
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ structural kinetics of picosecond laser-induced heating and fragmentation of colloidal gold spheres†
Anna Rosa
Ziefuss
a,
Stefan
Reich
b,
Sven
Reichenberger
a,
Matteo
Levantino
 c and
Anton
Plech
*b
c and
Anton
Plech
*b
aDepartment of Technical Chemistry I and Center for Nanointegration Duisburg-Essen CENIDE, University of Duisburg-Essen, Universitätsstrasse 7, D-45141 Essen, Germany
bInstitute for Photon Science and Synchrotron Radiation, Karlsruhe Institute of Technology, Postfach 3640, D-76021 Karlsruhe, Germany. E-mail: anton.plech@kit.edu
cEuropean Synchrotron Radiation Facility, 71, avenue des Martyrs, F-38043 Grenoble, France
First published on 14th February 2020
Abstract
Fragmentation of colloidal 54 nm gold nanoparticles by picosecond laser pulses is recorded by time-resolved X-ray scattering, giving access to structural dynamics down to a 80 ps resolution. Lattice temperature and energy dissipation have been quantified to verify that the maximum applied fluence of 1800 J m−2 heats up the particles close to boiling. Already within 30 ns, particles with significantly lower particle sizes of 2 to 3 nm are detected, which hints towards an ultrafast process either by a thermal phase explosion or Coulomb instability. An arrested growth is observed on a microsecond time scale resulting in a final particle size of 3–4 nm with high yield. In this context, the fragmentation in a NaCl/NaOH solution seems to limit growth by electrostatic stabilization of fragments, whereas it does not modify the initial product sizes. The laser-induced fragmentation process is identified as a single-step, instantaneous reaction.
A synthesis method that is mainly driven by physical mechanisms rather than chemical potential has a number of benefits, such as obtaining particles from a wide class of educt materials, a possible scale up of the process or particle suspensions that are less contaminated by reactants or byproducts than for instance in chemical synthesis. Such a process is the synthesis by laser ablation in liquids (LAL) that has gained wide popularity because of its straightforward applicability with limited technological efforts. Nevertheless, size definition has shown to be a delicate issue, as final sizes in LAL-produced nanoparticle batches will depend on a number of interrelated, hierarchical processes. While primary particle sizes are defined by the laser-induced educt decomposition, including phase explosion, evaporation or plasma-aided decomposition, further steps add to ripening and thus particle size growth. Particularly, very small particles (<3 nm) are difficult to obtain and stabilize. In noble metal systems, such as gold, these are labelled protoplasmonic. Their properties change from a quantum regime to a size-scaling regime and may offer a number of unique properties, such as permeability in biological systems1,2 or higher catalytic activity.3,4
While LAL has been intensively investigated5 and up-scaling routines have been defined,6 the tendency is to add a second process to reduce sizes and achieve a well-defined size definition by laser-induced fragmentation in liquids (LFL).7–9 In LFL, a laser pulse intersects the particle suspension such that the particles preferentially absorb energy and transiently pass through a high energy density state that ends in small fragments. The principal advantage of a separation of processes compared to LAL (where LFL would also happen parasitically in most cases) is that the optical properties, the distribution of particles in the suspension and the energy deposition can be well controlled. To that end, the process cascade would be uniform across the ensemble and decomposition would deliver a homogeneous final distribution.
The investigation of structural relaxation processes, particularly in metal nanoparticle excitation and LFL is a topic of current research, also in the view of nanophotonic applications. Energy absorption in the particles and the surrounding medium lead to a cascade of relaxations, partly reversible or irreversible. This can be harnessed in biological applications, including the hybridization with proteins, specifically with antibodies for in vitro and in vivo targeting or in nanoscale material processing.10–12 Transfection through cell membranes by transiently enhanced permeation1,13,14 has been described. A large number of studies have focused on the effect of laser-heated particles, in particular of gold for cell destruction.15–18
Fragmentation includes several levels of particle excitation both in energy and ultrafast time scales. Due to these complications the understanding of the mechanisms is far from being agreed to, despite many available theoretical models. Basically, the photo-excited particles would transform the energy into lattice-thermal degrees of freedom and electron emission within few picoseconds. A slower increasing lattice temperature, for instance in nanosecond excitation, could lead to a pure heating-melting-evaporation pathway, where the particle temperature essentially follows the binodal to reach melting and evaporation. Evaporated atoms and clusters would undergo a subsequent nucleated growth to form new, small particles.19 Growth can be limited by stabilization and/or kinetic arrest, which can be complicated by pre-existing surfactants in chemically synthesized particles.
Further, the importance of initial particle shape on the fragmentation efficiency of plasmonic nanoparticles has been discussed in literature. For instance, the effect of near-field enhancement even led to more exotic and specialized fragmentation, like near-field ablation,20 which has been discussed in case of rod-like Au nanoparticles.21 On the other hand, ultrashort-pulse excitation may lead to a non-equilibrium pathway in the phase diagram that eventually would allow to cross the spinodal line of the material.22 The consequence would be a violent explosion, or spinodal decomposition. This may directly lead to small particles with a distribution in sizes. A more exotic pathway has been discussed earlier, involving a massive electron emission23 (particularly in metal nanoparticles) that would destabilize a molten sphere to emit particles and clusters to reduce the Coulomb forces. This Coulomb instability (sometimes called ‘explosion’, referring to a related effect in molecules and clusters24) has been proposed based on observed laser-induced electron ejection9,25 and is still under debate.26–30 However, it should not be forgotten that finite-size effects not only encompass thermal properties, but also the spatial mode of fragmentation can be affected by geometry. It has been predicted in simulations that a strain-induced decomposition may be induced,29 if the photothermal excitation is faster than the particle acoustic eigenmodes. Recently, femtosecond-resolved diffraction experiments on single particles have witnessed such a behaviour.31
An approach that is able to shed light on the manifold of proposed reactions needs to resolve the caloric balance of the system (i.e. the observation of educts and products as function of energy input), and as well allow for a kinetic determination of structure formation on an ultrafast time scale. The first has been done repeatedly by observing the final particle population and transient optical spectroscopy.19,32–35 Meanwhile, it is difficult to get access to nanoscale structures on a time scale from picoseconds to microseconds.36–38 Here, we present results on time-resolved X-ray scattering (small and wide angle) on a surfactant-free gold nanoparticle suspension, excited by picosecond laser pulses of up to 1800 J m−2 in a time interval from 100 picoseconds to 100 microseconds to address temporal pathways of energy relaxation as well as the detection of product particles, including particles sizes, mass abundance and crystallinity.
