
Studies on the internal medium-range ordering and high pressure dynamics in modified ibuprofens†
Aldona
Minecka
 *a,
Ewa
Kamińska
*a,
Karolina
Jurkiewicz
bc,
Dawid
Heczko
a,
Barbara
Hachuła
d,
Wojciech
Pisarski
d,
Kamil
Kamiński
bc and
Marian
Paluch
bc
*a,
Ewa
Kamińska
*a,
Karolina
Jurkiewicz
bc,
Dawid
Heczko
a,
Barbara
Hachuła
d,
Wojciech
Pisarski
d,
Kamil
Kamiński
bc and
Marian
Paluch
bc
aDepartment of Pharmacognosy and Phytochemistry, Medical University of Silesia in Katowice, Faculty of Pharmaceutical Sciences in Sosnowiec, ul. Jagiellonska 4, 41-200 Sosnowiec, Poland. E-mail: aldona.minecka@med.sum.edu.pl; ekaminska@sum.edu.pl
bInstitute of Physics, University of Silesia, 75 Pulku Piechoty 1A, 41-500 Chorzow, Poland
cSilesian Center for Education and Interdisciplinary Research, University of Silesia, 75 Pulku Piechoty 1A, 41-500 Chorzow, Poland
dInstitute of Chemistry, University of Silesia, Szkolna 9, 40-006 Katowice, Poland
First published on 21st November 2019
Abstract
Broadband dielectric spectroscopy (BDS), combined with the X-ray diffraction (XRD) and Fourier transform infrared (FTIR) techniques, was used to study the dynamics of the primary (α) relaxation process and slow mode (SM), as well as structural properties and intermolecular interactions, in the methyl-, isopropyl-, hexyl-, and benzyl derivative of a well-known pharmaceutical, ibuprofen (IBU). Unexpectedly, the XRD and FTIR methods revealed the formation of medium-range ordering together with some molecular organization, which probably leads to the creation of small aggregates at the scale of several microns at lower temperatures. Moreover, high pressure dielectric experiments revealed that the SM (observed in the ambient pressure data) is not detected in the loss spectra of compressed IBU esters, which is consistent with the results reported previously for propylene carbonate and dioxolane derivatives. This finding can be interpreted as connected to either the comparable time scale of the structural dynamics and slow mode or suppression of the motions responsible for the latter process at elevated pressure. Additionally, it was found that the pressure coefficient of the glass transition temperature (dTg/dp) and activation volume (ΔV) change with molecular weight (Mw) in a non-monotonic way. It might be related to various chemical structures, conformations, and intermolecular interactions, as well as different architecture of supramolecular aggregates in the investigated compounds.
Introduction
During the last few decades, broadband dielectric spectroscopy (BDS) has become one of the most powerful tools to investigate intermolecular dynamics in various kinds of materials, including simple van der Waals, associating and ionic liquids, polymers, ceramics, etc., in a wide range of frequencies under different thermodynamic conditions or under nanometric spatial confinement. This technique allows one to follow the dynamics of supramolecular aggregates/associates formed via van der Waals interactions or H-bonds (HBs),1–13 chain diffusion in special type-A macromolecules,14,15 flip-flop rotations and tumbling reorientations in liquids crystals,16,17 and cooperative motions related to viscous flow as well as very local librations of dipole moments upon single measurements.18,19 Over the years, special attention has been paid to both understanding the relationship between relaxation processes observed in dielectric spectra of various materials,18,20–22 and explaining the molecular origin of the modes that are either slower or faster than the structural (primary, α) process (which governs the liquid to glass transition). In this context, one can mention comprehensive and systematic investigations on the molecular origin of Debye process, a special type of dielectric response, which is characterized by a simple exponential decay of the relaxation function (the slope of the low and high frequency side of the α-relaxation peak is equal to 1, and the Full Width at Half Maximum, FWHM, = 1.144),19 as well as a single relaxation time. Importantly, this process was observed in water,23 monohydroxy alcohols,1–9 secondary amides,10,11 ionic liquids,24 and in some active pharmaceutical ingredients (APIs), like etoricoxib25 or acetaminophen.26 In fact, over the years, monohydroxy alcohols became a model system to study the molecular origin and dynamics of Debye relaxation. Intensive investigations have revealed that this process is strictly related to the mobility of chain or ring-like associates that are formed between molecules connected via HBs. According to the transient chain model by Gainaru,27 one can postulate that molecules attaching to and detaching from the chain-like structures are responsible for that. The variation in the concentration of these supramolecular structures or counterbalance between them has a strong impact on the position as well as the dielectric strength of the Debye process.8,9,28,29 For example, the amplitude of this characteristic mode increases significantly with an increasing content of chain-like associates. It should also be added that during recent years, it has been considered that the Debye process is a characteristic feature of the dielectric response in monohydroxy alcohols, and a similar type of mobility cannot be detected by other experimental techniques dedicated to studying the molecular dynamics. However, current studies have clearly shown that this additional process may also be examined using the mechanical spectroscopy, nuclear magnetic resonance (NMR) and dynamic light scattering, DLS, (including photon correlation spectroscopy, PCS) methods.7,28,30–35 Thus, completely new opportunities to study the real molecular nature of Debye relaxation have appeared.Interestingly, recently, it has been shown that the slow mode (SM – a process that is observed at a lower frequency with respect to the α-relaxation), not always satisfying the criteria of Debye relaxation, can be detected in typical van der Waals liquids as well. Typical examples of those are derivatives of propylene carbonate (PC), and dioxolanes having alkyl chains of different lengths attached to the ring structure.12,13 Moreover, a similar situation has been found in modified ibuprofens.36 In this context, it is worth stressing that in the native ibuprofen (IBU), the presence of the low amplitude Debye process was linked with the dimers’ reorientation. However, further theoretical studies by Affouard and Correia,37 followed by experimental investigations by Adrjanowicz et al.,38 revealed that this process originates from the torsion rotations of polar carboxylic (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–H) groups. Therefore, it is more likely connected to the conformational variation between the synperiplanar (cis) and antiperiplanar (trans) position in the side chain, while the HBs only affect the dynamics and the time scale of this motion. Note that recently we have also examined the impact of various substituents (R) characterized by different molar mass on the activation barrier and time scale of the rotation around the OC–O–R moiety.36
C–O–H) groups. Therefore, it is more likely connected to the conformational variation between the synperiplanar (cis) and antiperiplanar (trans) position in the side chain, while the HBs only affect the dynamics and the time scale of this motion. Note that recently we have also examined the impact of various substituents (R) characterized by different molar mass on the activation barrier and time scale of the rotation around the OC–O–R moiety.36
In this paper, we have continued the investigations on the molecular dynamics of ibuprofen and its ester derivatives at high pressure with special attention put on the behavior of the structural and SM processes under these conditions. In this context, it is worth stressing that in the literature one can find a lot of articles reporting the investigation of the dynamics of the Debye process in monohydroxy alcohols at high compression, while there are only two studies focused on the examination of the impact of pressure on the dynamics of the SM in typical van der Waals liquids, i.e., PC and dioxolane derivatives.12,13 Herein, we show that analogously to the results reported for the two mentioned van der Waals systems, the SM in IBU esters is not observed at high pressure anymore. What is more, based on the collected diffractograms and infrared spectra, we demonstrate that some medium-range intermolecular arrangement occurs in these compounds. Additionally, we reveal a non-monotonic variation in the pressure coefficient of the glass transition temperature (dTg/dp), as well as the activation volume (ΔV), with respect to the molecular weight (Mw) that can be assigned to differences in the chemical structures, molecular conformations, and intermolecular interactions, as well as the possible formation of supramolecular aggregates in the examined systems.
Experimental section
Materials
Ibuprofen, IBU [(2-[4-(2-methylpropyl)phenyl]propanoic acid, which is a racemic mixture of (S)- and (R)-isomers)], a white crystalline powder, having purity >98% was supplied by Hubei Biocause Pharmaceutical Co. Ltd. and used as received. Methyl, isopropyl, hexyl and benzyl esters of ibuprofen: Met-IBU, methyl-2-[4-(2-methylpropyl)phenyl]propanoate; Iso-IBU, propan-2-yl-2-[4-(2-methylpropyl)phenyl]propanoate; Hex-IBU, hexyl-2-[4-(2-methylpropyl)phenyl]propanoate; and Ben-IBU, benzyl-2-[4-(2-methylpropyl)phenyl]propanoate, respectively, oily liquids of greater than 98% purity, have been synthesized for the purpose of this paper. The synthesis procedures, as well as NMR data of the obtained products, are presented in the ESI in ref. 36 and 38. The chemical formulas and molecular weights of the examined compounds are given in Table 2, whereas their chemical structures are presented in Scheme 1. | ||
| Scheme 1 The chemical structures of ibuprofen and its ester derivatives (image taken from ref. 36). | ||
Methods
Broadband dielectric spectroscopy (BDS)
Isobaric measurements of the dielectric permittivity ε*(ω) = ε′(ω) − iε′′(ω) were carried out using a Novo-Control Alpha dielectric spectrometer (Novocontrol Technologies GmbH & Co. KG, Hundsangen, Germany) over a frequency range from 1 × 10−2 to 3 × 106 Hz. The samples were placed between two stainless-steel electrodes (diameter: 20 mm, gap: 0.14 mm) and mounted on a cryostat. During the measurement, each sample was maintained under a dry nitrogen gas flow. The temperature was controlled by a Quatro System using a nitrogen gas cryostat, with stability better than 0.1 K.For dielectric measurements at elevated pressure, we used a high-pressure chamber with a special homemade flat parallel capacitor. Thin Teflon spacers were applied to maintain a fixed distance between the plates. The sample capacitor was sealed and covered carefully with Teflon tape to separate it from the silicon liquid. The pressure was measured using a Honeywell tensometric meter with a resolution of 1 MPa. The temperature was adjusted with a precision of 0.1 K using a refrigerated and heating Huber circulator. The complex dielectric permittivity was measured within a frequency range from 10−2 up to 106 Hz.
