
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferentiating the role of organic additives to assemble open framework aluminosilicates using INS spectroscopy†
Antony
Nearchou
a,
Jeff
Armstrong
 b,
Keith T.
Butler
ab,
Paul R.
Raithby
a and
Asel
Sartbaeva
*a
b,
Keith T.
Butler
ab,
Paul R.
Raithby
a and
Asel
Sartbaeva
*a
aUniversity of Bath, Department of Chemistry, Claverton Down, Bath, BA2 7AY, UK. E-mail: A.Sartbaeva@bath.ac.uk
bISIS Neutron and Muon Source, Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
First published on 11th June 2020
Abstract
Presently, there is little clarity concerning how organic additives control structure formation in the synthesis of zeolite catalysts. Such ambiguity is a major obstacle towards synthesis design of new bespoke zeolites with intended applications. Herein, we have applied inelastic neutron scattering (INS) spectroscopy to experimentally probe the nature of organic–framework interactions, which are crucial in understanding structure direction. With this technique we have studied the dynamics of 18-crown-6 ether, which can be used as an additive to direct the formation of four zeolites: Na-X, EMC-2, RHO and ZK-5. We observed significant softening of the 18-crown-6 ether molecule's dynamics upon occlusion within a zeolite host, with a strong influence on both the circular and radial vibrational modes. Furthermore, there is a strong correlation between the size/geometry of the zeolite framework cages and perturbations in the dynamics of the 18C6 oxyethylene chain. We propose that the approach used herein can be used to study other zeolites, and hence gain a more comprehensive view of organic–framework interactions.
Introduction
Zeolites are open framework aluminosilicates, known for their shape and size-selective molecular sieving properties.1,2 This selectivity can be used in conjunction with the introduction of active sites, allowing zeolites to be employed as catalysts in mainstream applications such as fluid catalytic cracking and methanol-to-hydrocarbon technology.3–5 However, their uses also extend to gas adsorption,6,7 water softening,8 sequestration of radionuclides9 and even emerging applications in drug delivery.10 Structurally they consist of corner sharing TO4 tetrahedra, where T = Si or Al. These tetrahedra are organised into geometric polyhedral shapes known as secondary building units (SBUs), which repeat throughout the zeolite's periodic framework structure.11The majority of zeolite syntheses require organic additives that promote the assembly of specific framework topologies.12,13 However, there is often no clarity on how organics influence the assembly process, which is a roadblock for endeavours to rationally synthesise new zeolites.14 Davis and Lobo15,16 propose that organic additives can be categorised into three groups based on their behaviour: space-filling species, organic structure directing agents (OSDAs) and true templates. Both OSDAs and templates are involved in the assembly of structural features, however templates are characterised by stronger organic–framework interactions which consequently imprint symmetry onto the zeolite structure. Alternatively, space-filling species are identified as merely occupying and stabilising the voids in the growing framework via weaker organic–framework interactions. Later studies have highlighted additional additive behaviours, such as ‘structure-blocking’ whereby the additive inhibits the cocrystallisation of impurity phases.17,18
Currently the use of organic additives is a necessity in the design and preparation of new zeolites for desired applications. Typically, synthesis design concerns computational calculations to determine which organic additives have an optimum geometric fit inside an existing or hypothetical framework.19–22 However, this approach only considers organic additives that behave as true templates, neglecting the other approaches available. In addition, there is little experimental work concerning the nature of the non-bonding van der Waals interactions between organic additives and the framework, let alone their importance in structure direction.
