
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-deposition of gas hydrates by pressurized thermal evaporation†
Stefan
Arzbacher
 *ab,
Nima
Rahmatian
ac,
Alexander
Ostermann
d,
Tobias M.
Gasser
b,
Thomas
Loerting
b and
Jörg
Petrasch
c
*ab,
Nima
Rahmatian
ac,
Alexander
Ostermann
d,
Tobias M.
Gasser
b,
Thomas
Loerting
b and
Jörg
Petrasch
c
aillwerke vkw Endowed Professorship for Energy Efficiency, Research Center Energy, Vorarlberg University of Applied Sciences, Hochschulstraße 1, Dornbirn 6850, Austria. E-mail: stefan.arzbacher@fhv.at; Tel: +43 5572 792 3803
bInstitute of Physical Chemistry, University of Innsbruck, Innrain 52c, Innsbruck 6020, Austria
cDepartment of Mechanical Engineering, Michigan State University, East Lansing, MI 48824, USA
dDepartment of Mathematics, University of Innsbruck, Technikerstraße 13, Innsbruck 6020, Austria
First published on 11th February 2020
Abstract
Gas hydrates are usually synthesized by bringing together a pressurized gas and liquid or solid water. In both cases, the transport of gas or water to the hydrate growth site is hindered once an initial film of hydrate has grown at the water–gas interface. A seemingly forgotten gas-phase technique overcomes this problem by slowly depositing water vapor on a cold surface in the presence of the pressurized guest gas. Despite being used for the synthesis of low-formation-pressure hydrates, it has not yet been tested for hydrates of CO2 and CH4. Moreover, the potential of the technique for the study of hydrate decomposition has not been recognized yet. We employ two advanced implementations of the condensation technique to form hydrates of CO2 and CH4 and demonstrate the applicability of the process for the study of hydrate decomposition and the phenomenon of self-preservation. Our results show that CO2 and CH4 hydrate samples deposited on graphite at 261–265 K are almost pure hydrates with an ice fraction of less than 8%. Rapid depressurization experiments with thin deposits (approx. 330 μm thickness) of CO2 hydrate on an aluminum surface at 265 K yield identical dissociation curves when the deposition is done at identical pressure. However, hydrates deposited at 1 MPa almost completely withstand decomposition after rapid depressurization to 0.1 MPa, while samples deposited at 2 MPa decompose 7 times faster. Therefore, this synthesis technique is not only applicable for the study of hydrate decomposition but can also be used for the controlled deposition of a super-preserved hydrate.
1 Introduction
Gas hydrates (hydrates for short) are non-stoichiometric crystalline inclusion compounds of water and gas molecules. The water molecules form hydrogen-bonded polyhedra around the gas molecules yielding different crystal structures, the most common of which are the cubic structure I (sI), the cubic structure II (sII), and the hexagonal structure H (sH).1Hydrates of natural gas are abundant in seafloor sediments and permafrost and thus, they constitute one of Earth's largest reservoirs of organic carbon.2 In addition to the potential use of hydrates in the transportation and storage of energy,3,4 hydrates can be utilized for carbon capture and storage,4 for firefighting,5 as a carbon source in the food industry6 or in refrigeration processes.7
After more than two centuries of hydrate research, many of the open questions are connected with time-dependent phenomena, such as hydrate nucleation and the kinetics of hydrate formation and decomposition.1 One of these open questions concerns the intriguing anomaly of self-preservation, i.e., the ability of hydrates of some guests (CH4, CO2, Kr, Ar, and others)8 to withstand decomposition at conditions outside their thermodynamic stability region for prolonged periods. The phenomenon is often observed at 1 atm in the temperature range 240–273 K and characterized by a strongly reduced rate of hydrate dissociation when compared to both lower or higher temperatures.9–18 A popular explanation for the reduction in dissociation rates is the formation of a layer of ice in the initial period of decomposition which covers the remaining hydrate.9,10 It is assumed that such a layer of ice, particularly if defect-free,15,19 can form an efficient diffusion barrier to the guest gas and thus prevent further decomposition. However, the complex relationship between the rate and the temperature of dissociation observed in rapid depressurization experiments cannot be fully explained by this argument.12 Similarly, a thin ice cover cannot readily explain the dependence of self-preservation on the type of guest gas,8 as well as the difference in dissociation behavior between temperature ramping and rapid depressurization experiments.12 Moreover, macroscopic defects like pores and cracks20 complicate the development of a comprehensive model of hydrate decomposition including the effect of self-preservation. Furthermore, the roles of mass and heat transport as well as hydrate composition have not been clarified yet, and more experiments are needed. Conclusive experiments are best done on samples with identical properties (e.g., composition, morphology, porosity). Since natural hydrate samples vary widely in their properties, hydrates produced in the laboratory under highly reproducible conditions offer the best route to experimental investigation of self-preservation.
Typically, hydrates are synthesized by bringing liquid water or ice into direct contact with a guest gas at pressures and temperatures inside the thermodynamic stability region of the respective hydrate. When starting with liquid water, hydrates are commonly formed in reactors containing a gas column placed on top of a water column.21 Alternatively, water is spray-injected into a reactor containing the gas,22–24 or the gas is bubbled into a water column within a reactor.25 When starting with ice, usually small ice particles with a high surface area are pressurized with the gas.26 In either case, the hydrate starts to grow at the interface forming a layer (cf.Fig. 1), which slows down the transport to the hydrate growth site as the layer thickness increases.1 Assuming that mass transfer is the rate limiting process, this initial hydrate layer drastically reduces the growth rate.
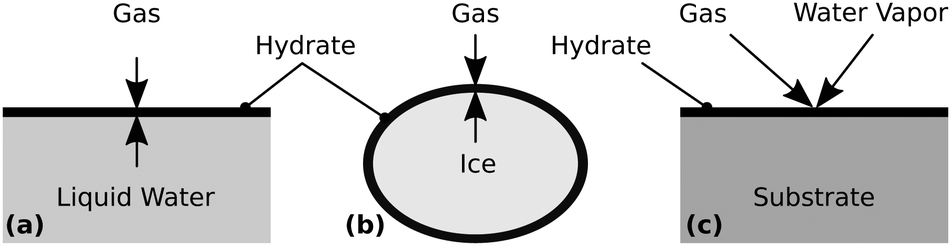 | ||
| Fig. 1 Schematic of hydrate layer formation at (a) the water–gas interface, (b) the ice–gas interface, or (c) by deposition from the gas phase. | ||
When starting with the guest gas and water vapor, the hydrate can be formed via aqueous solution nanodroplets27 or by deposition on a cold substrate (cf.Fig. 1). A popular non-equilibrium approach is the sequential- or co-deposition of water vapor and the gas as porous amorphous solid water in a vacuum chamber at T < 150 K. The gas is trapped in micropores and crystallizes into a hydrate upon heating.28–32 In a slightly different method, a hydrate of CO2 is grown epitaxially in a vacuum chamber using a molecular beam consisting of water vapor and the gas.33 Another route involves the separate humidification of a gas and a cooling chamber where the humid gas is crystallized as the hydrate.34 These processes either require a vacuum, which restricts the synthesis pressure, or a flowing mixture of gas and water in a system of pipes, which has a potential risk of clogging. Both disadvantages are circumvented by Cady's35–37 ingenious but seemingly forgotten slow condensation technique. To determine the composition of several hydrates, Cady evaporated liquid water of known mass in the presence of the pressurized guest gas and slowly condensed the vapor as a hydrate on a cold surface. Although Ceccotti38 previously used a similar process, Cady's approach offered less complexity and better control of hydrate nucleation.
A huge advantage of gas phase techniques is immediately evident: in contrast to the widely used methods involving transport in the condensed phase, mass transfer to the growth site here is not the limiting factor. That is, the rate of growth will not slow down substantially even if some hydrate has already formed.
