
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAttack of hydroxyl radicals to α-methyl-styrene sulfonate polymers and cerium-mediated repair via radical cations†
Tom M.
Nolte‡
 *ab,
Thomas
Nauser
b and
Lorenz
Gubler
a
*ab,
Thomas
Nauser
b and
Lorenz
Gubler
a
aElectrochemistry Laboratory, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland. E-mail: t.nolte@science.ru.nl
bEidgenössische Technische Hochschule (ETH) Zurich, Laboratory of Inorganic Chemistry, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland
First published on 23rd December 2019
Abstract
Both synthetic polymers (membranes, coatings, packaging) and natural polymers (DNA, proteins) are subject to radical-initiated degradation. In order to mitigate the deterioration of the polymer properties, antioxidant strategies need to be devised. We studied the reactions of poly(α-methylstyrene sulfonate), a model compound for fuel cell membrane materials, with different degrees of polymerization with OH˙ radicals as well as subsequent reactions. We observed the resulting OH˙-adducts to react with oxygen and eliminate H2O, the relative likelihood of which is determined by pH and molecular weight. The resulting radical cations can be reduced back to the parent molecule by cerium(III). This ‘repair’ reaction is also dependent on molecular weight likely because of intramolecular stabilization. The results from this study provide a starting point for the development of new hydrocarbon-based ionomer materials for fuel cells that are more resistant to radical induced degradation through the detoxification of intermediates via damage transfer and repair pathways. Furthermore, a more fundamental understanding of the mechanisms behind conventional antioxidants in medicine, such as ceria nanoparticles, is achieved.
Introduction
Oxidative stress is a critical lifetime-limiting factor for sulfonated polyaromatic proton exchange membranes (PEMs) in the polymer electrolyte fuel cell (PEFC). These ionomers are of interest to replace the widely used perfluoroalkylsulfonic acid (PFSA) membranes, owing to their much lower gas permeability, higher glass transition temperature, and potentially lower cost.1 Oxidative stress is created by the presence of radical species, such as HO˙, H˙ and HOO˙, which are formed during the operation of the fuel cell in the presence of H2, O2, and the noble metal catalyst.2The hydroxyl radical (HO˙) can be particularly detrimental to the polymer as it initiates degradation. HO˙ reacts with aromatic compounds with typical rate constants in the range of the diffusion limit of 109–1010 M−1 s−1.3,4 However, the nature of follow-up reactions, intermediates and the associated kinetics are rarely considered. The final result of HO˙ attack may be chain oxidation (e.g., hydroxylation), crosslinking, or chain fragmentation.5 These different mechanisms of polymer aging depend on the chemistry of the polymer. Thus, strategies to prevent aging ought to take these pathways into account (Fig. 1). For example, ‘repairing’ intermediates formed upon radical attack may be accomplished with suitable additives, i.e., antioxidants (e.g. in Fig. 1, reaction 11).6
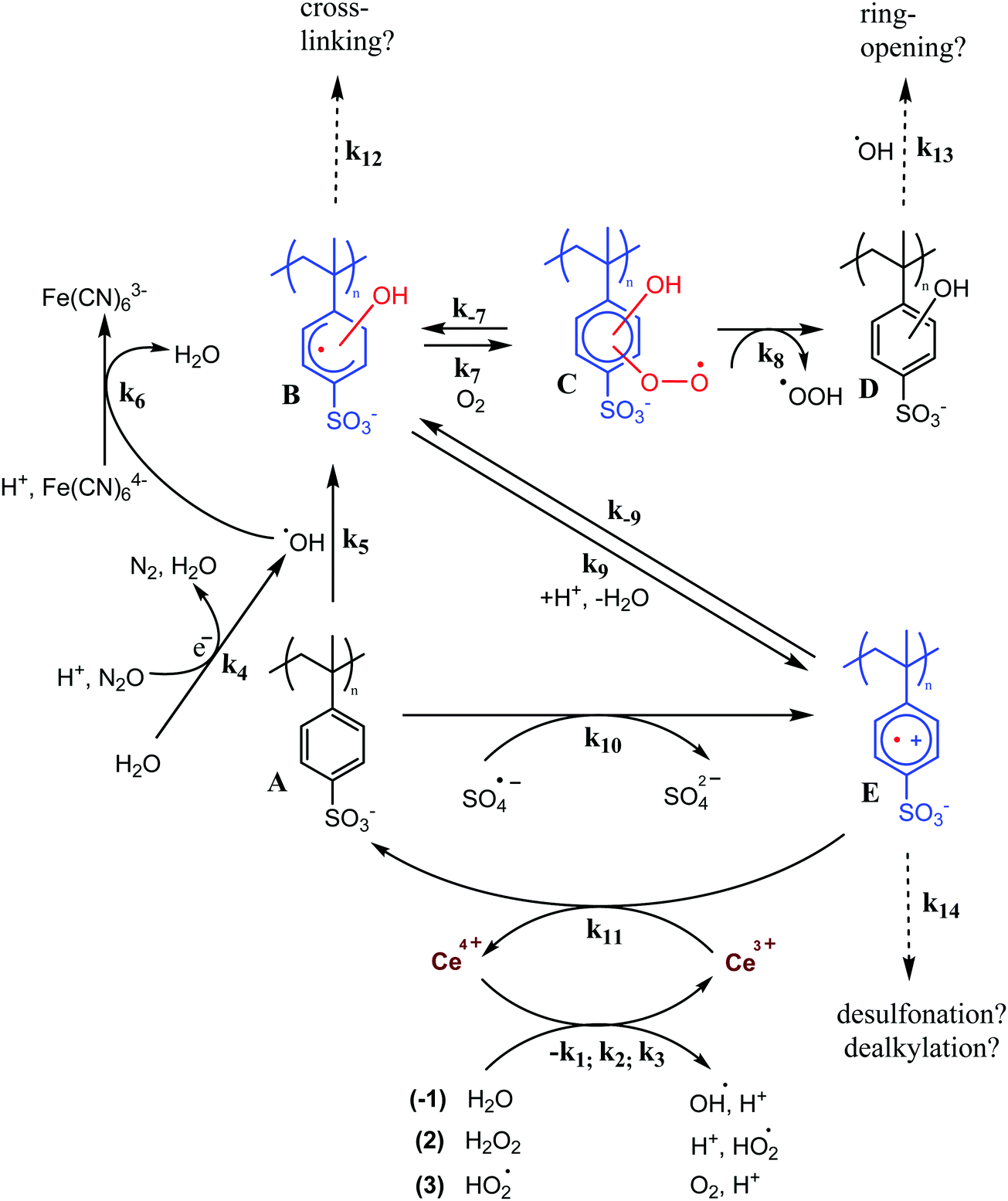 | ||
| Fig. 1 Reaction pathways considered in this study. Reaction between the PAMSS polymer/oligomer (compound A) with OH˙ (reaction 5), produces a hydroxycyclohexadienyl adduct (compound B). This adduct can undergo (acid-catalyzed) elimination of H2O (reaction 4) to produce a radical cation (compound E). Short-lived intermediates of PAMSS (≲1 ms) are indicated in color. The aim of this study is to determine whether Ce3+ is a competitive repair agent for the radical cation (reaction 11), in terms of side reactions (reaction 7, 8 and 12–14). | ||
There is an analogy to oxidative stress in living cells: here, it is the characteristic of imbalance between reactive oxygen species (ROS) generation and an organism's endogenous defenses. OH˙ is generated from ‘leakage’ of electrons along the cellular electron transport chain, and can react with cell constituents (DNA, proteins). As a consequence, oxidative stress is the basis of many serious diseases such as cancer. Nature has its way of detoxifying radicals, i.e., using vitamins and enzymes. When these endogenous mechanisms to combat oxidative stress fall short, we can consider treatment via synthetic alternatives. Ceria nanoparticles (CNPs) are promising inorganic antioxidants for many biomedical applications. CNPs have demonstrated antioxidant enzyme-mimetic activity, as well as the capacity to scavenge a variety of ROS in both cell and animal models. Concomitantly, a reduction in DNA damage (e.g. in lung cells) has been observed.7 Cerium ions or ceria particles are also used to mitigate degradation in PFSA-type fuel cell membranes8via direct scavenging of OH˙, and potentially H˙:9,10
Ce3+ + HO˙ + H+ → Ce4+ + H2O (k1 = 3 × 108 M−1 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3) 3) | (1) |
The antioxidant properties of CNPs are dependent, also, on the ability to undergo redox cycling between the valences Ce3+ and Ce4+ ions (as CeO2−δ) on the surface of CeO2 or Ce2O3 CNPs in aqueous solution.11 Radical scavenging by Ce3+ is very effective in PFSA membranes, because the lifetime of HO˙ is on the order of microseconds, thus with a relatively small concentration of Ce3+ of ∼0.1 M over 90% of HO˙ are quenched.5 The ratio of Ce4+ to Ce3+ is influenced by the chemistry of the medium. For example, H2O2 and HO2˙, which are also present in a fuel cell membrane,12 can reduce Ce4+ to Ce3+:13