Herein we have studied the zeolites ZK-5, RHO, EMC-2 and Na-X, which can all be prepared under similar synthetic conditions, using 18-crown-6 ether (18C6) as an organic additive.23–27 Interestingly, for zeolite EMC-2 the use of 18C6 is a necessity,27,28 however the other three zeolites can be prepared without the need of this, or any organic additive.29–31Fig. 1 displays the framework structure of these four zeolites, in addition to the supercage that the occluded 18C6 occupies.26,32 As can be seen, the host supercage differs, illustrating that the organic–framework interactions that drive the assembly process and the role of the additive must also be different. This case study adequately portrays the issue at hand, where the understanding of organic–framework interactions is lacking, hindering our appreciation of additive behaviour during crystallisation.
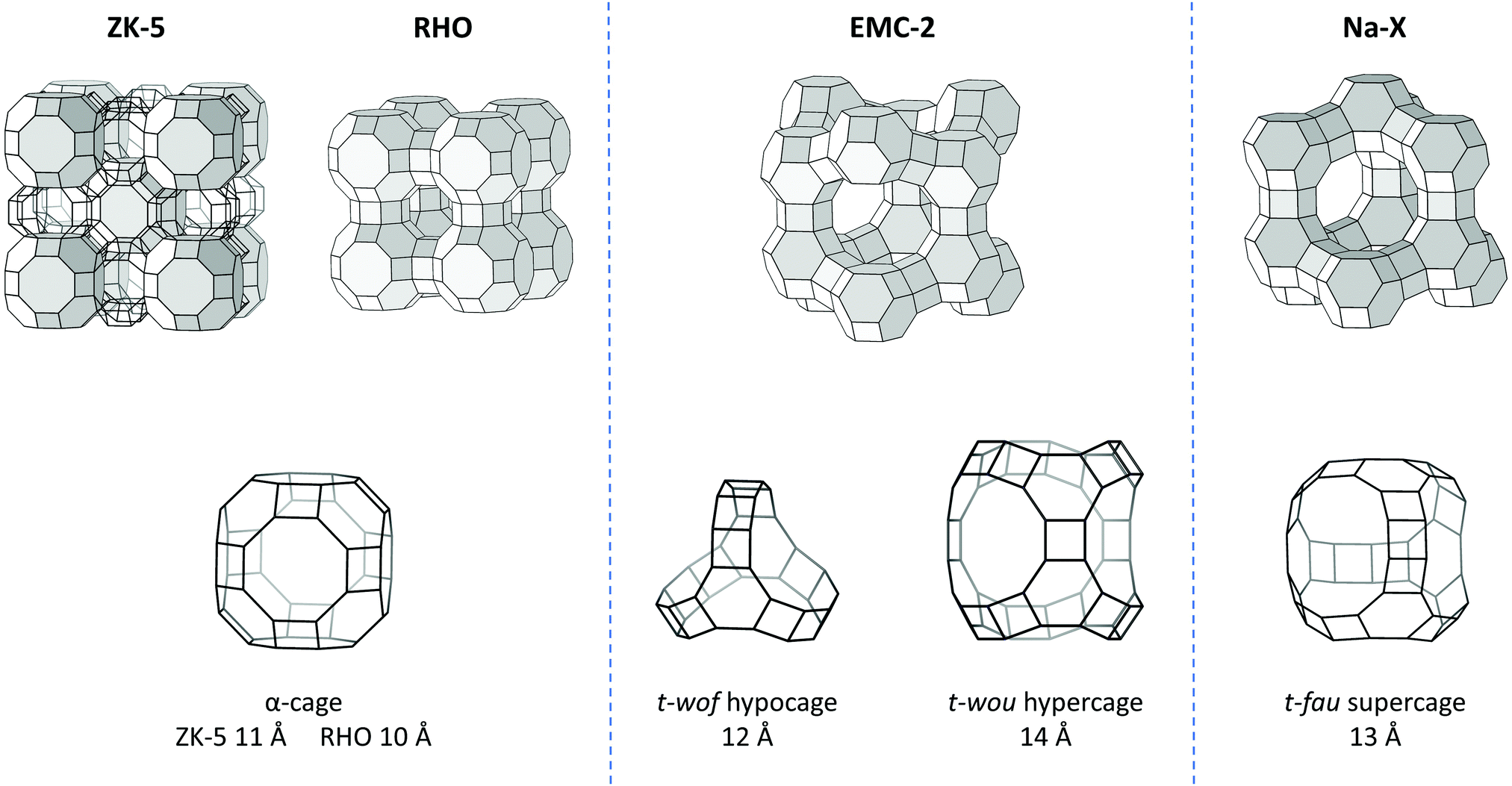 | ||
| Fig. 1 The framework structures of zeolites ZK-5, RHO, EMC-2 and Na-X, and the relevant supercages that the occluded 18C6 species occupy.26,32 Frameworks are shown as wireframes with O atoms removed and the nodes corresponding to a T (Si or Al) atom. Also shown is the approximate maximum internal diameter of the supercages. | ||
We have employed inelastic neutron scattering (INS) spectroscopy33,34 in order to probe the dynamics of the occluded 18C6 species and hence how the organic–framework interactions differ between the zeolites. This was achieved by comparison of the spectra of 18C6 in addition to the 18C6 containing (filled) zeolites and the calcined analogues where the additive has been removed (empty). Supplementing this, it was also considered that as metal cations are present in the synthesis process, they can conceivably coordinate to the central cavity of the 18C6 molecule, consequently influencing the molecular vibrations and organic–framework interactions. The relevant metal cations present during synthesis are the K+ cation for zeolite ZK-5, the Na+ cation for zeolites EMC-2 and Na-X, and a mix of both Cs+ and Na+ cations for zeolite RHO.
From studying these zeolites with INS spectroscopy, we report coherent differences in the organic–framework interactions which aid in understanding the behaviour of organic additives during crystallisation. Previously INS spectroscopy has been used to study the coordination of small organics in zeolites,35 however this is the first time that this technique has been employed to investigate additives used for zeolite synthesis.
Experimental
Sample preparation
The as-synthesised zeolite samples used for INS spectroscopy were prepared following the standard procedures reported by Chatelain et al.23–26 and used previously.27Table 1 contains the hydrogel batch compositions used to prepare each sample. To produce the batch hydrogels, sodium hydroxide (NaOH), potassium hydroxide (KOH), caesium hydroxide solution (50 wt% CsOH in water), strontium nitrate (Sr(NO3)2), 18-crown-6 ether (C12H24O6 18C6), sodium aluminate (NaAlO2), aluminium hydroxide (Al(OH)3), colloidal silica (LUDOX® HS-40, 40 wt% SiO2 in water) and deionised water were used. Additionally, deuterium oxide (D2O) was used to prepare the metal cation–crown complexes. Aside from the deionised water, the materials used were purchased from Sigma-Aldrich.| Zeolite | Al2O3 | Na2O | K2O | Cs2O | SrO | SiO2 | 18C6 | H2O |
|---|---|---|---|---|---|---|---|---|
| Na-X | 1 | 2.9 | 10 | 0.5 | 90 | |||
| EMC-2 | 1 | 2.0 | 10 | 0.5 | 87 | |||
| RHO | 1 | 1.8 | 0.3 | 10 | 0.5 | 100 | ||
| ZK-5 | 1 | 2.7 | 0.1 | 10 | 1.0 | 220 |
Zeolites Na-X, EMC-2 and RHO
A similar procedure was used to prepare zeolites Na-X, EMC-2 and RHO. In each case, the relevant metal hydroxides were first dissolved in deionised water alongside the 18C6. Following this, the sodium aluminate was added, and the solution stirred until homogeneous. Next, the colloidal silica was slowly poured into the solution under stirring, in order to prevent gelation.For zeolite Na-X the hydrogel was prepared in a Naglene Teflon FEP bottle. The hydrogel was aged at ambient conditions, under stirring for 4 hours. After aging, the bottle was sealed and placed into an oven at 100 °C for 8 days. The zeolite EMC-2 and RHO hydrogels were aged under stirring for 24 hours at ambient conditions. The gels were then transferred into Teflon cups within sealed stainless-steel autoclaves. For zeolite EMC-2 the autoclave was heated to 110 °C for 12 days. As for zeolite RHO, the autoclave was heated to 110 °C for 8 days.
After the appropriate duration of hydrothermal conditions, the respective crystallisation vessel was removed from the oven and cooled to ambient temperatures. The product was subsequently separated from the mother liquor using Buchner filtration, and washed with deionised water until the filtrate was of neutral pH. The white powder was then dried at 90 °C and ground.