Despite the advantages of Cady's slow condensation technique, to our knowledge, it is very rarely used. Due to his use of thin-walled glass reactors, Cady was restricted to the synthesis of hydrates with low formation pressures (e.g., Cl2, Br, Xe). Thus, slow condensation has not yet been tested for the synthesis of the two most prominent hydrates: those of CO2 and CH4. Since this technique allows, in principle, the formation of hydrates at any p–T condition inside the hydrate stability region and since it also scales well with the condenser surface area, it might prove beneficial for industrial applications. Moreover, we believe that the deposition of thin hydrate layers on a cooled surface at arbitrary pressures enables highly controlled formation and decomposition experiments.
Therefore, we report two advanced versions of the slow condensation technique to demonstrate that the process is suitable to synthesize hydrates of CO2 and CH4 as well as to show that Cady's method can be an important element in the study of hydrate decomposition and self-preservation in particular. One of the implementations allows us to capture changes in the morphology of the growing crystals by time-lapse micro-computed tomography (μCT). The other implementation is designed for the study of hydrate decomposition. Specifically, we investigate how the deposition synthesis conditions affect the degree of hydrate self-preservation. Pressure data from rapid depressurization experiments as well as powder X-ray diffraction (PXRD) profiles are used post-synthesis to quantify the bulk water to gas molar ratio and the crystallographic hydration number (mole fraction of water to gas in hydrate structure).
Due to the resemblance to the physical vapor deposition (PVD) technique of vacuum deposition by thermal vaporization,39 we henceforth refer to the process as co-deposition by pressurized thermal evaporation (PTE).
2 Experimental
Two different custom-built reactors are used for the synthesis of hydrate via co-deposition by PTE. In both reactors the water vapor is provided by a heated reservoir of liquid water within the reactor and naturally transported to the cooled growth site via advection and diffusion. During synthesis, the temperature at the hydrate growth site is kept below the hydrate equilibrium temperature at the reactor pressure. Therefore, hydrates are stable. The liquid water temperature is kept above the equilibrium temperature, making hydrates thermodynamically unfavorable. Deionized water, CH4 with purity more than 99.5% (Air Liquide), and CO2 with purity more than 99.7% (Air Liquide) are used for sample synthesis.2.1 Reactor R-CT
The reactor termed R-CT (cf.Fig. 2) is used for the in situ observation of the hydrate growth process with time-lapse micro-computed tomography (μCT). It resists pressures up to 10 MPa and can be used at temperatures as low as 180 K. For comparison, the largest pressure Cady36 used in his experiments is 0.42 MPa. The materials (glassy carbon and graphite) used in the reactor R-CT are transparent to X-rays and sufficiently heat conductive for cooling. The reactor's cylindrical shape avoids Feldkamp artifacts40 in the μCT scans. The hydrate growth site (cf.Fig. 2) is the wall of a graphite crucible (isostatically pressed; average grain size 10 μm, porosity 10%) placed in a gas-tight vessel made of glassy carbon (SIGRADUR G, HTW Germany). The reservoir of liquid water is enveloped by the growth site and sustained by a graphite rod at the center of the reactor. We use heat from the environment to evaporate the liquid water in the reservoir and cool the growth site thermoelectrically (Peltier element stack QC-17-1.4-3.7MS on QC-31-1.0-0-3.9MS, Quick-Ohm Germany) from the bottom of the reactor. The synthesis conditions result in a radial temperature field which drives the continuous transport of water vapor from the liquid water reservoir to the growth site. Temperatures T* and T° at the hydrate growth site, as well as Θ*, and Θ° at the liquid water reservoir, are derived from a calibrated thermocouple (K-type, d = 1 mm) positioned below the glassy carbon vessel and numerical simulations of the temperature field (cf. ESI†). The accuracy of the temperature T* is 0.2 K, that of T°, Θ*, and Θ° is estimated to be 1.0 K. A pressure transducer with a range of 0 to 10 MPa and an accuracy of 8 kPa (PXM459-100BGI, OMEGA Germany) is used for the synthesis experiments. For the rapid depressurization experiments, we use a transducer with a range of 0–700 kPa and an accuracy of 0.6 kPa (PXM459-007BGI, OMEGA Germany). The reactor R-CT is mounted on the manipulator of a lab-sized nanotom-m μCT system (GE Sensing & Inspection Technologies, Germany) operated at a tube voltage of 70 kV and a geometrical magnification factor of 16.7. Radiographs collected during the μCT measurements are used by GE's datos|x reconstruction software to compute the 3D image data with a voxel edge length of 6 μm.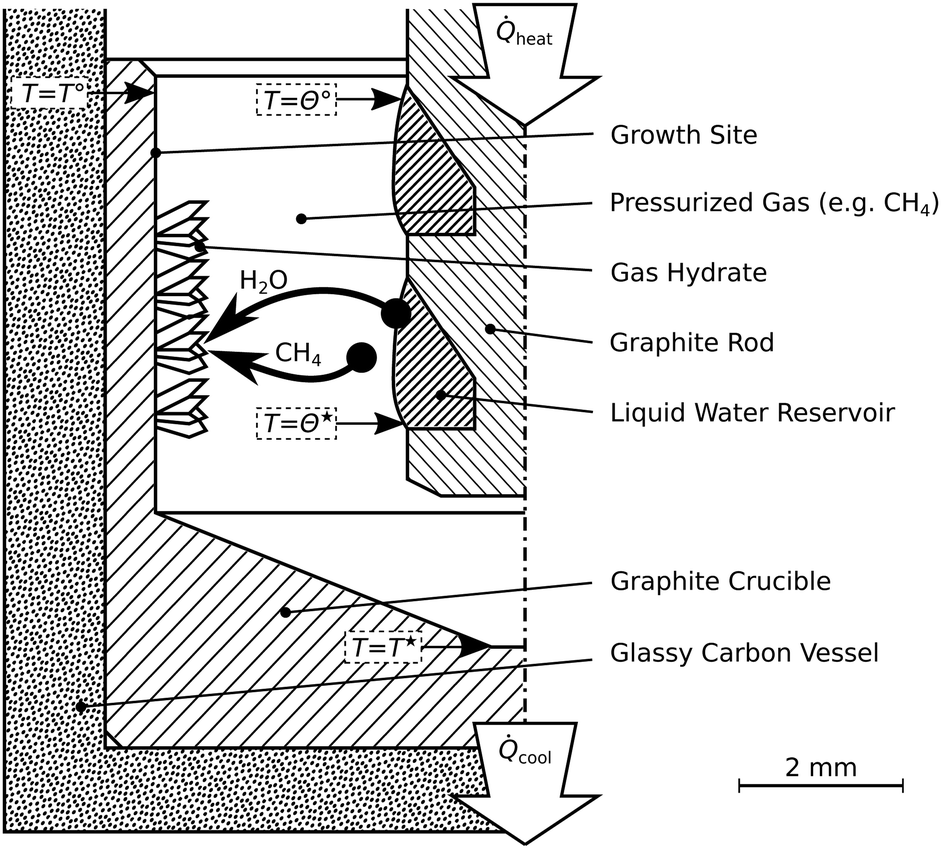 | ||
| Fig. 2 Schematic of the reactor R-CT. This reactor is used for the time-lapse micro-tomographic observation of hydrate synthesis by pressurized thermal evaporation. Synthesis temperatures labeled T*, T°, Θ*, and Θ° are summarized in Table 1. | ||
2.2 Reactor R-Vis
The second reactor allows visual inspection (cf.Fig. 3). It is termed R-Vis and employed to study the decomposition of hydrate layers subsequent to their synthesis at isothermal conditions. The maximum pressure in the reactor R-Vis is limited to 4 MPa due to the use of glass walls (ilmasil PN with wall thickness 15 mm, QSIL Germany). While the water vapor in the reactor R-CT is transported radially, from the reactor center to the reactor wall, the vapor in the reactor R-Vis is transported axially (cf.Fig. 3) from the aluminum base to the aluminum lid. The liquid water reservoir at the reactor base is evaporated using heat from electric heating cartridges. The cooling of the growth site at the top is done thermoelectrically (Peltier element QC-31-1.4-8.5MS, Quick-Ohm Germany). The temperatures T* and Θ* at the hydrate growth site and the liquid water reservoir, respectively, are measured with an accuracy of 0.2 K using calibrated thermocouples (K-type, d = 1 mm). The pressure sensors are the same as in the reactor R-CT.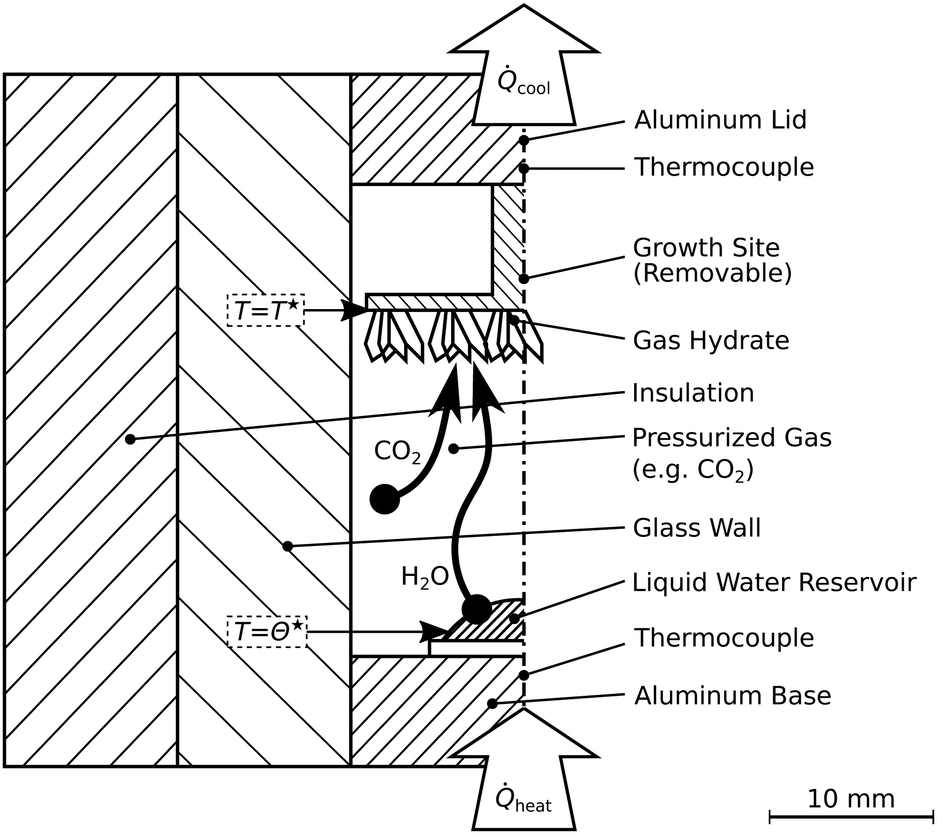 | ||