1 Experimental setup
1.1 Gold particle colloidal synthesis
Gold nanoparticles have been produced by pulsed laser ablation in liquids (LAL) by ablating a gold target in ultra-pure water (MilliQ, Millipore) in a batch chamber with a Nd:YAG laser (Ekspla, Atlantic Series, 10 ps, 1064 nm, 9.6 mJ, 100 kHz, 10 min), containing typically a multimodal size distribution.39–41 To get a well size-separated sample for the study, a centrifugal step (Hettich, 67.1 g, 70 min) had been added. The obtained pellet is left for one week to force coalescence processes42 and is subsequently diluted to 100 mg L−1 either in ultra-pure water or in an aqueous solution containing 0.3 mM NaOH and 0.3 mM NaCl each. These nanoparticles are surfactant-free. Exemplary TEM images of the educt particles can be found in Fig. 1 (top) together with extinction spectra (Thermo Scientific: Evolution 201 spectrometer) of the educt and product particles below.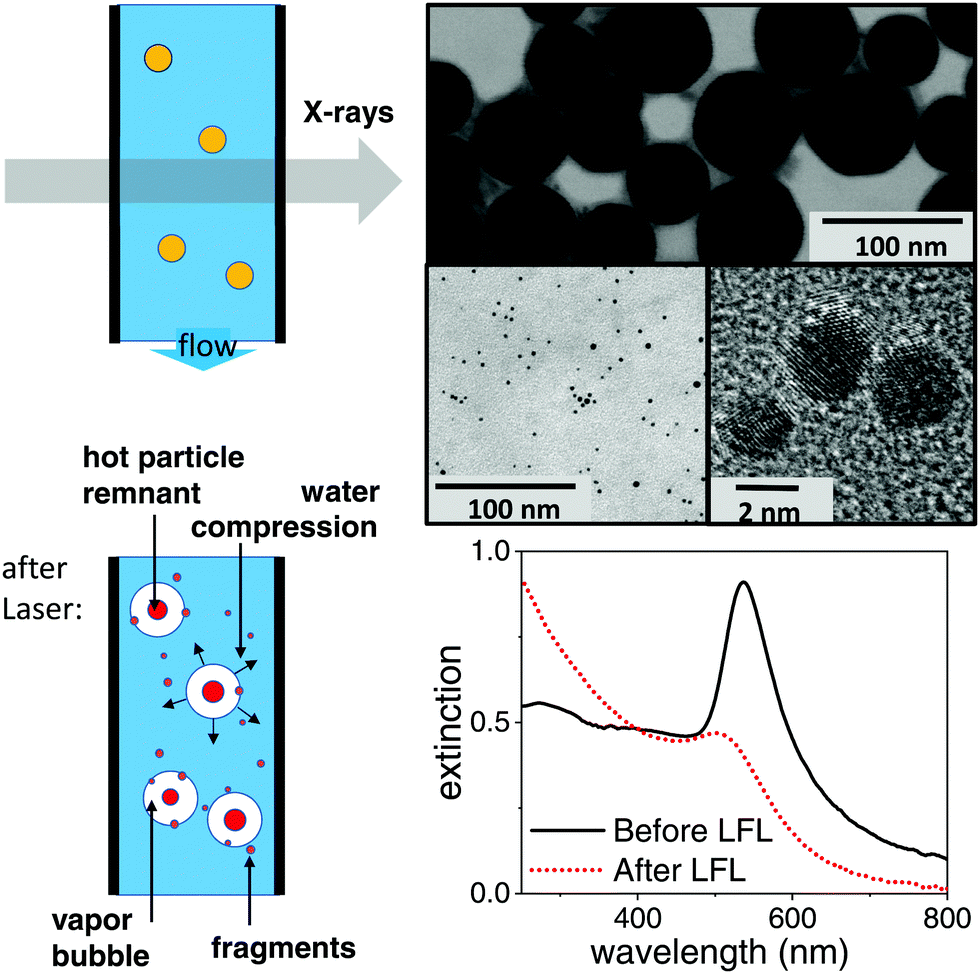 | ||
| Fig. 1 Left: Sketch of the dominant structure modifications of a metal nanoparticle photo-excitation in aqueous suspension. Possible relaxations include particle heating, fragmentation and vapour bubble formation. In an isochoric situation within the first tens of nanoseconds the expanding bubbles demand for a corresponding compression of the water bulk. Possible scenarios of particle destruction include a partial ablation for fragments around a remaining (size-reduced) parent particle or a complete explosion. Right: Transmission electron micrographs of the educt particles (top), product suspension (centre) together with the corresponding extinction spectra after LFL with 10 ps pulses at 300 J m−2. | ||
1.2 Ex situ analysis of fragmentation
Fragmentation for an analysis of the irradiated final particles has been done by a picosecond laser (Edgewave) with 10 ps pulses at 532 nm and 80 kHz. A fluence of 300 J m−2 was used. Fragmentation was performed in a free liquid jet as described earlier.41,43 Fragmentation has been performed either in ultra-pure water or in a solution of 0.3 mM NaCl and 0.3 mM NaOH.44 Transmission electron micrographs (TEM) have been acquired on a Zeiss EM 910 (80 kV). Size analysis of the Feret diameter has been done by the software ImageJ. The size distribution in suspension has been derived by an analytical disc centrifuge (ADC, CPS Instrument DC 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) at 24000 rpm. TEM images and extinction spectra after LFL are shown in Fig. 1 and the TEM histogram of product particles in the ESI.† No aggregates were detected by ADC. This is supported by the measured zeta potential. During LFL the zeta potential value of the nanoparticle colloids increased from −36.7 mV before to −75.5 mV after LFL, indicating high colloidal stability of the fragmented particles. Zeta potential measurements (Malvern Zetasizer Nano Z) were performed at a constant pH of 9.
000) at 24000 rpm. TEM images and extinction spectra after LFL are shown in Fig. 1 and the TEM histogram of product particles in the ESI.† No aggregates were detected by ADC. This is supported by the measured zeta potential. During LFL the zeta potential value of the nanoparticle colloids increased from −36.7 mV before to −75.5 mV after LFL, indicating high colloidal stability of the fragmented particles. Zeta potential measurements (Malvern Zetasizer Nano Z) were performed at a constant pH of 9.
1.3 In situ pulsed X-ray scattering of fragmentation
Time-resolved X-ray scattering has been performed at the beamline ID09 at the European Synchrotron Radiation Facility by a pump–probe setup at 1 kHz repetition rate. The suspension was pumped through a quartz capillary (Hilgenberg) of 0.35 mm diameter at a throughput of 2 ml min−1, such that the sample aliquot is replaced after each pulse pair, as indicated in Fig. 1. This assures single-pulse excitation of every part of the sample. The pump laser for fragmentation was a regenerative amplifier on Ti:Sa basis (Coherent Legend), whose fundamental at 800 nm and 2 ps pulse length has been converted into 532 nm light by an optical parametric amplifier (Light Conversion, TOPAS). The pulse length was thus reduced to ≤1 ps. The laser light at 532 nm was attenuated by a combination of polarizer (Glan Laser) and motorized waveplate retarder to control the fluence on the sample. Focusing to a 0.26 mm spot size (fwhm) was done by a 500 mm lens. The capillary was irradiated at an angle of 80 degrees, while the illuminated portion of the liquid was at the same time intersected by the X-ray beam.Pulsed X-rays at 15.2 keV for probing were produced by a single-line undulator45 and further monochromatized by a multilayer pair of ruthenium/B4C to a 0.17 keV bandwidth. The pulse train from the storage ring was reduced to 1 kHz by an ultrasonic chopper wheel.46 Scattered X-rays were detected by a CCD camera (charge coupled device) after conversion to visible light in a scintillator with fiber coupling (Rayonix 170). The transmitted X-ray beam was shielded from the detector by a millimetre-sized metal cylinder as beam stop. A short distance (0.12 m) between capillary and detector screen allowed to cover a solid angle of 60 degrees (wide-angle scattering, WAXS) to record different portions of the scattering yield, such as the liquid scattering from water, the powder rings from the suspended crystalline gold particles and also scattering at low angles related to mesoscopic structures (e.g. particle shapes). The small-angle scattering (SAXS) part could be further resolved to smaller angles by increasing the capillary-detector distance to 0.65 m. The images accumulated the scattering from typically 3000 pulses in SAXS and 30000 pulses in WAXS geometry.