Zeolite ZK-5
The hydrogel for zeolite ZK-5 was prepared using an alternative procedure. First the potassium hydroxide was dissolved in deionised water. To this solution the aluminium hydroxide was added, and the flask weighed before being heated to near 110 °C under stirring. After the solution became clear it was cooled to ambient temperature. The flask was then weighed and topped up with any water which evaporated during the heating process. In a separate vessel, the 18C6 and strontium nitrate were dissolved in a portion of deionised water. To this solution, the colloidal silica was added and stirred to ensure homogeneity. Following this, the alumina solution was rapidly poured into the silica solution to produce the hydrogel. The gel was subsequently aged under stirring for 30 minutes.The hydrogel was transferred to a Teflon cup within a stainless-steel autoclave and placed into a 150 °C oven for 5 days. After this time, the autoclave was removed from the oven and cooled to ambient temperatures. The product was then separated from the mother liquor using the same procedure as the other zeolites.
Metal cation–crown complexes
Isolated metal cation–crown complexes were prepared with Na+, K+ and Cs+ cations. These cations were selected, as they are the most likely candidates to coordinate to the 18C6 molecule during the synthesis of the relevant zeolites. For zeolites Na-X and EMC-2 this is the Na+ cation, for zeolite RHO it is both the Na+ and Cs+ cations, and for zeolite ZK-5 it is the K+ cation. To prepare the isolated complexes, a metal salt to 18C6 ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used to ensure that a cation was coordinated to every 18C6 molecule. The metal salt sources used were the same as those in the preparation of the zeolite samples.
:1 was used to ensure that a cation was coordinated to every 18C6 molecule. The metal salt sources used were the same as those in the preparation of the zeolite samples.
Preparation was conducted within a nitrogen atmosphere glove bag. For each complex, 3 g of 18C6 and the respective mass of metal hydroxide were dissolved in the minimum volume of deuterium oxide. The masses of metal hydroxide used were 0.92 g sodium hydroxide, 1.3 g potassium hydroxide and 6.8 g of the caesium hydroxide solution. Approximately 4.5 ml of deuterium oxide was used for dissolution, aside from the caesium hydroxide solution where the water content was sufficient. The solutions were then put under a nitrogen flow in the glove bag, to permit evaporation and the production of salt crystals via evaporative crystallisation. Subsequently, the salt was dissolved in deuterium oxide and evaporated once again. This was repeated for three cycles to remove as much non-deuterated water as possible.
Calcination and dehydration
A ‘filled’ and ‘empty’ analogue of each zeolite was prepared for INS spectroscopy. These terms referring to occluded 18C6 molecules being intact and removed respectively.The empty analogue was produced by calcining a portion of the as-synthesised zeolite sample. The samples were placed into a crucible within a tube furnace under air and heated at a ramp rate of 1 K min−1 to 450 °C for 6 hours. During this heating regime, the program was halted at 100 °C, 200 °C and 300 °C for 1 hour each. After calcination, the furnace was cooled at 1 K min−1 to 200 °C, held static for 1 hour and then allowed to cool to ambient conditions.
Both the empty and filled (as-synthesised) analogues of the zeolite were dehydrated. This was achieved by heating the samples under vacuum within a tube furnace. The heating cycle consisted of heating at a ramp rate of 1 K min−1 to 200 °C for 6 hours and involved a static stage at 100 °C for 1 hour. After dehydration the furnace was cooled to 100 °C at a rate of 1 K min−1, held static for 1 hour and then allowed to cool to ambient conditions.
The metal cation–crown complexes and 18-crown-6 ether (from Sigma Aldrich) were also dehydrated, however this was achieved under vacuum at ambient temperature overnight. Heating was not used to avoid melting or sublimation of the organic samples. Following dehydration, both the zeolites and organic samples were transported under vacuum to an inert argon glove box. The samples were then sealed into glass vials under argon gas.
Thermogravimetry
A Setaram Setsys Evolution TGA 16/18 instrument was used to determine the 18C6 and water mass% content in the four zeolites via thermogravimetry. The samples were first purged with air at a flow rate of 20 ml min−1 at 30 °C for 5 minutes. The samples were then heated under air flow from 30 °C to 600 °C at a ramp rate of 5 K min−1. For both zeolites Na-X and EMC-2 there was an overlap of the water desorption and 18C6 decomposition events. In order to separate these events, intermediate stages of static temperature were introduced to the ramping segments. These were 130 °C for 60 min for zeolite Na-X, and 180 °C for 20 min for EMC-2. An average mass% was calculated from three separate scans.Inelastic neutron scattering spectroscopy
Analysis of the samples with inelastic neutron scattering (INS) spectroscopy was achieved on the TOSCA indirect geometry spectrometer at ISIS Neutron and Muon Source, Didcot, UK.36 The dehydrated samples were prepared for analysis within a glove box under an inert argon atmosphere. The samples were first loaded into aluminium foil sachets, with the loaded sample mass recorded. The sachets were then sealed between two aluminium plates, using indium wire as a seal to keep the sample under an inert atmosphere. INS spectra were recorded within the energy transfer range −20 to 8050 cm−1, at a temperature of 10 K for 5–7 hours.Subsequent data analysis, visualisation and normalisation was achieved using the Mantid software.37 The INS spectra were normalised to the mass of sample loaded into the aluminium foil sachets – corresponding to the amount of sample in the neutron beam path. For the zeolite samples, the spectra were further normalised to the mass of 18C6 present in the sample.