| Fig. 3 Schematic of the reactor R-Vis. This reactor allows visual access and is designed for the synthesis of thin hydrate layers and subsequent depressurization experiments. Synthesis temperatures labeled T* and Θ* are summarized in Table 1. | ||
2.3 Powder X-ray diffraction
The crystal structure of the hydrates is analyzed using PXRD at 20 K. A Bruker D8 Advance powder diffractometer equipped with a PheniX Helium Cryostat (Oxford Cryosystems, UK) is used for the post-synthesis measurements with Cu-Kα1 radiation (40 kV, 40 mA) in Θ/2Θ scanning mode. The PXRD measurements were done in the 2Θ range from 5–90° with a step width of 0.02°. The software GSAS-II41 is used to analyze the PXRD profiles and determine the phase fractions of hexagonal ice and structure I hydrate. Experimental details on the PXRD measurements and the refinement procedure are provided in the ESI.†2.4 Procedure in reactor R-CT
The course of a synthesis experiment in the reactor R-CT is described exemplarily for CH4 hydrate and shown in Fig. 4. After loading the liquid water into the reactor (see ESI,† for details on the loading procedure), the reactor is pressurized with CH4 from a gas cylinder and then cooled to the synthesis conditions. While during synthesis the temperature is kept constant, the pressure must be restored several times mainly to compensate for the inevitable permeation of CH4 through the gaskets of the tightly closed reactor. As determined in a test series, this permeation results in a pressure leak of approx. 15 kPa h−1 per 1 MPa reactor pressure (for CO2 the rate is approx. 10 kPa h−1 MPa−1). This large leakage rate is the consequence of experimental limitations associated with size restrictions and the use of glassy carbon as a material. These limitations make the use of metal gaskets impractical. In consequence, O-rings (PTFE and FKM) are used, which, in combination with the small surface to volume ratio of the reactor, result in large pressure drops because of leakage. The pressure drop caused by the uptake of CH4 by the growing hydrate is small when compared with the leakage rate and is estimated to account for approx. 5 kPa h−1 (for CO2 the uptake rate is approx. 7 kPa h−1). Clearly, this relatively small uptake rate (with respect to the leakage rate) makes the direct and accurate determination of gas uptake by the hydrate difficult. Therefore, the uptake of gas by the hydrate is not taken into consideration for the quantitative analysis of hydrate composition or growth dynamics. However, the measured leakage rates are still small enough to be neglected in decomposition experiments carried out at significantly lower pressures (approx. 0.1 MPa) and over shorter periods. Hence, the release of gas from the hydrate can be measured with high accuracy. The restoration of pressure is done with CH4 at ambient temperature. Thus, a small amount of heat is introduced into the reactor. Although additional heat might disturb the synthesis for a short time, it is of no effect in the long run. μCT scans and X-ray radiographs of the reactor are used to quantify the amount of liquid water in the reservoir. Once all liquid water is consumed by the hydrate, the synthesis is terminated by depressurization. When the guest gas CH4 is used, T* is decreased to 243 K before the reactor is opened (and hence depressurized) to avoid rapid hydrate decomposition. Contrary, T* is kept constant upon the depressurization in the case of CO2, because a reduction in temperature would result in the liquefaction of the gas. Either one of two procedures is carried out after depressurization: (1) the reactor is closed again to measure the gas released by the decomposing hydrate or (2) the reactor is immersed in liquid nitrogen to extract the hydrate sample for structure analysis by scraping it off the graphite crucible.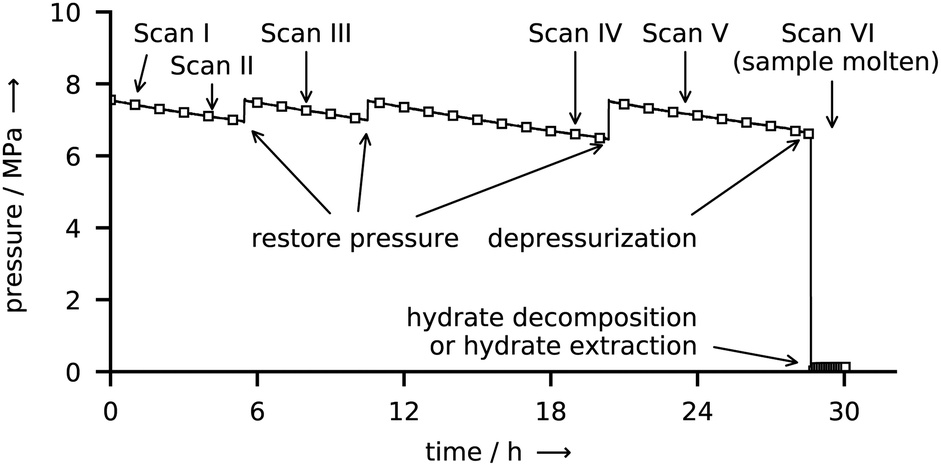 | ||
| Fig. 4 Gauge pressure and ending times of six μCT scans during a CH4 hydrate synthesis experiment. | ||
2.5 Procedure in reactor R-Vis
All experiments in the reactor R-Vis are started by loading 100 μL of water on the aluminum base of the reactor at ambient temperature. After flushing with CO2, the valves are closed and the reactor is pressurized to either 1 MPa or 2 MPa absolute pressure. Within 5 min after pressurization, the liquid water reservoir is heated to 303 K and the growth site is cooled to 265 K. Both temperatures are then kept constant for hydrate synthesis. During that period, the pressure drops from 2.0 MPa to 1.8 MPa or from 1.0 MPa to 0.9 MPa, respectively. A big part of this pressure drop is caused by the permeation of CO2 across the O-rings (FKM), which are chosen as gaskets for reasons of practicability and reactor dimensions. A series of dry (i.e., experiments with no water in the reactor) runs is used to distinguish the pressure drop due to leakage and that due to the uptake of gas by the hydrate (cf. Fig. S3 in the ESI†). The pressure drop in the reactor is reproducible in both dry and wet (i.e., experiments with water in the reactor) runs. Approximately (70 ± 20) kPa of pressure drop are due to the uptake by the growing hydrate. Yet, due to the large relative uncertainty, the gas uptake rate can only be used to indicate a trend but not for any quantitative analysis. After 20 h of synthesis no liquid water is left and a thin and uniform layer (mean thickness approx. 330 μm) of hydrate has formed on the growth site (cf. Fig. S9 in the ESI†). While keeping the temperature constant, the reactor is depressurized to 0.1 MPa by opening and closing a pressure relief valve within 5 s. Subsequently, the reactor pressure is monitored at a frequency of 1 Hz for 2 h. During the first hour of pressure monitoring, all temperatures are kept at the values used for hydrate synthesis. This allows the investigation of the hydrate decomposition dynamics at isothermal conditions. The second hour of monitoring is started by triggering the complete decomposition of the hydrate sample. To this end, the temperature T* is first raised at 5 K min−1 until it reaches 295 K, and then kept constant for the rest of the experiment. The purpose of the second hour of monitoring is solely to completely decompose the gas hydrate and to measure the total amount of gas released upon hydrate decomposition.2.6 Sample composition terminology
