
Hydrogen polarity of interfacial water regulates heterogeneous ice nucleation
Mingzhe
Shao
 abc,
Chuanbiao
Zhang†
b,
Chonghai
Qi
d,
Chunlei
Wang
d,
Jianjun
Wang
c,
Fangfu
Ye
b and
Xin
Zhou
*b
abc,
Chuanbiao
Zhang†
b,
Chonghai
Qi
d,
Chunlei
Wang
d,
Jianjun
Wang
c,
Fangfu
Ye
b and
Xin
Zhou
*b
aCollege of Light Industry Science and Engineering, Tianjin University of Science and Technology, Tianjin, China
bSchool of Physical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China. E-mail: xzhou@ucas.ac.cn
cInstitute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
dInstitute of Applied Physics, Chinese Academy of Sciences, Shanghai, China
First published on 22nd November 2019
Abstract
Using all-atomic molecular dynamics (MD) simulations, we show that the structure of interfacial water (IW) induced by substrates characterizes the ability of a substrate to nucleate ice. We probe the shape and structure of ice nuclei and the corresponding supercooling temperatures to measure the ability of IW with various hydrogen polarities for ice nucleation, and find that the hydrogen polarization of IW even with the ice-like oxygen lattice increases the contact angle of the ice nucleus on IW, thus lifting the free energy barrier of heterogeneous ice nucleation. The results show that not only the oxygen lattice order but the hydrogen disorder of IW on substrates are required to effectively facilitate the freezing of top water.
1 Introduction
Water freezing on various material surfaces is of importance in wide-ranging fields1–5 such as cloud seeding, frost heaving, and cell preserving. So far, the microscopic mechanisms of the key process of freezing on substrates, heterogeneous ice nucleation, still remains elusive.6–9 It has been proposed that the lattice match of crystal surfaces of substrates to that of ice promotes water freezing, and has been regarded as the reason that silver iodide (AgI) greatly promotes ice nucleation.10,11 However, the lattice matching mechanism has often been questioned recently, since some materials with almost the same lattice to that of ice crystals, such as cuprous bromide (CuBr) and barium fluoride (BaF2), do not facilitate ice nucleation.12–19 Besides, recent experiments and molecular dynamics (MD) simulations show that many aspects of substrates, such as proton ordering,20 charge,21,22 sign of the charge,23 electric field,24 hydrophobicity,25,26 amphoterism,27 and the morphology of surfaces,9,28 can profoundly affect ice nucleation together, in cooperation or in competition. It is very hard to give a general picture to describe the mechanism of ice nucleation on these various surfaces.While the various kinds of aspects of different materials are hard to universally describe, all these different substrates induce atomic rearrangement of the first layers of interfacial water (IW), and regulate the freezing of top water. It has found that the molecules of the first layers of the interfacial liquid on substrates rearrange and these first layers dominate supercooling,29,30 wetting,31–34 adsorption20 and evaporation35 of the top liquids. It would be desirable to investigate the correlation between the atomic reconstruction of IW and freezing of water on various kinds of substrates, which might provide a universal understanding about the effects of various kinds of substrates on ice nucleation.
Recently, a number of MD simulations focusing on the rearrangement of IW have already revealed its significance on affecting ice nucleation. It was found that the oxygen atoms of the first layer of IW on β-AgI were almost perfectly ice-like, and promoted the formation of an ice-like lattice in the next water layers thus freezing all of the top water.16,17 The polarity of the substrate was also found to be crucial in heterogeneous ice nucleation as well, and a charge-induced α-alumina surface suppresses ice nucleation upon it irrespective of the sign of the surface charge.20,36 Moreover, the adsorption energy of a polar monomer on the ice surface exhibits a strong correlation with the IW orientation. These recognitions emphasize the role of IW in heterogeneous ice nucleation, and inspires our interest in pursuing a more detailed perception of the specific effects of transitional and rotational order of IW. To our best knowledge, there is no report on the hydrogen rearrangement influence on ice nucleation with oxygen lattice interference eliminated up to now.
In this article, based on all-atomic MD simulations, we found that the formation of an ice-like oxygen lattice of IW alone is not sufficient to aid ice nucleation, and the hydrogen polarization of IW induced by substrates also sensitively regulates freezing of the top water. This hydrogen dominating ice nucleation will generate fresh insight into the molecular mechanism of heterogeneous ice nucleation, and a broader perspective on the lattice matching mechanism as well.
2 Models
2.1 Simulation details
We investigate the ice freezing process upon polarized IW with or without substrates (AgI). We simulate, at most, 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 rigid TIP4P/ice water molecules37,38 employing the GROMACS 4.6.2 software package. The model parameters of the substrates are the same as that in ref. 17. To polarize IW on AgI, we change equally the charges of the cation and anion of the AgI substrate while keeping the substrate charge neutral. Although the change is unphysical without corresponding to any real substrate, it is reasonable for our goal to generate a modeling IW surface with the ice-like oxygen lattice but different hydrogen polarity for testing the possible effects on ice nucleation. The NVT ensembles with Nose–Hoover thermostat and periodic boundary conditions (PBC) in only (x, y) directions are applied, while two resilient walls are induced in the z-direction and far from the water and substrates. The integration time step is 2 fs.
800 rigid TIP4P/ice water molecules37,38 employing the GROMACS 4.6.2 software package. The model parameters of the substrates are the same as that in ref. 17. To polarize IW on AgI, we change equally the charges of the cation and anion of the AgI substrate while keeping the substrate charge neutral. Although the change is unphysical without corresponding to any real substrate, it is reasonable for our goal to generate a modeling IW surface with the ice-like oxygen lattice but different hydrogen polarity for testing the possible effects on ice nucleation. The NVT ensembles with Nose–Hoover thermostat and periodic boundary conditions (PBC) in only (x, y) directions are applied, while two resilient walls are induced in the z-direction and far from the water and substrates. The integration time step is 2 fs.
In the simulations, the Coulombic interaction is calculated based on the particle mesh Ewald method with slab correction.39 It has been noted that the water structures for this model of AgI with the slab correction are very likely unphysical.40 However, in this work, we apply the AgI-like substrate to achieve the polarized IW rather than to direct study the ice nucleation on the real AgI surfaces. The possible effects of the unphysical water structure is not key for our purpose. We generated the polarized IW without the AgI-like substrate to verify this in this work.
For probing the freezing of water, we count the number of maximal ice clusters during a 50 ns regular MD simulation, following the method of Dellago and Doye;41–43 here a molecule is identified as ice or water based on its orientational order,44 and we group hydrogen-bonded ice molecules as clusters.