The data acquisition was repeated both for a given delay between X-ray pulses and laser pulses and for a negative delay (−2 ns) in order to isolate signal changes that were linked to photo-excitation. At a (negative) delay of −2 ns the laser pulse followed X-ray scattering, such that only a non-excited sample was probed. The time resolution was given by the X-ray pulse length (about 80 ps) rather than the timing jitter between laser and X-rays (<10 ps). The average fluence is calculated from the intensity distribution of the laser pulses across the X-ray probed central area (0.04 × 0.06 mm2).
1.4 Data analysis
The 2D X-ray scattering images were subjected to standard image corrections (dark current, flat field, solid angle coverage of different pixels and masking invalid pixels) and integrated azimuthally to get a scattering function S(q) with q being the scattering vector q = 4·π/λ·sin(2Θ/2), with X-ray wavelength λ and the solid angle of scattering 2Θ.The scattering is dominated by the liquid scattering of water and the capillary scattering. The latter is removed by the process of taking differences ΔS(q) = S(q)τ − S(q)−2ns between the scattering at delay τ and the negative delay. The liquid scattering is only modified slightly, if the liquid undergoes changes in volume and/or temperature.47 In the difference curve the very weak change in bond angle in such a situation is prominently visible as smooth difference features, as indicated in Fig. 2. As this change in scattering is stationary (only scales with amplitude of change in T and V, but does not shift) in the linear approximation, it can serve to quantify the amount of change. In particular the derivatives dS/dT|p and dS/pV|T are known.47,48 By using the thermal expansion coefficient of water we can translate the signal height into change in volume of the liquid phase. The basic assumption here is that once vapour bubbles form around the excited nanoparticles (and vapour contributing only as background to the scattering curve), the bulk of water needs to be compressed to accommodate the bubbles. After some 100 nanoseconds the liquid column may react by a macroscopic expansion. In this scenario, the total bubble volume can be derived directly (open circles in Fig. 2b).
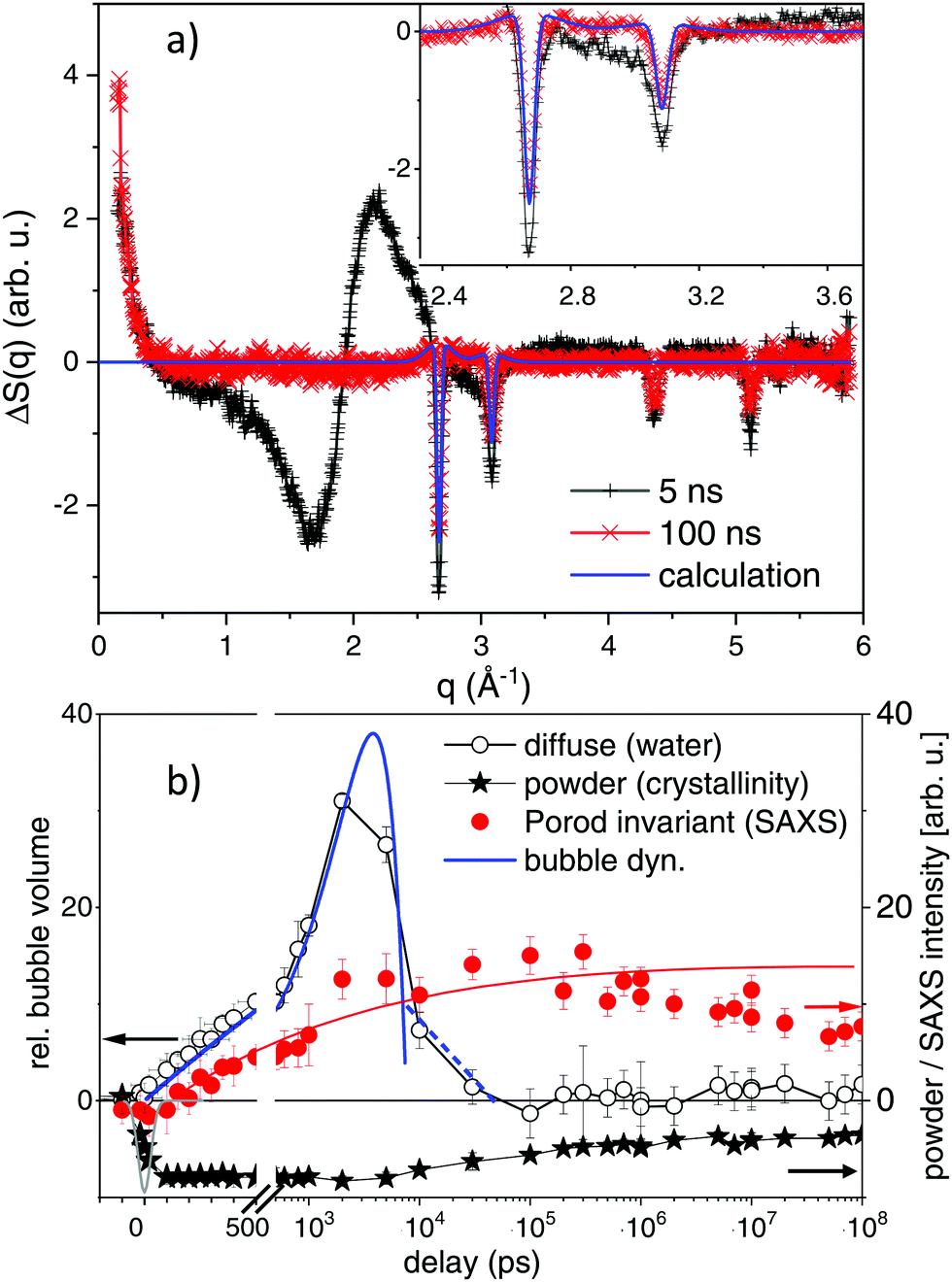 | ||
| Fig. 2 (a) Selected difference scattering curves after excitation at maximum fluence (1800 J m−2). Black crosses mark the signal at a delay of 5 ns with large excursions around the liquid water peak of 2.2 Å−1. The 100 ns delay is shown by red crosses. The inset magnifies the small-signal region together with a line plot of a simulation of powder scattering, see text. (b) Observables for time-dependent transients: diffuse scattering signal for water changes, SAXS Porod invariant (<0.3 Å−1) reflecting formation of new, small particles and the loss in powder intensity of the (111) reflection, showing particle melting and recrystallization. The thick blue line indicates parabolic kinetic trace indicative of bubble dynamics together with possible rebound stages (dashed line). The thin red line is a guide to the eye for the SAXS signal evolution. The grey (Gaussian) line around zero delay indicates the temporal resolution of the X-ray pulses. | ||
Gold particles crystallized in an fcc structure give rise to lattice planes that diffract X-rays. The corresponding powder peaks (particles are randomly oriented in the suspension) are seen in the difference scattering in Fig. 2a as sharp negative peaks, as order is lost during photo excitation. The shift in peak position reflects thermal expansion, while the peak intensity is related to the amount of remaining crystalline material and to a lesser extent to thermal-vibration induced intensity reduction (Debye–Waller factor). Here, we mainly analyse the peak intensity, as due to the X-ray bandwidth the resolution of crystalline grain size is limited to below 14 nm. The integral over the (111) peak in Fig. 2a is displayed as crosses in Fig. 2b.