We use two terms for sample composition to distinguish between the sample (comprised of an ice and a hydrate phase) and the hydrate phase. The water to gas molar ratio, i.e., the number n in CO2·nH2O or in CH4·nH2O, here always refers to the sample and includes the excess water in ice form. The term “hydration number” is used only to denote the water to gas molar ratio of the pure hydrate phase.3 Results and discussion
Results are divided into two subsections. The first subsection demonstrates the use of the process for the synthesis of the gas hydrates of CO2 and CH4. The second subsection shows exemplarily for CO2 hydrate how the process can be applied to study hydrate decomposition at well-defined isothermal conditions. Table 1 summarizes the p–T conditions used for sample synthesis in the entire set of experiments.| Gas in reactor | CO2 in R-CT | CH4 in R-CT | CO2 in R-Vis |
|---|---|---|---|
| a Temperatures are according to Fig. 2 and 3 and described in detail in the ESI. b The large uncertainties in pressure during synthesis are due to leakage of gas in a small confinement and not due to measurement error. | |||
| Pressureb (MPa) | 2.2 ± 0.1 | 7.1 ± 0.6 | 0.95 ± 0.05 or 1.90 ± 0.10 |
| T* (K) | 260.5 ± 0.2 | 261.0 ± 0.2 | 265.0 ± 0.2 |
| T° (K) | 263.5 ± 1.0 | 264.5 ± 1.0 | — |
| Θ* (K) | 289.5 ± 1.0 | 285.5 ± 1.0 | 303 ± 0.2 |
| Θ° (K) | 290.0 ± 1.0 | 286.5 ± 1.0 | — |
3.1 Hydrate co-deposition by PTE under μCT Monitoring
The gas hydrates of CH4 and CO2 are formed by slow co-deposition of water and gas in the pressurized reactor R-CT. Exemplarily, we only show the morphology of the CH4 hydrate deposit, since the morphology of the CO2 hydrate appears identical in the μCT scans. | ||
| Fig. 5 Exemplary 3D renderings of μCT scans performed during a CH4 hydrate synthesis experiment. | ||
 | ||
| Fig. 6 Hydrate crystals grown on the graphite crucible before being scraped off for post-synthesis X-ray analysis under liquid nitrogen. | ||
Fig. 7 shows p–T data in the tightly closed reactor after the rapid depressurization at the end of a CH4 hydrate synthesis experiment. The temperature is first raised from 243 K to 263 K within 3 min and then raised at a rate of 1 K min−1 to 285 K. In the initial period after depressurization, the pressure increases steadily at a rate of roughly 3 kPa min−1. When a temperature of 268 K is reached the pressure increase rate changes abruptly to 34 kPa min−1 before it drops to only 0.2 kPa min−1 at 271 K and above. A pressure increase in the closed reactor is caused by the thermal expansion of gas and by the release of gas from the decomposing hydrate. Thus, the observed pressure curve points towards an initial period of hydrate decomposition followed by the abrupt and complete dissociation of the hydrate between 268 K and 271 K (i.e., between 8 min and 11 min). At temperatures above 271 K, the increase of pressure is caused by the thermal expansion of the gas alone. The p–T data at the beginning and upon the completion of hydrate decomposition is used to derive bounds of the released gas mass and of the water to gas molar ratio (see Section S5.4 of the ESI,† for a detailed derivation). In total, the CH4 hydrate sample with a water mass of 50 mg releases 5.6–6.8 mg of CH4 during decomposition yielding a water to gas molar ratio of 6.6–7.9. In an analogous experiment (see ESI,† Fig. S5) a release of 50.6–52.4 mg of CO2 and a water to gas molar ratio of 7.0–7.3 are determined for a CO2 hydrate sample with a water mass of 150 mg. Note that the computed bounds are conservative estimates since they do not account for the gas which might be released during the short period of depressurization. Hence, the actual water to gas molar ratios are smaller or equal to the numbers reported above.
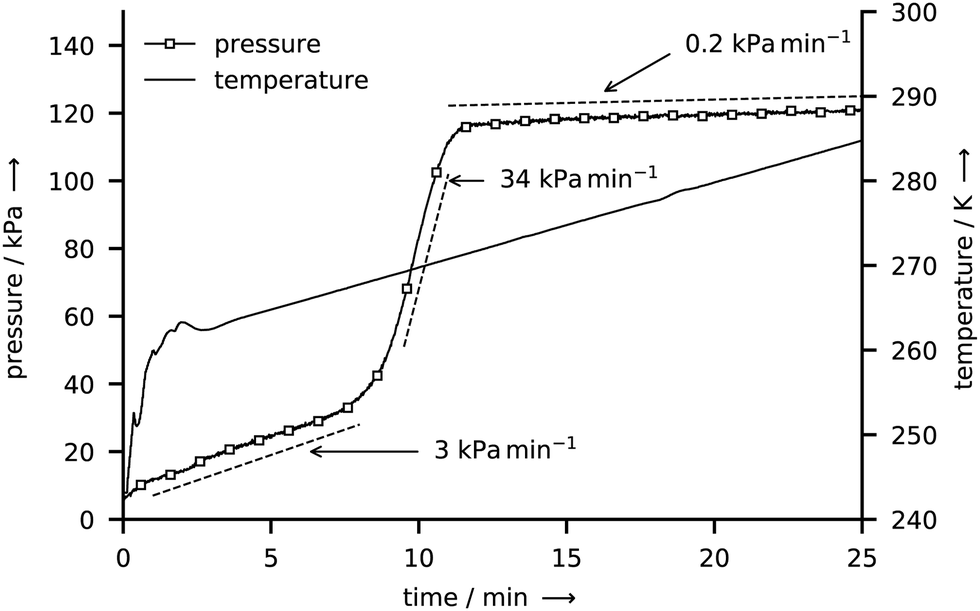 | ||
| Fig. 7 Gauge pressure and temperature T* during the decomposition of a CH4 hydrate sample after rapid depressurization. | ||
Approximately 100 mg of sample is collected at a rate of approx. 4 g per hour per square meter of growth site for both CO2 and CH4 hydrate by repeating the synthesis experiment several times. After each experiment, the hydrate is scraped off the graphite crucible under liquid nitrogen and then stored in liquid nitrogen before the structure analysis is performed. The PXRD profiles (cf.Fig. 8) both exhibit intense Bragg peaks at angles corresponding to the sI crystal structure and only minor Bragg peaks at angles corresponding to hexagonal ice. The refined mass fractions for the hexagonal ice phase are (6.3 ± 0.4)% for the CH4 hydrate sample and (7.3 ± 0.2)% for the CO2 hydrate sample (see Section S5.5 of the ESI,† for details on the refinement procedure). These mass fractions represent upper boundaries. The real amount of hexagonal ice in the samples is lower or equal to the one represented in the diffractograms since traces of hexagonal ice may deposit on top of the hydrate sample from the humid air during sample transfer. When the amount of water in the hexagonal ice is subtracted from the water mass of the samples, bounds for the hydration number follow from the water to gas molar ratios and are found to be 6.1–7.4 for the CH4 hydrate and 6.3–6.5 for the CO2 hydrate.