X-ray diffraction (XRD)
X-ray diffraction measurements were performed on a Rigaku-Denki D/MAX RAPID II-R diffractometer (Rigaku Corporation, Tokyo, Japan) equipped with a rotating Ag anode, an incident beam (002) graphite monochromator, and an image plate in the Debye–Scherrer geometry. The pixel size was 100 μm × 100 μm. The beam width at the sample was 0.3 mm. Measurements were carried out for the sample-filled and empty capillaries, and the background intensity was then subtracted. The collected two-dimensional diffraction patterns were converted into one-dimensional intensity data versus the scattering vector, Q.Fourier transform infrared spectroscopy (FTIR)
FTIR measurements of IBU derivatives were carried out using a Nicolet iS50 FTIR spectrometer (Thermo Scientific) equipped with a DLaTGS-KBr detector. The absorption spectra were collected in the range 4000–400 cm−1, with a spectral resolution of 4 cm−1. A total of 32 scans were averaged for each sample. The spectra were recorded at room temperature (T = 293 K) and at the glass transition temperature: Tg = 184 K (Met-IBU), 180 K (Iso-IBU), 172 K (Hex-IBU) and 200 K (Ben-IBU), using a liquid nitrogen-cooled stage (Linkam THMS600 stage; Linkam Scientific Instruments Ltd, Surrey, UK). The cooling rate of the samples was equal to 10 K min−1, while the temperature stabilization accuracy was 0.1 K. A small drop of the compound was squeezed between a pair of CaF2 windows, and then a thick film of the sample was placed inside the cooling stage.Density measurements
The density (d) of the studied substances was measured with an Anton Paar DMA 5000M densitometer with an uncertainty not worse than 0.0001 g cm−3 at T = 293 K.Results and discussion
As mentioned in the introduction, the presence of an additional relaxation process in IBU derivatives, slower than the structural one, is most likely related to synperiplanar (cis) to antiperiplanar (trans) torsion rotations of the carbonyl moiety.37,38 However, in several previous studies, it was demonstrated that the slow mode (SM) could also be observed in the loss spectra of a few low molecular weight substances that form aggregates and/or are characterized by medium-range ordering.13,39–41 This phenomenon was well manifested in the X-ray diffractogram, where at low scattering vector, on the left side of the main diffraction peak, a so-called “pre-peak” has been detected. To verify whether any signs of medium-range order can also be found in the case of IBU derivatives (pure van der Waals systems), we have carried out similar diffraction measurements at T = 293 K.Fig. 1a presents the diffractograms collected for the crystalline and supercooled IBU, as well as supercooled Met-IBU, Iso-IBU, Hex-IBU, and Ben-IBU. As can be seen, the diffraction patterns of the supercooled API and its esters show two common significant features, a pronounced principal broad maximum and a pre-peak at smaller scattering vectors. The main maximum of the diffraction patterns arises from the short-range order between neighboring molecules. From the main-peak position around Q = 1.25 Å−1, an average spatial repeat distance between molecules of approximately 5 Å can be estimated. The position of the pre-peak at around Q = 0.45 Å−1 involves a spatial distance d = 2π/Q of approximately 14 Å, much longer than that, which may be attributed to the averaged nearest intermolecular distance. It indicates the presence of intermediate-range order. To get a better insight into the nature of this potential order, a comparison with the diffraction pattern of the crystalline API reference is useful (Fig. 1a). The diffraction pattern of IBUcryst is characterized by the first (100) Bragg peak, whose position coincides very well with that of the pre-peak. The crystal structure of IBU is monoclinic P21/c with four molecules per unit cell.42 The performed Rietveld refinement of crystalline ibuprofen at 293 K (details in the ESI†) provided the values of the lattice parameters a = 14.5779(9) Å, b = 7.8676(0) Å, c = 10.6836(3) Å, and β = 99.7°. These four molecules in the unit cell form two dimers, each comprising a chiral molecule H-bonded to its mirror image across a center of inversion. The H-bonds, as well as the methyl groups of the IBU molecules, form planes, as shown in Fig. 1b. The pre-peak related to the repeating distance 14 Å matches the length 14.6 Å, which is roughly equal to that separating such planes along the a-axis. Therefore, the pre-peak in supercooled IBU can correspond to the clusters of molecules locally associated with HBs.
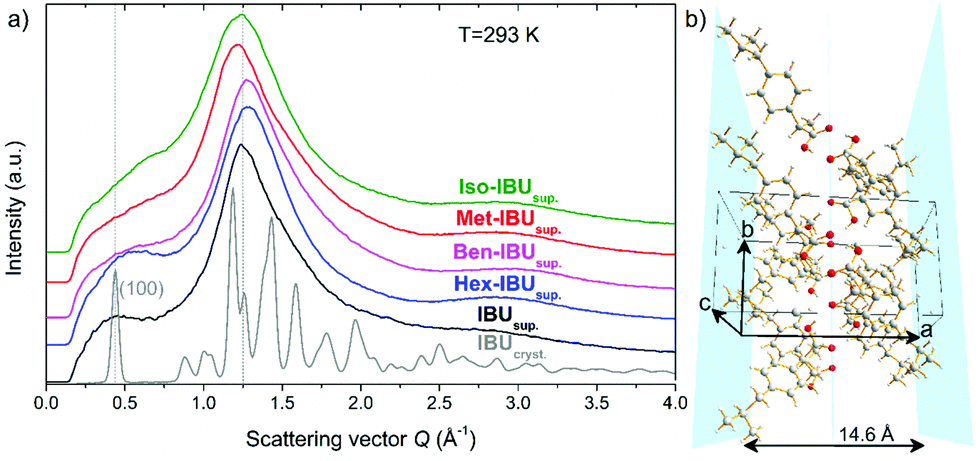 | ||
| Fig. 1 X-ray diffraction patterns collected at T = 293 K for the crystalline and supercooled IBU (IBUcryst. and IBUsup., respectively), as well as supercooled methyl, isopropyl, hexyl, and benzyl derivatives of this pharmaceutical: Met-IBUsup., Iso-IBUsup., Hex-IBUsup., and Ben-IBUsup. (a). A fragment of the crystalline structure of IBU, characterized by planes of H-bonds and methyl groups (marked in blue), unit cell axes are marked in black (b). | ||
The studied API derivatives show very similar diffraction peaks to supercooled IBU, slightly shifted in the positions. For Met-IBU, Iso-IBU, and Ben-IBU, the pre-peak is less pronounced than for IBU and Hex-IBU. It is worth noting that a similar pre-peak feature was observed in the case of primary alcohols40,41 and was interpreted as related to the creation of H-bonded supramolecular clusters. Based on this knowledge and our direct experimental observations, one can suppose that the formation of small supramolecular clusters occurs in the examined herein IBU-based systems. The pre-peak may be a signature of pair correlations between these associated molecular assemblies. Since further insight into the potential nature of the molecular organization of IBU esters can be gained from FTIR measurements, we have also performed such experiments in a wider temperature range (Troom–Tg).
The measured spectra, presented in Fig. 2 and Fig. S2 (ESI†), show the absorption bands characteristic for IBU derivatives. As illustrated, the position of most bands and their intensities change slightly with decreasing temperature. In the range of 3600–2600 cm−1, at T = 293 K, strong peaks attributed to CH3, CH2, and CH stretching vibrations are visible. At lower T, the increase in the intensity of a band between 3400 and 3100 cm−1 is noticeable; the integrated absorbance in this region increases continuously during cooling. Such a scenario may indicate the growing role of interactions of the HB type. However, simultaneous analysis of the temperature dependence of the position of CO (potential H-bond acceptor) and C–H (potential H-bond donor) vibrations along with a constant shape of these bands ruled out such a possibility. In this context, it is worth emphasizing that small red shifts of the CO (ca. 4 cm−1) stretching vibration band positions upon lowering the temperature were observed. Interestingly, a similar spectral effect has been reported for phenyl salicylate (salol).43 In this particular case, the authors considered the conformational variation as well as the delocalization of electrons in the ester moiety and varying stiffness of the CO unit to be responsible for that. Therefore, the same explanation can be used for the examined IBU esters, especially when we take into account the data reported in ref. 44 and the change in the spectral features of bands occurring in the frequency range 1400–1200 cm−1 – Fig. S2 (ESI†) – (assigned to the bending vibrations of the aliphatic (Met, Iso, and Hex) or aromatic (Ben) units of IBU derivatives), indicating conformational reorganization of IBU derivatives upon cooling.45 To explain the peculiar increase in the intensity of the band at higher wavenumbers (>3000 cm−1), further optical studies at lower temperatures have been carried out. These measurements revealed clear turbidity of the sample occurring during the temperature drop. This effect was the most prominent in the case of Hex-IBU. One can suppose that the decrease in IR radiation transmission, observed in the spectral range of 3400–3100 cm−1 (which corresponds to a wavelength of ca. 3 μm), is most likely related to the increase in the scattering of photons on objects having a size comparable to the wavelength. As a consequence, the turbidity of the sample at lower T is noted. Therefore, infrared investigations supported by the optical studies performed on IBU derivatives indicated some structure reorganization, which probably leads to the formation of multiple aggregates on the scale of several microns by these materials.
