
Direct investigation of chalcogen bonds by multinuclear solid-state magnetic resonance and vibrational spectroscopy†

Abstract
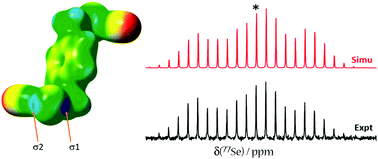
We report a multifaceted experimental and computational study of three self-complementary chalcogen-bond donors as well as a series of seven chalcogen bonded cocrystals. Bis(selenocyanatomethyl)benzene derivatives were cocrystallized with various halide salts (Bu4NCl, Bu4NBr, Bu4NI) and nitrogen-containing Lewis bases (4,4′-bipyridine and 1,2-di(4-pyridyl)ethylene). Three new single-crystal X-ray structures are reported. 77Se solid-state nuclear magnetic resonance spectroscopic study of a series of cocrystals establishes correlations between the NMR parameters of selenium and the local ChB geometry. For example, the 77Se isotropic chemical shift generally decreases on cocrystal formation. Diagnostic 13C chemical shifts are also described. In addition, all the chalcogen bonded cocrystals and pure tectons are investigated by Raman and IR spectroscopy techniques. Characteristic red shifts of the NC–Se stretching band upon cocrystal formation on the order of 10 to 20 cm−1 are observed, which provides a distinct signature of the chalcogen bond involving selenocyanates. The 125Te chemical shift tensor and X-ray structure of chalcogen-bonded tellurocyanatomethylbenzene are also reported. Insights into the connection between the electronic structure of the chalcogen bond and the experimentally measured 77Se chemical shift tensors are afforded through a natural localized molecular orbital density functional theory analysis. For the systems studied here, the lack of a very strong a correlation between experimental and DFT-computed 77Se chemical shift tensors leads to the conclusion that many structural features likely influence their ultimate values; however, computations on model systems reveal that the ChB alone produces consistent and predictable effects (e.g., the chalcogen chemical shift decreases as the chalcogen bond is shortened).
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