Small angle scattering changes due to several effects: once by the formed vapour bubbles37,49 and secondly by size changes of the laser-fragmented gold particles. A loss of intensity at scattering vector q ≪ 0.01 Å−1 is found, if the parent particles are destroyed, while an increase at q > 0.015 Å−1 is observed, if new fragments appear (see Fig. 2a). The integration of S(q)·q2 (Porod invariant) from 0.015 Å−1 to 0.5 Å−1 serves as a measure for the total mass of small fragments (filled bullets in Fig. 2b). Overall, the observables in Fig. 2b allow for disentangling the temporal evolution of particle crystallinity, size and bubble formation, as discussed in Section 2.2.
2 Results and discussion
The understanding of photo-reactions in gold nanoparticles requires the definition of the thermal and structural pathway. In particular, phase transitions and dissipative channels are of importance. The concept that the laser light is preferentially absorbed in the nanoparticles embedded in the medium is central. The energy is quickly transformed into heat (except for non-thermal relaxations, such as near-field effects20 or electron,25 light, and shock wave emission). The temperature increase of the particles and subsequently the medium leads to structural changes, such as particle melting, bubble formation in water or particle decomposition. Quantifying the contribution of each process is not trivial. In the following we discuss a caloric and a kinetic verification of the state of matter.2.1 Caloric prediction of phase behaviour
In a caloric approach the absorbed energy is compared to the latent heat of the particles, including heat capacities and heat of melting and fusion.19,28,34,50 This can be done by (i) an estimation of the absorbed energy from incident fluence and particle-related optical properties. The extinction cross section for spherical particles can be derived by Mie theory.51,52 Alternatively, (ii) the absorbed energy can be estimated from the extinction of the suspension and a known particle mass density in the suspension. In this case, however, absorption needs to be detailed out, as scattering may contribute to extinction but not to energy uptake.Both approaches have been used in literature. A direct access to temperature may also be available through (iii) recording temperature-dependent properties, such as elastic moduli53,54 or thermal expansion.50 This breaks down for plastic or irreversible relaxations, such as melting or decomposition. Nevertheless, it is possible to nominally estimate the energy uptake by extrapolating the slope of e.g. thermal expansion versus applied fluence, given that the absorption process (within the pulse length) is shorter than the onset of irreversible relaxations (e.g. melting). Further care has to be taken to not neglect transient loss mechanisms, such as intrapulse extinction change through transient bleaching or other energy dissipation.
Dissipation can happen via heat conduction or liquid-vapour formation. Heat transfer calculations based on continuum models are successful below bubble formation.37,50,55 Bubble formation isolates the particles thermally from the liquid, which requires numerical modelling and assumptions about lifetimes or effective transfer coefficients.56–60 In the present experiment, the pulse length of 1 ps assures that laser energy is absorbed before major structural relaxations and dissipation occur (the latter is slower than the 80 ps observation delay), such that the absorbed energy is localized in the particle.
We have used a laser fluence of 1800 J m−2 in the X-ray probed portion of the liquid jet, which afforded a pulse energy after the OPA of 160 μJ. The direct access to mean particle temperature of the crystalline fraction consists in the in situ analysis of the powder scattering from the gold fcc lattice. The intensity will drop and finally vanish at the melting transition, while the position of the peaks (for instance the (111) reflection) will shift towards lower angles, reflecting lattice expansion. The lattice expansion at 80 ps delay together with peak intensity is shown in Fig. 3 as function of fluence. With a 80 ps time resolution and cooling times of about 0.3–1 ns50,61 the observed expansion within the time resolution is expected to record the maximal value and represent the most strongly heated state. At the same time, local thermal equilibrium is supposed to be reached. We observe that the expansion initially scales linearly with fluence, showing a slope of (8.5 ± 0.8) K (J m−2)−1. Around 80–120 J m−2 the slope strongly decreases, while the scattering intensity drops. The extrapolated linear expansion would reach the value of 1.78%,62 which is the maximum lattice expansion of gold at the melting point. Melting point depression is only expected to amount for about 10 K,63 while the (initial) lattice expansion of gold nanoparticles is found not to deviate strongly from the bulk value.64 It should be noted that an onset of melting removes the hottest particles from observation such that the apparent temperature is lower than the full ensemble average. This can explain the reduction of the slope above 80 J m−2. Consequently, the transition to complete melting is manifested at a fluence F above 120 J m−2. One has to take into account that the real transition even in an individual particle may be smeared out in fluence due to the finite size and distribution of the absorbed energy between lattice heating and (surface) melting.
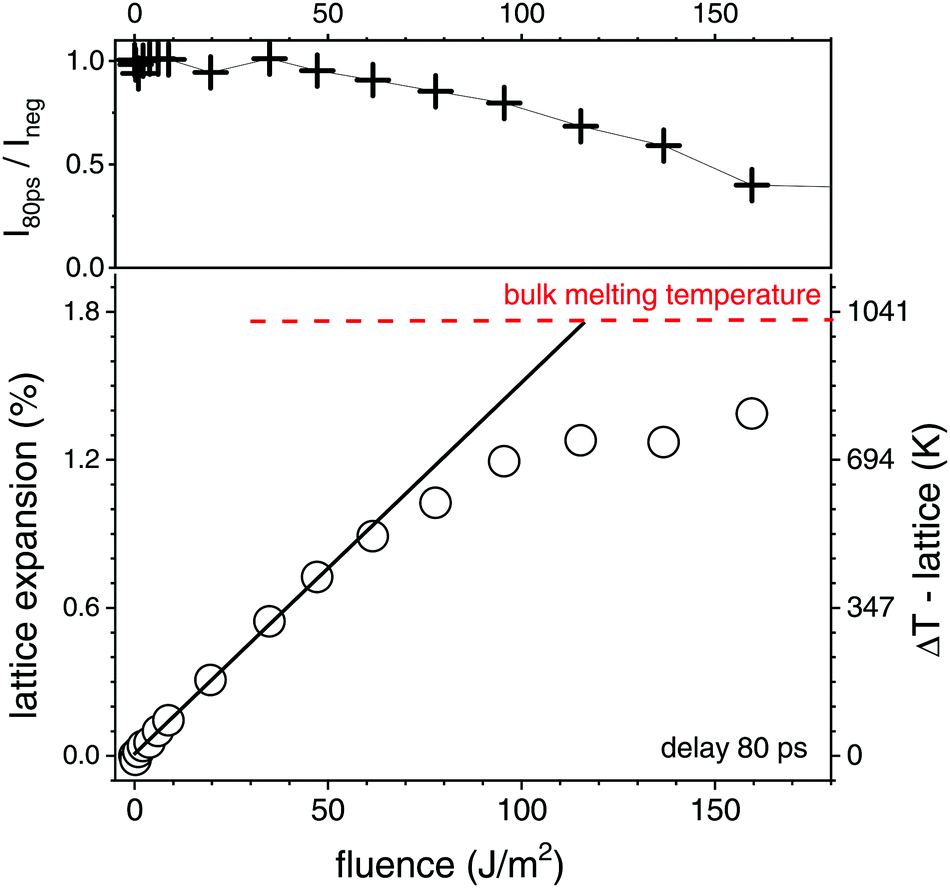 | ||
| Fig. 3 Top: Change of the intensity of the (111) powder peak at delay τ = 80 ps as function of laser fluence relative to the non excited suspension (τ = −2 ns). Bottom: Lattice expansion (correlated to temperature increase ΔT) as function of fluence at τ = 80 ps, the size of the open circles represents the error margin. The horizontal dashed line marks the maximum expansion at the melting point of gold. The full line indicates the initial slope of the expansion versus fluence. | ||
A calculation of expected heating ΔT as function of fluence according to approach (i) requires to calculate the change in temperature via:
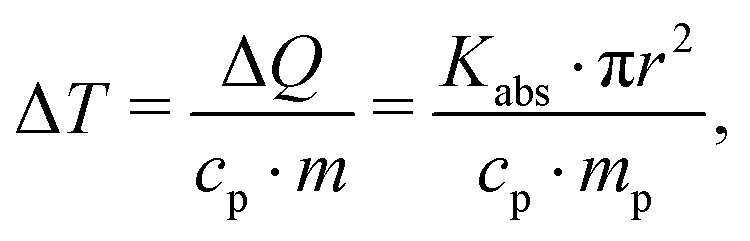 | (1) |
Altogether, we can secure the general caloric scale for the excitation of the gold nanoparticle suspension, noting that melting is observed above 120 J m−2. Using the experimentally determined slope of 8.5 K (J m−2)−1, fully melted particles are expected above 165 J m−2, while full evaporation may be achieved above 2000 J m−2.34 This, however, does not include nonlinearity in absorption (such as bleaching or plasma formation69) or possible loss mechanisms, such as heat dissipation during particle overheating, additional scattering or bubble formation. We note that the applied maximum laser fluence of 1800 J m−2 is barely able to fully evaporate the particles, in particular with unquantified loss mechanisms. Thus, a pure evaporation mechanism is unlikely.
