graciani and V. F. Ximenes, PLoS One, 2013, 8, e76849 CrossRef CAS PubMed.
- K. Baler, O. A. Martin, M. A. Carignano, G. A. Ameer, J. A. Vila and I. Szleifer, J. Phys. Chem. B, 2014, 118, 921–930 CrossRef CAS PubMed.
- D. V. Sotnikov, A. N. Berlina, V. S. Ivanov, A. V. Zherdev and B. B. Dzantiev, Colloids Surf., B, 2019, 173, 557–563 CrossRef CAS.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G. W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- H. Ohshima, T. W. Healy and L. R. White, J. Colloid Interface Sci., 1982, 90, 17–26 CrossRef CAS.
- H. Ohshima, J. Colloid Interface Sci., 2001, 239, 587–590 CrossRef CAS PubMed.
- N. Ouadah, T. Doussineau, T. Hamada, P. Dugourd, C. Bordes and R. Antoine, Langmuir, 2013, 29, 14074–14081 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp06371d |
| This journal is © the Owner Societies 2020 |