Density functional theory (DFT) calculations
In order to identify the vibrational modes of the 18C6 molecule, the modes were calculated in the quantum-mechanics-based Gaussian software.38 The input data used were the crystal structures of the isolated 18C6 and thiocyanate metal cation–crown complexes of sodium, potassium and caesium in the literature, converted to molecular clusters for the simulations.39–43 For each species, the geometry was optimised before calculation of the vibrational modes. We used the B3LYP44,45 functional and a triple zeta Gaussian basis set, optimised at the one-electron exact two-component level.46 From the basis set exchange47 (label x2c-TZVPall) was used for Cs and the 6-31G* basis set was used for all other atoms. The simulated INS spectra were calculated from the Gaussian output data with the AbINS algorithm.48 Visualisation of the vibrational modes and production of the 18C6 molecule graphics were achieved using the Avogadro (version 1.2.0) software.49Results and discussion
18-crown-6 ether – previous work
Table 2 lists the energy ranges at which the vibrational modes of 18-crown-6 ether (18C6) are anticipated, as reported by Fukuhara et al.50 As shown, this includes the conventional vibrations of discrete diatomic bonds and groups, in addition to the lower energy skeletal vibrations of the molecule. These skeletal vibrations correspond to a mixture of C–C and C–O bond torsions and bends, which are divided into radial and circular modes. The radial modes refer to vibrations where the oxyethylene chain is moving out of the molecular plane and breathing multiaxially. Alternatively, the circular modes correlate to vibrations where the oxyethylene chain remains in the molecular plane.| ν/cm−1 | Vibrational assignment |
|---|---|
| <70 | Breathing modes, out of the plane of the ring |
| >200 | CCO deformation and COC bending |
| <300 | Radial modes, C–C and C–O torsions |
| 300–600 | Circular modes |
| <600 | Skeletal bending and torsions |
| 820–1160 | C–O and C–C stretching and CH2 rocking – sensitive to oxyethylene chain conformation |
| 1220–1310 | CH2 twisting |
| 1330–1440 | CH2 wagging |
| 1440–1500 | CH2 bending/scissoring |
| 2800–3000 | CH2 antisymmetric and symmetric vibrations of the oxyethylene group |
In addition to the vibrational modes, the conformation of the 18C6 molecule needs to be considered. Previously it has been shown that the 18C6 oxyethylene chain is inherently flexible, capable of adapting a series of possible conformations which can influence the vibrational modes observed in the low energy skeletal region. Fig. 2 displays the two most energetically stable conformers of the 18C6 oxyethylene chain, as determined by calculations.50 These are the D3d and Ci conformations, which are observed in polar solvents and the solid crystal respectively.39,40,50,51 Furthermore, the presence of a metal cation coordinated to the 18C6 molecule will also influence the conformation of the oxyethylene chain based on both the size of the cation, and the electrostatic environment.41–43,51,52
 | ||
| Fig. 2 The conformation of the 18C6 oxyethylene chain in the D3d and Ci symmetries. | ||
Metal cation–crown complexes
Fig. 3 displays the INS spectra (0–1600 cm−1) of 18C6 and the metal cation–crown complexes. For 18C6 the energies of the observed vibrational modes have been labelled, in addition to the anticipated energy ranges of specific oxyethylene chain vibrations according to Fukuhara et al.50 in Table 2. There is a good match-up of the 800–1600 cm−1 spectral region between the complexes and 18C6, which corresponds to discrete bond vibrations within the oxyethylene chain. This congruence confirms that atomic connectivity of the 18C6 oxyethylene chain is sustained upon complex formation.In the <700 cm−1 region there is an excellent match-up of the spectra for the Na+ and K+ cation–crown complexes to the isolated 18C6. These three samples show a vibrational mode at 416 cm−1, which is a characteristic circular mode for the Ci conformation – shown in Fig. 2.50,52 This is anticipated for the isolated 18C6, which is reported to be in this conformation in the solid state crystal structure.39 However, Dobler41 and Seiler42 previously reported that that the oxyethylene chain is bent and in the D3d conformation for the Na+ and K+ thiocyanate complexes respectively. Ab initio calculations by Glendening et al.40 on the hydrated non-thiocyanate complexes are consistent with Dobler and Seiler, with the elucidation that in the Na+ complex the oxyethylene chain is in the C1 conformation. This contrasts our observations herein, with our complexes which have been prepared in water with hydroxide sources. Closer inspection of the INS spectrum of the K+ complex in Fig. 3 shows low intensity peaks at 280 and 360 cm−1, not seen in the isolated 18C6 sample. These vibrations are characteristic circular modes for the D3d symmetry.50 This suggests that some of the K+ complex molecules are adopting the D3d symmetry, agreeing with the observations seen in other potassium crown ether systems.
 | ||
| Fig. 3 INS spectra of 18C6 and the metal cation–crown complexes in the 0–1600 cm−1 region. The spectrum in black corresponds to the isolated 18C6, red to the Na+ complex, green to the K+ complex and blue to the Cs+ complex. The spectra have been normalised to the mass of samples loaded onto the spectrometer. | ||
The <700 cm−1 region of the INS spectrum of the Cs+ complex shows significant differences to the isolated 18C6. Most notable is the absence of the 404, 416 and 452, 464 cm−1 doublets, and the appearance of new singlet vibrational modes at 321 and 587 cm−1, where doublets are observed in the 18C6 spectrum. Combined with the presence of a vibrational mode at 280 cm−1, these observations confirm that the oxyethylene chain is in the D3d conformation. This agrees with the crystal structure of the thiocyanate Cs+ complex,43 in addition to the ab initio calculations of the isolated 18C6 molecule by Glendening et al.40 Furthermore, the breathing mode region (<200 cm−1) shows peak broadening and substantial loss of coherence compared to the isolated 18C6. This is characteristic of disorder, whereby there are a range of local environments for the relevant atoms involved in the vibration, thus creating a distribution of peaks. Such disorder is rationalised by the position of the large Cs+ cation above the molecular plane,40,43 outside of the crown ether cavity, and smearing the vibrational modes.
The vibrational modes corresponding to the Ci and D3d conformations were identified using DFT calculations. Tables 3 and 4 list the calculated vibrational modes for the isolated Ci symmetry 18C6 molecule and D3d symmetry Cs+ complex, respectively. Also included are the peaks observed from the INS spectra of these two samples, and the assigned type of vibrational mode. These tables show a good agreement between the calculated and experimental datasets, confirming that the oxyethylene chain in each sample is in the assigned conformation. The ESI† contains a full list of the DFT calculated vibration modes and a comparison of the experimental and AbINS48 simulated INS spectra, which further support our assignments.