2.2 Hydrogen polarity
Normal ice can have a different hydroxyl orientation arrangement even with the same oxygen lattice. For example, there are 16 different H-bond ordering schemes in the same ice Ih lattice.45 Order parameters were presented to describe special arrangements of OH bonds.20,46 In this work, we define an order parameter to generally describe the hydrogen polarity of IW, i.e., the asymmetry of hydroxyl directions,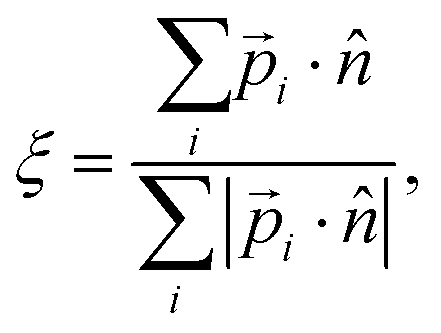 | (1) |
![[p with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0070_20d1.gif) i is the dipole moment of the water molecule and
i is the dipole moment of the water molecule and ![[n with combining circumflex]](https://www.rsc.org/images/entities/i_char_006e_0302.gif) is the normal vector of the IW surface. ξ = 0 refers to ordinary ice, the proton-disordered hexagonal ice phase ice Ih, while ξ = 1 refers to the ice XI,47–51 the proton-ordered form of ice Ih. An alternative (more intuitive) order parameter to describe the hydrogen polarity is to bring a direct recognition of the rotation of IW as below:
is the normal vector of the IW surface. ξ = 0 refers to ordinary ice, the proton-disordered hexagonal ice phase ice Ih, while ξ = 1 refers to the ice XI,47–51 the proton-ordered form of ice Ih. An alternative (more intuitive) order parameter to describe the hydrogen polarity is to bring a direct recognition of the rotation of IW as below: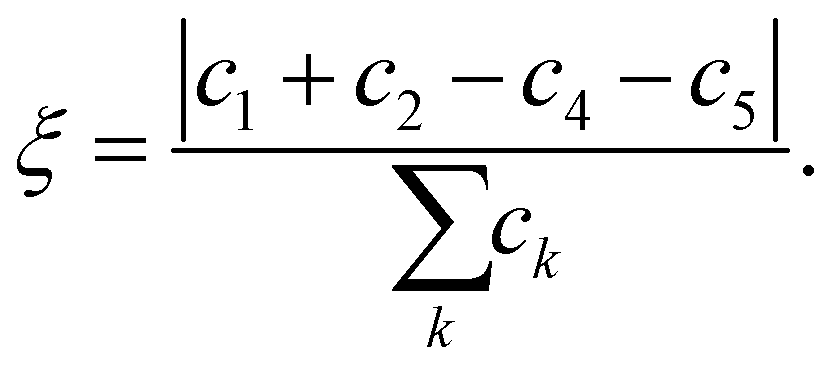 | (2) |
Here water molecules in ice are classified into five types according to the direction of its OH bond, and the numbers (or concentrations) of the five types of water are denoted as ck, k = 1,…,5, respectively, see Fig. 1. For example, as the ordered form of ice Ih, ice XI (ξ = 1) only has type 1 and 2 water molecules. There is no significant statistical difference between these two hydrogen polarity definitions in our cases.
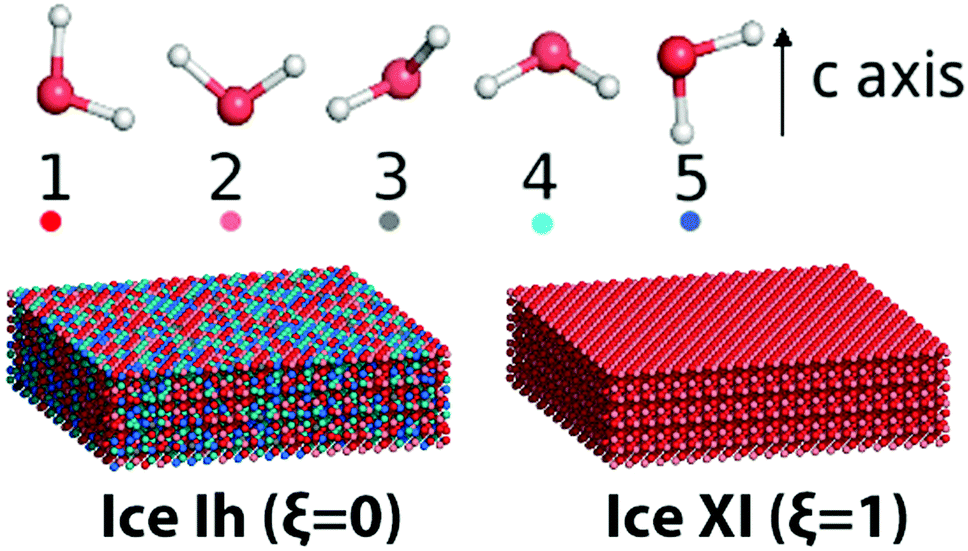 | ||
| Fig. 1 Five kinds of water molecules in ice with different OH directions. A guide view for different ice structures with the same lattice but different hydrogen polarity is also presented. | ||
On AgI substrates,17 we verify the formation of the IW layer with almost perfect ice lattice, and the freezing of water on the surfaces happens within 10 nanoseconds at 260 K. To study if IW with ice-like lattice but various polarizations facilitates the freezing of top water, we apply soft harmonic springs (k = 50 kcal mol−1 Å−2) to constrain the oxygen atoms of the first IW layer to keep its ice-like lattice. Additional charge (by directly changing the charge of the cation and anion of the AgI substrate) is applied to polarize the IW. The ice lattice structure remains and the hydrogen polarity increases from 0 to 0.3 while increasing the charge of the ions of AgI from 0.3e to 1.8e. The original AgI has the ion charge 0.6e, where e is the basic unit of charge, equal to that of a proton.
In order to emphasize the role of IW rather than the substrates, we also generate a pure polarized ice slab (without any another substrate surface) as the IW with ice-like lattice. Many different methods can be applied to generate this kind of polarized IW. For example, we may fix oxygens of the regular ice Ih and apply a strong electric field to get a polarized ice slab. In this work, we follow another way, first relaxing the hydrogens of some layers of ice XI at a higher temperature to achieve a few layers of ice with layer-varied partial polarity, then applying an electric field (one between −0.8 and 1.2 V nm−1, along the c axis) which makes the polarity of every ice layer be the same as any desired value between 0 to 0.7. The obtained ice-like slab is applied as the substrate by constraining all of its oxygen and hydrogen positions for checking the effect on ice nucleation on IW.