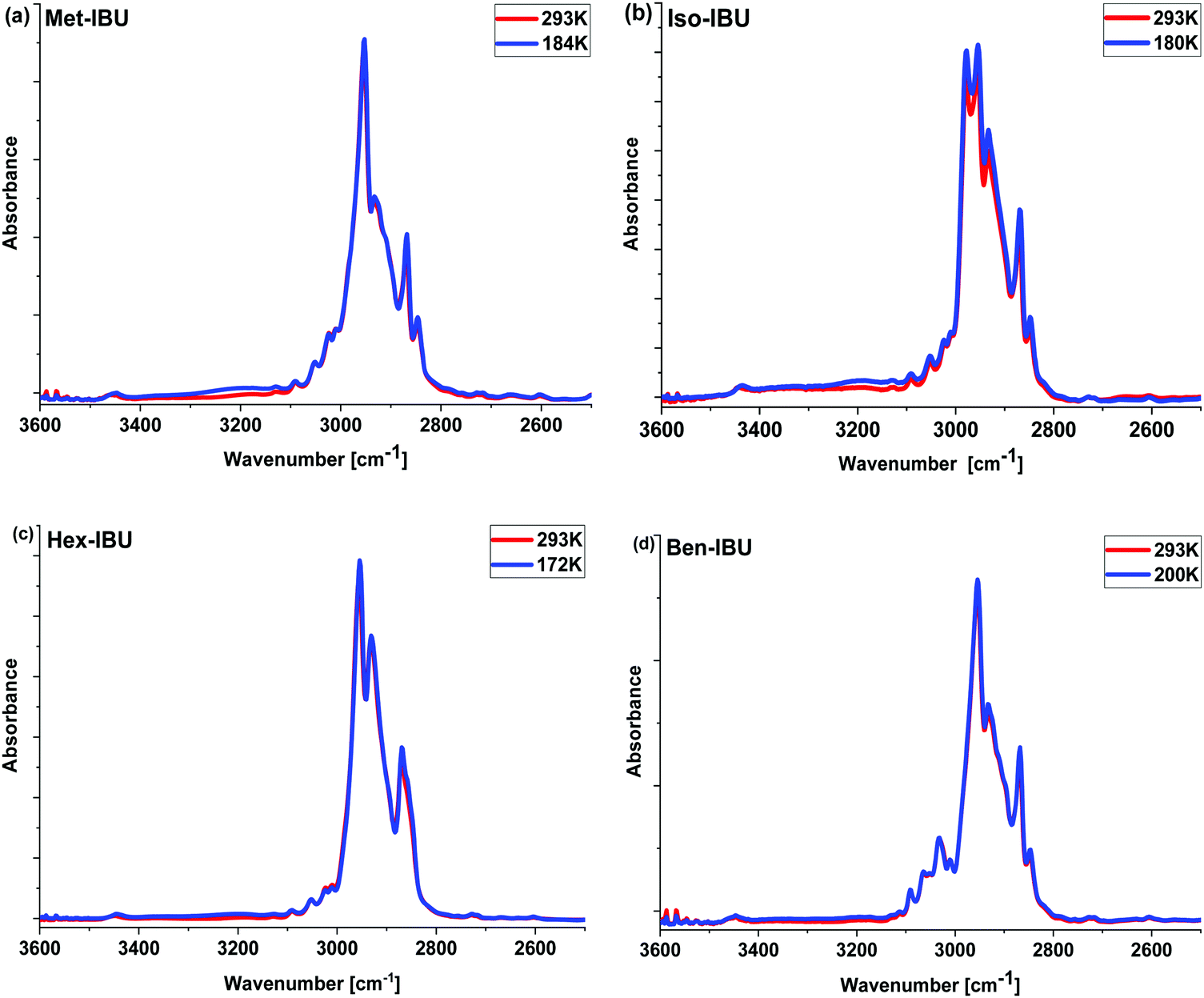 | ||
| Fig. 2 Infrared spectra of the investigated esters measured at T = 293 K and the glass transition temperature (Tg) of each sample in the 3600–2500 cm−1 frequency range. | ||
The unusual temperature behavior observed in the FTIR spectra of the examined compounds motivated us to perform additional X-ray diffraction (XRD) measurements at low temperature (T = 223 K). Results obtained for one of the IBU derivatives (Hex-IBU) are presented in Fig. 3. As can be seen, the main peak position clearly shifts to greater values due to cooling, indicating smaller distances between molecules and denser packing due to less thermal motion. There is also an increase in the intensity of the main peak at lower T caused by attenuated thermal movement of molecules like the Debye–Waller factor predicts. Surprisingly, the position of the pre-peak does not change significantly with decreasing temperature. Such behavior may originate from different geometrical properties of supramolecular clusters, and it leads to the assumption that the molecules exhibit various compressibility in different directions and medium-range structuring beyond the usual short-range order.
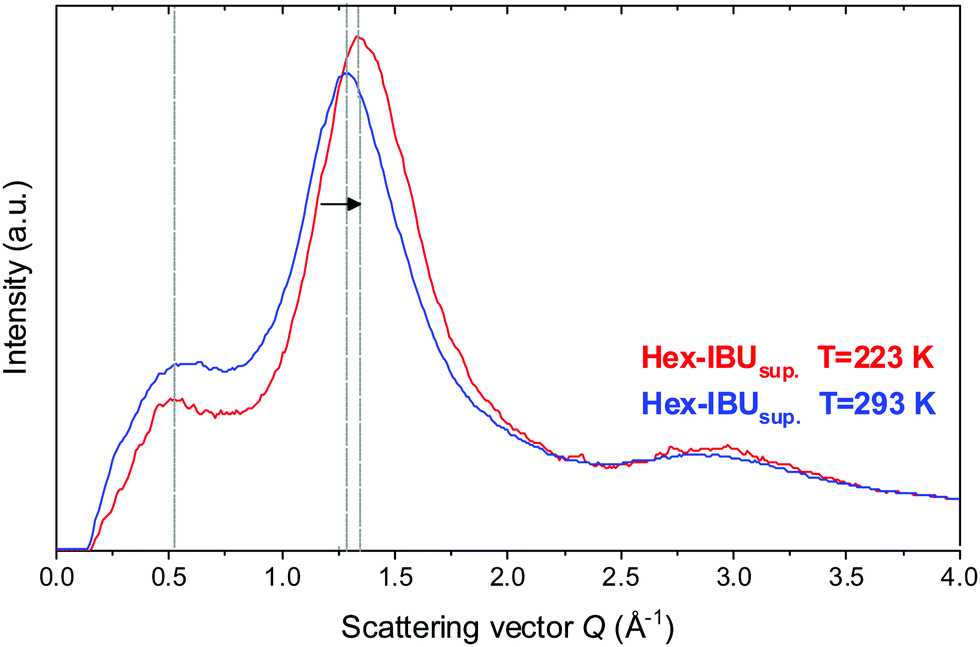 | ||
| Fig. 3 Comparison of the X-ray diffraction patterns for the supercooled hexyl derivative of ibuprofen (Hex-IBUsup) at T = 293 K and T = 223 K. | ||
Thus, both methods used hitherto (XRD and FTIR) revealed the occurrence of medium-range intermolecular organization in the considered IBU derivatives that seems to be enhanced due to the formation of local structuring on the micron-scale at low temperatures.
Considering the results described above, one could suppose that there might be a possible connection between the pre-peak and slow mode in IBU/IBU esters. However, this issue is very complex and is a matter of intensive debate. In this context, one can recall the monohydroxy alcohols, where both the pre-peak and SM were detected, while in the case of metatoluidine46 and Ben-IBU a medium range order was found, but there was no additional slower relaxation process in the loss spectra. In this place, it is also worth adding that the temperature evolution of the relaxation times of the Debye process in native IBU followed VFT-like behavior, while those of the SM in IBU esters were rather of Arrhenius type.36 Although, it must be stressed that since the SM has been covered by the α-relaxation relatively fast, it was observed only in a very narrow range of temperatures and frequencies. Consequently, we were not able to estimate the temperature dependences of τSM around Tg. Therefore, at the moment, it is difficult to settle unquestionably what is the real molecular origin of the SM in IBU and its derivatives and whether the same molecular motions underlie this process in the investigated compounds.
In the next step, we have carried out additional extensive high pressure dielectric measurements on the considered IBU esters. One of the aims of these investigations was to find out whether similarly to the case of dioxolane and PC derivatives the slow relaxation process will be suppressed at high compression.12,13 In Fig. 4, representative dielectric loss spectra of two selected esters: Met-IBU (panels (a) and (b)) and Ben-IBU (panels (c) and (d)), measured at the indicated temperatures (T > Tg) and pressures (p > pg; where pg is a glass transition pressure) are presented. Analogous spectra registered for Iso-IBU and Hex-IBU are shown in Fig. S3 (ESI†), while for native IBU in Fig. 1 in ref. 47. In the supercooled liquid state of all examined esters, a well-pronounced structural (primary, α)-process originating from the cooperative motions of the molecules is observed. As illustrated, the maxima of α-loss peaks are shifting toward lower frequencies with increasing pressure (panels (b) and (d)), and decreasing temperature (panels (a) and (c)). It should be stressed that due to the strong contribution of dc conductivity to the measured data, we decided to subtract it from the loss spectra.