| Ce4+ + H2O2 → Ce3+ + HO2˙ + H+ (k2 = 1 × 106 M−1 s−18,14) | (2) |
| Ce4+ + HO2˙ → Ce3+ + O2 + H+ (k3 = 2.7 × 106 M−1 s−114) | (3) |
An intermediate formed as a result of OH˙ attack on arylenes is the aromatic radical cation (Ar˙+) (compound E, Fig. 1), produced by acid-catalyzed elimination of H2O (reaction 9, Fig. 1) from the OH-adduct (compound B, Fig. 1).15,16 Since proton exchange membrane fuel cells operate under acidic conditions, elimination of H2O is usually fast.15–17 The redox potential E° of Ce4+/Ce3+ of 1.44 V18 is favorable as compared to E°(Ar˙+/Ar) = 2.0–2.4 V.19 In previous work using poly(α-methylstyrene sulfonate) (PAMSS) oligomers as model aromatic compound, we found that the lifetime of the radical cation increases with the degree of polymerization, potentially due to π–π interactions, and does not produce benzyl radicals due to the presence of the α-methyl group.20 PAMSS is well soluble in water making it a viable candidate to study the kinetics of its reaction with Ce3+ in aquo. PAMSS represents a constituent of a fuel cell membrane. Though other model compounds representing polyarylene type polymers (e.g. polysulfones) are conceivable, they are beyond the current scope.
In the work reported here, we studied the interaction (reaction 11, Fig. 1) between Ce3+ and the aromatic radical cation of PAMSS (compound E) to investigate whether cerium could function as a regenerative antioxidant for repairing aryl-type polymers. To determine whether the repair pathway is viable we compared the regeneration kinetics with a potential side reaction, prominently, the reaction of the HO-adduct (compound B) with O2 (reactions 7/−7 and 8, Fig. 1). The degree of polymerization of PAMSS was varied from 1 to 1700 in order to study the effect of Ar˙+ lifetime and redox properties. The results of this study are of high relevance to material scientists and engineers looking to improve the durability of hydrocarbon based fuel cell membranes, as well as for medicinists aiming to elucidate and optimize antioxidant mechanisms.
Materials and methods
PAMSS oligomers/polymers (>95% sulfonation degrees) with molecular weights Mw of 2660, 14600, 73800 and 354000 Da, and polydispersity indices of <1.5, <1.2, <1.2 and <1.2, respectively, were supplied by PSS (Polymer Standards Service, Mainz, Germany). A ‘monomeric unit’ of PAMSS, 4-tert-butyl-benzenesulfonate, was used to represent a ‘degree of polymerization’ of 1. Ce3+ was added as a salt, Ce2(SO4)3, obtained from Sigma-Aldrich (≥99.99% purity). Water from a Millipore-Q system was used to prepare solutions that were saturated with O2, N2O, or Ar depending on the reaction studied. Samples were gas saturated in Schlenk-tubes which were repeatedly evacuated to 10 mbar and refilled (a minimum of 3 repeats) with the desired gas. The solutions were transferred from a gas-tight syringe (10 ml, Hamilton, SampleLock, Bonaduz, Switzerland) to the measurement cell via a syringe pump. Acidic pH was adjusted with H2SO4 (95-97% purity), while solutions at pH 7 were buffered with 0.1 mM phosphate buffer (NaH2PO4·H2O, >99% purity), unless stated otherwise. Experiments were carried out at room temperature (25 °C). tert-Butanol was obtained from Merck (Darmstadt, Germany) to scavenge OH˙ when needed.
Pulse radiolysis
Pulse radiolysis experiments were carried out using a Febetron 705 (Titan Systems Corp., presently L-3 Communications, San Leandro, CA, USA), equipped with an optical detection system. For details see Nauser et al. (2008).21 Irradiations were performed in a 6 cm quartz cell (Hellma, Müllheim, Germany) with <50 ns pulses of 2 MeV electrons. The dose was measured using a thiocyanate dosimeter.Production of OH˙ radicals
Pulse irradiation of water results in the formation of primary species with yields G(OH˙) (primary yield (molecules per 100 eV), pH 7), G(eaq−) and G(H˙) of 2.7, 2.65 and 0.6,22–24 respectively, whereby G = 1 equals to 0.1036 μmol of a species generated species per 1 J kg−1 absorbed energy. The solutions were saturated with N2O (22 mM) to increase the OH˙ yield and reduce side reactions. The solvated electron, eaq−, reacts with N2O to yield additional OH˙:| N2O + eaq− + H2O → N2 + OH˙ + OH− (k4 = 9.1 × 109 M−1 s−123) | (4) |
Reaction of OH˙ with PAMSS
Reaction of OH˙ with an aromatic unit (Fig. 1, reaction 5) produces a hydroxycyclohexadienyl adduct (Fig. 1, compound B).20,25 Here, we quantified the rate constants for the reaction between the polymers and OH˙:| OH˙ + PAMSS → PAMSS(–OH)˙ (k5) | (5) |
PAMSS radicals have relatively low extinction coefficients and side-reactions might be involved at high concentrations of radicals. To bypass and minimize this, we studied the reaction with OH˙ via competition with Fe(CN)64−:
| OH˙ + Fe(CN)64− → OH− + Fe(CN)63− (k6 = 1.05 × 1010 M−1 s−1) | (6) |
At relatively low dose (∼10 Gy), the concentration of Fe(CN)64− was ∼50 μM and the concentration of PAMSS was varied. Under N2O saturated conditions the radiation chemical yield (G) of OH˙ is ∼5.6 × 10−7 mol J−1 and is unaffected by the PAMSS concentration (dilute solutions, ≤100 μM). Thereby, the absorbance at 420 nm (Fe(CN)63−, ε420 = 1040 M−1 cm−1) is solely affected by the PAMSS concentration via competition for OH˙. The absorption at 420 nm, virtually constant after ∼5 μs, was measured.
Control experiments were performed using different dose/Fe(CN)64− ratios, as well as direct observation (details in the ESI†).