2.2 Kinetic identification of structural pathways
In a kinetic approach, structure relaxations may be identified in a time-resolved manner by observing abrupt (discontinuous) changes of properties, such as probe light absorption.28,70 A well-documented case is the appearance of vapour bubbles on heated nanoparticles, which changes the particle extinction as well as adding light scattering pathways.37,68In optical measurements different relaxation pathways can be correlated to the changes in the extinction, mainly around the plasmon resonance. However, specific transients (shift of the plasmon resonance due to changes in the particle electron density or refractive index of the liquid)33,68,71,72 mix with unspecific transients, such as scattering from particles and bubbles70 or aggregated nanoparticles. In X-ray scattering, due to separation of length scales in reciprocal space, different structural reorganization can be discerned. The first discussed feature is the lattice expansion and loss of crystallinity seen at the gold powder scattering. In Fig. 2a scattering difference curves of the excited suspension at given delay are displayed in a wide range of scattering vector q. The loss of crystallinity is seen as sharp dips at several q positions, related to the different gold fcc reflections. The angular resolution is slightly lower than required to resolve the Scherrer width of the educt due to the finite bandwidth of the X-rays, but sufficient for quantifying the peak shift. The scattering difference at the (111) peak at 2.67 Å−1 is negative at 1800 J m−2 due to the complete melting at 5 ns. This is still the case at 100 ns, while at this delay a positive contribution at the sides of the peak appears. This can be modelled by assuming additional powder scattering from crystalline particles with the same lattice parameter, but much broader Scherrer width (indicated a bold line in the inset of Fig. 2a). An estimate of the crystalline size from the Scherrer width returns about 2 nm in diameter. The kinetics of the powder scattering intensity loss in Fig. 2b shows that the powder peaks are lost within the time resolution of the experiment to only partly reappear after some 100 to 500 ns. Most of the intensity is lost throughout the probed delay range, indicating liquefied or fragmented particles.
A prominent part of scattering change is the broad modulation around 1.7, 2.2 and 3 Å−1, which is related to the position of the (twin) liquid water peak. It has been shown earlier that this can be interpreted as thermodynamic change of the water phase,47 in the present case a water compression following vapour bubble expansion.48,49,65 The kinetics show a rise and decay within a 10 ns interval after excitation, which is compatible with a parabolic (ballistic) growth and recess known from bubble dynamics theory.73 Such bubble dynamics can show multiple oscillations, which in the present case can not be temporally resolved due to the differences in lifetimes between individual bubbles in the ensemble.49,60,68 Yet, it appears that most of the bubble signal has decayed within 30 ns. It can be inferred that when bubbles have vanished the transiently hot nanomaterial (product and educt particles) would cool down within 1–2 ns.50,55
Most interestingly in view of the envisaged fragmentation, a prominent SAXS signal starts to appear at low q (<0.3 Å−1). This holds 3 contributions, SAXS from the appearing bubbles,49 a loss in scattering, if the large educt particles are destroyed and a positive scattering contribution for newly formed small product particles. The achieved lower resolution limit in q, however, gives limited access to the educt particles and bubbles. Due to their large size, the corresponding scattering is strongest below 0.015 Å−1,74 which is close to our lower limit (0.01 Å−1). Therefore, the main part of SAXS is positive in Fig. 2b, being related to formation of new product particles.
Within the first 10 ns the Porod invariant, as a measure for the total particle mass of particles below about 15 nm, increases monotonously, similar to bubble growth at early delay (see Fig. 2b). It is difficult to quantify the exact magnitude of the invariant while vapour bubbles are still present. Nevertheless, after bubbles have vanished after 10–30 ns, the SAXS signal persists and tends to grow slightly until a delay of 1 μs. Afterwards, it decays slightly, but one should be aware that the flow in the capillary transports the excited plug out of the probed region.
Overall, particle melting, disintegration (persistent loss of crystallinity) and bubble formation are seen in the kinetic traces. Energy dissipation is almost complete after 10–20 ns of bubble lifetime, which may prevent more rapid thermal dissipation. Therefore, the time window of violent particle modifications is limited to that interval, which is corroborated by the stationary Porod invariant after this delay and the characterization as single-step process.41
2.3 Fragmentation
The scenario for fragmentation through evaporation as opposed to decomposition via spinodal decay and Coulomb instability would be a complete particle atomization, followed by nucleated growth of atom clusters. A direct appearance of product particles of different sizes, respectively in spinodal or Coulomb decomposition75 would take place. The Coulomb instability suggests that the creation of daughter particles of some nanometre size would be an efficient pathway to lower the surface charge density of the excited (molten) particles.9,25,29,76 A current study reports on the observation of Coulomb fission in silica-shell gold particles, that show a signature of small product droplets ejected after femtosecond excitation.30 A recent structural resolution of gold particle excitation and fragmentation in air on a substrate has shown a stress-mediated explosion at relatively low fluence just above melting.31 Such a scenario may not be distinguishable from spinodal decomposition with the fragment size depending on the level of overheating.29 Stronger overheating will produce smaller fragments together with evaporation.At the earliest delay of 30 ns, which is not mixed any more with bubble signal in SAXS, we already observe a distinct scattering distribution below 0.5 Å−1. This scattering persists and changes shape only slightly for increasing delay. Fig. 4a shows the scattering distribution as Kratky representation at 1 μs. Qualitatively, maxima in this plot mark distinct size fractions (1–3 nm and 10–25 nm), whose diameters scale inversely with the corresponding scattering vector. A simulation of the scattering has been done by a reverse Monte Carlo approach,77 optimizing a freely adjustable distribution of spheres in a large size interval. The resulting mass-weighted histogram shows a predominant population of 2–3 nm particles, accompanied by larger particles (Fig. 4b) at a delay of 1 μs for the gold colloid with NaCl/NaOH. The distribution is fairly narrow, also for earlier times. This would be even more peaked at 2–3 nm in a size-weighted distribution as often presented for ex situ characterization. A comparison to the size analysis by ADC and TEM of the final irradiated particles shows a comparable distribution, which is shifted slightly towards larger particles in TEM (see ESI†). This could be induced by drying of the colloid when preparing the TEM grid, sensitivity of the (Feret) diameter in TEM on anisotropy during deposition78 or selective detection of larger (higher-contrast) particles. Fig. 4c shows the temporal evolution of the small particle fraction with the delay. The hydrodynamic diameter of post-synthesis analysis by ADC (ESI†) fits the in situ size distribution quite well. We find fragments of 2 nm diameter both in pure water and aqueous NaCl/NaOH directly after bubble disappearance. The diameter undergoes a growth phase with the largest particles being observed at the longest analyzed delay of 10 μs. It seems that fragments in NaCl/NaOH solution do not grow as strongly as in pure water on the microsecond time scale, limited by the statistical significance. For delays <1 μs the particle size evolution seems not to be affected by the electrolyte content, whereas later the electrolytes appear to quench growth. Further studies need to clarify this transition and the time scale of size quenching. Nevertheless, it should be emphasized that the dominant fragment nanocluster population of 2–3 nm is quantitatively resolved already after 30 ns and is qualitatively indicated on a sub-nanosecond scale. This should be put in perspective to the early expansion phase of the fragmented particles and bubble. Therefore, we expect that fragmentation at a fluence close to complete evaporation of the gold particles would produce clusters of finite initial size together with atoms and ions as opposed to a pure evaporation and condensation process. Our present level of structure resolution does not yet allow to conclude on the quantitative partition between phase explosion, charge instability or evaporation.