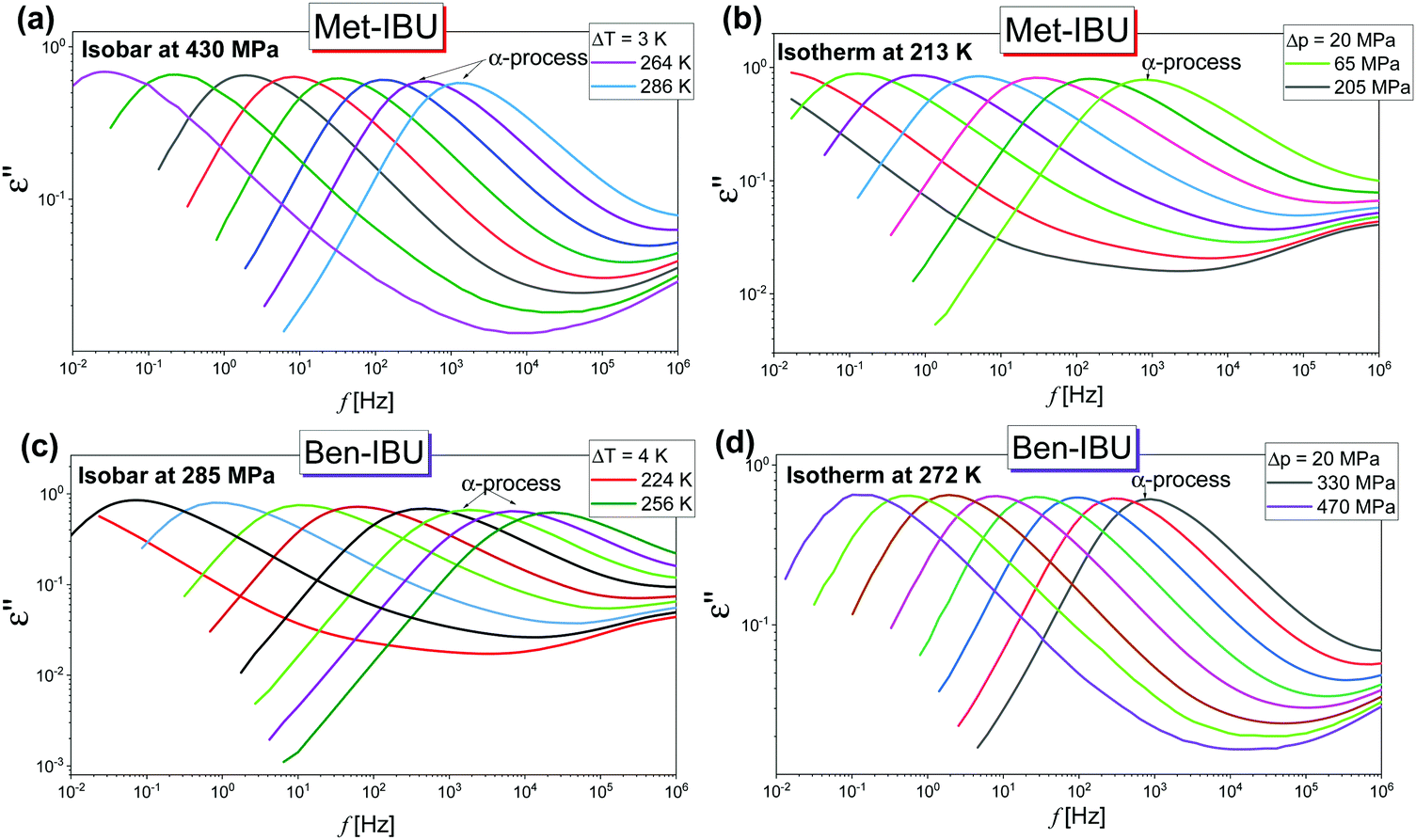 | ||
| Fig. 4 Representative dielectric loss spectra measured for two IBU derivatives: Met-IBU (panels (a) and (b)) and Ben-IBU (panels (c) and (d)) under the indicated T and p conditions. | ||
As a further part of our investigations, we have compared dielectric spectra collected at ambient and elevated pressure close to the glass transition points (the maxima of α-loss peaks near 1 Hz). Fig. 5 shows the normalized dielectric data recorded for each ester. As can be observed, the shape of the structural relaxation remains constant independent of the T and p conditions. It indicates that the Temperature–Pressure-Superposition (TPS) rule is satisfied. Such behavior is typical for van der Waals liquids.48,49 However, it should be noted that Adrjanowicz et al.47 and Bras et al.50 reported that the mentioned rule is also fulfilled for the native IBU. It was explained by the tendency of this API to form dimeric structures, whose population is weakly affected by density fluctuations. What is more, similar observations were also found for other low-molecular-weight substances creating small H-bonded supramolecular aggregates (dimeric, trimeric, etc.).51–55 It is quite unusual with regard to the general observation that for most associated liquids, due to the variation in the strength and population of HBs under different thermodynamic conditions, the TPS rule breaks down.56–58
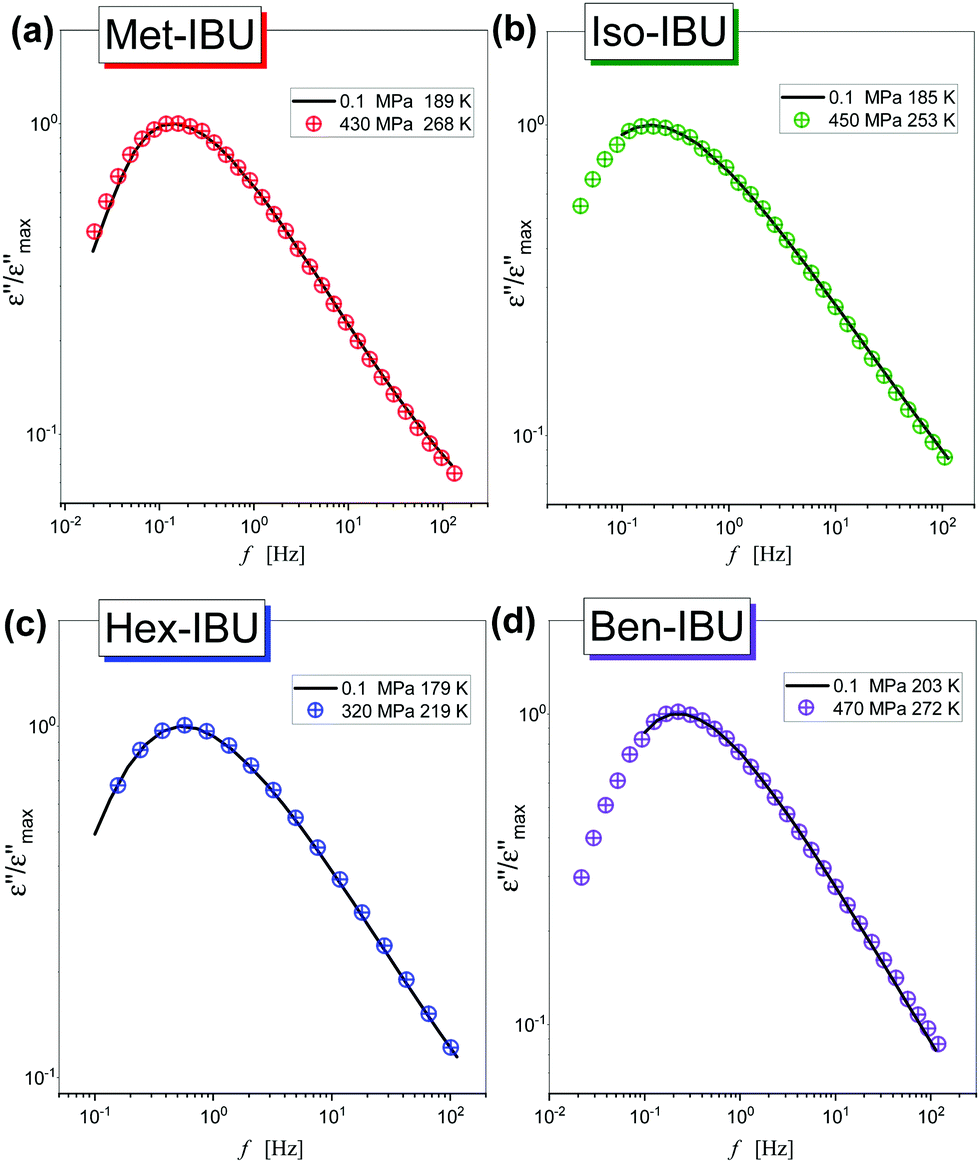 | ||
| Fig. 5 A comparison of the shape of structural relaxation peaks measured under various thermodynamic conditions, near Tg, for Met-IBU (a), Iso-IBU (b), Hex-IBU (c), and Ben-IBU (d). The spectra were normalized with respect to the maximum of dielectric loss (εmax′′). | ||
Further, we have focused on the behavior of an additional slower relaxation process (SM) at higher compression. For this purpose, the normalized dielectric loss spectra registered for Met-, Iso-, and Hex-IBU under various thermodynamic conditions, for which the maximum of the structural process was around fmax ≈ 1 MHz, were compared. Herein, it should be mentioned that our ambient pressure data36 indicated that the SM in the examined compounds is observed only when the structural relaxation times are very short (τα < 10−4 s). With lowering temperature, the slow and α modes tend to merge. Consequently, in the vicinity of Tg, only the structural process is visible at p = 0.1 MPa. Additionally, these studies revealed that the position (and shape) of the SM depends on the chemical structure of the investigated compound – it shifts toward lower frequencies with increasing size of the substituent attached to the carboxyl group. However, this relaxation is not present in the benzyl derivative of IBU due to the low mobility of this material. At first sight (Fig. 6), it can be noticed that at high pressures the SM disappears in all the considered IBU esters. Interestingly, the obtained results are consistent with the data published for a series of PC and dioxolane derivatives,12,13 where the additional slow relaxation also vanished at high compression. To better illustrate the discussed phenomenon, we fitted the loss spectra collected at elevated pressure (Fig. 6) by one Havriliak–Negami (HN) function with an additional term describing the conductivity contribution:
 | (1) |