2.3 Critical ice nucleus on substrate
Usually, freezing on IW with a larger ξ is less likely to happen, and it is difficult to directly simulate this process since too long a MD time is required. Sanz et al.52 developed an efficient indirect MD method to study the homogeneous ice nucleation of bulk water. They detected the corresponding supercooled temperature of a preset spheric ice nucleus instead of directly seeking the corresponding critical nucleus at a preset temperature. The main difficulty in expanding the method to heterogeneous nucleation is that the shape and structure of the critical ice nucleus, such as the contact angles and crystalline surface of the nucleus on substrates, are unknown.Pedevilla and coworkers presented a heterogeneous seeding approach (HSEED)53 to overcome the difficulty, which enables the assessment of the ice nucleation ability upon crystalline substrates. The HSEED approach presets multiple possible ice nuclei, e.g., five initial combinations of crystal polytype and face exposed to the substrate: Ih(001), Ih(100), Ih(110), Ic(001), and Ic(111), and calculates the adsorption energy of water molecules on the nuclei, to pinpoint which nucleus is most likely to form on substrates.
Here, we present a different implementation to achieve the suitable ice nucleus on a substrate by gradually adjusting the shape, crystal polytype, position and face of the ice nucleus on the substrate through a repeated melting and growing process: (1) we preset a sphere-cap (or a spherical) hexagonal ice (Ih) nucleus and locate it nearby the substrate then immerse in supercooled water as the initial conformation; (2) we choose a few supercooled temperatures within a (small) range where the ice nucleus is predicted to be able to grow obviously at its low limit but melt obviously at the high limit. Then we simulate the system from the initial conformation for a segment of time, e.g., 10 ns, at each of these temperatures, respectively, to adjust the ice nucleus; (3) we choose one from these trajectories where the size and shape of the ice nucleus was most obviously adjusted (but not completely melting out or growing up too large). The final conformation is resetting the chosen trajectory as a new initial conformation to repeat the step (2) and (3). The corresponding temperature and a few of its neighbor values were applied as the new simulation temperatures to adjust the ice nucleus. The steps were repeated a few times, until the shape and size of the ice nucleus had already changed a lot and did not change obviously any more. Thus we achieved a suitable ice nucleus on the substrate, and the middle value of the two neighboring temperatures where the ice nucleus grows and shrinks respectively during the final simulation segment is the corresponding temperature where the ice nucleus is critical.
3 Results
3.1 Direct simulation of nucleation on AgI-like substrates
The hydrogen polarity of IW, which measures the out-of-plane asymmetry of hydroxyl directions,20,45,54 sensitively affects the formation of the ice nucleus, even while the oxygens of IW form an almost perfect ice-like lattice.The freezing processes upon IW with different hydrogen polarities ξ are shown in Fig. 2. For small ξ, an ice-like IW layer can quickly form with the ice Ih lattice within 10 nanoseconds at T = 260 K, and grow fast, similar to previous results.16,17 However, as ξ increases by increasing the charge of the cation and anion of the AgI substrate, the formation of ice clusters becomes difficult. The hydrogen bond network of IW is fractured when ξ > 0.2, and water on top cannot freeze within the 50 ns, even though the IW remains the same ice-like oxygen lattice by constraining these oxygen atoms.
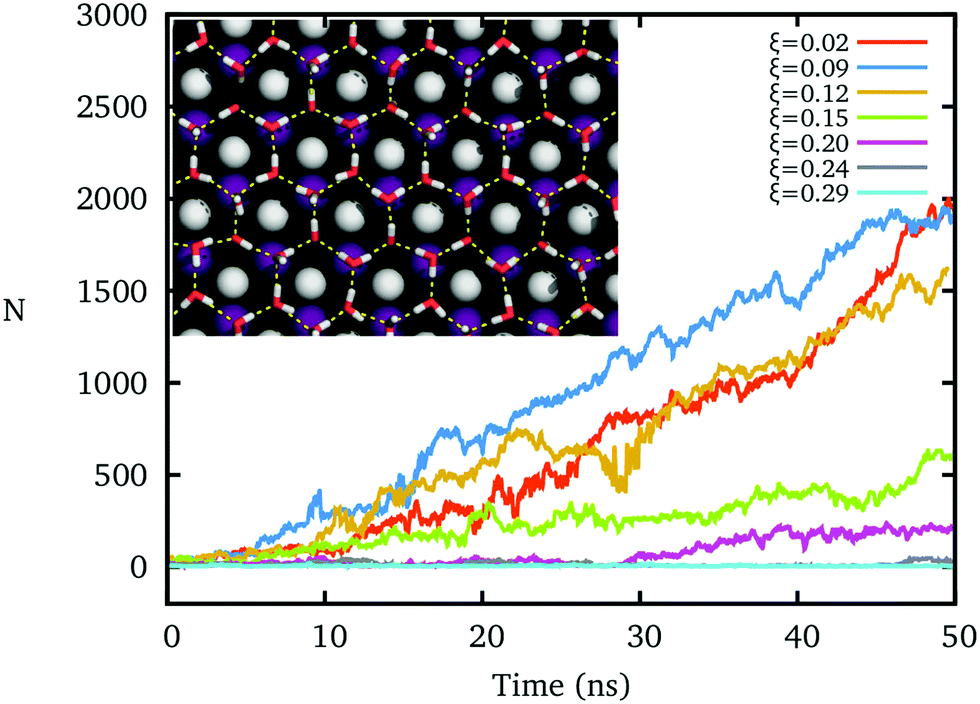 | ||
| Fig. 2 The forming and growing of an ice nucleus upon the AgI-like substrates is directly simulated. The hydrogen polarity (ξ) of interfacial water is induced by the applied charges on the cation and anion of the substrate. T = 260 K. Inset: the ice-like first layer of interfacial water on the AgI-like substrates. Silver: white; iodine: purple; the red sphere is the oxygen of water, and the small white sphere is the hydrogen of water. | ||
As shown in the inset of Fig. 3a, while ξ = 0.09 (the corresponding charge of ions is about 0.6e, similar to that of the original AgI), the hydroxyl groups of IW almost equally point to the two sides of IW, where ice nucleation is easy. But when ξ = 0.21 (the corresponding charge of the ions is about 1.4e, similar to that of BaF2), more hydroxyl groups point toward the substrate and fewer hydroxyl groups point to the reverse direction, the water side, where the ice nucleation is difficult. Thus, the IW with ice-like lattice can be hydrogen ordered20,45,54 (large ξ) or hydrogen disordered (small ξ) providing different abilities to facilitate ice nucleation of top water.