| ν obs/cm−1 | ν calc/cm−1 | Vibrational assignment | ν obs/cm−1 | ν calc/cm−1 | Vibrational assignment |
|---|---|---|---|---|---|
| 37 | Breathing | 404 | 406 | Circular | |
| 45 | Breathing | 416 | 419 | Circular | |
| 59 | 56, 58 | Breathing | 452 | 450 | Circular |
| 75 | 68 | Radial | 464 | 463 | Circular |
| 83 | 82 | Radial | 524 | 526 | Circular |
| 87 | Radial | 539 | 543 | Circular | |
| 94 | 98 | Radial | 570 | 576 | Circular |
| 101 | 101, 102 | Circular | 582 | 588 | Circular |
| 110 | 114 | Circular | 834 | 840, 841 | C–O/C–C str CH2 rock |
| 120 | Circular | 864 | 884, 886 | C–O/C–C str CH2 rock | |
| 131 | Circular | 890 | 904 | C–O/C–C str CH2 rock | |
| 147 | 145, 153 | Circular | 938 | 916 | C–O/C–C str CH2 rock |
| 164 | 160 | Circular | 986 | 969 | C–O/C–C str CH2 rock |
| 171 | 180 | Circular | 1054 | 1065, 1067 | C–O/C–C str CH2 rock |
| 195 | 184, 203 | Circular | 1096 | 1091 | C–O/C–C str CH2 rock |
| 226 | 213 | Circular | 1142 | 1152 | C–O/C–C str CH2 rock |
| 234 | Circular | 1229 | 1197 | CH2 twist | |
| 268 | 262, 263 | Circular | 1273 | 1283 | CH2 twist |
| 312 | 304, 308 | Circular | 1338 | 1336 | CH2 wag |
| 322 | Circular | 1378 | 1373, 1374 | CH2 wag | |
| 350 | 341, 351 | Circular | 1449 | 1461, 1463 | CH2 bend |
| ν obs/cm−1 | ν calc/cm−1 | Vibrational assignment | ν obs/cm−1 | ν calc/cm−1 | Vibrational assignment |
|---|---|---|---|---|---|
| 237 | 239 | Circular | 837 | 842 | C–O/C–C str, CH2 rock |
| 253 | 257 | Circular | 871 | 887 | C–O/C–C str, CH2 rock |
| 264 | 258 | Circular | 929 | 931 | C–O/C–C str, CH2 rock |
| 283 | 272 | Circular | 953 | 950 | C–O/C–C str, CH2 rock |
| 321 | 315 | Circular | 1085 | 1083 | C–O/C–C str, CH2 rock |
| 360 | 359 | Circular | 1112 | 1109 | C–O/C–C str, CH2 rock |
| 529 | 528, 534 | Circular | 1140 | 1142 | C–O/C–C str, CH2 rock |
| 548 | 549 | Circular | 1247 | 1262 | CH2 twist |
| 587 | 585 | Circular | 1279 | 1279 | CH2 twist |
| 1364 | 1387 | CH2 wag | |||
| 1413 | 1414, 1415 | CH2 wag |
These findings suggest that the zeolites prepared with Na+ or K+ cations will have no significant influence on the occluded 18C6 species vibrations which are due to the coordinated cation. Therefore, the zeolite framework is anticipated to be responsible for any perturbations to the vibrational modes observed. This includes zeolites Na-X, EMC-2 and ZK-5. Concerning zeolite RHO the synthesis hydrogel contains a mixture of Na+ and Cs+ cations, the latter of which is shown to modify the oxyethylene chain conformation and dynamics. However, the synthesis hydrogel contains a greater concentration of Na+ to Cs+ cations, so we anticipated that the Na+ complex is the most likely candidate to be present in the zeolite.
The zeolites
Table 5 contains the 18C6 mass% content of the four zeolites, as determined by thermogravimetry, which were used to normalise the INS spectra. The table also includes the name of the supercage that the 18C6 species occupies, as well as the approximate diameter of these cages.53 The INS spectra (0–1600 cm−1) of the 18C6 containing (filled) and empty zeolite samples are shown in Fig. 4. By direct comparison of the filled and empty zeolite spectra the 18C6 species vibrational modes can be visualised more easily. The 800–1600 cm−1 region of the filled zeolites show a good match-up with the isolated 18C6, demonstrating that the molecule is intact after zeolite crystallisation. The lower energy edge of this range displays broadening of the CH2 rocking vibrations in the filled zeolites; zeolite Na-X in particular. The 820–1160 cm−1 region is known to be sensitive to the conformation of the oxyethylene chain,50 which we anticipate is the cause of this spectral broadening.| Zeolite | Occupied supercage | 18C6 content mass% | Max. internal diameter of supercage/Å | Max. diameter of an occluded sphere/Å |
|---|---|---|---|---|
| ZK-5 | α-cage | 2.94 (0.12) | 11 | 10.61 |
| RHO | α-cage | 7.32 (0.09) | 10 | 10.37 |
| Na-X | t-fau | 10.2 (0.11) | 13 | 11.18 |
| EMC-2 | t-wof, t-wou | 15.2 (0.07) | 12, 14 | 11.49 |
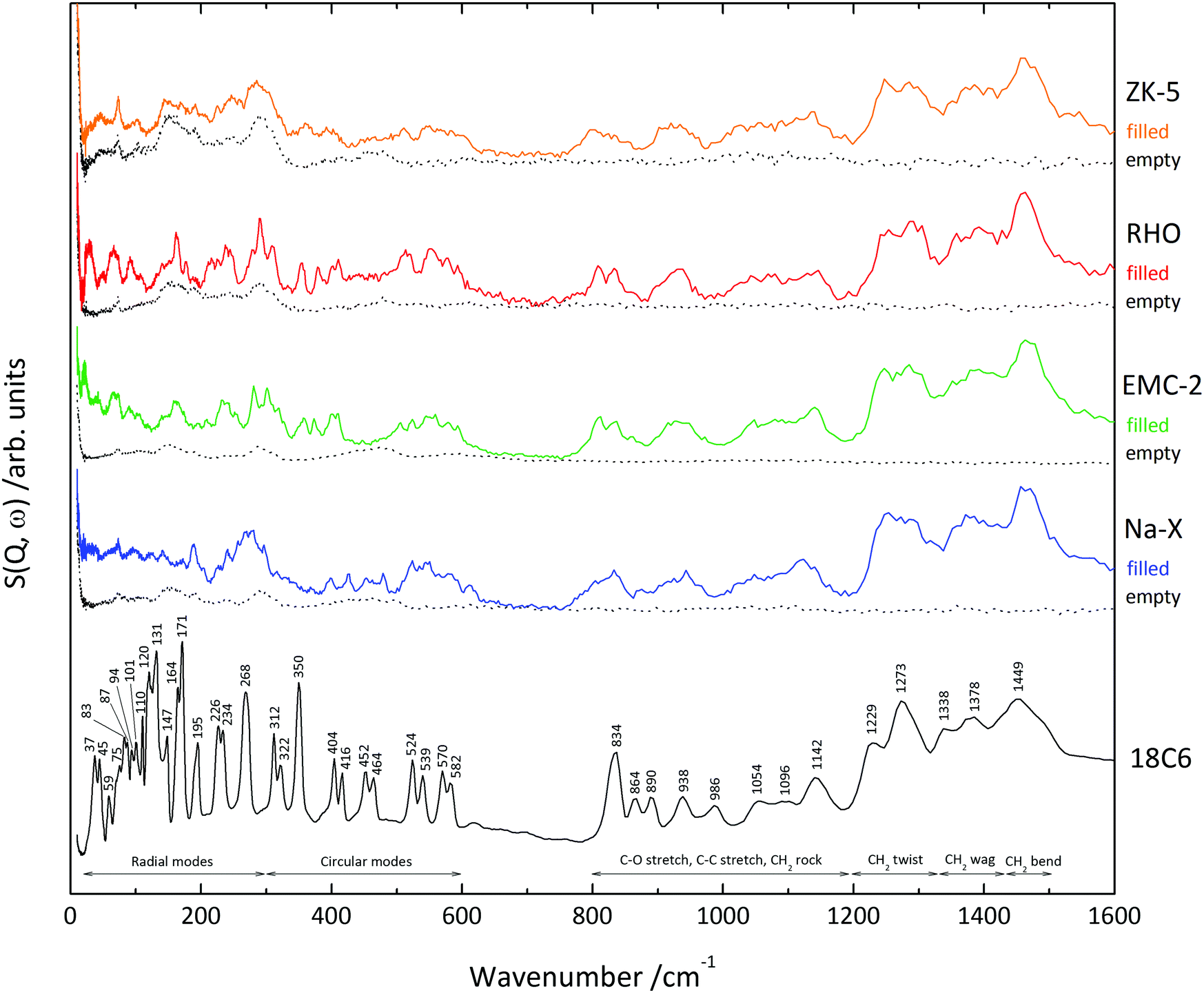 | ||