![[small omega, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0da.gif) is an angular frequency ( = 2πf), while α and γ are the shape parameters representing the breath and asymmetry of given relaxation peaks. In this context, one can add that for α and γ = 1, we have a Debye response function. As a result, satisfactory fits (solid red lines) that perfectly described the experimental data were achieved only in the case of Iso-IBU and Hex-IBU (Fig. 6b and c). For the methyl derivative of IBU (Fig. 6a), the generated fit was of much poorer quality. Thus, just like in the case of ambient pressure data, two HN functions had to be used to describe the measured spectra correctly. It means that in Met-IBU there is a very small contribution from the slow mode to the dielectric spectra, but due to similar time scales of both processes (α-relaxation and SM), it is not possible to separate them. One can add that similar effects have been noted for monohydroxy alcohols, where due to weak separation of the structural and slow modes, only a single process was observed at elevated pressure.2,3,8,9
is an angular frequency ( = 2πf), while α and γ are the shape parameters representing the breath and asymmetry of given relaxation peaks. In this context, one can add that for α and γ = 1, we have a Debye response function. As a result, satisfactory fits (solid red lines) that perfectly described the experimental data were achieved only in the case of Iso-IBU and Hex-IBU (Fig. 6b and c). For the methyl derivative of IBU (Fig. 6a), the generated fit was of much poorer quality. Thus, just like in the case of ambient pressure data, two HN functions had to be used to describe the measured spectra correctly. It means that in Met-IBU there is a very small contribution from the slow mode to the dielectric spectra, but due to similar time scales of both processes (α-relaxation and SM), it is not possible to separate them. One can add that similar effects have been noted for monohydroxy alcohols, where due to weak separation of the structural and slow modes, only a single process was observed at elevated pressure.2,3,8,9
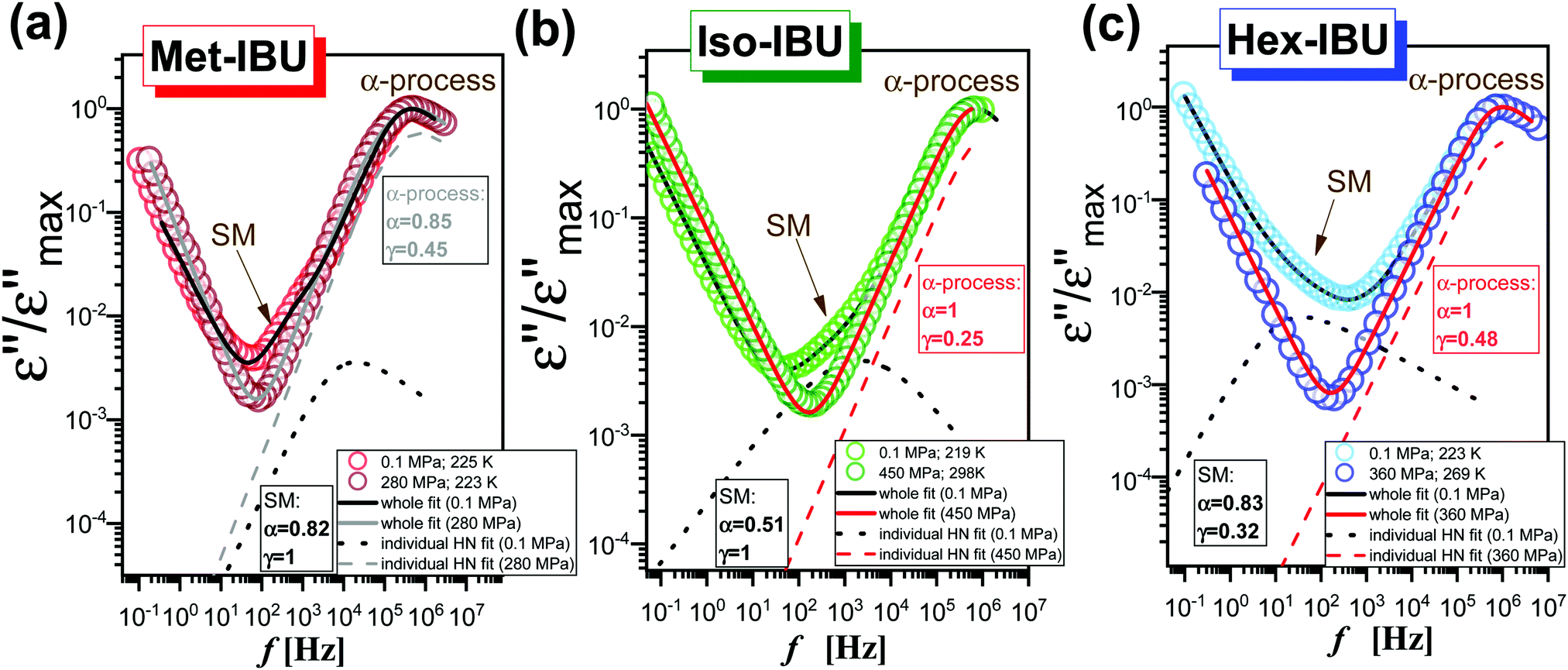 | ||
| Fig. 6 A comparison of dielectric loss spectra collected under different T and p conditions for the same τα (the maxima of α-loss peaks near 1 MHz) for Met-IBU (a), Iso-IBU (b) and Hex-IBU (c). The spectra were normalized with respect to the maximum of dielectric loss (εmax′′). The red and black (as well as grey) solid lines represent, respectively, the fits of one and two HN functions with an additional term describing the conductivity contribution, whereas the dashed and dotted lines are individual HN fits for, respectively, α-relaxation (high pressure spectra) and SM (ambient pressure spectra). | ||
To fully characterize the molecular dynamics of all the studied IBU esters, structural relaxation times, τα, obtained from the analysis of dielectric data using eqn (1) (recalculated from τHN according to the formula in ref. 19) have been plotted versus T and p. The smooth surfaces formed by these data within the ranges of the investigated pressures and temperatures were subsequently fitted to the modified Avramov equation:59
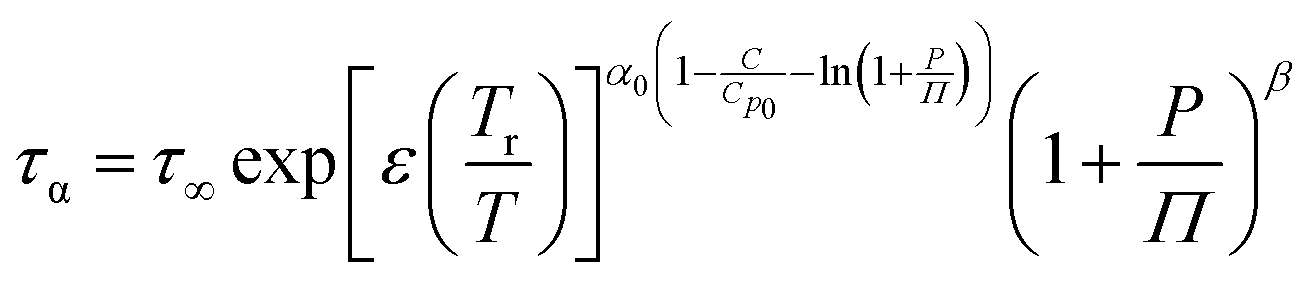 | (2) |
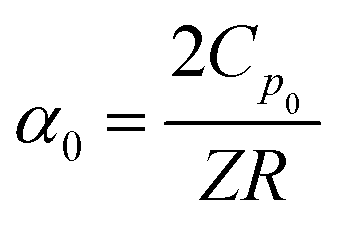 | (3) |
 | (4) |
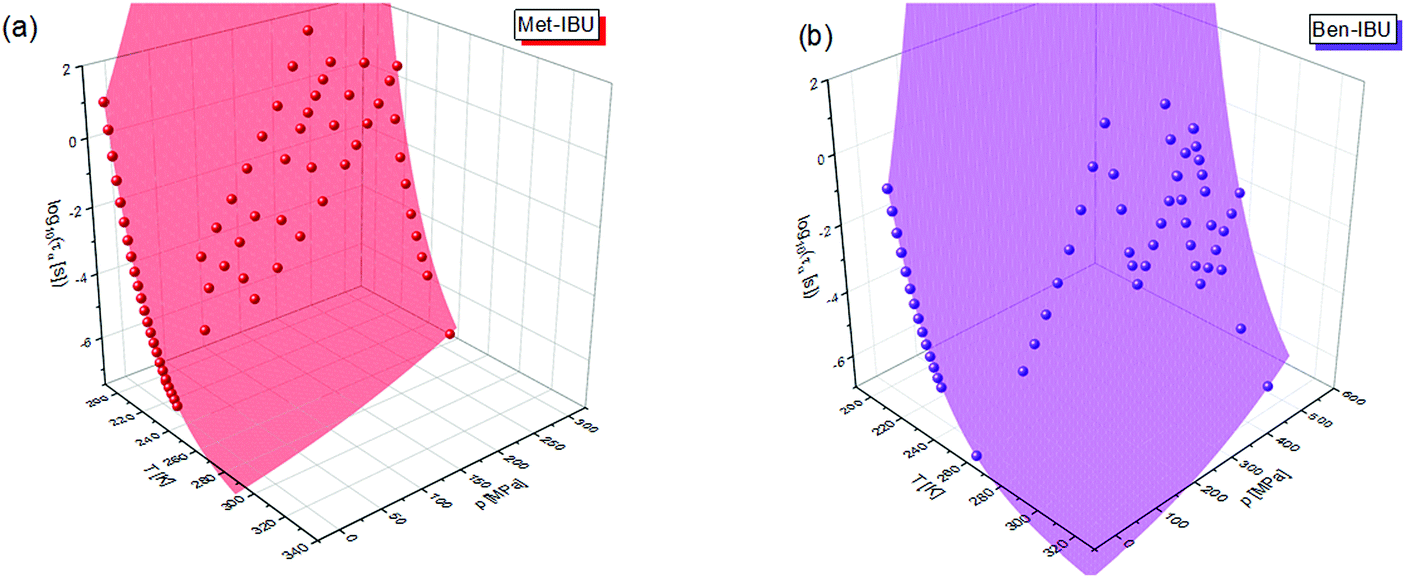 | ||
| Fig. 7 α-Relaxation times of two representative IBU esters: Met-IBU (a) and Ben-IBU (b) plotted versus temperature (T) and pressure (p). Red and violet areas represent surface fits to eqn (2). | ||