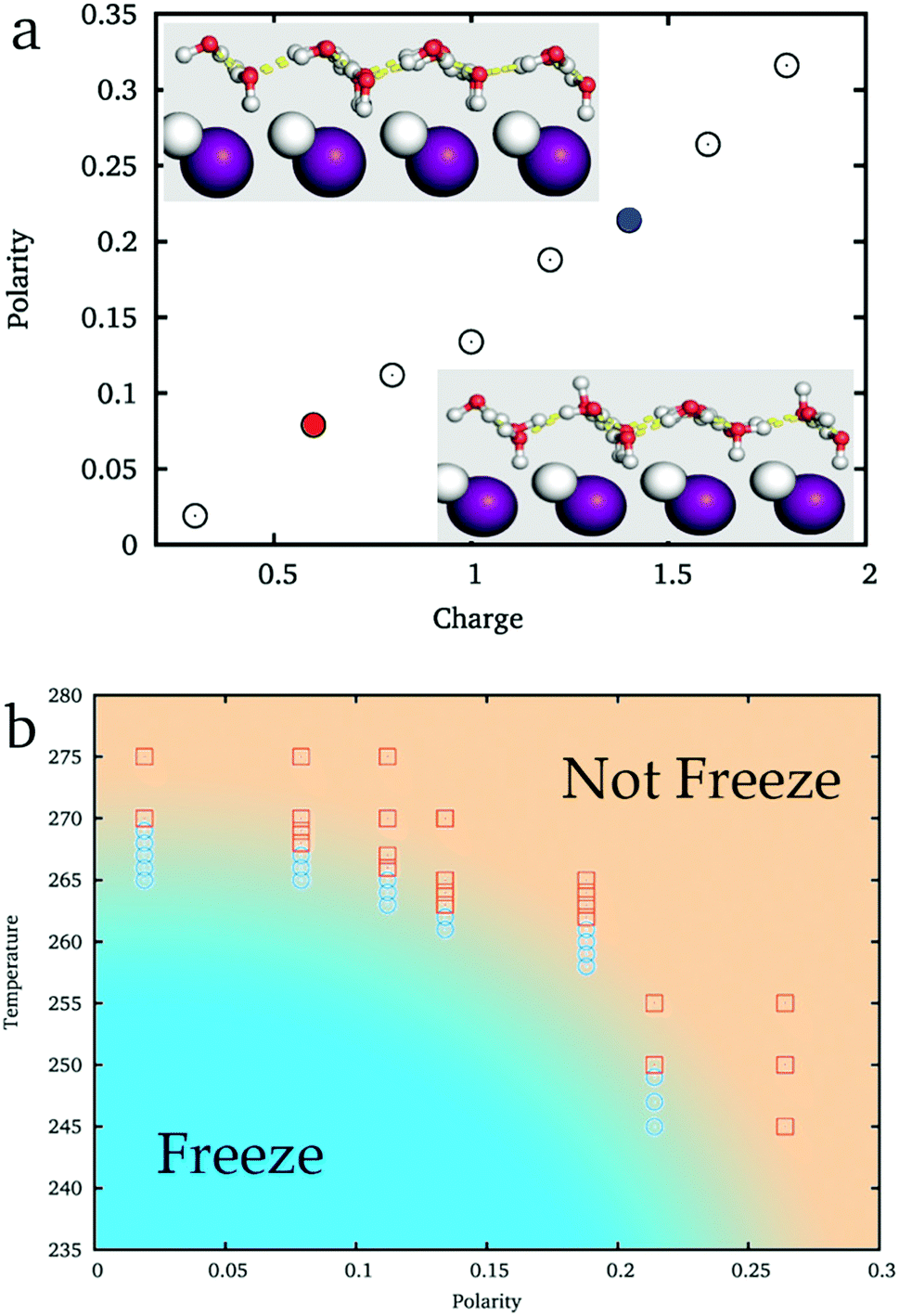 | ||
| Fig. 3 (a) The hydrogen polarity of interfacial water varies as the ionic charge of the AgI-like surface. A local view revealing the distinct difference between the hydrogen ordered and disordered IW on substrates, shown while ξ = 0.08 (bottom-right) and ξ = 0.21 (top-left). Silver: white; iodine: purple; the red sphere is the oxygen of water, and the small white sphere is the hydrogen of water. (b) The approximate phase diagram of the freezing temperature and hydrogen polarity of interfacial water. Each point is obtained by a 50 ns standard MD simulation from an initial state of liquid. | ||
By simulating 50 ns each from initial liquid water at various temperatures, we get an approximate phase diagram of water freezing on substrates with the ice-like lattice but hydrogen polarized IW, see Fig. 3b. The freezing temperature of water abruptly decreases with the increasing ξ of IW. When ξ ≈ 0, water freezes at 269 K, approaching the melting temperature of the applied TIP4P/ice water model, 272 K. On increasing the hydrogen polarity of IW, when ξ ≈ 0.09, water is found to freeze around 265 K, and when ξ ≈ 0.21 the freezing temperature decreases to about 250 K.
3.2 Simulation by preseting ice nucleus upon polarized IW
For detecting the relation between the structure of IW and the freezing of top water, we check the ice nucleation on the IW with ice-like lattice but various different hydrogen polarities without applying any other substrate surface (such as AgI).As shown in Fig. 4, in the absence of AgI-like substrates, the freezing of supercooled water is also found to become more difficult as the hydrogen polarity of the ice-like polarized IW increases. At 265 K, water freezes on the ice-like polarized IW layers (usually 4–6 layers) with ξ = 0.10, but does not form an ice cluster at ξ = 0.18 during 20 ns regular MD simulations. We also carried out simulations at 255 K, where water freezes at ξ = 0.10 and ξ = 0.18, but not at ξ = 0.43 within 20 ns. This result indicates that the heterogeneous free energy barrier of ice nucleation is obviously dependent on the hydrogen polarity of the ice-like IW. This result is in agreement with the phase diagram of ice nucleation on AgI-like substrates shown in Fig. 3b.