| Fig. 4 INS spectra of 18C6 and the filled zeolite samples in the 0–1600 cm−1 region. The spectrum in black corresponds to the isolated 18C6, orange to zeolite ZK-5, red to RHO, green to EMC-2 and blue to Na-X. The block dotted spectra correspond to the empty zeolites as labelled. The spectra have been normalised to the mass of samples loaded onto the spectrometer, and the mass% content of 18C6. | ||
In the <700 cm−1 skeletal region of the spectra it is apparent that the 18C6 vibrational modes display broadening in the filled zeolite samples. This softening of the skeletal motions is due to the molecule's confinement within the framework supercages. The vibrational mode at approximately 416 cm−1 is present for all four zeolites, demonstrating that the occluded 18C6 species is in the Ci conformation. This confirms that any coordinated metal cations to the 18C6 do not change the symmetry of the oxyethylene chain within the zeolites. Furthermore, the presence of this characteristic mode in zeolite RHO suggests it is the Na+ complex present in the supercage, not the Cs+ complex, as we anticipated.
Zeolites RHO and EMC-2 present coherent peaks throughout the skeletal range, however for zeolites ZK-5 and Na-X the spectral peaks correlated to the occluded 18C6 are less clear. The reason for this incoherence in zeolite ZK-5 is due to the low 18C6 mass content of 2.94%, as shown in Table 5, which is below the sensitivity of the spectrometer. This is confirmed by the excellent match-up below 300 cm−1 for the empty and filled zeolite ZK-5 analogues, meaning the modes observed in this region correspond solely to the zeolite framework. Concerning zeolite Na-X, the spectra illustrate that the occluded 18C6 species displays broad, low intensity vibrational modes in the 0–200 cm−1 region. Interestingly, zeolite Na-X has the second greatest 18C6 mass content (10.2%) of the four zeolites, meaning that these low intensity peaks are not attributed to low quantities of organic material as with ZK-5. Rather, this broadening in the low energy region is indicative of dynamic disorder. As this is occurring in the low energy phonon and rotation region,54 we suggest that the 18C6 species is tumbling within the spherical t-fau supercage that it occupies.
The radial modes can only be identified clearly in zeolites RHO and EMC-2, where they are seen to shift to a higher wavenumber. In this case, it is the physical confinement of the 18C6 species by the framework, which restricts the molecular breathing motions, so a greater energy is required to activate the vibrational modes. This is comparable to observations seen by Jobic et al. for the out-of-plane C–H bending modes of benzene occluded in zeolite NaY.55 Although the radial modes cannot be observed in the spectra for zeolite ZK-5, such an energy shift is expected as the 18C6 molecule occupies an α-cage as it does in zeolite RHO.26
The circular modes in zeolites RHO, EMC-2 and ZK-5 are generally observed to shift to a lower wavenumber – including the characteristic Ci conformation 416 cm−1 vibration. We suggest that this decrease in energy is due to van der Waals and electrostatic interactions between the zeolite framework and oxyethylene chain. For zeolite Na-X there is little change to the circular modes, indicating there is an absence of these organic–framework interactions seen in the other zeolites. This is congruent with our proposal that the 18C6 species is tumbling in the zeolite Na-Xt-fau cavity.
Although this is the general trend with the circular modes, some exceptions are observed. The 350 cm−1 vibration is seen to shift to a higher energy in zeolites RHO, EMC-2 and ZK-5, but is absent in Na-X. This vibrational mode is shown in Fig. 5, and is primarily characterised by a C–C torsion about the gauche OCCO group, leading to movement of the oxyethylene chain above and below the molecular plane. Despite being a circular mode, it is this radial characteristic which leads to the relevant zeolites confining the vibration, so a greater energy for excitation is required; as with the radial modes. Alternatively, the absence of the mode in zeolite Na-X is anticipated to be due to the dynamic disorder which leads to this specific vibration becoming smeared.