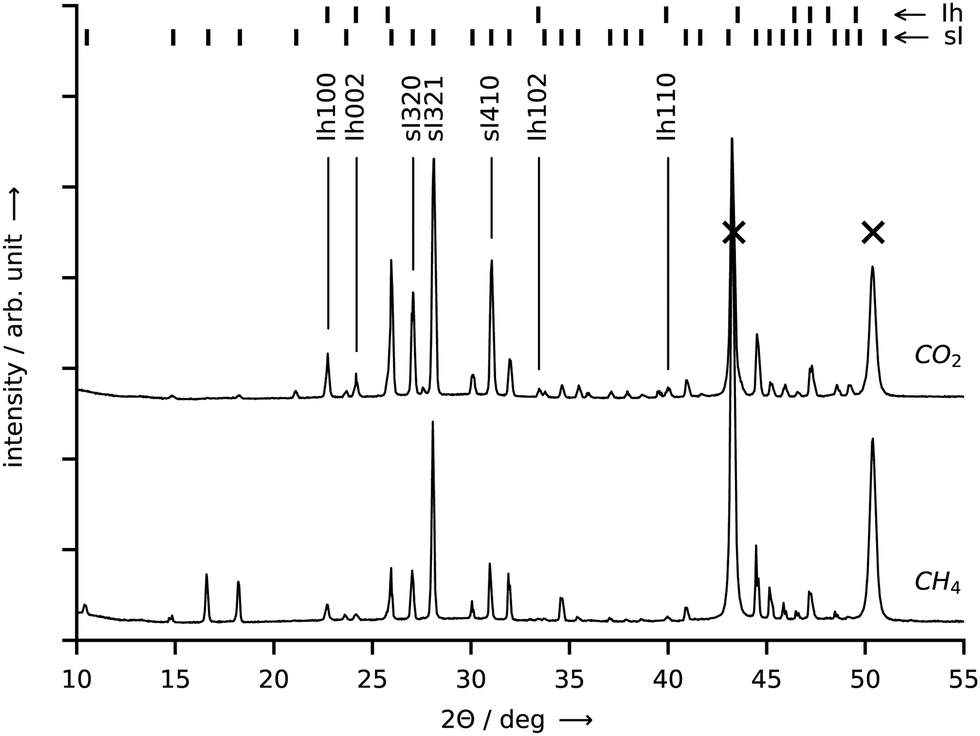 | ||
| Fig. 8 PXRD patterns obtained at 20 K from samples of CO2 hydrate (top curve) and CH4 hydrate (bottom curve). Both hydrates were synthesized in the reactor R-CT at the conditions listed in Table 1. Bragg peaks labelled with “sI” denote cubic structure I hydrate, “Ih” denotes hexagonal ice, and “X” denotes the sample holder. Cu-Kα1 radiation (λ = 1.5406 Å) was employed. Temperature resolved PXRD results in the range 80–300 K are provided in the ESI.† | ||
For CO2 hydrate, values ranging from 5.64 to 8.55 are listed in three compilations of hydration numbers.44–46 The scatter in the values is presumably caused by different conditions (pressure, temperature, dissolved gas concentration) during hydrate formation and by the use of different techniques for the measurement of the hydration numbers.44 Values close to the optimum of 5.75 (sI hydrate) are reported at formation pressures which are several times the equilibrium pressure.45 At formation pressures similar to ours (1 to 2 MPa), the majority of values lies in the range between 6 and 7, which reflects the difficulty of filling the small cages of the hydrate, while the large cages are almost fully occupied.47 In the case of CH4 hydrate, most reported experimental values for the hydration number are approx. 6.0 over a wide range of temperatures.46,48–53 This corresponds to cage occupancies for small and large cages of 90% and more.46 With increasing synthesis pressure, the hydration number of CH4 hydrate decreases slightly and approaches 5.75.52 Hence, while our result for the CO2 hydrate agrees well with the literature, it seems that we overestimate the hydration number for the CH4 hydrate.
3.2 Decomposition dynamics of co-deposited hydrate
Two series of experiments are conducted in the reactor R-Vis to study the effect of the formation pressure on CO2 hydrate decomposition. Except for different formation pressures, both series of experiments are identical. Besides, as expressed by identical trends in the gas uptake data (see Fig. S3 in the ESI†), the formation kinetics seem to be very similar as well. At both formation pressures, the gas uptake rate is largest at the beginning of the formation experiments and then gradually decreases until it vanishes after approximately 19 h of formation. This trend can be explained by the gradually decreasing surface area of the liquid water reservoir (i.e., the liquid water droplet).Subsequent to their formation, the depressurization destabilizes the hydrates, which start to decompose and release their gas. The accompanying increase in reactor gauge pressure is shown in Fig. 9. After one hour of hydrate decomposition at 265 K the temperature at the growth site is raised to 295 K at a rate of 5 K min−1. A vigorous release of gas accompanied by a subtle change in sample appearance from an opaque lusterless to a lustrous bright white is observed between approximately 271 K and 273 K. No changes in the sample morphology and no signs of liquid water are visible during that period of strong gas release. Both, the gas release and the change in color point towards the complete decomposition of the hydrate to gas and ice. The complete decomposition of hydrates slightly below the melting point of ice has been observed before20 and also reported by other authors.12,45,54 First signs of sample melting occur only later at approximately 274 K and above. The deviation from the melting point of ice can be explained by thermal lag due to the temperature ramping. After 1 h at 295 K, the reactor pressure equilibrates and the experiments are terminated.