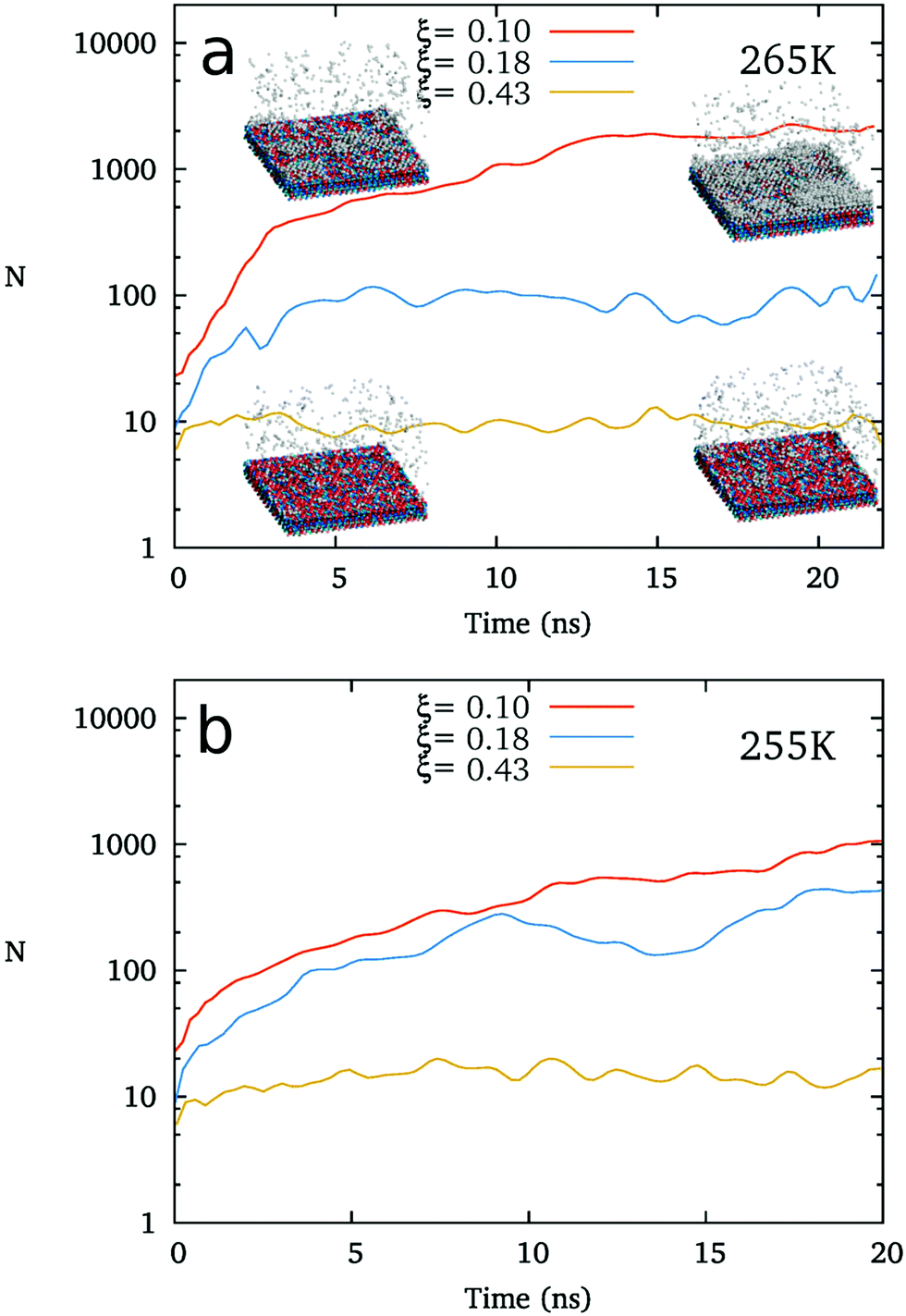 | ||
| Fig. 4 The heterogeneous nucleation upon ice-like interfacial water at (a) 265 K and (b) 255 K. | ||
We preset ice nuclei on the polarized IW with various ξ to get the critical ice nuclei and the corresponding temperatures. Fig. 5 illustrates the whole simulation scheme on the ice-like IW with ξ = 0.16. The ice nucleus is sufficiently adjusted to change its shape and its size (initial 2000, growing to about 3000, and finally back 2350 molecules) after 6 × 10 ns simulations at a few preset temperatures. Then we simulate the final 10 ns trajectories at a few neighboring temperatures to find the ice nucleus shrinks at 259 K but grows at 258 K. Thus we get the middle temperature T = 258.5 K where the ice nucleus is thought to be critical.
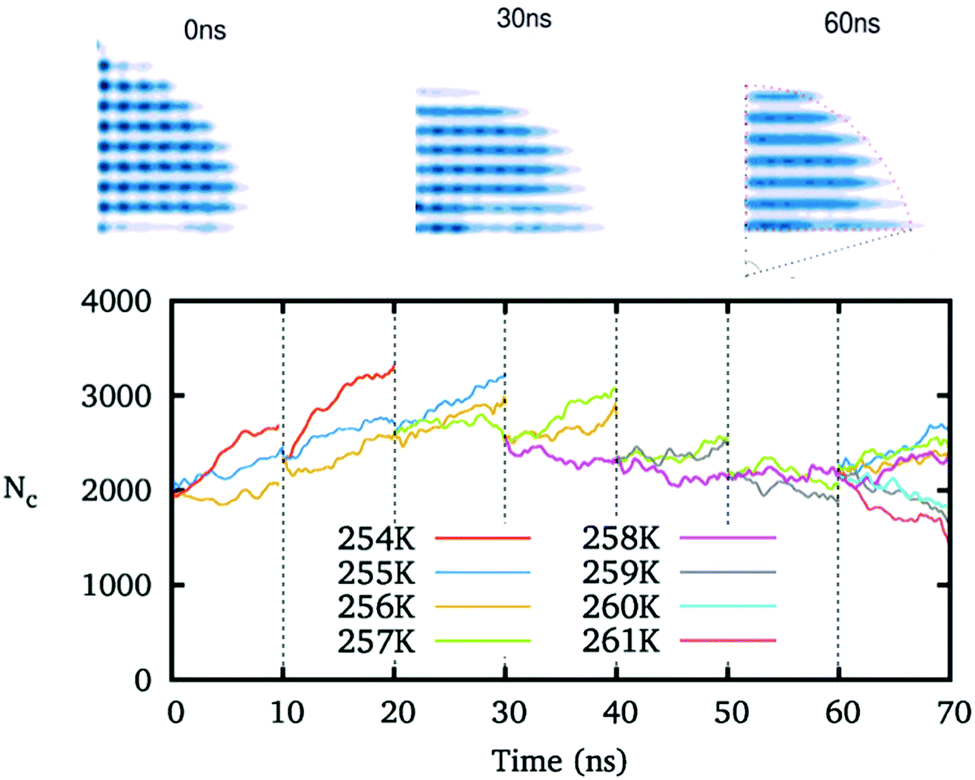 | ||
| Fig. 5 The time evolution of the shape and size of the ice nucleus during the simulations adjusting the critical ice nucleus on IW with ξ = 0.16. Top: the outlines of the section of half an ice nucleus at the simulation time t = 0, 30 and 60 ns are shown. Bottom: the number of water molecules inside the ice nucleus versus the simulation time. A series of simulations at different temperatures are applied to obviously adjust the preset ice nucleus to suit the applied IW surfaces. | ||
By extracting the outlines of the final ice nuclei from the average density of ice nuclei ρ = 0.5ρI, we find that all the critical ice nuclei are approximately sphere-caps, as expected from classical nucleation theory (CNT), except for a small deviation in the first layers for the small and large ξ cases, see Fig. 6. Here ρI is the density of bulk ice, about 0.906 g cm−3 in this model.