 | ||
| Fig. 5 Graphics showing the 350 and 452/464 cm−1 circular vibrational modes of the 18C6 oxyethylene chain in the Ci conformation. The yellow arrows demonstrate the primary movements. Videos of these vibrations can be found alongside the ESI.† | ||
Furthermore, the degenerate 452 and 464 cm−1 circular modes are observed in zeolite Na-X but absent in the other three zeolites. Like the 350 cm−1 mode, this vibration involves a C–C torsion about the gauche OCCO group which in this case causes a creasing of the oxyethylene chain with radial character, as shown in Fig. 5. In zeolites RHO and ZK-5 it is believed that this mode is not observed due to considerable softening from the smaller volume of the α-cage compared to the t-fau cavity in Na-X (Table 5). The fact that these two modes are present in zeolite Na-X illustrates what little influence any organic–framework interactions have on the 18C6 dynamics. This agrees with our previous findings from high-pressure X-ray diffraction.56
In contrast, for zeolite EMC-2 the supercages are more voluminous than the t-fau cavity (Table 5), so the rationale of size fails to explain the softening of the vibrational modes. Instead, we suspect the softening is due to the elliptical geometry of the zeolite EMC-2 supercages,13 providing a degree of geometric match-up and stronger organic–framework interactions to the 18C6 species. This is supported by the crystal structure of zeolite EMC-2, which shows that the occluded 18C6 species are perpendicular to the c axis, aligned with the cage geometry.32 Such a focus on geometrical match-up between the 18C6 species and the zeolite EMC-2 supercages is congruent with the understanding that 18C6 behaves as a true template during the crystallisation process.57–60
Conclusions
Overall, it is seen that there are substantial changes to the dynamics of the 18C6 oxyethylene chain upon occlusion within a zeolite host. Such changes are dominated by the characteristics of the framework supercages that the 18C6 species occupies. In zeolites EMC-2, RHO and ZK-5 there is softening of the vibrational modes that express significant radial characteristics. Such softening is rationalised by either the geometry of the cage (EMC-2) or the size (RHO and ZK-5), stressing the significance of organic–framework interactions in these zeolites. Alternatively, in zeolite Na-X there is evidence of dynamic disorder, suggesting that the occluded 18C6 species is tumbling within the t-fau supercage. This highlights that the organic–framework interactions have little impact on dynamics of the 18C6 oxyethylene chain, agreeing with our previous findings.These observations provide valuable insights into how different framework geometries interact with an organic additive, which can be indicative of its role in synthesis. We propose that the approach herein can be applied to understand the importance of organic–framework interactions in other zeolitic systems. For example, MFI-type zeolites such as silicalite-1 and ZSM-5 used in the petrochemical industry can be prepared using a variety of organic additives.61–63 It would therefore be beneficial to observe the differing nature of the organic–framework interactions.
Author contributions
The original manuscript was prepared and written by A. N. with input from all co-authors. Samples preparation and data analysis was performed by A. N. The DFT calculations were performed by K. T. B. The INS spectra were obtained using the TOSCA instrument at ISIS Neutron and Muon Source, collected by A. N. and A. S. who were assisted by J. A. All authors were involved in the data interpretation and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. thanks the Royal Society for funding. Both A. N. and P. R. R. thank the EPSRC for funding (EP/K004956/1). The INS spectra herein were collected on the TOSCA instrument at ISIS Neutron and Muon Source, Didcot, UK. We thank the STFC for accepting our research proposal.Notes and references
- M. E. Davis, Ind. Eng. Chem. Res., 1991, 30, 1675–1683 CrossRef CAS.
- D. W. Breck, Zeolite molecular sieves: structure, chemistry and use, Krieger, 1984 Search PubMed.
- B. M. Weckhuysen and J. Yu, Chem. Soc. Rev., 2015, 44, 7022–7024 RSC.
- E. Vogt and B. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- H. W. Langmi, A. Walton, M. M. Al-Mamouri, S. R. Johnson, D. Book, J. D. Speight, P. P. Edwards, I. Gameson, P. A. Anderson and I. R. Harris, J. Alloys Compd., 2003, 356, 710–715 CrossRef.
- J. C. Fisher, R. V. Siriwardane and R. W. Stevens Jr, Ind. Eng. Chem. Res., 2011, 50, 13962–13968 CrossRef CAS.
- S. Wang and Y. Peng, Chem. Eng. J., 2010, 156, 11–24 CrossRef CAS.
- H. Yeritsyan, A. Sahakyan, V. Harutyunyan, S. Nikoghosyan, E. Hakhverdyan, N. Grigoryan, A. Hovhannisyan, V. Atoyan, Y. Keheyan and C. Rhodes, Sci. Rep., 2013, 3, 2900 CrossRef PubMed.
- M. G. Rimoli, M. R. Rabaioli, D. Melisi, A. Curcio, S. Mondello, R. Mirabelli and E. Abignente, J. Biomed. Mater. Res., Part A, 2008, 87, 156–164 CrossRef PubMed.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Amsterdam, 6th edn, 2007 Search PubMed.
- M. D. Oleksiak and J. D. Rimer, Rev. Chem. Eng., 2014, 30, 1–49 CAS.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/, accessed 09/01/16, 2016.
- P. Guo, J. Shin, A. G. Greenaway, J. G. Min, J. Su, H. J. Choi, L. Liu, P. A. Cox, S. B. Hong and P. A. Wright, Nature, 2015, 524, 74 CrossRef CAS PubMed.
- R. F. Lobo, S. I. Zones and M. E. Davis, Inclusion Chemistry with Zeolites: Nanoscale Materials by Design, 1995, pp. 47–78 Search PubMed.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- P. A. Cox, J. L. Casci and A. P. Stevens, Faraday Discuss., 1997, 106, 473–487 RSC.
- S. I. Zones and Y. Nakagawa, Microporous Mater., 1994, 2, 543–555 CrossRef CAS.
- D. W. Lewis, D. J. Willock, C. R. A. Catlow, J. M. Thomas and G. J. Hutchings, Nature, 1996, 382, 604–606 CrossRef CAS.
- D. W. Lewis, C. M. Freeman and C. R. A. Catlow, J. Phys. Chem., 1995, 99, 11194–11202 CrossRef CAS.
- J. Dhainaut, T. J. Daou, A. Chappaz, N. Bats, B. Harbuzaru, G. Lapisardi, H. Chaumeil, A. Defoin, L. Rouleau and J. Patarin, Microporous Mesoporous Mater., 2013, 174, 117–125 CrossRef CAS.