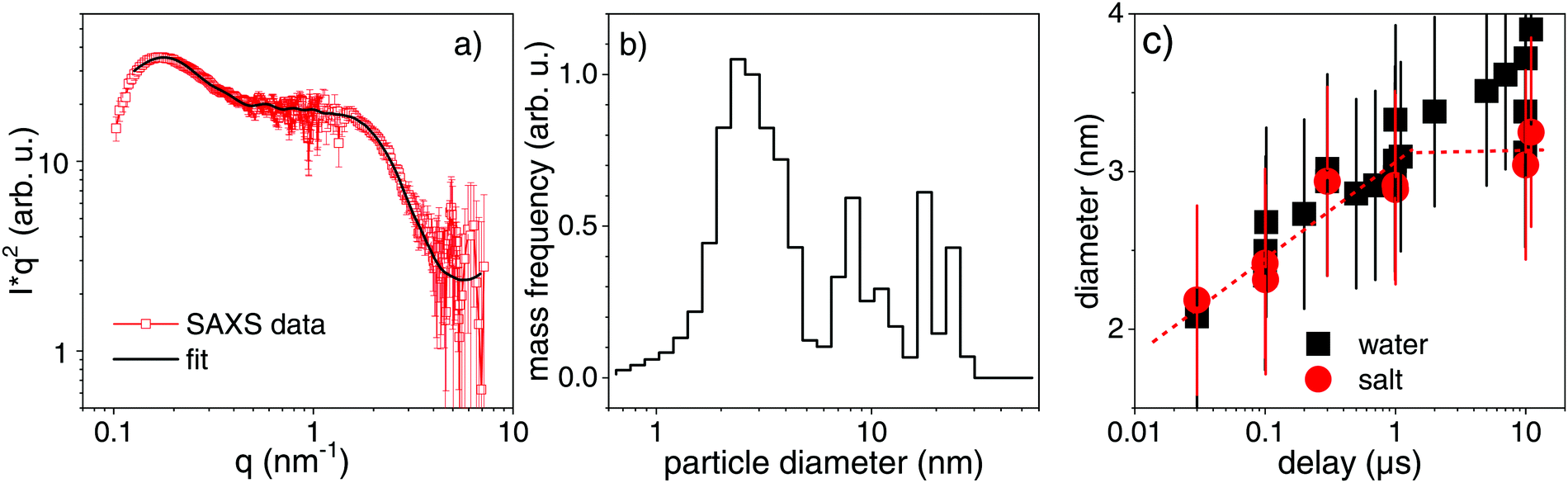 | ||
| Fig. 4 (a) Scattering distribution at low angles (SAXS) at a delay of 1 μs together with a simulation according to a reverse Monte Carlo optimization. (b) Size distributions as derived from the SAXS simulation at 1 μs. (c) Evolution of the particle size as function of delay for the dominant size fraction. Black squares represent fragmentation in pure water and red bullets fragmentation in a micromolar (300 μM) NaCl/NaOH solution. The symbol size indicates the error in mean size determination, while the error bars reflect the root mean square distribution width. | ||
Besides, these early fragments tend to grow in a limited fashion, in particular, if micromolar concentrations of anions are present. Recently, Ziefuß et al. studied the role of anions during LFL and found synergism between specific halide anions and basic pH on yielding narrow nanocluster size distributions.44 The profile of powder scattering in Fig. 2 as well suggests that apart from the scattering loss due to molten educt particles the additional scattering on the wings of the powder peaks indicates that smaller fragments (<2 nm) have formed and have rapidly crystallized as soon as getting in contact with water.
Both Coulomb instability and spinodal decomposition above the boiling point would deliver small product particles as a single-step, instantaneous (on the scale of the local dynamics) reaction. These mechanisms are yet not discriminated. A pure (surface) evaporation process is unlikely here due to the fast onset of product signal. Earlier reports have inspected the emerging fragments by electron microscopy and have found small spherical clusters similar to our observations as well as fingering structures.9,79 This fingering could be a result of a beginning phase explosion with a fast crystallization as soon as the fragments get in contact to the liquid due to the faster cooling of smaller structures.
The notion of a single-step, instantaneous process has important implications. First, a single-step process is capable of producing the desired fragment particles in one laser irradiation shot. Therefore, processing in a flow reactor or jet41 is achievable by one passage for an optimized throughput. Secondly, the results show that nanoparticles are produced instantaneously, i.e. not requiring a certain delay or incubation time, notwithstanding limited ripening. In an evaporation and growth process, the diffusion and coalescence of fragmented species would dictate the quality of the fragment output. This would be disturbed when adding surface charge delivering additives for preventing ripening.44 In the instantaneous process of educt particle explosion, the product particles are directly formed and could be size-quenched, if desired. The process would also be biased by external parameters, such as educt concentration, if further secondary processes would define the products. This would limit the generalization of the description and definition of the process.
3 Conclusions
The calorics and kinetics of structure formation during laser-induced fragmentation of spherical gold particles in aqueous suspension, respectively containing a minute amount of ions, has been investigated by direct time-resolved and structural access in situ. The results are based on a laser-pump and X-ray-probe setup that allows to irradiate each volume element by a single laser pulse by employing a jet flow. At a fluence of 1800 J m−2, which is close to full evaporation of the educt particles, an almost complete transformation into 2–3 nm particles could be observed. On a time scale of up to 10 μs, a slight growth was observed, quenched by ion adsorption. Above 10 μs, trends indicate further limited particle growth but was not resolved by the experiment. In turn, the adsorption process of anions on the nanoparticle surface improving the colloidal stability has to occur already on time scales below 1 μs.Note that energy losses also occur via dissipation by heat transfer into water and subsequent water boiling and probably non-linear plasmon bleaching during the laser pulse duration. Although the fluence is compatible to an evaporation process followed by a secondary particle nucleation and growth, we could quantify product particles of 2 nm as early as 30 ns after excitation. The given time scale is very short for effective diffusional growth from atoms. Therefore, it is concluded that the fragmentation should include direct fragmentation into nm sized products. Yet, we can not distinguish between different phenomena, such as stress mediation, phase explosion or Coulomb instability.