log10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) τ∞ τ∞ |
T r | α 0 | C/Cp0 | Π | β | |
|---|---|---|---|---|---|---|
| IBU | −9.90 | 224.7 | 6.71 | 0.041 | 271.79 | 1.77 |
| Met-IBU | −9.43 | 184.1 | 7.31 | 0.063 | 439.14 | 2.71 |
| Iso-IBU | −9.51 | 180.1 | 7.29 | 0.001 | 253.40 | 2.24 |
| Hex-IBU | −8.51 | 172.2 | 6.83 | 0.081 | 282.02 | 1.66 |
| Ben-IBU | −9.20 | 199.9 | 7.97 | 0.001 | 471.76 | 3.27 |
Afterward, we calculated the pressure coefficient of the glass transition temperature (dTg/dp) for the examined IBU derivatives, as well as the native API. For this purpose, Tg (defined herein as the temperature at which τα is equal to 100 s), determined under different pressure conditions, was plotted versus pg in panel (a) of Fig. 8. Subsequently, we applied the following expression, which was derived from eqn (2):60
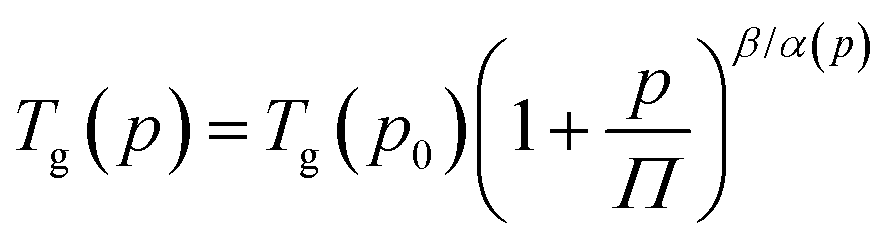 | (5) |
 | (6) |
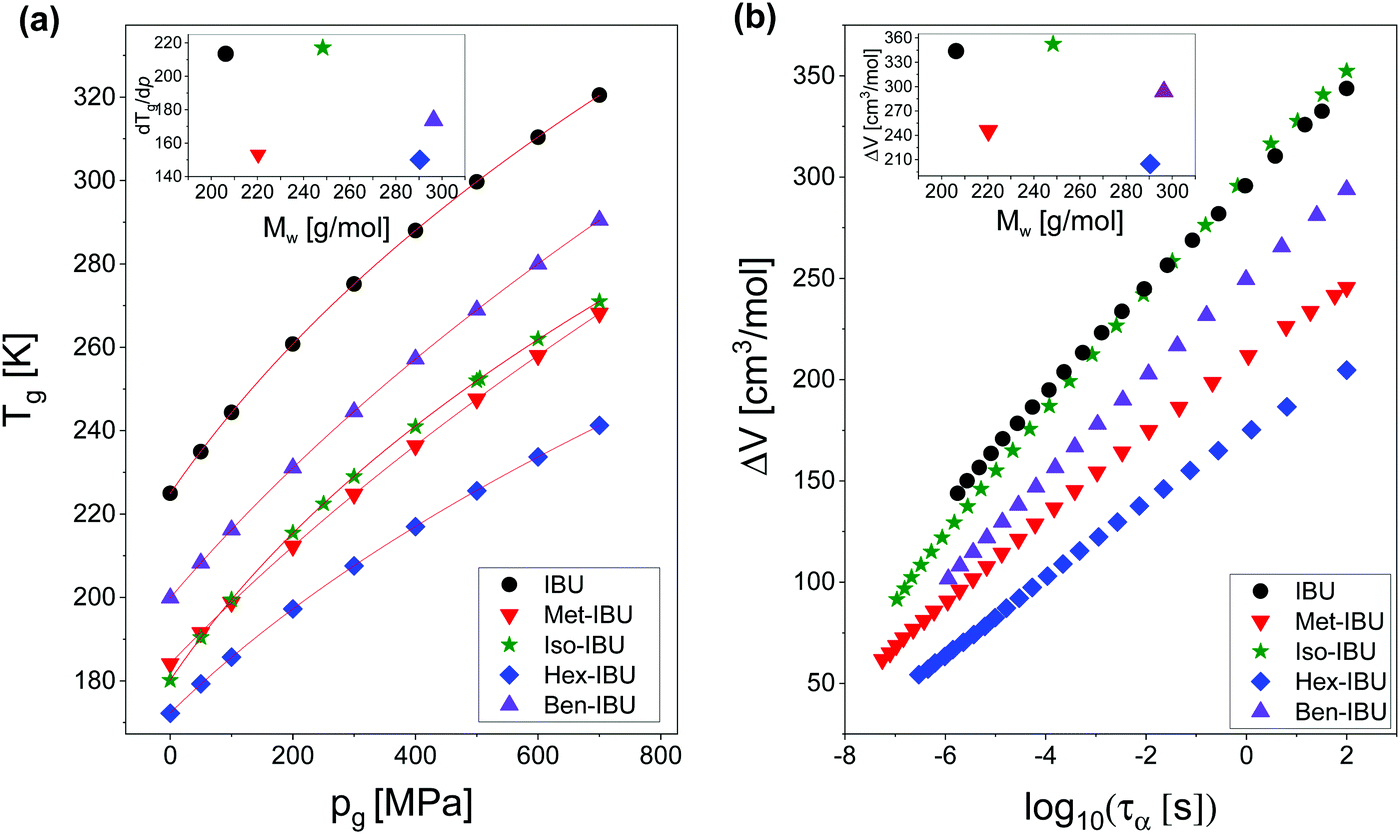 | ||
| Fig. 8 T g plotted versus pg for IBU and its derivatives (a). The solid lines are the best fits to eqn (5). Activation volume (ΔV) calculated for the studied esters as a function of the structural relaxation time, τα (b). The insets present dTg/dp and ΔV values plotted versus molecular weight (Mw) of the investigated substances. Data for native IBU were taken from ref. 47. | ||
The obtained values of dTg/dp are presented in Table 2. Additionally, they were plotted versus molecular weight in the inset to Fig. 8a. As illustrated, for all samples, the structural dynamics is sensitive to pressure. However, there is no clear connection between the dTg/dp values and Mw of the examined compounds. In this context, one can mention that in the case of H-bonded systems, the pressure coefficient of the glass transition temperature increases monotonically (e.g., polypropylene glycols, PGs48) or varies only slightly (e.g., polyalcohols61) with the molecular weight of the sample. After removing the HBs, one can rather expect the monotonic dependence of dTg/dp vs. Mw. However, such a scenario is not observed for the considered IBU esters. As can be seen in the inset to Fig. 8a, the sensitivity of the native IBU and its isopropyl derivative (Iso-IBU) to compression is greater than the other esters. For IBU, the high value of this parameter can be explained by the fact that this API forms dimers connected by strong H-bonds. In turn, it is difficult to interpret the obtained high value of dTg/dp in the case of Iso-IBU.
| Molecular formula | Molecular weight, Mw [g mol−1] | dTg/dp [K GPa−1] | Density, d [g cm−3] | Molar volume, Vm [cm3 mol−1] | Activation volume, ΔV [cm3 mol−1] | |
|---|---|---|---|---|---|---|
| IBU | C13H18O2 | 206.28 | 214.0 | 1.030 | 200.3 | 343.8 |
| Met-IBU | C14H20O2 | 220.31 | 156.0 | 0.970 | 227.1 | 245.5 |
| Iso-IBU | C16H24O2 | 248.36 | 217.0 | 0.935 | 265.6 | 352.3 |
| Hex-IBU | C19H30O2 | 290.44 | 148.4 | 0.930 | 312.3 | 204.7 |
| Ben-IBU | C20H24O2 | 296.40 | 173.9 | 1.015 | 292.0 | 293.8 |
As a final point of our studies, we estimated the activation volume, ΔV, for all the studied herein compounds. This parameter describes the sensitivity of the structural dynamics to pressure changes and in the light of transition state theory62 is defined as the difference between the volume occupied by a molecule in activated and non-activated states (Va − Vs).