- R. Pophale, F. Daeyaert and M. W. Deem, J. Mater. Chem. A, 2013, 1, 6750–6760 RSC.
- T. Chatelain, J. Patarin, R. Farre, O. Petigny and P. Schulz, Zeolites, 1996, 17, 328–333 CrossRef CAS.
- T. Chatelain, J. Patarin, M. Soulard, J. L. Guth and P. Schulz, Zeolites, 1995, 15, 90–96 CrossRef CAS.
- T. Chatelain, J. Patarin, E. Fousson, M. Soulard, J. L. Guth and P. Schulz, Microporous Mater., 1995, 4, 231–238 CrossRef CAS.
- T. Chatelain, J. Patarin, E. Brendle, F. Dougnier, J. L. Guth and P. Schulz, Stud. Surf. Sci. Catal., 1997, 105, 173–180 CrossRef.
- A. Nearchou, P. R. Raithby and A. Sartbaeva, Microporous Mesoporous Mater., 2018, 255, 261–270 CrossRef CAS.
- A. Nearchou, M.-L. U. Cornelius, J. M. Skelton, Z. L. Jones, A. B. Cairns, I. E. Collings, P. R. Raithby, S. A. Wells and A. Sartbaeva, Molecules, 2019, 24, 641 CrossRef CAS PubMed.
- A. Nearchou and A. Sartbaeva, CrystEngComm, 2015, 17, 2496–2503 RSC.
- L. Zhang and Y. Huang, J. Porous Mater., 2015, 22, 843–850 CrossRef CAS.
- H. Robson, Verified Syntheses of Zeolitic Materials, Elsevier Science B.V., Amsterdam, 2nd edn, 2001 Search PubMed.
- C. Baerlocher, L. B. McCusker and R. Chiappetta, Microporous Mater., 1994, 2, 269–280 CrossRef CAS.
- S. F. Parker, F. Fernandez-Alonso, A. J. Ramirez-Cuesta, J. Tomkinson, S. Rudić, R. S. Pinna, G. Gorini and J. F. Castañon, J. Phys.: Conf. Ser., 2014, 554, 012003 CrossRef.
- M. Zanetti, S. Bellissima, L. del Rosso, F. Masi, M. Chowdhury, A. De Bonis, L. Di Fresco, C. Scatigno, J. Armstrong and S. Rudić, Phys. B, 2019, 562, 107–111 CrossRef CAS.
- A. Seel, A. Sartbaeva, A. Rammirez-Cuesta and P. Edwards, Phys. Chem. Chem. Phys., 2010, 12, 9661–9666 RSC.
- R. S. Pinna, S. Rudić, S. F. Parker, J. Armstrong, M. Zanetti, G. Škoro, S. P. Waller, D. Zacek, C. A. Smith and M. J. Capstick, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 896, 68–74 CrossRef CAS.
- O. Arnold, J.-C. Bilheux, J. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. F. Leal and M. Gigg, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- J. t. Dunitz and P. Seiler, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2739–2741 CrossRef.
- E. D. Glendening, D. Feller and M. A. Thompson, J. Am. Chem. Soc., 1994, 116, 10657–10669 CrossRef CAS.
- M. Dobler, J. Dunitz and P. Seiler, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2741–2743 CrossRef.
- P. Seiler, M. Dobler and J. Dunitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2744–2745 CrossRef.
- M. Dobler and R. Phizackerley, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2748–2750 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- P. Pollak and F. Weigend, J. Chem. Theory Comput., 2017, 13, 3696–3705 CrossRef CAS PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- K. Dymkowski, S. F. Parker, F. Fernandez-Alonso and S. Mukhopadhyay, Phys. B, 2018, 551, 443–448 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- K. Fukuhara, K. Ikeda and H. Matsuura, J. Mol. Struct., 1990, 224, 203–224 CrossRef CAS.
- A. Al-Kahtani, N. Al-Jallal and A. El-Azhary, Spectrochim. Acta, Part A, 2014, 132, 70–83 CrossRef CAS PubMed.
- H. Takeuchi, T. Arai and I. Harada, J. Mol. Struct., 1986, 146, 197–212 CrossRef CAS.
- M. Foster, I. Rivin, M. Treacy and O. D. Friedrichs, Microporous Mesoporous Mater., 2006, 90, 32–38 CrossRef CAS.
- G. Kearley, Nucl. Instrum. Methods Phys. Res., Sect. A, 1995, 354, 53–58 CrossRef CAS.
- H. Jobic and A. Fitch, Studies in Surface Science and Catalysis, Elsevier, 1997, vol. 105, pp. 559–566 Search PubMed.
- A. Nearchou, M.-L. U. Cornelius, Z. L. Jones, I. E. Collings, S. A. Wells, P. R. Raithby and A. Sartbaeva, R. Soc. Open Sci., 2019, 6, 182158 CrossRef PubMed.
- O. Terasaki, T. Ohsuna, V. Alfredsson, J. O. Bovin, D. Watanabe, S. W. Carr and M. W. Anderson, Chem. Mater., 1993, 5, 452–458 CrossRef CAS.
- T. Ohsuna, O. Terasaki, V. Alfredsson, J. O. Bovin, D. Watanabe, S. W. Carr and M. W. Anderson, Proc. R. Soc. A, 1996, 452, 715–740 CAS.
- S. L. Burkett and M. E. Davis, Microporous Mater., 1993, 1, 265–282 CrossRef CAS.
- E. J. P. Feijen, K. Devadder, M. H. Bosschaerts, J. L. Lievens, J. A. Martens, P. J. Grobet and P. A. Jacobs, J. Am. Chem. Soc., 1994, 116, 2950–2957 CrossRef CAS.
- E. M. Flanigen, J. Bennett, R. Grose, J. Cohen, R. Patton, R. Kirchner and J. Smith, Nature, 1978, 271, 512–516 CrossRef CAS.
- W. Xu, J. Dong, J. Li, J. Li and F. Wu, J. Chem. Soc., Chem. Commun., 1990, 755–756 RSC.
- S. Sang, F. Chang, Z. Liu, C. He, Y. He and L. Xu, Catal. Today, 2004, 93, 729–734 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05798f |
| This journal is © the Owner Societies 2020 |