Nevertheless, the picosecond laser fragmentation process is classified as a single-step, instantaneous reaction. This represents an important aspect for the application as a high-throughput method for production of small, protoplasmonic gold particles in an upscaled thoughput.
Conflicts of interest
The authors declare that no conflicts of interest exist.Acknowledgements
The research is funded by the Helmholtz Association under the topic “from matter to materials and life” and German Science foundation under contract BA 3580/22-1. The ESRF is acknowledged for provision of beamtime and funding. Support by M. Sander is gratefully acknowledged. AP acknowledges the support from the DFG within the Heisenberg fellowship. We would like to thank L. Zhigilei for fruitful discussions. Jurij Jakobi (U. Duisburg-Essen) is acknowledged for the provision of the TEM micrographs.Notes and references
- C. Yao, X. Qu, Z. Zhang, G. Hüttmann and R. Rahmanzadeh, J. Biomed. Opt., 2009, 14, 054034 CrossRef PubMed.
- Y. Zhang, C. Zhang, C. Xu, X. Wang, C. Liu, G. I. N. Waterhouse, Y. Wang and H. Yin, Talanta, 2019, 200, 432–442 CrossRef CAS PubMed.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X.-S. Du, S. Yang, J. Chai and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS PubMed.
- S. Reichenberger, G. Marzun, M. Muhler and S. Barcikowski, ChemCatChem, 2019, 11, 4489–4518 CrossRef CAS.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- R. Streubel, S. Barcikowski and B. Goekce, Opt. Lett., 2016, 41, 1486–1489 CrossRef CAS PubMed.
- F. Mafuné, J. y. Kohno, Y. Takeda and T. Kondow, J. Phys. Chem. B, 2002, 106, 8555–8561 CrossRef.
- S. Inasawa, M. Sugiyama, S. Noda and Y. Yamaguchi, J. Phys. Chem. B, 2006, 110, 3114–3119 CrossRef CAS PubMed.
- S. Hashimoto, T. Katayama, K. Setoura, M. Strasser, T. Uwada and H. Miyasaka, Phys. Chem. Chem. Phys., 2016, 18, 4994–5004 RSC.
- A. Podlipensky, A. Abdolvand, G. Seifert and H. Graener, Appl. Phys. A: Mater. Sci. Process., 2005, 80, 1647–1652 CrossRef CAS.
- Z. Peng, T. Walther and K. Kleinermanns, J. Phys. Chem. B, 2005, 109, 15735–15740 CrossRef CAS PubMed.
- V. Resta, J. Siegel, J. Bonse, J. Gonzalo, C. N. Afonso, E. Piscopiello and G. V. Tenedeloo, J. Appl. Phys., 2006, 100, 084311 CrossRef.
- C. Yao, R. Rahmanzadeh, E. Endl, Z. Zhang, J. Gerdes and G. Hüttmann, J. Biomed. Opt., 2005, 10, 064012 CrossRef PubMed.
- J. Krawinkel, U. Richter, M. L. Torres-Mapa, M. Westermann, L. Gamrad, C. Rehbock, S. Barcikowski and A. Heisterkamp, J. Nanobiotechnol., 2016, 14, 2 CrossRef PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed.
- C. M. Pitsillides, E. K. Joe, X. Wei, R. R. Anderson and C. P. Lin, Biophys. J., 2003, 84, 4023–4032 CrossRef CAS PubMed.
- N. S. Abadeer and C. J. Murphy, J. Phys. Chem. C, 2016, 120, 4691–4716 CrossRef CAS.
- A. Takami, H. Kurita and S. Koda, J. Phys. Chem. B, 1999, 103, 1226–1232 CrossRef CAS.
- A. Plech, V. Kotaidis, M. Lorenc and J. Boneberg, Nat. Phys., 2006, 2, 44–47 Search PubMed.
- G. González-Rubio, P. Díaz-Núñez, A. Rivera, A. Prada, G. Tardajos, J. González-Izquierdo, L. Bañares, P. Llombart, L. G. Macdowell, M. A. Palafox, L. M. Liz-Marzán, O. Peña-Rodriguez and A. Guerrero-Martínez, Science, 2017, 358, 640–644 CrossRef PubMed.
- M. E. Povarnitsyn, T. E. Itina, P. R. Levashov and K. V. Khishchenko, Phys. Chem. Chem. Phys., 2013, 15, 3108–4114 RSC.
- P. V. Kamat, M. Flumiani and G. V. Hartland, J. Phys. Chem. B, 1998, 102, 3123–3128 CrossRef CAS.
- T. Döppner, T. Fennel, T. Diederich, J. Tiggesbäumker and K. H. Meiwes-Broer, Phys. Rev. Lett., 2005, 94, 013401 CrossRef PubMed.
- K. Yamada, Y. Tokumoto, T. Nagata and F. Mafuné, J. Phys. Chem. B, 2006, 110, 11751–11756 CrossRef CAS PubMed.
- S. Hashimoto, D. Werner and T. Uwada, J. Photochem. Photobiol., C, 2012, 13, 28–54 CrossRef CAS.
- T. E. Itina, K. Gouriet, L. V. Zhigilei, S. Noël, J. Hermann and M. Sentis, Appl. Surf. Sci., 2007, 253, 7656–7661 CrossRef CAS.
- M. Strasser, K. Setoura, U. Langbein and S. Hashimoto, J. Phys. Chem. C, 2014, 118, 25748–25755 CrossRef CAS.
- L. Delfour and T. E. Itina, J. Phys. Chem. C, 2015, 119, 13893–13900 CrossRef CAS.
- J. M. Voss, P. K. Olshin, R. Charbonnier, M. Drabbels and U. J. Lorenz, ACS Nano, 2019, 13, 12445–12451 CrossRef CAS PubMed.
- Y. Ihm, D. H. Cho, D. Sung, D. Nam, C. Jung, T. Sato, S. Kim, J. Park, S. Kim, M. Gallagher-Jones, Y. Kim, R. Xu, S. Owada, J. H. Shim, K. Tono, M. Yabashi, T. Ishikawa, J. Miao, D. Y. Noh and C. Song, Nat. Commun., 2019, 10, 2411 CrossRef PubMed.
- S. Link, Z. L. Wang and M. A. El-Sayed, J. Phys. Chem. B, 2000, 104, 7867–7870 CrossRef CAS.
- D. Werner, A. Furube, T. Okamoto and S. Hashimoto, J. Phys. Chem. C, 2011, 115, 8503–8512 CrossRef CAS.
- A. Pyatenko, H. Wang, N. Koshizaki and T. Tsuji, Laser Photonics Rev., 2013, 7, 596–604 CrossRef CAS.
- D. Werner and S. Hashimoto, Langmuir, 2013, 29, 1295–1302 CrossRef CAS PubMed.
- C.-Y. Ruan, Y. Murooka, R. K. Raman and R. A. Murdick, Nano Lett., 2007, 7, 1290–1296 CrossRef CAS PubMed.
- A. Siems, S. A. L. Weber, J. Boneberg and A. Plech, New J. Phys., 2011, 13, 043018 CrossRef.