Reaction of hydroxycyclohexadienyl radicals with oxygen
Reaction of O2 with hydroxycyclohexadienyl radicals (Fig. 1, reaction 7) produces an Ar–OH(–O2) radical adduct (Fig. 1, compound C):| PAMSS(–OH)˙ + O2 ⇌ PAMSS(–OH)(–O2)˙ (k7/k−7) | (7) |
The addition of O2 to hydroxycyclohexadienyl radicals was studied by varying the O2 concentration, and observing the effect on the kinetic trace of hydroxycyclohexadienyl radicals. Mixtures of N2O/O2 were used and the concentration of O2 in the samples was determined via the partial pressure of O2 and its solubility in water (1.25 mM at 1.0 atm). O2 concentrations ranged from 0 to 1.25 mM. Doses applied were ∼40 Gy. All experiments reported were reproduced at least five times.
The characteristic λmax for hydroxycyclohexadienyl-type radicals is ∼325 nm, Fig. 2. However, other species, among which the Ar–OH(–O2) radical adduct, absorbs also in this spectral range (Fig. 2B). Moreover, the addition of oxygen is reversible25 with an equilibrium constant K on the order of ≥103 M−1,25 because the reverse reaction is relatively slow (reaction −7, Fig. 1, with k−7 = 4.5(±0.9) × 103 s−1 as an upper limit for poly(styrene sulfonate) (PSS)26).
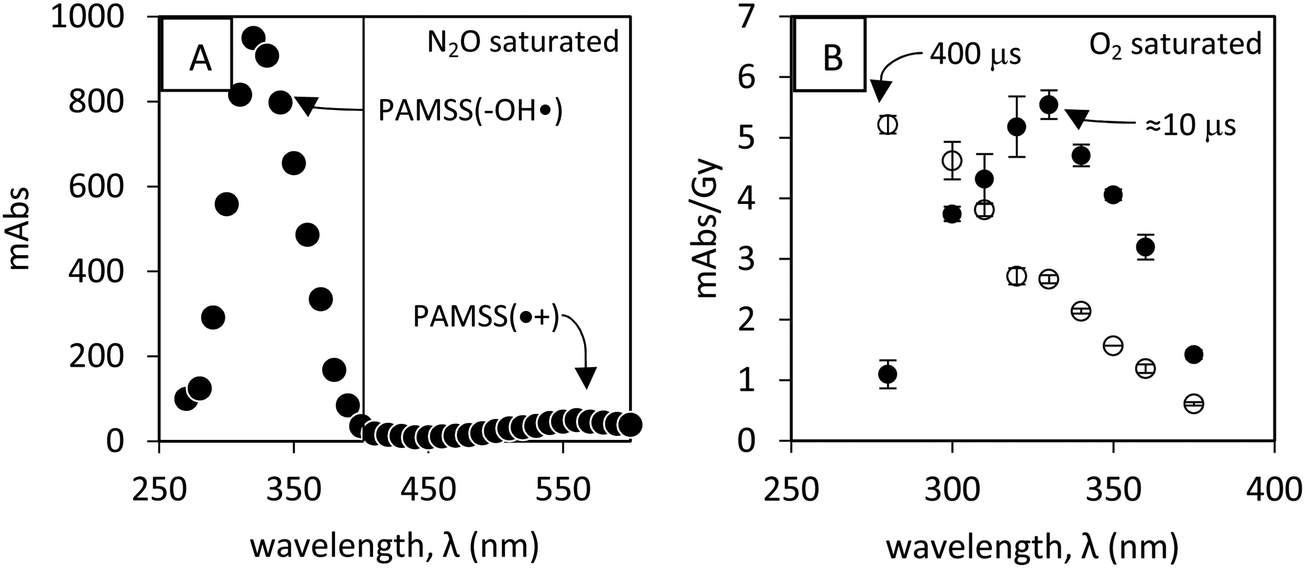 | ||
| Fig. 2 (A) Spectrum of 5 μM poly(α-methylstyrene sulfonate) Mw = 354000 Da. Dose ∼15 Gy, N2O saturated, pH ∼ 7. The absorption was highest within the time interval of the first 10 μs after the pulse. (B) Spectrum of 32.7 μM poly(α-methylstyrene sulfonate) Mw = 14600 Da. Dose ∼50 Gy, O2 saturated (1.25 mM), pH ∼ 7. Highest within 10 μs (filled symbols, see Fig. S8-1A, ESI†) and 400 μs (open symbols) after the pulse. Absorbances <280 nm are omitted because optimization of spectral resolution at the expense of incident light intensity resulted in noisy and inaccurate absorbance. | ||
The addition reaction was monitored at λ = 360 nm, chosen empirically such that both the relative absorption of the OH˙-adduct (relative to the Ar–OH(–O2) adduct) and absolute absorption are maximized (Fig. S4B, ESI†). Based on earlier results25,26 we identified the decay within ∼10–20 μs of the absorption maximum. The kinetic traces were fitted with pseudo-first order functions. This analytical setup minimized the need of fitting to equilibria or taking into account side reactions, such as ˙OOH elimination (reaction 8, Fig. 1, with k8 = 2.7(±0.3) × 103 s−1 for PSS18) to form a stable hydroxylated product (compound D). Thus, we can fit kinetic traces for the OH˙-adduct with minimal interference.25
Control experiments were performed using gas mixtures of Argon/O2 and N2O/O2 under low doses (∼10 Gy) to characterize the relevance of the yield of OH-adducts (details in Fig. S8, ESI†) on the apparent rate constants.
Production of radical cations and reaction with Ce3+
Under N2O/O2 conditions and pH ∼7 the yield of radical cations from OH˙-adducts (e.g. via reaction 9, Fig. 1) is low: 0.1–1 μM for Mw = 354000 Da (Fig. 2A, λmax ≅ 560 nm), and below our detection limit for smaller polymer weights. When the radical cation was produced at pH ∼7 via elimination from the PAMSS-OH˙ adduct (Mw = 354000 Da), no reaction between the radical cation and Ce3+ was observed.
Rate constants for the elimination of OH−/H2O have previously been identified as ∼104 s−1 for a range of aryl-type OH˙-adducts,16 whereas for the reaction between radical cation and H2O (reaction −9, Fig. 1) values differ 101–10716 (k−9 = 1–2 × 104 s−1, Fig. 5 and Fig. S10–S12, ESI†). Thus, simultaneous production of radical cations via elimination (reaction 9, Fig. 1) from the PAMSS-OH˙ adduct (Fig. 2A) might obscure the depletion via reaction with Ce3+ (reaction 11, Fig. 1).
Bypassing the H2O/OH− elimination from PAMSS-OH˙ route, radical cations were produced by electron transfer to sulfate radicals (reaction 10, Fig. 1):
| PAMSS + SO4˙− → PAMSS˙+ + SO42− (k10 = 0.6–1.0 × 109 M−1 s−120) | (8) |
Potassium peroxodisulfatesulfate (50 mM) was added to argon-saturated solutions at pH ∼ 2 (adjusted with H2SO4). Dose was ∼100 Gy, and tBuOH (100 mM) was added to scavenge the primary OH˙ radicals. Concentrations of PAMSS were used according to the degree of polymerization (e.g. 4.5 mM for PAMSS-354000). We varied the Ce3+ concentration to obtain pseudo first-order rate constants for the reaction:
| Ce3+ + PAMSS˙+ → Ce4+ + PAMSS (k11) | (9) |
Results and discussion
Reaction of OH˙ with PAMSS
The introduction of PAMSS to a solution containing Fe(CN)63− led to a decrease in the absorption recorded at 420 nm (i.e. reaction between Fe(CN)63− and OH˙). This is indicative of a reaction between OH˙ and PAMSS (i.e. a competition for OH˙). Taking into account reaction 6 (Fig. 1, Fe(CN)63− + OH˙) we fitted the data (blue lines and triangles in Fig. 3) to obtain k5 = 5.7(±0.2) × 1011 M−1 s−1 for the bimolecular reaction rate constant between PAMSS-354000 (the polymer with the highest Mw) and OH˙ (Fig. 1, reaction 5). By extension, we obtained the following data for the series of oligomers/polymers with different molecular weight, given in Table 1.