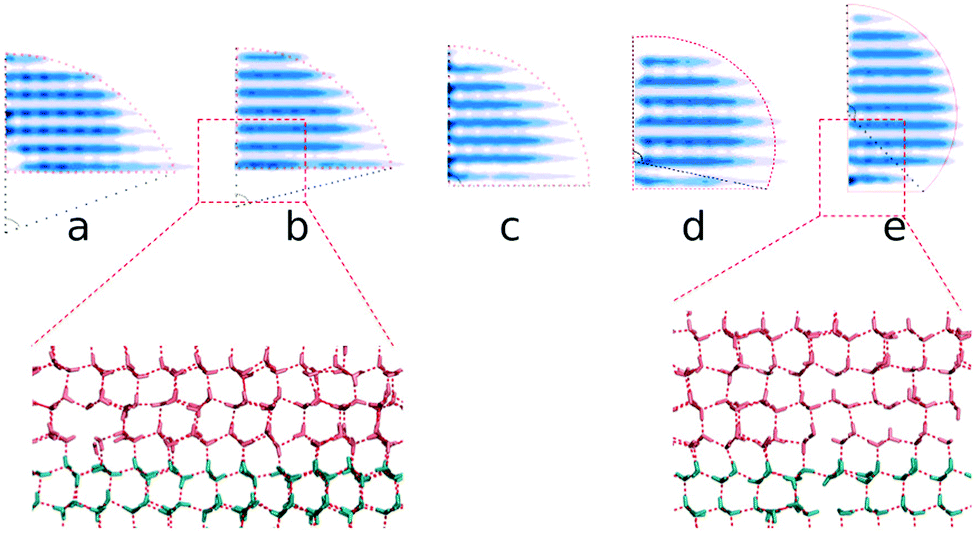 | ||
| Fig. 6 Top: the obtained critical ice nuclei on the ice-like polarized IW surfaces with ξ = 0.14, 0.16, 0.27, 0.39 and 0.53 in (a)–(e), respectively. All nuclei are sphere-caps, here only the right half of their section is shown (the whole nucleus can be imaged by rotational symmetry). Bottom: the hydrogen-bond connection between atoms in the ice nucleus (red) and that in polarized IW (blue) with ξ = 0.16 and 0.53, respectively. The liquid water molecules are omitted for clarification. | ||
The critical temperatures of heterogeneous nuclei (about 2000 molecules) upon IW with different polarities is estimated. As shown in Table 1, the corresponding temperature of the critical ice nuclei with similar size is obviously dependent on the polarization of the IW.
| No. of iterations | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| ξ = 0.14 | 263 ± 2 | 263 ± 1 | 263 ± 1 | 262 ± 1 | 262 ± 1 | 262 ± 1 | 262 ± 1 |
| ξ = 0.16 | ∼255 | >256 | ∼257 | 257 ± 1 | 258 ± 1 | 258 ± 1 | 258 ± 1 |
| ξ = 0.27 | ∼255 | >256 | ∼256 | 257 ± 1 | 258 ± 1 | 257 ± 1 | 257 ± 1 |
| ξ = 0.39 | ∼255 | ∼255 | ∼255 | 255 ± 1 | 255 ± 1 | 255 ± 1 | 255 ± 1 |
| ξ = 0.53 | ∼255 | ∼255 | ∼254 | ∼254 | 253 ± 1 | 252 ± 1 | 252 ± 1 |
From the simulations, we have the size Nc, radius R, the (apparent) contact angle θ of the sphere-cap critical nucleus, the corresponding (supercooled) temperature and the free energy barrier ΔG of nucleation, shown in Table 2. Here the free energy barriers are estimated from the CNT, 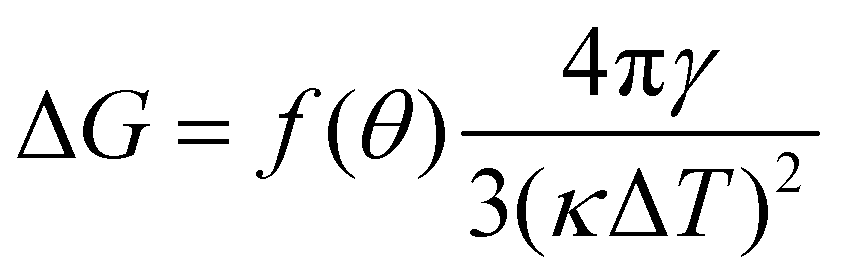 . Here
. Here 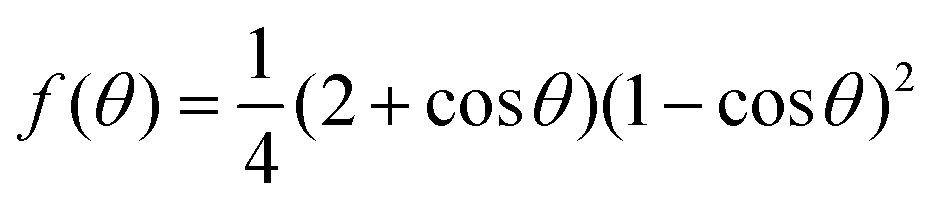 is the shape factor of a spherical cap.
is the shape factor of a spherical cap.
| ξ | θ | f(θ) | R | T c (K) | ΔG |
|---|---|---|---|---|---|
| 0.14 | 72 | 0.28 | 10.0 | 262 ± 1 | 50 |
| 0.16 | 78 | 0.35 | 8.8 | 258 ± 1 | 32 |
| 0.27 | 90 | 0.50 | 7.7 | 257 ± 1 | 40 |
| 0.39 | 98 | 0.60 | 6.9 | 255 ± 1 | 37 |
| 0.53 | 134 | 0.94 | 6.1 | 252 ± 1 | 42 |
In Fig. 7, we find that the inverse of the radius R of a critical nucleus is proportional to the corresponding supercooled temperature, 1/R ≈ κΔT, with κ ≈ 0.03 nm−1 K−1. The result is in good agreement with the expectation of CNT, and 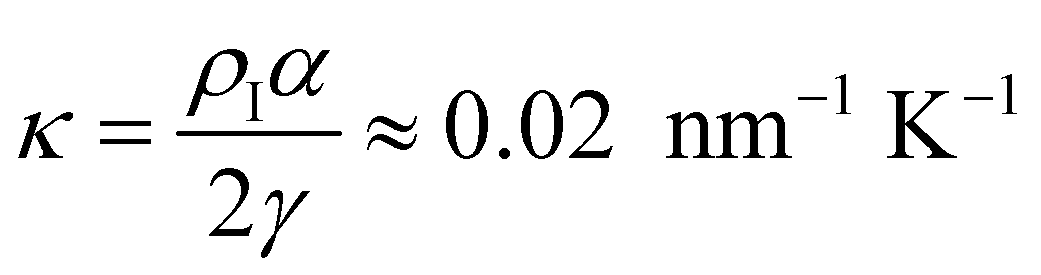 . Here
. Here 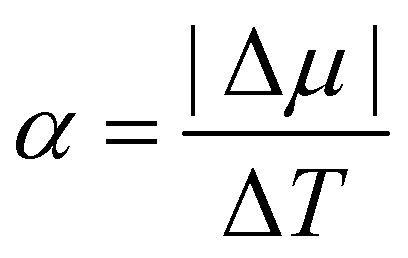 is about 0.0043 kcal mol−1 K−1,52 the ice-water surface tension γ was chosen as a typical value of about 26 mN m−1, and Δμ is the chemical potential difference between ice and water.
is about 0.0043 kcal mol−1 K−1,52 the ice-water surface tension γ was chosen as a typical value of about 26 mN m−1, and Δμ is the chemical potential difference between ice and water.