ΔV can be easily calculated from the dependence logταversus p using the following expression:
 | (7) |
However, herein, the values of ΔV (defined according to eqn (7)) under various T and ambient pressure conditions were obtained directly from the surface fits of the Avramov approach. Next, they were plotted versus structural relaxation time in Fig. 8b (main panel). As illustrated, ΔV increases with τα, which is typical behavior for low- (H-bonded, van der Waals) and high-molecular weight compounds.55,63–65 It is due to the fact that with increasing size of cooperatively reorienting molecules upon cooling towards Tg, a larger space is required for such motions.
Since in many papers published so far, a strong interdependence between the activation volume (ΔV) and molecular weight (Mw), as well as molar volume (Vm), was found for some low- and high-molecular weight glass formers,54,63,66 we decided to test this relationship in the case of the examined IBU derivatives. In the inset to Fig. 8b, the dependence of ΔV (calculated at p = 0.1 MPa at Tg) versus molecular weight (Mw) is presented. As illustrated, the activation volume, like the pressure coefficient of the glass transition temperature, changes with Mw in a non-monotonic way. Interestingly, the obtained behavior is different from that reported in the literature for various types of materials: van der Waals (e.g., BMPC, and BMMPC)48 and H-bonded (e.g., series of polyalcohols,61 and PPGs and their derivatives67) systems, as well as polymers (e.g., polymethylphenylsiloxanes68), where the systematic increase of ΔV with increasing Mw was observed. To understand the unusual dependency between the activation volume and molecular weight in IBU esters, we decided to compare ΔV with the values of molar volume (Vm) calculated for each of the examined compounds using the following formula:  . Interestingly, as illustrated in Table 2, the values of ΔV and Vm are similar for Met-IBU and Ben-IBU. In turn, there is a significant discrepancy between both parameters for the native IBU as well as its hexyl and isopropyl derivatives (ΔV − Vm = 143.5, 107.6, and 86.7 cm3 mol−1, respectively). In the case of IBU, it can be easily explained by the fact that this compound forms strong H-bonded dimeric aggregates.50 Therefore, the real molecular volume of such dimeric structures is more or less two times higher with respect to the single IBU molecules. In turn, one can suppose that the quite large disagreement between ΔV and Vm for Hex-IBU and Iso-IBU (especially for the first one) may be due to the enhanced ability to form supramolecular structures as revealed from XRD investigations on these compounds. However, it must be stressed that IBU and its esters should be considered as five different molecules, which differ from each other not only in the chemical structures, but also molecular interactions, conformations and possibly the architecture of aggregates. Therefore, it is very difficult to expect a monotonic variation of dTg/dp and ΔV versus Mw, as is usually reported for polymers.
. Interestingly, as illustrated in Table 2, the values of ΔV and Vm are similar for Met-IBU and Ben-IBU. In turn, there is a significant discrepancy between both parameters for the native IBU as well as its hexyl and isopropyl derivatives (ΔV − Vm = 143.5, 107.6, and 86.7 cm3 mol−1, respectively). In the case of IBU, it can be easily explained by the fact that this compound forms strong H-bonded dimeric aggregates.50 Therefore, the real molecular volume of such dimeric structures is more or less two times higher with respect to the single IBU molecules. In turn, one can suppose that the quite large disagreement between ΔV and Vm for Hex-IBU and Iso-IBU (especially for the first one) may be due to the enhanced ability to form supramolecular structures as revealed from XRD investigations on these compounds. However, it must be stressed that IBU and its esters should be considered as five different molecules, which differ from each other not only in the chemical structures, but also molecular interactions, conformations and possibly the architecture of aggregates. Therefore, it is very difficult to expect a monotonic variation of dTg/dp and ΔV versus Mw, as is usually reported for polymers.
Conclusions
In this paper, three experimental techniques: BDS, XRD, and FTIR, were applied to further characterize the molecular dynamics, as well as the internal structure and intermolecular interactions in four esters of IBU, at a wide range of temperatures and pressures. Interestingly, XRD studies revealed medium-range ordering and some molecular organization in the examined systems. Moreover, FTIR investigations, supported by optical measurements, indicated the reorganization of the internal structure of the molecules and the formation of small aggregates having a size around 3 μm. Further extensive high pressure dielectric experiments showed that the slow mode, which was detected in the loss spectra measured for each IBU derivative at ambient pressure, could not be observed for the compressed samples anymore. The obtained results are consistent with the data published earlier for a series of propylene carbonate and dioxolane derivatives.12,13 Thus, one can suppose that either the time scale of both processes: SM and α-relaxation, becomes comparable or the motions which are responsible for the former mode are suppressed. Furthermore, we found an unusual behavior of the two coefficients, i.e., dTg/dp and ΔV, calculated from high pressure dielectric experiments for all the examined systems. Unexpectedly, in contrast to other low- and high molecular weight systems,48,61,67,68 these parameters in the case of IBU derivatives changed with the molecular weight in a non-monotonic way. This peculiar behavior is probably due to changes in the intermolecular interactions, conformations and architecture of supramolecular aggregates, determined by their different chemical structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M., E. K. and D. H. are thankful for the financial support from the National Science Center within the framework of the Sonata BIS project (Grant No. DEC-2016/22/E/NZ7/00266). We are grateful to Dr Monika Geppert-Rybczyńska for the density measurements.References
- C. Gainaru, S. Kastner, F. Mayr, P. Lunkenheimer, S. Schildmann, H. J. Weber, W. Hiller and A. Loidl, Phys. Rev. Lett., 2011, 107, 118304 CrossRef CAS PubMed.
- S. Pawlus, M. Paluch and M. Dzida, J. Phys. Chem. Lett., 2010, 1, 3249–3253 CrossRef CAS.
- S. Pawlus, S. Klotz and M. Paluch, Phys. Rev. Lett., 2013, 110, 173004 CrossRef CAS PubMed.
- S. Bauer, K. Burlafinger, C. Gainaru, P. Lunkenheimer, W. Hiller, A. Loidl and R. Böhmer, J. Chem. Phys., 2013, 138, 094505 CrossRef CAS PubMed.
- L. P. Singh, C. Alba-Simionesco and R. Richert, J. Chem. Phys., 2013, 139, 144503 CrossRef PubMed.
- T. Büning, J. Lueg, J. Bolle, C. Sternemann, C. Gainaru, M. Tolan and R. Böhmer, J. Chem. Phys., 2017, 147, 234501 CrossRef PubMed.
- T. Hecksher and B. Jakobsen, J. Chem. Phys., 2014, 141, 101104 CrossRef PubMed.
- S. Pawlus, M. Wikarek, C. Gainaru, M. Paluch and R. Böhmer, J. Chem. Phys., 2013, 139, 064501 CrossRef CAS PubMed.
- M. Wikarek, S. Pawlus, S. N. Tripathy, A. Szulc and M. Paluch, J. Phys. Chem. B, 2016, 120, 5744–5752 CrossRef CAS PubMed.
- C. Gainaru, S. Bauer, E. Vynokur, H. Wittkamp, W. Hiller, R. Richert and R. Böhmer, J. Phys. Chem. B, 2015, 119, 15769–15779 CrossRef CAS PubMed.
- L. M. Wang and R. Richert, J. Chem. Phys., 2005, 123, 054516 CrossRef PubMed.
- M. Rams-Baron, A. Jedrzejowska, M. Dulski, K. Wolnica, K. Geirhos, P. Lunkenheimer and M. Paluch, Phys. Chem. Chem. Phys., 2018, 20, 28211 RSC.
- A. Jedrzejowska, S. Hensel-Bielowka, K. Koperwas, K. Jurkiewicz, K. Chmiel, J. Jacquemin, D. Kruk and M. Paluch, J. Chem. Phys., 2019, 150, 044504 CrossRef PubMed.
- K. Adachi and T. Kotaka, Prog. Polym. Sci., 1993, 18, 585–622 CrossRef CAS.
- M. Tarnacka, A. Talik, E. Kaminska, M. Geppert-Rybczyńska, K. Kaminski and M. Paluch, Macromolecules, 2019, 52, 3516–3529 CrossRef CAS.
- Z. Wojnarowska, M. Paluch, P. Wlodarczyk, L. Hawelek, R. Wrzalik, J. Zioło, M. Wygledowska-Kania, B. Bergler-Czop, L. Brzezinska-Wcislo and P. Bujak, Phys. Rev. E, 2011, 83, 051502 CrossRef CAS PubMed.
- M. Tarnacka, K. Adrjanowicz, E. Kaminska, K. Kaminski, K. Grzybowska, K. Kolodziejczyk, P. Wlodarczyk, L. Hawelek, G. Garbacz, A. Kocot and M. Paluch, Phys. Chem. Chem. Phys., 2013, 15, 20742–20752 RSC.
- Relaxation and Diffusion in Complex Systems, ed. K. L. Ngai, Springer, New York, 2011 Search PubMed.
- Broadband Dielectric Spectroscopy, ed. F. Kremer and A. Schönhals, Springer, Berlin, 2003 Search PubMed.