 | ||
| Fig. 3 The determination of the reaction rate constant between PAMSS and OH˙ via competition with Fe(CN6)3−. The Fe(CN6)3− concentration was approximately 100 μM. Dose ∼25 Gy. Lines denote fitting based on competition kinetics. | ||
| Polydispersity index | Molecular weight, Mw (Da) | k 5 (M−1 s−1) | k 5/nd | Source |
|---|---|---|---|---|
|
a 4-tert-Butyl-benzenesulfonate.
b Using a Fe(CN)63−:OH˙ ratio of ≥5:1. Controls using ratios of >10:1 gave 3.1(±0.2) × 1010 M−1 s−1 for PAMSS-14600 and 6.9(±0.9) × 1011 M−1 s−1 for PAMSS-354000.
c Dockheer et al. evaluated the kinetics via direct observation of the OH-adduct.
d Whereby n was taken as the ratio between the molecular weights (Mw) between the polymer and monomer.
|
||||
| 1 | 214a | 4.9(±0.1) × 109b |
4.9(±0.1) × 109 | This work, c.k. |
| <1.5 | 2660 | 1.3(±0.2) × 1010b |
9.4 × 108 | This work, c.k. |
| n/a | 2640 | 2.0 × 1010 | 1.5 × 109 | Dockheer et al.c20 |
| n/a | 6400 | 2.5(±0.5) × 1010 | 7.7 × 108 | Dockheer et al.c20 |
| <1.2 | 14600 |
2.8(±0.2) × 1010b |
3.8 × 108 | This work, c.k. |
| <1.2 | 73800 |
9.4(±0.3) × 1010b |
2.5 × 108 | This work, c.k. |
| <1.2 | 354000 |
5.7(±0.2) × 1011b |
3.2 × 108 | This work, c.k. |
| <1.2 | 354000 |
5.8(±0.3) × 1011 | 3.2 × 108 | This work, direct obs. |
The apparent yield of adducts (absorption at 360 nm) under N2O atmosphere is less than twice that obtained under O2 (3.2 and 5.9 abs Gy−1, resp.), i.e. G(N2O)/G(O2)= 1.86 ± 0.04 (Fig. 4A). The ratio in yields of the primary radicals, G(OH˙, N2O) + G(H˙, N2O) = 0.622 and G(OH˙, O2) + G(H˙, O2) = 0.342, i.e. G(N2O)/G(O2) = 1.82, is similar. We infer that the absorption corresponds to 5–10% PAMSS-H˙ adducts. Nevertheless, measurement of k5via direct observation produced the same value as obtained via competition kinetics (Table 1), Fig. S1 (ESI†). The suite of control studies indicated the stability of the method (Table 1).
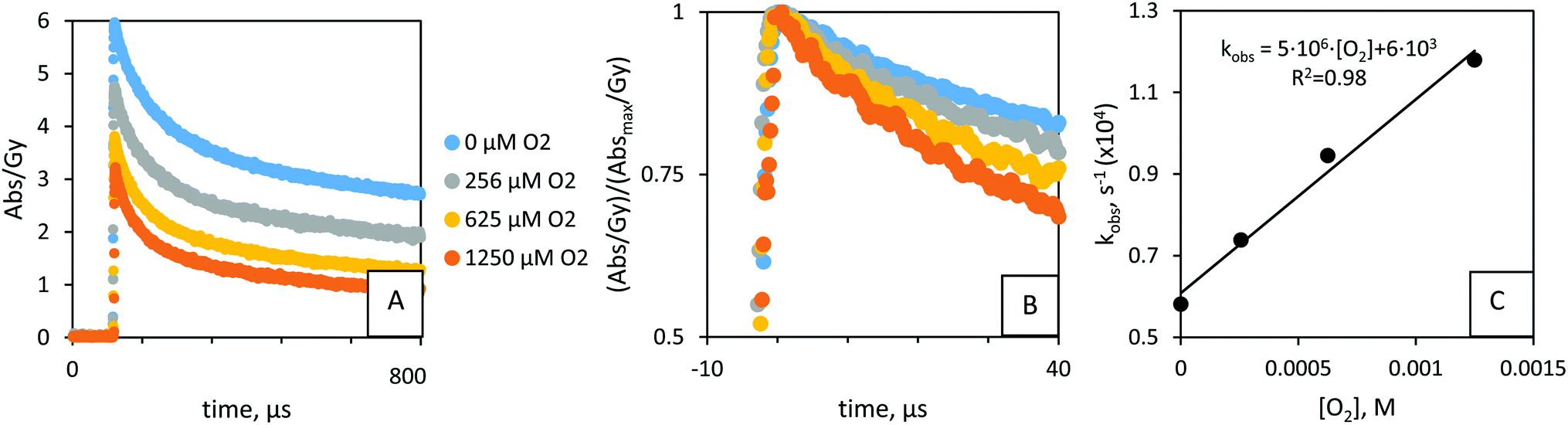 | ||
| Fig. 4 (A) Kinetic traces for the reaction between (decay of) PAMSS-14600(–OH˙) adduct and O2, monitored at 360 nm. Time-averaged. 32.7 μM PAMSS-14600, ∼25 °C. pH ∼ 7; 5 replicates per data point. Colors show the concentration of O2 in μM. (A) N2O/O2 solutions., ∼40 Gy. Long time interval. (B) N2O/O2 solutions. ∼40 Gy. Short time interval; normalized by the absorption at t0 (∼2 μs). (C) N2O/O2 solutions. ∼40 Gy. Pseudo 1st order rate constants versus O2 concentration. k7 = 4.8(±0.4) × 106 M−1 s−1. | ||
We obtained larger values for k5 for larger molecular weights. However, when we express k5 per monomer unit, there is a notable decrease. The data match earlier observations by, e.g., Dockheer et al.20 for a more limited range of Mw (Table 1); we find our values either slightly lower or equal to what has been reported.
The influence of size could be expressed as k5 ∼ [n]0.60±0.08 (k5 ∼ [n]0.57 including Dockheer et al. data,20Fig. 6), wherein [n] is the degree of polymerization. Variation in the polydispersity index (PDI) between the polymers is not expected to influence the relationships obtained in this study (since PDI < 1.2). The “reaction exponent”, here 0.60 ± 0.08, is sometimes defined as θ:27,28
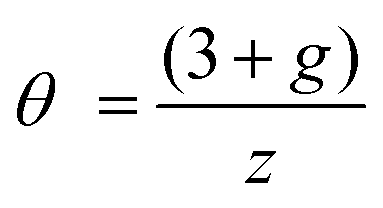 | (10) |
In case the polymer dynamics are ‘Rouse-like’ (single un-entangled chain; no significant interactions between chain segments) z = 4. In turn, g = 0 represents a non-interacting electron–electron system (the correlation hole29 is screened out). Then, the reaction exponent is (3 + 0)/4 = 0.75. Indeed, k5 values are near the “diffusion control”: k5 ∼ [n]0.60, i.e. 0.60 ± 0.08 ≲ 0.75. Thus, kr is largely independent of the reactivity of the reaction site.