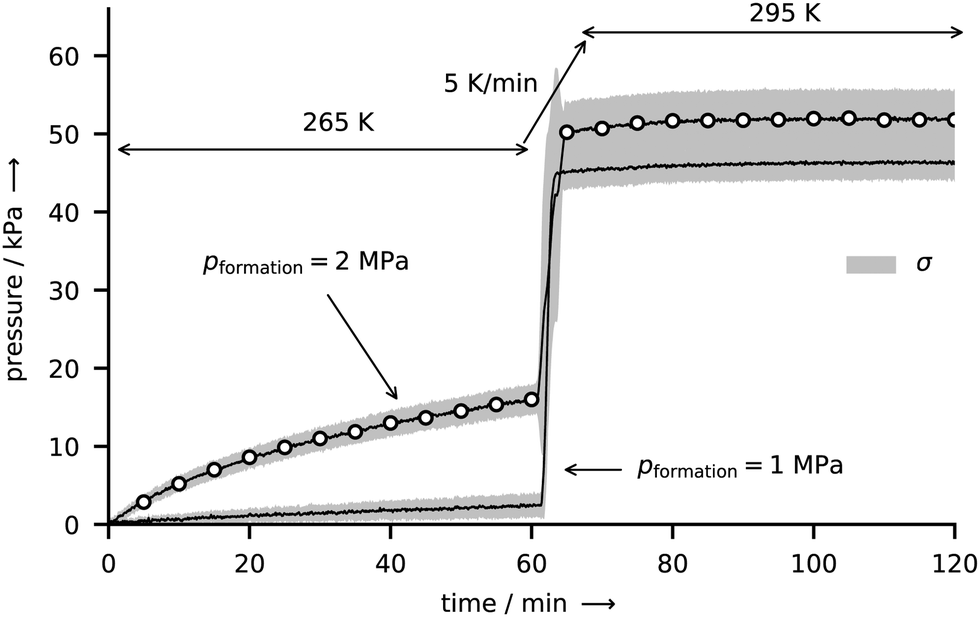 | ||
Fig. 9 CO2 hydrate decomposition as expressed by the release of gas and the accompanied increase in pressure in the closed reactor R-Vis immediately after rapid depressurization. The temperature program is indicated by the arrows (265 K for 1 h, heating to 295 K at 5 K min−1, 295 K for 1 h). Each curve represents the mean decomposition pressure ![[p with combining macron]](https://www.rsc.org/images/entities/i_char_0070_0304.gif) derived from five identical decomposition experiments. The curve marked with open circles results from samples formed at 265 K and 2 MPa. The solid line refers to the formation condition 265 K and 1 MPa. Both curves are enveloped by a gray sleeve representing values in the range [ − σ, + σ], where σ denotes the standard deviation of the five respective experiments. Note that the pressure data have been corrected by the effects of thermal gas expansion and degassing of CO2 from the gaskets using series of dry runs at both 1 MPa and 2 MPa formation pressure (cf. Fig. S4 in the ESI†). derived from five identical decomposition experiments. The curve marked with open circles results from samples formed at 265 K and 2 MPa. The solid line refers to the formation condition 265 K and 1 MPa. Both curves are enveloped by a gray sleeve representing values in the range [ − σ, + σ], where σ denotes the standard deviation of the five respective experiments. Note that the pressure data have been corrected by the effects of thermal gas expansion and degassing of CO2 from the gaskets using series of dry runs at both 1 MPa and 2 MPa formation pressure (cf. Fig. S4 in the ESI†). | ||
Three key observations can be made from Fig. 9, as described in the following paragraphs.
This variation in end pressures within one set of experiment is of stochastic nature and can be explained using the period of nucleation observed in the initial stage of deposition. The visual inspection of the hydrate growth site of reactor R-Vis suggests that the water vapor is initially condensed as micrometer-sized liquid droplets. Within approx. 30 min, these droplets nucleate (either as hydrate or ice) almost simultaneously, as can be inferred from a change in appearance from transparent to opaque. The remarkable simultaneous nucleation of dispersed liquid droplets has been observed before, however, an explanation has remained elusive.43 The crystals formed upon nucleation later act as seed crystals for the further deposition process and grow with time. Hence, the composition of the initial deposit has a determining influence on the fraction of ice and hydrate in the sample and thus on the end pressure.
The small difference in mean end pressure between the two sets of experiment can be due to two factors. A deterministic factor is the dependence of the hydrate cage occupancy Θ on the pressure p. The statistical theory of van der Waals and Platteeuw1,55 suggests that Θ follows the Langmuir isotherm
 | (1) |
For a positive constant C, Θ(p) grows strictly monotonically with p. Hence, a higher formation pressure results in a larger cage occupancy (i.e., more of the hydrogen-bonded polyhedra are occupied by a guest molecule) and thus in a larger end pressure. Although it seems established that higher pressures during synthesis yield CO2 hydrate with higher cage occupancies,45,56,57 the Langmuir isotherm could not be confirmed for the small cages of the hydrate.47 Particularly at p–T conditions similar to ours, the hydration number of CO2 hydrate seems to be only weakly sensitive to synthesis pressure (at 273 K a hydration number of 6.9 and 6.3 is reported for 1.5 MPa and 6.0 MPa, respectively).47
The second factor is again the period of nucleation. An increase in formation pressure directly raises the driving force for hydrate nucleation.1 At the same time, however, an increase in formation pressure can also indirectly affect nucleation via the change of mass and heat transfer in the reactor. The impact of these factors has not yet been determined, though. Although the available number of experiments is insufficient for a proper statistical analysis, on average, induction times for nucleation are shorter (approx. 10 min) for 2 MPa than for 1 MPa (approx. 30 min) formation pressure. The enhanced nucleation at 2 MPa formation pressure might therefore result in a larger hydrate fraction in the sample and thus in a larger end pressure.
In general, nucleation is of major importance for the co-deposition of hydrates by PTE, particularly at temperatures where liquid water can exist in stable or supercooled form. For instance, Cady35,36 noticed the incomplete conversion to hydrate at temperatures close to the ice point on several occasions and attributed it to a failure in nucleation. To ensure better nucleation, Cady often used dry ice (195 K) to cool the hydrate growth site at the beginning of his experiments. Poor nucleation can also explain the comparably large water to gas molar ratios (larger than 13.5; see Table S4 in the ESI†) found for CO2 hydrates formed in the reactor R-Vis. For comparison, the ratios for CO2 hydrates in the reactor R-CT are less than 7.3. Since the synthesis conditions for CO2 hydrate are similar in both reactors, cage occupancies and hydration number are expected to be similar as well. Therefore, the hydration number cannot account for such a large difference in the water to gas molar ratios. Thus, the difference arises mainly from the ice fraction of the samples. This implies either that the slightly lower temperature in the reactor R-CT improves nucleation, or that hydrate nucleation works better on the graphite surface in reactor R-CT than on the aluminum growth site of reactor R-Vis. We measure a contact angle of 50–60° for water on both the machined graphite and the aluminum surface using photographs of 5 μL droplets on the surfaces. Accordingly, the reduction of the free energy barrier, which has to be surmounted for the formation of a nucleus of critical size,1 is similar on both growth sites. Therefore, the higher synthesis temperature is more likely to be the reason for the deteriorated nucleation on the aluminum growth site. However, an aging effect of the aluminum surface was observed in a series of test runs. While the synthesis of hydrates works well on a fresh aluminum (oxide) surface, it becomes more and more difficult as the surface ages. After roughly two weeks, nucleation at 265 K and both 1 MPa and 2 MPa becomes almost impossible. This aging effect can be the result of a wettability transition from a fresh hydrophilic aluminum oxide surface to an aged and hydrophobic surface. The formation of the hydrophobic surface is presumably caused by the adsorption of organic species from air.58 In terms of classical nucleation theory, the change in contact angle implies a transition from a low to a high energy barrier, which must be overcome to form nuclei of critical size.1
The good agreement between the dissociation curves within single sets of experiments is a direct result of the fine control of experimental conditions and shows a strength of the applied synthesis procedure. The sample mass is well defined by the known amount of loaded water. Similarly, the sample-gas interface area is directly related to the area of the growth site, since the sample is deposited uniformly on that. Moreover, the good thermal contact between the thin layer of sample and the cooled growth site allows the efficient transport of heat and ensures isothermal conditions throughout the sample. The small variations between the results are presumably mainly due the stochastic nature of hydrate nucleation and are assumed to decrease with lower synthesis temperatures or higher synthesis pressures.