- M. Mierzwa, S. Pawlus, M. Paluch, E. Kaminska and K. L. Ngai, J. Chem. Phys., 2008, 128, 044512 CrossRef CAS PubMed.
- R. Böhmer, G. Diezemann, B. Geil, G. Hinze, A. Nowaczyk and M. Winterlich, Phys. Rev. Lett., 2006, 97, 135701 CrossRef PubMed.
- S. Capaccioli, K. L. Ngai, M. Shahin Thayyil and D. Prevosto, J. Phys. Chem. B, 2015, 119, 8800–8808 CrossRef CAS PubMed.
- R. Buchner, J. Barthel and J. Stauber, Chem. Phys. Lett., 1999, 306, 57–63 CrossRef CAS.
- P. J. Griffin, A. P. Holt, Y. Wang, V. N. Novikov, J. R. Sangoro, F. Kremer and A. P. Sokolov, J. Phys. Chem. B, 2014, 118, 783–790 CrossRef CAS PubMed.
- M. Rams-Baron, Z. Wojnarowska, M. Dulski, A. Ratuszna and M. Paluch, Phys. Rev. E, 2015, 92, 022309 CrossRef CAS PubMed.
- H. J. Kwon, T. H. Kim, J. H. Ko and Y. H. Hwang, Chem. Phys. Lett., 2013, 556, 117–121 CrossRef CAS.
- C. Gainaru, R. Meier, S. Schildmann, C. Lederle, W. Hiller, E. A. Rössler and R. Böhmer, Phys. Rev. Lett., 2010, 105, 258303 CrossRef CAS PubMed.
- C. Gainaru, R. Figuli, T. Hecksher, B. Jakobsen, J. C. Dyre, M. Wilhelm and R. Böhmer, Phys. Rev. Lett., 2014, 112, 098301 CrossRef CAS PubMed.
- L. P. Singh and R. Richert, Phys. Rev. Lett., 2012, 109, 167802 CrossRef PubMed.
- C. Gainaru, M. Wikarek, S. Pawlus, M. Paluch, R. Figuli, M. Wilhelm, T. Hecksher, B. Jakobsen, J. C. Dyre and R. Böhmer, Colloid Polym. Sci., 2014, 292, 1913–1921 CrossRef CAS.
- C. Lederle, W. Hiller, C. Gainaru and R. Böhmer, J. Chem. Phys., 2011, 134, 064512 CrossRef PubMed.
- C. Gainaru, R. Meier, S. Schildmann, C. Lederle, W. Hiller, E. A. Rössler and R. Böhmer, Phys. Rev. Lett., 2010, 105, 258303 CrossRef CAS PubMed.
- Y. Wang, P. J. Griffin, A. Holt, F. Fan and A. P. Sokolov, J. Chem. Phys., 2014, 140, 104510 CrossRef PubMed.
- J. Gabriel, F. Pabst, A. Helbling, T. Böhmer and T. Blochowicz, Phys. Rev. Lett., 2018, 121, 035501 CrossRef CAS PubMed.
- J. Gabriel, F. Pabst and T. Blochowicz, J. Phys. Chem. B, 2017, 121, 8847–8853 CrossRef CAS PubMed.
- A. Minecka, E. Kaminska, D. Heczko, M. Tarnacka, I. Grudzka-Flak, M. Bartoszek, A. Zięba, R. Wrzalik, W. E. Śmiszek-Lindert, M. Dulski, K. Kaminski and M. Paluch, J. Chem. Phys., 2018, 148, 224505 CrossRef CAS PubMed.
- F. Affouard and N. T. Correia, J. Phys. Chem. B, 2010, 114, 011397–011402 CrossRef CAS PubMed.
- K. Adrjanowicz, K. Kaminski, M. Dulski, P. Wlodarczyk, G. Bartkowiak, L. Popenda, S. Jurga, J. Kujawski, J. Kruk, M. K. Bernard and M. Paluch, J. Chem. Phys., 2013, 139, 111103 CrossRef CAS PubMed.
- S. Sarkar and R. N. Joarder, J. Chem. Phys., 1993, 99, 2032–2039 CrossRef CAS.
- K. S. Vahvaselkä, R. Serimaa and M. Torkkeli, J. Appl. Crystallogr., 1995, 28, 189–195 CrossRef.
- A. K. Karmakar, P. S. R. Krishna and R. N. Joarder, Phys. Lett. A, 1999, 253, 207–210 CrossRef CAS.
- N. Shankland, A. J. Florence, P. J. Cox, D. B. Sheen, S. W. Love, N. S. Steward and C. C. Wilson, Chem. Commun., 1996, 855–856 RSC.
- P. Papadopoulos, W. Kossack and F. Kremer, Soft Matter, 2013, 9, 1600–1603 RSC.
- A. Minecka, E. Kaminska, M. Tarnacka, A. Talik, I. Grudzka-Flak, K. Wolnica, M. Dulski, K. Kaminski and M. Paluch, Phys. Chem. Chem. Phys., 2018, 20, 30200–30208 RSC.
- Infrared and Raman characteristic group frequencies. Tables and charts, ed. G. Socrates, John Wiley and Sons, Ltd, Chichester, 3rd edn, 2001 Search PubMed.
- D. Morineau and C. Alba-Simionesco, J. Chem. Phys., 1998, 109, 8494 CrossRef CAS.
- K. Adrjanowicz, Z. Wojnarowska, M. Paluch and J. Pionteck, J. Phys. Chem. B, 2011, 115, 4559–4567 CrossRef CAS PubMed.
- Molecular Dynamics of Glass-Forming Systems: Effects of Pressure, ed. G. Floudas, M. Paluch, A. Grzybowski, K. L. Ngai, Springer-Verlag, Berlin, Heidelberg, 2011, ch. 2 Search PubMed.
- K. L. Ngai, R. Casalini, S. Capaccioli, M. Paluch and C. M. Roland, J. Phys. Chem. B, 2005, 109, 17356–17360 CrossRef CAS PubMed.
- A. R. Bras, J. P. Noronha, A. M. Antunes, M. M. Cardoso, A. Schönhals, F. Affouard, M. Dionisio and N. T. Correia, J. Phys. Chem. B, 2008, 112, 11087–11099 CrossRef CAS PubMed.
- K. Adrjanowicz, K. Kaminski, M. Tarnacka, K. Szutkowski, L. Popenda, G. Bartkowiak and M. Paluch, Phys. Chem. Chem. Phys., 2016, 18, 10585–10593 RSC.
- G. Szklarz, K. Adrjanowicz, M. Dulski, J. Knapik and M. Paluch, J. Phys. Chem. B, 2016, 120, 11298–11306 CrossRef CAS PubMed.
- M. Romanini, M. Barrio, R. Macovez, M. D. Ruiz-Martin, S. Capaccioli and J. L. Tamarit, Sci. Rep., 2017, 7, 1346 CrossRef PubMed.
- D. Heczko, E. Kamińska, A. Minecka, A. Dzienia, K. Jurkiewicz, M. Tarnacka, A. Talik, K. Kamiński and M. Paluch, J. Chem. Phys., 2018, 148, 204510 CrossRef PubMed.
- E. Kaminska, M. Tarnacka, K. Jurkiewicz, K. Kaminski and M. Paluch, J. Chem. Phys., 2016, 144, 054503 CrossRef CAS PubMed.
- M. Paluch, S. Casalini, S. Hensel-Bielowka and C. M. Roland, J. Chem. Phys., 2002, 116, 9839 CrossRef CAS.
- S. Hensel-Bielowka, M. Paluch and K. L. Ngai, J. Chem. Phys., 2005, 123, 014502 CrossRef CAS PubMed.
- C. M. Roland, R. Casalini and M. Paluch, Chem. Phys. Lett., 2003, 367, 259–264 CrossRef CAS.
- I. Avramov, A. Grzybowski and M. Paluch, J. Non-Cryst. Solids, 2009, 355, 733–736 CrossRef CAS.
- I. Avramov, J. Volcanol. Geotherm. Res., 2007, 160, 165–174 CrossRef CAS.
- M. Paluch, K. Grzybowska and A. Grzybowski, J. Phys.: Condens. Matter, 2007, 19, 205117 CrossRef.
- Theory of unimolecular reactions, ed. N. B. Slater, Comell University Press, Ithaca, New York, 1959 Search PubMed.
- K. Kaminski, S. Pawlus, K. Adrjanowicz, Z. Wojnarowska, P. Wlodarczyk and M. Paluch, J. Phys.: Condens. Matter, 2012, 24, 065105 CrossRef CAS PubMed.
- M. Paluch, J. Gapinski, A. Patkowski and E. W. Fischer, J. Chem. Phys., 2001, 114, 8048 CrossRef CAS.
- M. Paluch, S. Pawlus, A. Grzybowski, K. Grzybowska, P. Włodarczyk and J. Zioło, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 052501 CrossRef PubMed.
- G. Floudas, C. Gravalides, T. Reisinger and G. Wegner, J. Chem. Phys., 1999, 111, 9847 CrossRef CAS.
- A. Talik, M. Tarnacka, A. Dzienia, E. Kaminska, K. Kaminski and M. Paluch, Macromolecules, 2019, 52, 5658–5669 CrossRef CAS.
- S. Pawlus, S. J. Rzoska, J. Ziolo, M. Paluch and C. M. Roland, Rubber Chem. Technol., 2003, 76, 1106–1115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04886c |
| This journal is © the Owner Societies 2020 |