Decay of hydroxycyclohexadienyl radicals
The first order decay of PAMSS-OH˙ seems to be a function of the yield of OH˙-adducts (Fig. S3 and S8, ESI†). If we assume the yield of OH˙-adducts is 8 times larger under 40 Gy and N2O, as compared to under 10 Gy and argon, we derive based on Fig. S3 and S8 (ESI†) a slope (rate constant) on the order of 5(±1) × 102 M−1 s−1 (Fig. S3, ESI†). The intercept is 2.2(±0.5) × 103 s−1 (Fig. S3, ESI†). The half-life of the PAMSS-OH˙ adduct (as observable at ∼325 nm) generally increased with MW (e.g. kobs = 1.4(±0.3) × 104 s−1 and 6.1(±0.3) × 103 s−1 for MW = 2660 and 14600, resp.), though quantification requires the relative (effective) concentration of OH-adducts for different MW. PAMSS-OH adducts seem to be more stable as the MW increases.
A neighboring monomer might interact with the radical site in such a way that its intrinsic reactivity is lowered, or the coiling of the polymer chain may decrease its accessibility. Values for propagation in radical polymerisations are typically 103±1 M−1 s−1,30i.e. slow due to steric crowding about the radical center. Based on this, we consider the value 5(±1) × 102 M−1 s−1 to correspond to reaction 12 (crosslinking and/or disproportionation, k12) in Fig. 1.
Protonation of OH˙-adducts occurs with a rate constant of k = 1–2 × 109 M−1 s−116 implying a pseudo 1st order rate constant of 1–2 × 102 s−1 at pH 7, whereas the elimination of OH− occurs with a rate on the order ∼104 s−1 for a range of aryl-type OH˙-adduct monomers.16 Thus, the value of 2.2(±0.5) × 103 s−1 might characterize reaction 9 (either elimination of OH− or acid-catalysed elimination of H2O, k9) of PAMSS-14600(–OH˙) as denoted in Fig. 1, reaction 9/−9.
Reaction of hydroxycyclohexadienyl radicals with oxygen
Upon exposure of the hydroxycyclohexadienyl radicals to O2 we observed a shift to shorter wavelengths over time (Fig. 2B). ∼400 μs after the pulse, there was no apparent contribution from the OH-adduct (as seen at λmax = 325 nm) to the absorbance anymore (Fig. 2B, see also ESI†). The shift to shorter wavelength for reaction products under O2 atmosphere (Fig. 2B and Fig. S4–S6, ESI†) has been observed in related studies20,25 and is attributed to O2-radical adducts.The yield of OH˙-adducts varied for different ratios of N2O/O2 and between pulses (Fig. 4A); the concentration of OH˙-adducts under N2O/O2 mixtures was directly proportional to the yield of OH˙, see Fig. 4A and Fig. S7 (ESI†). This was taken into account when evaluating the pseudo-first order reaction with O2 (reaction 7, Fig. 1):24 we consider the relative concentrations of OH˙-adducts. Thus, Fig. 4A was corrected for the adduct yields (maximum absorbance, at ∼2 μs), to give Fig. 4B.
Control studies indicated the stability of the method (Fig. S8, ESI†): H˙ is quenched by PAMSS, rather than O2. There is no difference between the k7 values obtained from series under argon or N2O. Therefore, PAMSS-14600(–H˙) adducts react slower with O2 than do PAMSS-14600(–OH˙) adducts and do not contribute significantly to the result. Different initial yields of PAMSS-14600(–OH˙) adducts (at t = 2 μs), do not appear to affect the results (Fig. 4C and Fig. S8, ESI†). Thus, in the range of 2 μs to 10–20 μs, there is no measurable equilibration or elimination of OOH˙ yet. Compared to 1.25 × 10−3 M for O2 (in O2 saturated solutions), the experimental series may also yield superoxide, O2˙− (Fig. S8-1A, ESI†), in 10 μM (higher-end estimation). In O2 saturated solutions (∼1.25 mM) G(OH˙) ∼ 2.8 × 10−7 mol J−1 and G(O2˙−) ∼ 3.4 × 10−7 mol J−1. The reaction between PAMSS-OH and O2 is thermodynamically favored (compared to that with O2˙−) based on the energies of their frontier orbitals:31 |ESOMO(O2) − ESOMO(PAMSS-OH˙)| < |ESOMO(O2˙−) − ESOMO(PAMSS-OH˙)|, i.e. ΔE ∼ 0.4 eV < ΔE ∼ 0.9 eV (in-house calculation32–34). The influence of O2˙− has not been reported in related studies (under high O2 concentrations25). Thus, the reaction between O2˙− and PAMSS-OH is of minor importance, and the rate constants for addition of O2 obtained in this study can be considered accurate.
From fitting we obtained a rate constant k7 = 4.8(±0.4) × 106 M−1 s−1 for the reaction between PAMSS-14600(−OH˙) and O2 (see ESI† for other Mw) in Fig. 4C. The reaction of a carbon-centered radical with O2 to form the corresponding peroxyl radical generally proceeds with a rate constant on the order of 109 M−1 s−1.35 However, because of electron delocalization in allylic and dienylic C-centered radicals, oxygen binds relatively weakly, which results in slower and reversible O2 addition. These notions are in agreement with the data obtained. Other factors explaining the relatively low rate constants include steric hindrance (tert-butyl) and polarity (sulfate) but need to be studied in more detail.
There appeared to be an influence of molecular weight, as a k7 of 2.2(±0.3) × 107 M−1 s−1 was found for the 4-tert-butyl-benzenesulfonate (–OH˙) adduct. The data are in the same order as was found for PSS-1100(–OH˙): k = 3.0(±0.5) × 107.26 For the reaction between the OH˙-adducts and O2, k7 scales with n: k7 ∼ [n]−0.26±0.07. PSS data26 might be included in the analysis if no significant influence by the extra methyl group is expected on the reaction thermochemistry, relative to the effect of Mw (Fig. 6). The influence of polymer size on the reaction between PAMSS(–OH˙) and O2 (−0.26 ± 0.07) is different (2σ) from for the reaction between PAMSS and OH˙ (0.60 ± 0.08). Since there is only 1 reactive site on the polymer, no relationship with n (k7 ∼ [n]0) should be expected if diffusive or quantum-chemical effects are absent. Instead, the polymer dynamics i.e. diffusive pathway (z) and/or quantum-chemical properties (g) (eqn (10)) are affected (<0) by the breaking of aromaticity by OH˙ to produce the hydroxycyclohexadienyl radical. It is worth noting that hydroxycyclohexadienyl radicals are relatively electron-rich and non-planar (Fig. 1, compound B), which would disfavor π–π interaction.