- J. N. Clark, L. Beitra, G. Xiong, D. M. Fritz, H. T. Lemke, D. Zhu, M. Chollet, G. J. Williams, M. M. Messerschmidt, B. Abbey, R. J. Harder, A. M. Korsunsky, J. S. Wark, D. A. Reis and I. K. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7444–7448 CrossRef CAS PubMed.
- S. Reich, P. Schönfeld, P. Wagener, A. Letzel, S. Ibrahimkutty, B. Gökce, S. Barcikowski, A. Menzel, T. dos Santos Rolo and A. Plech, J. Colloid Interface Sci., 2017, 489, 106–113 CrossRef CAS PubMed.
- S. Reich, A. Letzel, A. Menzel, N. Kretzschmar, B. Gökce, S. Barcikowski and A. Plech, Nanoscale, 2019, 11, 6962–6969 RSC.
- A. R. Ziefuß, S. Reichenberger, C. Rehbock, I. Chakraborty, M. Gharib, W. J. Parak and S. Barcikowski, J. Phys. Chem. C, 2018, 122, 22125–22136 CrossRef.
- S. Jendrzej, B. Gökce, V. Amendola and S. Barcikowski, J. Colloid Interface Sci., 2016, 463, 299–307 CrossRef CAS PubMed.
- M. Lau and S. Barcikowski, Appl. Surf. Sci., 2015, 348, 22–29 CrossRef CAS.
- A. R. Ziefuß, S. Barcikowski and C. Rehbock, Langmuir, 2019, 35, 6630–6639 Search PubMed.
- A. Plech, R. Randler, A. Geis and M. Wulff, J. Synchrotron Radiat., 2002, 9, 287–292 CrossRef CAS PubMed.
- M. Cammarata, L. Eybert, F. Ewald, W. Reichenbach, M. Wulff, P. Anfinrud, F. Schotte, A. Plech, Q. Kong, M. Lorenc, B. Lindenau, J. Räbiger and S. Polachowski, Rev. Sci. Instrum., 2009, 80, 15101 CrossRef PubMed.
- M. Cammarata, M. Lorenc, T. K. Kim, J. H. Lee, Q. Y. Kong, E. Pontecorvo, M. L. Russo, G. Schiró, A. Cupane, M. Wulff and H. Ihee, J. Chem. Phys., 2006, 124, 124504 CrossRef CAS PubMed.
- A. Plech, V. Kotaidis, M. Lorenc and M. Wulff, Chem. Phys. Lett., 2005, 401, 565–569 CrossRef CAS.
- V. Kotaidis and A. Plech, Appl. Phys. Lett., 2005, 87, 213102 CrossRef.
- A. Plech, V. Kotaidis, S. Grésillon, C. Dahmen and G. von Plessen, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 195423 CrossRef.
- G. Mie, Ann. Phys., 1908, 4, 377–445 CrossRef.
- P. Laven, 2005, http://www.philiplaven.com/mieplot.htm.
- G. V. Hartland, M. Hu and J. E. Sader, J. Phys. Chem. B, 2003, 107, 7472–7478 CrossRef CAS.
- H. Petrova, J. P. Juste, I. Pastoriza-Santos, G. V. Hartland, L. M. Liz-Marzán and P. Mulvaney, Phys. Chem. Chem. Phys., 2006, 8, 814–821 RSC.
- O. M. Wilson, X. Hu, D. G. Cahill and P. V. Braun, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 224301 CrossRef.
- S. Merabia, S. Shenogin, L. Joly, P. Keblinski and J.-L. Barrat, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15113–15118 CrossRef CAS PubMed.
- J. Lombard, T. Biben and S. Merabia, Phys. Rev. Lett., 2014, 112, 105701 CrossRef PubMed.
- K. Metwally, S. Mensah and G. Baffou, J. Phys. Chem. C, 2015, 119, 28586–28596 CrossRef CAS.
- J. Lombard, T. Biben and S. Merabia, Nanoscale, 2016, 8, 14870–14876 RSC.
- A. Dagallier, E. Boulais, C. Boutopoulos, R. Lachaine and M. Meunier, Nanoscale, 2017, 9, 3023–3032 RSC.
- V. Kotaidis, C. Dahmen, G. von Plessen, F. Springer and A. Plech, J. Chem. Phys., 2006, 124, 184702 CrossRef CAS PubMed.
- Y. S. Touloukian, R. K. Kirby, R. E. Taylor and P. D. Desai, Thermal expansion – Metallic elements and alloys, IFI Plenum, New York, Thermodynamical properties of matter edition, 1975, vol. 12 Search PubMed.
- P. Buffat and J.-P. Borel, Phys. Rev. A: At., Mol., Opt. Phys., 1976, 13, 2287–2298 CrossRef CAS.
- A. Plech, B. Krause, T. Baumbach, M. Zakharova, S. Eon and H. Bracht, Nanomaterials, 2019, 9, 501 CrossRef PubMed.
- A. Plech, S. Ibrahimkutty, S. Reich and G. Newby, Nanoscale, 2017, 9, 17284–17292 RSC.
- M. Magnozzi, R. Proietti Zaccaria, D. Catone, P. O'Keeffe, A. Paladini, F. Toschi, A. Alabastri, M. Canepa and F. Bisio, J. Phys. Chem. C, 2019, 123, 16943–16950 CrossRef CAS.
- X. Zhang, C. Huang, M. Wang, P. Huang, X. He and Z. Wei, Sci. Rep., 2018, 8, 10499 CrossRef PubMed.
- T. Katayama, K. Setoura, D. Werner, H. Miyasaka and S. Hashimoto, Langmuir, 2014, 30, 9504–9513 CrossRef CAS PubMed.
- E. Boulais, R. Lachaine and M. Meunier, J. Phys. Chem. C, 2013, 117, 9386–9396 CrossRef CAS.
- E. Lukianova-Hleb, Y. Hu, L. Latterini, L. Tarpani, S. Lee, R. A. Drezek, J. H. Hafner and D. O. Lapotko, ACS Nano, 2010, 4, 2109–2123 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 Search PubMed.
- G. V. Hartland, Phys. Chem. Chem. Phys., 2004, 6, 5263–5274 RSC.
- M. S. Plesset and A. Prosperetti, Annu. Rev. Fluid Mech., 1977, 9, 145–185 CrossRef CAS.
- A. Plech, V. Kotaidis, K. Istomin and M. Wulff, J. Synchrotron Radiat., 2007, 90, 022905 Search PubMed.
- F. Giammanco, E. Giorgetti, P. Marsili and A. Giusti, J. Phys. Chem. C, 2010, 114, 3354–3363 CrossRef CAS.
- U. Näher, S. Bjørnholm, S. Frauendorf, F. Garcias and C. Guet, Phys. Rep., 1997, 285, 245–320 CrossRef.
- B. R. Pauw, J. S. Pedersen, S. Tardif, M. Takata and B. B. Iversen, J. Appl. Crystallogr., 2013, 46, 365–371 CrossRef CAS PubMed.
- A. Letzel, B. Gökce, A. Menzel, A. Plech and S. Barcikowski, Appl. Surf. Sci., 2018, 435, 743–751 CrossRef CAS.
- S. Inasawa, M. Sugiyama and Y. Yamaguchi, J. Phys. Chem. B, 2005, 109, 9404–9410 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05202j |
| This journal is © the Owner Societies 2020 |