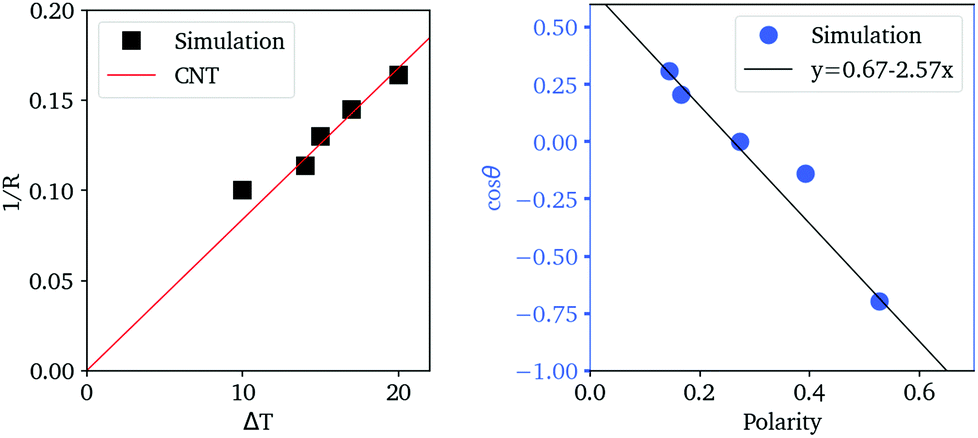 | ||
| Fig. 7 Left, the inverse radius of ice nuclei versus supercooling temperatures. The line is fitted based on the classic nucleation theory. Right, the relation between contact angles of ice nuclei versus the hydrogen polarity of IW. | ||
The cosine of contact angle is found to be linearly related to ξ in the whole range 0 < ξ < 1, as shown in Fig. 7. From the Young's equation, we have
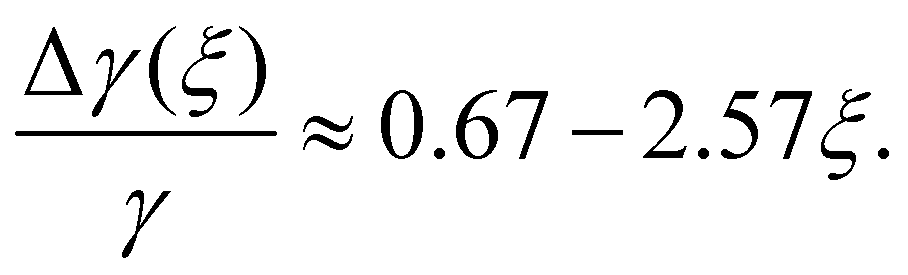 | (3) |
Considering the fact that the IW with ξ = 0 is similar to the normal hexagonal ice, we have γice,IW(ξ) = γ(δ1 + k1ξ +…), while γwater,IW(ξ) = γ(1 − δ2 + k2ξ +…). Here both δ1 and δ2 are small positive values, and k1 > k2 > 0, since liquid water is more flexible than an ice nucleus in terms of rearranging its conformations on IW. Therefore, we have, δ1 + δ2 ≈ 0.33, and k1 − k2 = 2.57. The higher order dependence of surface tensions on ξ seem very small (or cancel each other) even when ξ approaches unity, where the completely polarized IW distorts the lattice of both itself and the growing ice nucleus to avoid dangling hydrogen bonds. It is a little surprising that the macroscopic-level picture based on surface tension describes well the ice nucleation happening on the nanometer scale. Identifying the physical reason behind this is interesting for the next studies.
4 Conclusions
In this work, interfacial water (IW) with an ice-like lattice but various different hydrogen polarities is found to have different effects in promoting water freezing. Some previous studies27,55–57 have noted the importance of the directions of the hydrogen bonds of IW on freezing of top water. The studies on ice formation on kaolinite27,55,56 and hydrophobic metal surfaces57 revealed that the substrate determined the orientation of the first contact water layer and significantly affected the adsorption energy, density profile and structures of adjacent liquid water, thus meaning the proton order is absolutely important in understanding heterogeneous ice nucleation. Our results systematically verify this effect and present a general perspective that the structure of IW, not only the oxygen lattice but the hydrogen disorder, can provide a fingerprint to understand heterogeneous ice nucleation. Only considering the lattice matching of substrates with ice may be insufficient to understand the effect of substrates on ice nucleation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is under the financial support of the NSFC grant with no. 11574310, 11674345 and 21733010. C.-L. Wang thanks the support of the Youth Innovation Promotion Association, CAS. M.-Z. Shao thanks the support of Beijing National Laboratory for Molecular Sciences (BNLMS).References
- A. Hudait, D. R. Moberg, Y. Qiu, N. Odendahl, F. Paesani and V. Molinero, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8266–8271 CrossRef PubMed.
- J. Russo, F. Romano and H. Tanaka, Nat. Mater., 2014, 13, 733–739 CrossRef CAS PubMed.
- B. J. Murray, D. O'Sullivan, J. D. Atkinson and M. E. Webb, Chem. Soc. Rev., 2012, 41, 6519–6554 RSC.
- Y.-C. C. Liou, A. Tocilj, P. L. Davies and Z. Jia, Nature, 2000, 406, 322–324 CrossRef CAS PubMed.
- T. Inada, T. Koyama, F. Goto and T. Seto, J. Phys. Chem. B, 2012, 116, 5364–5371 CrossRef CAS PubMed.
- T. Bartels-Rausch, Nature, 2013, 494, 27–29 CrossRef CAS PubMed.
- E. B. Moore and V. Molinero, Nature, 2011, 479, 506–508 CrossRef CAS.
- L. Kaufmann, C. Marcolli, B. Luo and T. Peter, Atmos. Chem. Phys., 2017, 17, 3525–3552 CrossRef CAS.
- Z. Zhang and X. Y. Liu, Chem. Soc. Rev., 2018, 47, 7116–7139 RSC.
- B. Vonnegut, Jpn. J. Appl. Phys., 1947, 18, 593–595 CrossRef CAS.
- B. Vonnegut and H. Chessin, Science, 1971, 174, 945–946 CrossRef CAS PubMed.
- H. E. Swanson, Natl. Bur. Stand. Circ., 1954, 539, 73 Search PubMed.
- P. L. Davies, Trends Biochem. Sci., 2014, 39, 548–555 CrossRef CAS PubMed.
- L. Lupi, A. Hudait and V. Molinero, J. Am. Chem. Soc., 2014, 136, 3156–3164 CrossRef CAS PubMed.
- A. Reinhardt and J. P. Doye, J. Chem. Phys., 2014, 141, 084501 CrossRef PubMed.