Decay of radical cations
In argon-saturated conditions and pH ∼ 2, SO4˙− radicals were produced from the reaction between peroxodisulfate and the solvated electron. SO4˙− radicals oxidize PAMSS to produce a radical cation observable at 560 nm. We detected radical cations for 2600 ≥ Mw ≥ 354000, whereas for the 4-tert-butyl-benzenesulfonate (i.e. the monomer unit) no radical cations could be observed. The half-life of the radical cation was found to be ∼40 to ∼100 μs depending on the chain length (Fig. 5). The influence of size was reported earlier by Dockheer et al. for a more limited range of Mw.20
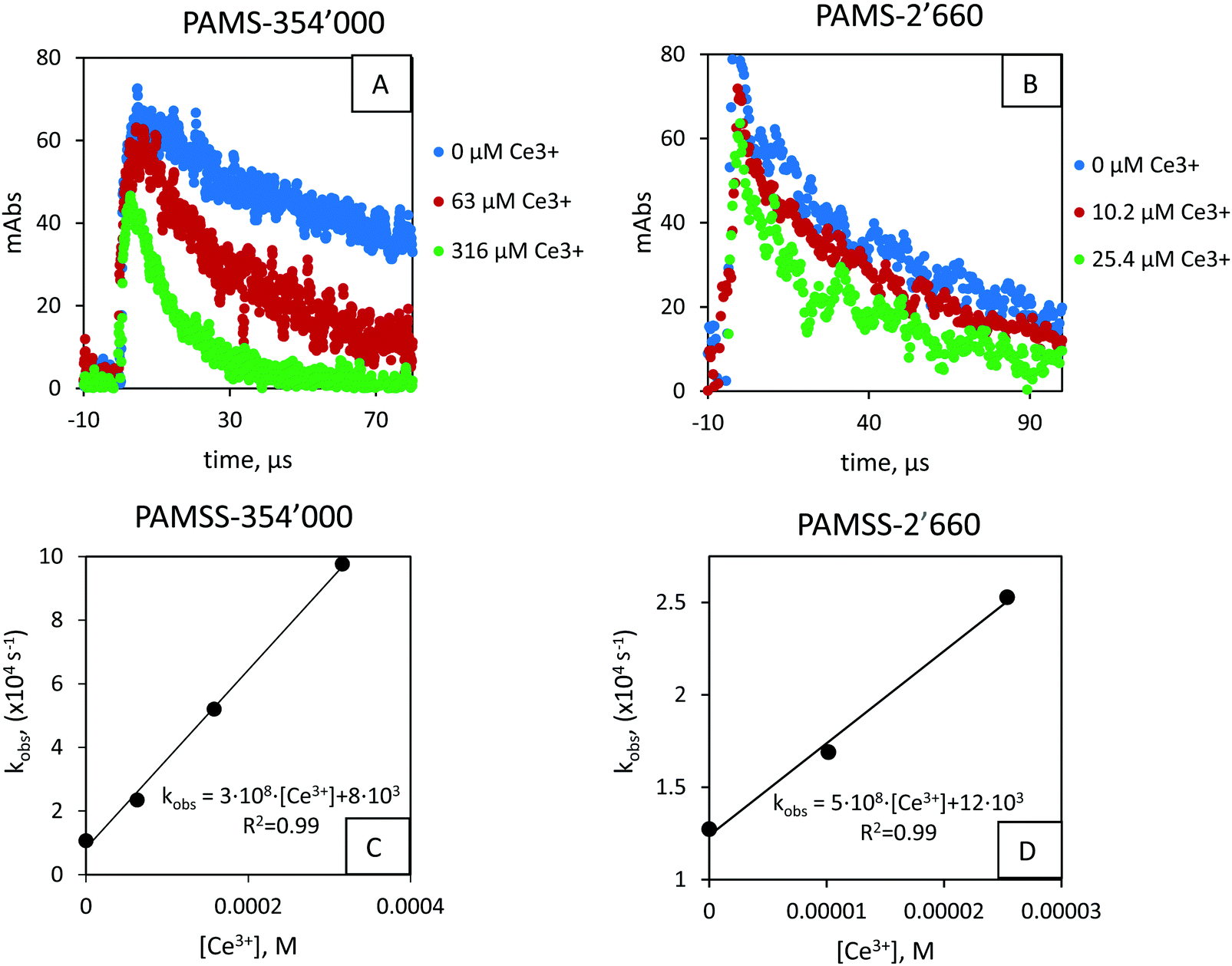 | ||
| Fig. 5 (A and B) Kinetic traces for PAMSS-354000 and PAMSS-2660, resp. for various Ce3+ concentrations. For visibility, only 3 kinetic traces are shown per graph. (C and D) Pseudo first-order rate constants versus Ce3+ concentration. k11 = 2.8(±0.2) × 108 M−1 s−1 and k11 = 5.0(±0.4) × 108 M−1 s−1 for PAMSS-354000 and PAMSS-2660, resp. Data for MW = 14600 and MW = 73800 can be found in the ESI.† | ||
The radical cations showed longer lifetimes upon increasing the molecular weight of the polymer (Fig. 6 and Fig. S10 and S11, ESI†), with reaction exponents (k ∼ [n]0.09±0.03) similar to those for the reaction between the radical cation and Ce3+ (0.12 ± 0.01). Extrapolation of the data recorded in absence of O2 (under Ar), using a power function in Fig. 6 results in k = 1.6 × 104 s−1 for the first-order decay of the 4-tert-butylbenzenesulfonate radical cation (∼40 μs half-life). This is relatively low compared to simple arenes, or even tert-butyl-benzene cations (105–106 s−116). The mode of decay by the radical cations is uncertain. The α-methyl group prevents formation of a benzyl radical via a proton elimination (splitting) reaction and, instead, elimination of the sulfate was suggested20 or dealkylation (scission), Fig. 1. The cation might react intramolecularly with a nearby monomer,16 requiring a k of 106–107 s−1. This is unlikely based on thermodynamic grounds, and it would likely result a shift in λmax (for a similar radical cation), which was not observed. A radical–radical reaction would involve rate constants in the range of 108–109 M−1 s−1 (diffusion-limited, no thermodynamic arguments). This mechanism does not explain the size dependence observed and the decay that is still observed at very low concentrations, ∼104 s−1. Rate constants for addition of H2O to the radical cation (i.e. the backward reaction −9, Fig. 1) to form the water adduct are on the order of 102–103 M−1 s−1.16 The decay observed in acidic aqueous solution is given by k = 6 × 103–6 × 104 s−1. Addition of H2O to the radical cation would occur via direction of the lone pair into the SOMO of the radical cation. The cation, and by extension the energy of this SOMO, could be subject to intramolecular π–π interaction. As a result, the interaction can lower the rate of H2O addition. Taking the molar concentration of H2O for a dilute solution, k−9 would be 2(±1) × 106 M−1 s−1.
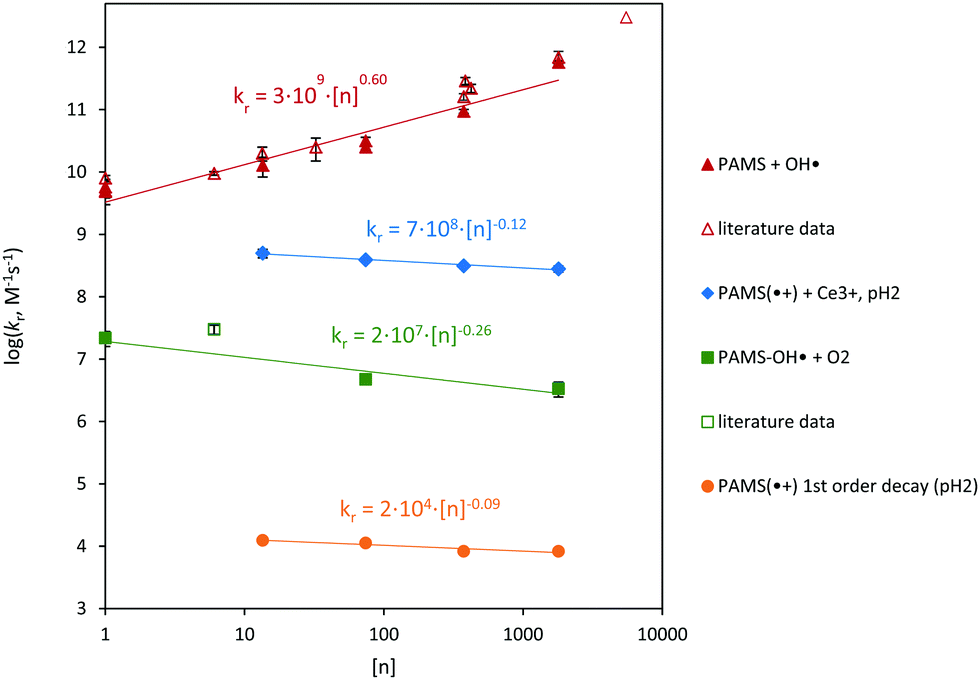 | ||
| Fig. 6 Combined results for the reactions studied. Rate constants are plotted versus the degree of polymerisation, [n]. Literature data refer to Behar (1991),37 Behar and Rabani (1988),38 Bhardwaj et al. (2001)39 and Dockheer et al. (2010)26 for PSS and PAMSS, as well as control experiments. ‘Reaction exponents’: θ = 0.60 ± 0.08 for PAMSS + OH˙, θ = −0.12 ± 0.01 for PAMSS˙+ + Ce3+, θ = −0.26 ± 0.07 for PAMSS(−OH˙) + O2, and θ = −0.09 ± 0.02 for the first order decay of PAMSS˙+. | ||
Reaction of radical cations with Ce3+
Excluding the 4-tert-butylbenzenesulfonate (n = 1), the radical cations produced from the oligomers/polymers were exposed to Ce3+. For all molecular weights, a decrease in the lifetime of the radical cation was observed with increasing Ce3+ concentration. The influence of Ce3+ was attributed to the reduction of the radical cation back to the starting compound (reaction 11, Fig. 1). Minor bleaching was observed in some cases (Fig. S11A, ESI†), potentially due to reaction −1 (cf.Fig. 1). From the plot of pseudo-first order reaction rates versus concentration, reaction rate constants were obtained, e.g. for the PAMSS-14600 radical cation in Fig. 5 we obtained k11 = 3.9(±0.2) × 108 M−1 s−1 (see ESI† for other Mw).