In general, hydrates of CO2 exhibit very low rates of dissociation upon the rapid depressurization from stability pressures to 0.1 MPa in the temperature range of 240–271 K, which is explained by the effect of self-preservation.15,45 According to the decomposition temperature of 265 K and considering the low rates of dissociation (more than two thirds of hydrate outlast 60 min outside the stability region), we conclude that all of our own samples are self-preserved as well. However, samples formed at 1 MPa seem to be better preserved than those formed at 2 MPa. The resulting two distinct dissociation curves are hard to explain using the popular argument of a diffusion limiting layer of ice, which forms at the free surface of the hydrate in the initial phase of decomposition. This is because the type of guest molecule as well as temperature and pressure are identical between sets of experiments during the dwell time at 265 K. It is possible though that the microstructure, which is assumed to be an important ingredient of the self-preservation phenomenon,15 is different between sample sets and thus at the origin of the differing dissociation behavior. Differences in the microstructure can be, for instance, due to the initial phase of deposition. Since different induction times are observed between the two sets of samples, the initial structure for further deposition might be different as well. Besides, differences in heat and mass transfer during deposition can lead to different microstructure, too.
An alternative explanation can be derived from the end pressures of the two experiment sets. Because the end pressures depicted in Fig. 9 overlap, we can rule out a relation between the two dissociation behaviors and the bulk molar to water ratio. Nevertheless, differences in the hydration number as well as differences in the sample ice fraction can both have an influence on the decomposition behavior of the hydrate. A difference in hydration numbers means a difference in hydrate cage occupancies and implies different chemical potentials. Such a difference in chemical potential leads to differing driving forces for decomposition which can additionally affect the microstructural component of self-preservation.15 Since an ice fraction larger than 7 wt% is found in the PXRD investigations of the CO2 hydrate samples produced in the reactor R-CT under similar conditions, we infer that the samples produced in the reactor R-Vis also exhibit a considerable amount of ice. This assumption is further substantiated by the significantly larger water to gas molar ratio found in the samples of reactor R-Vis (>13 compared to <8; see Tables S3 and S4 in the ESI†). Consequently, a variation in the ice fraction of the samples can have severe effects on the mass transfer of gas from the decomposing hydrate, regardless of the existence of an ice shield covering the hydrate. An increase in ice fraction can for instance change the sample structure from “ice embedded in a hydrate matrix” to “hydrate embedded in an ice matrix”, a structure which can preserve the gas for many months.17 The ice fraction of the samples is likely to be evenly distributed due to the stochastic nature of nucleation. Yet, the two dissociation rates are deterministic. This suggests rather that the hydration number or the microstructure are responsible for the differences in decomposition dynamics.
4 Conclusions
As evidenced by PXRD and p-T data, we successfully synthesized CO2 and CH4 cubic structure I clathrate hydrate at temperatures of 261–265 K and corresponding stability pressures by advancing a rediscovered deposition technique, which makes use of the thermal evaporation of liquid water in a reactor pressurized with the guest gas. The redesign of the reactor extends the pressure range accessible to 10 MPa. This allows us to not only study more exotic guest gases, but also gases relevant on Earth, namely CO2 and CH4, via this gas phase deposition process. We propose its utilization for hydrates of further guests, no matter the required formation pressure.Our samples display facets on macroscopic crystals with a side length of up to 1 mm forming side by side on the deposition site. When deposited on graphite, the samples exhibit a hydration number under 7.4 and contain less than 8 wt% of free water in the form of hexagonal ice. The deposition on aluminum yields larger fractions of free water in the sample and water to gas molar ratios above 13.5. The difference in sample composition is explained with the initial stage of deposition, which is characterized by the heterogeneous nucleation of hydrate and ice from water droplets condensed on the hydrate growth site.
At the conditions used in this study, hydrate deposition via pressurized thermal evaporation (PTE) is a very slow process with formation rates of the order of milligrams of hydrate per hour and square centimeter deposition site area. This is particularly slow compared to spray injection or stirred reactors where hydrate formation rates of the order of grams per minute and more are achieved in reactors of sizes similar to ours.22,59 Nevertheless, due to the scalability with deposition site area as well as due to the absence of a lower boundary for formation temperature, the process might still prove beneficial for hydrate-based applications (e.g., gas separation). Besides, the choice of other synthesis conditions (temperature field, partial pressures) might speed up the deposition process. More important in the context of the present work, however, is that the deposition of hydrates by PTE offers precise control of mass transport via the reactor temperature field and the partial pressures of water and gas. This allows the formation of thin hydrate deposits of reproducible composition over a wide range of p–T conditions (below and above the freezing point of water) and facilitates the study of hydrate decomposition kinetics at well-controlled initial conditions (pressure, temperature, sample mass and surface area). In two series of deposition-decomposition experiments, we demonstrate this capability of the process for the first time in a study of the decomposition dynamics of self-preserved CO2 hydrates. Our results show that differences in deposition conditions can have severe effects on the subsequent decomposition kinetics. Hydrate samples deposited at 265 K and a pressure of 1 MPa exhibit an extra high degree of self-preservation, termed super-preservation. Super-preservation is expressed by a dissociation rate 85% lower than that of self-preserved samples formed at the same temperature and a pressure of 2 MPa, while the bulk water to gas molar ratio of super-preserved samples is only 12% above that of the self-preserved ones. Although the physical chemistry behind this remarkable reduction in dissociation rate is not clear yet, the deterministic formation of super-preserved hydrates of CO2via the PTE process is possible already.
However, future work is needed to understand the cause for different sample compositions as well as their influence on the decomposition kinetics. That involves the determination of the relation between synthesis conditions, hydrate growth rate and hydrate composition, as well as the careful analysis of hydrate nucleation at the initial phase of deposition. Moreover it will be interesting to examine, whether super-preservation is related to the recently observed barriers to decomposition, which were explained similarly with variations in sample composition.20 Such work will not only result in a better understanding of the synthesis procedure on its own, but it can also improve our understanding of hydrate formation, decomposition and self-preservation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Austrian Research Promotion Agency FFG (project TomoFuMa, No. 839070, and project EARLYSNOW) and the Austrian Science Fund FWF (project I1392) for financial support.References
- E. D. Sloan and C. A. Koh, Clathrate Hydrates of Natural Gases, CRC Press, Boca Raton, FL, USA, 3rd edn, 2007 Search PubMed.
- Z. R. Chong, S. H. B. Yang, P. Babu, P. Linga and X.-S. Li, Appl. Energy, 2016, 162, 1633–1652 CrossRef.
- H. P. Veluswamy, A. Kumar, Y. Seo, J. D. Lee and P. Linga, Appl. Energy, 2018, 216, 262–285 CrossRef CAS.
- P. Englezos and J. D. Lee, Korean J. Chem. Eng., 2005, 22, 671–681 CrossRef CAS.
- T. Hatakeyama, E. Aida, T. Yokomori, R. Ohmura and T. Ueda, Ind. Eng. Chem. Res., 2009, 48, 4083–4087 CrossRef CAS.
- T. Peters, J. Smith and J. Brisson, J. Food Eng., 2010, 100, 669–677 CrossRef CAS.
- L. Fournaison, A. Delahaye, I. Chatti and J.-P. Petitet, Ind. Eng. Chem. Res., 2004, 43, 6521–6526 CrossRef CAS.
- S. Takeya and J. A. Ripmeester, Angew. Chem., 2008, 120, 1296–1299 CrossRef.
- D. Davidson, S. Garg, S. Gough, Y. Handa, C. Ratcliffe, J. Ripmeester, J. Tse and W. Lawson, Geochim. Cosmochim. Acta, 1986, 50, 619–623 CrossRef CAS.
- Y. Handa, J. Chem. Thermodyn., 1986, 18, 891–902 CrossRef CAS.
- V. S. Yakushev and V. Istomin, in Physics and Chemistry of Ice, ed. N. Maeno and T. Hondoh, Hokkaido Univ. Press, Sapporo, Japan, 1992, pp. 136–140 Search PubMed.