- G. Fraux and J. P. K. Doye, J. Chem. Phys., 2014, 141, 216101 CrossRef PubMed.
- S. A. Zielke, A. K. Bertram and G. N. Patey, J. Phys. Chem. B, 2014, 119, 9049–9055 CrossRef PubMed.
- P. Conrad, G. E. Ewing, R. L. Karlinsey and V. Sadtchenko, J. Chem. Phys., 2005, 122, 064709 CrossRef PubMed.
- G. Bullock and V. Molinero, Faraday Discuss., 2013, 167, 371–388 RSC.
- Z. Sun, D. Pan, L. Xu and E. Wang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13177–13181 CrossRef CAS PubMed.
- M. Gavish, J.-L. Wang, M. Eisenstein, M. Lahav and L. Leiserowitz, Science, 1992, 256, 815–818 CrossRef CAS PubMed.
- L. Wilen, Science, 1993, 259, 1469–1470 CrossRef CAS PubMed.
- D. Ehre, E. Lavert, M. Lahav and I. Lubomirsky, Science, 2010, 327, 672–675 CrossRef CAS PubMed.
- J. Y. Yan and G. N. Patey, J. Phys. Chem. A, 2012, 116, 7057–7064 CrossRef CAS PubMed.
- S. J. Cox, S. M. Kathmann, B. Slater and A. Michaelides, J. Chem. Phys., 2015, 142, 184704 CrossRef PubMed.
- S. J. Cox, S. M. Kathmann, B. Slater and A. Michaelides, J. Chem. Phys., 2015, 142, 184705 CrossRef PubMed.
- X. L. Hu and A. Michaelides, Surf. Sci., 2007, 601, 5378–5381 CrossRef CAS.
- M. Fitzner, G. C. Sosso, S. J. Cox and A. Michaelides, J. Am. Chem. Soc., 2015, 137, 13658–13669 CrossRef CAS PubMed.
- T. U. Schülli, R. Daudin, G. Renaud, A. Vaysset, O. Geaymond and A. Pasturel, Nature, 2010, 464, 1174–1177 CrossRef PubMed.
- A. L. Greer, Nature, 2010, 464, 1137–1138 CrossRef CAS PubMed.
- C. Wang, H. Lu, Z. Wang, P. Xiu, B. Zhou, G. Zuo, R. Wan, J. Hu and H. Fang, Phys. Rev. Lett., 2009, 103, 137801 CrossRef PubMed.
- G. A. Kimmel, N. G. Petrik, Z. Dohnálek and B. D. Kay, Phys. Rev. Lett., 2005, 95, 166102 CrossRef PubMed.
- C. Zhu, H. Li, Y. Huang, X. C. Zeng and S. Meng, Phys. Rev. Lett., 2013, 110, 126101 CrossRef PubMed.
- P. Guo, Y. Tu, J. Yang, C. Wang, N. Sheng and H. Fang, Phys. Rev. Lett., 2015, 115, 186101 CrossRef PubMed.
- R. Wan, C. Wang, X. Lei, G. Zhou and H. Fang, Phys. Rev. Lett., 2015, 115, 195901 CrossRef PubMed.
- A. Abdelmonem, E. H. Backus, N. Hoffmann, M. A. Sánchez, J. D. Cyran, A. Kiselev and M. Bonn, Atmos. Chem. Phys., 2017, 17, 7827–7837 CrossRef CAS.
- J. L. F. Abascal, E. Sanz, R. García Fernández and C. Vega, J. Chem. Phys., 2005, 122, 234511 CrossRef CAS PubMed.
- C. Vega, E. Sanz and J. L. F. Abascal, J. Chem. Phys., 2005, 122, 114507 CrossRef CAS PubMed.
- I. C. Yeh and M. L. Berkowitz, J. Chem. Phys., 1999, 111, 3155–3162 CrossRef CAS.
- T. Sayer and S. J. Cox, Phys. Chem. Chem. Phys., 2019, 21, 14546–14555 RSC.
- W. Lechner and C. Dellago, J. Chem. Phys., 2008, 129, 114707 CrossRef PubMed.
- A. Reinhardt, J. P. K. Doye, E. G. Noya and C. Vega, J. Chem. Phys., 2012, 137, 194504 CrossRef PubMed.
- J. Russo and H. Tanaka, J. Chem. Phys., 2016, 145, 211801 CrossRef PubMed.
- H. Tanaka, Eur. Phys. J. E: Soft Matter Biol. Phys., 2012, 35, 113 CrossRef PubMed.
- T. K. Hirsch and L. Ojama, J. Phys. Chem. B, 2004, 108, 15856–15864 CrossRef CAS.
- D. Pan, L. M. Liu, G. A. Tribello, B. Slater, A. Michaelides and E. Wang, Phys. Rev. Lett., 2008, 101, 155703 CrossRef PubMed.
- C. G. Salzmann, P. G. Radaelli, A. Hallbrucker, E. Mayer and J. L. Finney, Science, 2006, 311, 1758–1761 CrossRef CAS PubMed.
- C. G. Salzmann, P. G. Radaelli, E. Mayer and J. L. Finney, Phys. Rev. Lett., 2009, 103, 105701 CrossRef PubMed.
- Z. Raza, D. Alfè, C. G. Salzmann, J. Klimeš, A. Michaelides and B. Slater, Phys. Chem. Chem. Phys., 2011, 13, 19788–19795 RSC.
- X. Fan, D. Bing, J. Zhang, Z. Shen and J.-L. Kuo, Comput. Mater. Sci., 2010, 49, S170–S175 CrossRef.
- P. Geiger, C. Dellago, M. Macher, C. Franchini, G. Kresse, J. Bernard, J. N. Stern and T. Loerting, J. Phys. Chem. C, 2014, 118, 10989–10997 CrossRef CAS PubMed.
- E. Sanz, C. Vega, J. R. Espinosa, R. Caballero-Bernal, J. L. F. Abascal and C. Valeriani, J. Am. Chem. Soc., 2013, 135, 15008–15017 CrossRef CAS PubMed.
- P. Pedevilla, M. Fitzner, G. C. Sosso and A. Michaelides, J. Chem. Phys., 2018, 149, 072327 CrossRef PubMed.
- H. Nada, J. Phys. Chem. B, 1997, 5647, 6163–6166 CrossRef.
- S. J. Cox, Z. Raza, S. M. Kathmann, B. Slater and A. Michaelides, Faraday Discuss., 2013, 167, 389–403 RSC.
- B. Glatz and S. Sarupria, Langmuir, 2018, 34, 1190–1198 CrossRef CAS PubMed.
- D. T. Limmer, A. P. Willard, P. Madden and D. Chandler, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4200–4205 CrossRef CAS.
Footnote |
| † Current address: College of Physics and Electronic Engineering, Heze University, Heze 274015, China. |
| This journal is © the Owner Societies 2020 |