As a validation exercise, experiments were conducted for a lower PAMSS (73800) and higher persulfate concentrations, as well as under higher pH (Fig. S12, ESI†). The same value for k11 was obtained for the series when fitting the kinetic traces over a short time regime, indicating the stability of the method. Nevertheless, the observed rate constants are higher at pH ∼ 3, as compared to pH ∼ 2. An offset of 2.9(±0.2) × 104 s−1 between regressions for pH ∼ 3 and pH ∼ 2 was obtained. The offset is partially attributed to the acid–base equilibrium (reaction, 9/−9, Fig. 1) (see previous section).
The influence of molecular weight on the reaction between the PAMSS radical cation and Ce3+ was considered. The ‘reaction exponent’ is −0.12 ± 0.01, i.e. k11 ∼ [n]−0.12±0.01. The value is similar to that observed for the reaction between the radical cation and H2O (previous section), −0.09 (Fig. 6). The negative values support the interpretation that the thermochemistry of the radical cation is affected intramolecularly. Intramolecular charge–radical stabilization has already been confirmed on a fundamental level: chemical reactivity is controlled by radicals flanking the charged groups or by charged groups flanking the radicals.36 For the radical cations, too, a significant stabilization is implied, which may be expressed as θ < 0, eqn (10). The radical cation is initially produced on the outer ‘surface’ of the polymer coil with a high degree of polymerization (diffusion limited). Quantum-chemical stabilization (g, eqn (10)) can be a π-donation into the outer-surface cation hole (i.e. the electron deficiency), effectively delocalizing (transferring) the cation hole to center of the polymer. It is conceivable that the polymer dynamics (z, eqn (10)) are also affected, for example, through bridging between polymer segments to facilitate radical stabilization.
Extrapolation of the data resulted in k11 = 6.4(±0.7) × 108 M−1 s−1 for the reaction between 4-tert-butyl-benzenesulfonate radical cation and Ce3+ (Fig. 6). No literature data could be found for reactions of Ce3+ with aromatic radical cations. However, the ferrous ion (Fe2+) reacts with (reduces) the radical cation of anisole with a rate constant of k = 6 × 108 M−1 s−1 (pH 1.0) and with pseudocumene and isodurene radical cations with k = 6(±1)107 M−1 s−1 (pH 2.5–3.5).16 These reactions are dependent on the ionic strength of the solution and, potentially, the polarity of the cation (Nolte et al., in prep.).32
Conclusion
The reaction between PAMSSS and OH˙ borders the diffusion-limit, resulting in the formation of hydroxycyclohexadienyl, i.e. OH radical adducts, Ar(–OH˙). These adducts can react with O2, kr = 3 × 106–7 M−1 s−1, depending on the molecular weight, to produce O2 adducts. Even though Ar(–OH˙) reacts with O2, the effectiveness of this reaction depends on the thermodynamic equilibrium of H2O elimination/addition, which is a function of pH. Under acidic conditions, the OH-adduct eliminates H2O to form a radical cation. The polymeric radical cation was shown to react with Ce3+ with rate constants of 3–5 × 108 M−1 s−1 influenced by intramolecular stabilization, likely including radical–charge (π–π) interaction. Radical cations could not be observed for the monomeric compound (4-tert-butyl-benzenesulfonate), for which no intramolecular stabilization is possible, potentially also due to the fast backward reaction (H2O addition). The competition between the reaction pathways involving Ce3+ (the repair reaction) or O2 (as an indicator for irreversible damage) can be fine-tuned by modifying the size and structure of PAMSSS through its effect on both steric/clustering and redox properties, with the ‘repair’ reaction with Ce3+ generally being more efficient (compared to O2) for shorter polymer chains.The results of this study show that repair and stabilization of polymeric radicals can be achieved using cerium ions and neighboring groups via suitable thermochemical and kinetic interactions. This has ramifications for the design of durable arylene type fuel cell membranes and polymeric materials in general. The results also provide a basis for more fundamentally understanding the mechanisms behind conventional antioxidants in medicine, such as ceria nanoparticles, and represent a starting point for improvement of additives that detoxify radicals or intermediates formed therefrom, e.g., via damage transfer or repair pathways.
Conflicts of interest
The authors have no conflicts of interest to disclose.Acknowledgements
Funding by the Swiss National Science Foundation (SNSF) is gratefully acknowledged (grant no. 175493). Personal discussions with A. J. Hendriks (RU) aided the interpretation of our results, and were greatly appreciated.References
- L. Gubler, et al., Prospects for Durable Hydrocarbon-Based Fuel Cell Membranes, J. Electrochem. Soc., 2018, 165(6), F3100–F3103 CrossRef CAS.
- M. Danilczuk, F. D. Coms and S. Schlick, Visualizing Chemical Reactions and Crossover Processes in a Fuel Cell Inserted in the ESR Resonator: Detection by Spin Trapping of Oxygen Radicals, Nafion-Derived Fragments, and Hydrogen and Deuterium Atoms, J. Phys. Chem. B, 2009, 113, 8031–8042 CrossRef CAS PubMed.
- G. V. Buxton, et al., Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (˙OH/˙O−) in Aqueous Solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- T. M. Nolte and A. M. J. Ragas, A review of quantitative structure–property relationships for the fate of ionizable organic chemicals in water matrices and identification of knowledge gaps, Environ. Sci.: Processes Impacts, 2017, 19(3), 221–246 RSC.
- L. Gubler and W. H. Koppenol, Hydrocarbon Proton Exchange Membranes, in The Chemistry of Membranes Used in Fuel Cells: Degradation and Stabilization, ed. S. Schlick, John Wiley & Sons, 2018, pp. 107–138 Search PubMed.
- L. Gubler, T. Nolte and T. Nauser, Antioxidant Strategies for Hydrocarbon-Based Membranes, ECS Trans., 2018, 86(13), 369–379 CrossRef CAS.
- L. Rubio, et al., Antioxidant and anti-genotoxic properties of cerium oxide nanoparticles in a pulmonary-like cell system, Arch. Toxicol., 2016, 90(2), 269–278 CrossRef CAS PubMed.
- F. D. Coms, H. Liu and J. E. Owejan, Mitigation of Perfluorosulfonic Acid Membrane Chemical Degradation Using Cerium and Manganese Ions, ECS Trans., 2008, 16(2), 1735–1747 CAS.