- L. A. Stern, S. Circone, S. H. Kirby and W. B. Durham, J. Phys. Chem. B, 2001, 105, 1756–1762 CrossRef CAS.
- A. Falenty, W. F. Kuhs, M. Glockzin and G. Rehder, Energy Fuels, 2014, 28, 6275–6283 CrossRef CAS.
- S. Takeya, T. Ebinuma, T. Uchida, J. Nagao and H. Narita, J. Cryst. Growth, 2002, 237-239, 379–382 CrossRef CAS.
- A. Falenty and W. F. Kuhs, J. Phys. Chem. B, 2009, 113, 15975–15988 CrossRef CAS PubMed.
- S. Takeya, W. Shimada, Y. Kamata, T. Ebinuma, T. Uchida, J. Nagao and H. Narita, J. Phys. Chem. A, 2001, 105, 9756–9759 CrossRef CAS.
- V. A. Istomin, V. S. Yakushev, N. A. Makhonina, V. G. Kwon and E. M. Chuvilin, Gas Industry of Russia, 2006, 4, 16–27 Search PubMed.
- V. P. Melnikov, L. S. Podenko, A. N. Nesterov, A. O. Drachuk, N. S. Molokitina and A. M. Reshetnikov, Dokl. Chem., 2016, 466, 53–56 CrossRef CAS.
- W. F. Kuhs, G. Genov, D. K. Staykova and T. Hansen, Phys. Chem. Chem. Phys., 2004, 6, 4917–4920 RSC.
- S. Arzbacher, N. Rahmatian, A. Ostermann, B. Massani, T. Loerting and J. Petrasch, Phys. Chem. Chem. Phys., 2019, 21, 9694–9708 RSC.
- H. P. Veluswamy, A. Kumar, R. Kumar and P. Linga, Appl. Energy, 2017, 188, 190–199 CrossRef CAS.
- R. Ohmura, S. Kashiwazaki, S. Shiota, H. Tsuji and Y. H. Mori, Energy Fuels, 2002, 16, 1141–1147 CrossRef CAS.
- S. Matsuda, H. Tsuda and Y. H. Mori, AIChE J., 2006, 52, 2978–2987 CrossRef CAS.
- T. Kobayashi, N. Imura, R. Ohmura and Y. H. Mori, Energy Fuels, 2007, 21, 545–553 CrossRef CAS.
- Y.-T. Luo, J.-H. Zhu, S.-S. Fan and G.-J. Chen, Chem. Eng. Sci., 2007, 62, 1000–1009 CrossRef CAS.
- D. K. Staykova, W. F. Kuhs, A. N. Salamatin and T. Hansen, J. Phys. Chem. B, 2003, 107, 10299–10311 CrossRef CAS.
- J. P. Devlin and I. A. Monreal, J. Phys. Chem. A, 2010, 114, 13129–13133 CrossRef CAS PubMed.
- J. E. Bertie and J. P. Devlin, J. Chem. Phys., 1983, 78, 6340–6341 CrossRef CAS.
- D. Blake, L. Allamandola, S. Sandford, D. Hudgins and F. Freund, Science, 1991, 254, 548–551 CrossRef CAS PubMed.
- A. Hallbrucker, Angew. Chem., Int. Ed. Engl., 1994, 33, 691–693 CrossRef.
- H. Nakayama, D. D. Klug, C. I. Ratcliffe and J. A. Ripmeester, Chem. – Eur. J., 2003, 9, 2969–2973 CrossRef CAS.
- C. Mitterdorfer, M. Bauer and T. Loerting, Phys. Chem. Chem. Phys., 2011, 13, 19765 RSC.
- F. Fleyfel and J. P. Devlin, J. Phys. Chem., 1991, 95, 3811–3815 CrossRef CAS.
- R. D. Bee and A. M. Palmer, EU Pat., EP0355875B1, 1993 Search PubMed.
- G. H. Cady, J. Phys. Chem., 1981, 85, 3225–3230 CrossRef CAS.
- G. H. Cady, J. Phys. Chem., 1983, 87, 4437–4441 CrossRef CAS.
- G. H. Cady, J. Phys. Chem., 1985, 89, 3302–3304 CrossRef CAS.
- P. J. Ceccotti, Ind. Eng. Chem. Fundam., 1966, 5, 106–109 CrossRef CAS.
- D. M. Mattox, in Handbook of Physical Vapor Deposition (PVD) Processing, ed. W. Andrew, Elsevier Science, Norwich, N.Y., Oxford, 2010 Search PubMed.
- L. A. Feldkamp, L. C. Davis and J. W. Kress, J. Opt. Soc. Am. A, 1984, 1, 612 CrossRef.
- B. H. Toby and R. B. V. Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- A. Bogdan, M. J. Molina, H. Tenhu, E. Mayer and T. Loerting, Nat. Chem., 2010, 2, 197–201 CrossRef CAS PubMed.
- P. Servio and P. Englezos, AIChE J., 2003, 49, 269–276 CrossRef CAS.
- T. Uchida, Waste Manage., 1998, 17, 343–352 CrossRef.
- S. Circone, L. A. Stern, S. H. Kirby, W. B. Durham, B. C. Chakoumakos, C. J. Rawn, A. J. Rondinone and Y. Ishii, J. Phys. Chem. B, 2003, 107, 5529–5539 CrossRef CAS.
- R. Sun and Z. Duan, Geochim. Cosmochim. Acta, 2005, 69, 4411–4424 CrossRef CAS.
- W. F. Kuhs, B. Chazallon, A. Klapproth and F. Pauer, Rev. High Pressure Sci. Technol., 1998, 7, 1147–1149 CrossRef CAS.
- G. K. Anderson, J. Chem. Thermodyn., 2004, 36, 1119–1127 CrossRef CAS.
- J. A. Ripmeester and C. I. Ratcliffe, J. Phys. Chem., 1988, 92, 337–339 CrossRef CAS.
- Y. Handa, J. Chem. Thermodyn., 1986, 18, 915–921 CrossRef CAS.
- A. K. Sum, R. C. Burruss and E. D. Sloan, J. Phys. Chem. B, 1997, 101, 7371–7377 CrossRef CAS.
- S. Circone, S. H. Kirby and L. A. Stern, J. Phys. Chem. B, 2005, 109, 9468–9475 CrossRef CAS PubMed.
- T. Uchida, T. Hirano, T. Ebinuma, H. Narita, K. Gohara, S. Mae and R. Matsumoto, AIChE J., 1999, 45, 2641–2645 CrossRef CAS.
- L. A. Stern, S. Circone, S. H. Kirby and W. B. Durham, Can. J. Phys., 2003, 81, 271–283 CrossRef CAS.
- J. H. van der Waals and J. C. Platteeuw, in Advances in Chemical Physics, ed. I. Prigogine, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1959, vol. 2, pp. 1–57 Search PubMed.
- R. W. Henning, A. J. Schultz, V. Thieu and Y. Halpern, J. Phys. Chem. A, 2000, 104, 5066–5071 CrossRef CAS.
- K. A. Udachin, C. I. Ratcliffe and J. A. Ripmeester, J. Phys. Chem. B, 2001, 105, 4200–4204 CrossRef CAS.
- J. Long, M. Zhong, H. Zhang and P. Fan, J. Colloid Interface Sci., 2015, 441, 1–9 CrossRef CAS PubMed.
- H. P. Veluswamy, A. J. H. Wong, P. Babu, R. Kumar, S. Kulprathipanja, P. Rangsunvigit and P. Linga, Chem. Eng. J., 2016, 290, 161–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Section S1: custom-built reactor R-CT. Section S2: thermodynamic equilibrium conditions. Section S3: loading procedures. Section S4: PXRD experimental details. Section S5: complementary results. See DOI: 10.1039/c9cp04735b |
| This journal is © the Owner Societies 2020 |