- L. M. Kohan, J. Meesungnoen, S. Sanguanmith, R. Meesat and J. Jay-Gerin, Radiolysis of the ceric-cerous sulfate dosimeter at elevated temperatures: Monte Carlo simulations, Recent Res. Dev. Phys. Chem., 2014, 11, 15–27 Search PubMed , (ISBN: 978-81-7895-608-4).
- E. Collinson, F. S. Dainton and J. Kroh, The radiation chemistry of aqueous solutions II. Radical and molecular yields for tritium β-particles, Proc. R. Soc. London, Ser. A, 1962, 265, 422 CAS.
- A. Karakoti, et al., Redox-active radical scavenging nanomaterials, Chem. Soc. Rev., 2010, 39(11), 4422–4432 RSC.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, Radical (HO˙, H˙ and HO˙) Formation and Ionomer Degradation in Polymer Electrolyte Fuel Cells, J. Electrochem. Soc., 2011, 158(7), B755–B769 CrossRef CAS.
- B. C. Nelson, et al., Antioxidant Cerium Oxide Nanoparticles in Biology and Medicine, Antioxidants, 2016, 5(2), 15 CrossRef PubMed.
- B. H. J. Bielski, et al., Reactivity of HO2˙/O2˙− radicals in aqueous-solution, J. Phys. Chem. Ref. Data, 1985, 14(4), 1041–1100 CrossRef CAS.
- P. Oneill, S. Steenken and D. Schultefrohlinde, Formation of Radical Cations of Methoxylated Benzenes by Reaction with Oh Radicals, Tl2+, Ag2+, and So4− in Aqueous-Solution – an Optical and Conductometric Pulse-Radiolysis and Insitu Radiolysis Electron-Spin Resonance Study, J. Phys. Chem., 1975, 79(25), 2773–2779 CrossRef CAS.
- J. Holcman, Formation and reactions of radical cations of substituted benzenes in aqueous media. A pulse radiolysis study, Roskilde, Denmark: Risø National Laboratory. Risø-M, No. 1947, 1977.
- S. Steenken and P. Neta, Transient phenoxyl radicals: Formation and properties in aqueous solutions, in The Chemistry of Phenols, ed. Z. Rappoport, John Wiley & Sons, 2003, p. 1115 Search PubMed.
- L. Gubler, Radiation Grafted Membranes for Polymer Electrolyte Fuel Cells, Departement Chemie und Angewandte Biowissenschaften, Eidgenössische Technische Hochschule, Zürich, 2016, DOI:20.500.11850/190017.
- M. Jonsson, et al., Redox Chemistry of Substituted Benzenes – the One-Electron Reduction Potentials of Methoxy-Substituted Benzene Radical Cations, J. Phys. Chem., 1993, 97(43), 11278–11282 CrossRef CAS.
- S. M. Dockheer, L. Gubler and W. H. Koppenol, Reactions of the tetraoxidosulfate(˙−) and hydroxyl radicals with poly(sodium alpha-methylstyrene sulfonate), Phys. Chem. Chem. Phys., 2013, 15(14), 4975–4983 RSC.
- T. Nauser, et al., Reversible Intramolecular Hydrogen Transfer between Cysteine Thiyl Radicals and Glycine and Alanine in Model Peptides: Absolute Rate Constants Derived from Pulse Radiolysis and Laser Flash Photolysis, J. Phys. Chem. B, 2008, 112(47), 15034–15044 CrossRef CAS PubMed.
- R. H. Schuler, L. K. Patterson and E. Janata, Yield for the Scavenging of OH˙ Radicals in the Radiolysis of N2O-Saturated Aqueous-Solutions, J. Phys. Chem., 1980, 84(16), 2088–2089 CrossRef CAS.
- C. von Sonntag, The Chemical Basis of Radiation Biology, Taylor & Francis, London, 1987 Search PubMed.
- R. H. Schuler, A. L. Hartzell and B. Behar, Track Effects in Radiation-Chemistry – Concentration-Dependence for the Scavenging of OH˙ by Ferrocyanide in N2O-Saturated Aqueous-Solutions, J. Phys. Chem., 1981, 85(2), 192–199 CrossRef CAS.
- X. W. Fang, et al., Reversibility in the Reaction of Cyclohexadienyl Radicals with Oxygen in Aqueous-Solution, Chem. – Eur. J., 1995, 1(7), 423–429 CrossRef CAS.
- S. M. Dockheer, et al., Damage to fuel cell membranes. Reaction of HO˙ with an oligomer of poly(sodium styrene sulfonate) and subsequent reaction with O2, Phys. Chem. Chem. Phys., 2010, 12(37), 11609–11616 RSC.
- B. Oshaughnessy, Effect of Concentration on Reaction-Kinetics in Polymer-Solutions, Macromolecules, 1994, 27(14), 3875–3884 CrossRef CAS.
- B. Friedman and B. Oshaughnessy, Kinetics of Intermolecular Reactions in Dilute Polymer-Solutions and Unentangled Melts, Macromolecules, 1993, 26(21), 5726–5739 CrossRef CAS.
- J. des Cloizeaux, J. Phys., 1980, 41, 223–238 CrossRef CAS.
- K. Matyjaszewski, Typical features of radical polymerization, in Controlled and living polymerizations: from mechanisms to applications, ed. K. Matyjaszewski and A. H. E. Müller, Wiley-VCH, Weinheim, 2009, ch. 3.2, pp. 105–106 Search PubMed.
- G. Klopman, Chemical Reactivity and Concept of Charge- and Frontier-Controlled Reactions, J. Am. Chem. Soc., 1968, 90(2), 223–234 CrossRef CAS.
- T. M. Nolte, T. Nauser, L. Gubler and W. J. G. M. Peijnenburg, Thermochemical unification of molecular descriptors to predict radical hydrogen abstraction with low computational cost, Phys. Chem. Chem. Phys., 2019 Search PubMed , submitted.
- T. M. Nolte and W. J. G. M. Peijnenburg, Aqueous-phase photooxygenation of enes, amines, sulfides and polycyclic aromatics by singlet (a1Δg) oxygen: prediction of rate constants using orbital energies, substituent factors and quantitative structure–property relationships, Environ. Chem., 2018, 14(7), 442–450 CrossRef.
- J. J. P. Stewart, MOPAC, Stewart Computational Chemistry, Colorado Springs, CO, USA, 2016 Search PubMed.
- D. Minakata, et al., Development of Linear Free Energy Relationships for Aqueous Phase Radical-Involved Chemical Reactions, Environ. Sci. Technol., 2014, 48(23), 13925–13932 CrossRef CAS PubMed.
- T. Mazur and B. A. Grzybowski, Theoretical basis for the stabilization of charges by radicals on electrified polymers, Chem. Sci., 2017, 8(3), 2025–2032 RSC.
- D. Behar and B. Behar, Pulse-Radiolysis Studies of Aminobenzenesulfonates – Formation of Cation Radicals, J. Phys. Chem., 1991, 95(19), 7552–7556 CrossRef CAS.
- D. Behar and J. Rabani, Pulse-Radiolysis of Poly(Styrenesulfonate) in Aqueous-Solutions, J. Phys. Chem., 1988, 92(18), 5288–5292 CrossRef CAS.
- Y. K. Bhardwaj, et al., Radiation effect on poly(p-sodium styrene sulphonate) of different degrees of polymerization in aqueous solution: pulse radiolysis and steady state study, Radiat. Phys. Chem., 2001, 62(2–3), 229–242 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05454e |
| ‡ Current address: Department of Environmental Science, Institute for Water and Wetland Research, Radboud University Nijmegen, 6500 GL Nijmegen, The Netherlands. |
| This journal is © the Owner Societies 2020 |