
Pivotal role of the redox-active tyrosine in driving the water splitting catalyzed by photosystem II†
Shin
Nakamura
a,
Matteo
Capone
b,
Daniele
Narzi
c and
Leonardo
Guidoni
 *d
*d
aDepartment of Biochemical Sciences “A. Rossi Fanelli”, University of Rome “Sapienza”, P.le Aldo Moro 5, 00185, Rome, Italy
bDepartment of Information Engineering, Computational Science, and Mathematics, Università dell’Aquila, L’Aquila, 67100, Italy
cInstitute of Chemical Sciences and Engineering Ecole Polytechnique Federale de Lausanne Av. F.-A. Forel 2, 1015 Lausanne, Switzerland
dDepartment of Physical and Chemical Science, Università dell’Aquila, L’Aquila, 67100, Italy. E-mail: leonardo.guidoni@univaq.it
First published on 28th November 2019
Abstract
Photosynthetic water oxidation is catalyzed by the Mn4Ca cluster in photosystem II (PSII). The nearby redox-active tyrosine (YZ) serves as a direct electron acceptor of the Mn4Ca cluster and it forms a low-barrier H-bond (LBHB) with a neighboring histidine residue (D1-His190). Experimental evidence indicates that YZ oxidation triggers changes in the hydrogen bonding network that precede proton abstraction from the Mn4Ca cluster. In order to characterize such changes, we compare ab initio molecular dynamics simulations of different states of the catalytic cycle of PSII with dynamics of isolated tyrosine models (namely, p-cresol) in different oxidation states. The systematic comparison of the H-bond networks in different simulated systems suggests that the YZ oxidation leads to a water hydration pattern which is more similar to that of the neutral p-cresol rather than that of the p-cresol anion. Our simulations also reveal the twofold nature of the interactions between YZ and the Mn4Ca cluster. Firstly, the YZ oxidation triggers rapid structural changes of the H-bond pattern in the proximity of the cluster which have been observed to propagate on the ps time scale on the Ca2+ hydration shell up to other water molecules in the proximity of the cluster. Secondly, it is clear that YZ interacts with the Mn4Ca cluster also through Coulombic interactions mediated by CP43-Arg357 through the remaining positive charge of the 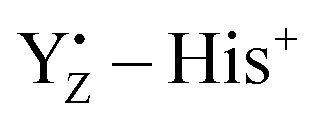 pair. Our results are able to identify, for the first time, the structural rearrangements guided by the oxidation of YZ necessary for the evolution of the water splitting reaction in PSII. Based on these findings, we propose a mechanism of structural changes which is functional towards the progression of the catalytic cycle in PSII.
pair. Our results are able to identify, for the first time, the structural rearrangements guided by the oxidation of YZ necessary for the evolution of the water splitting reaction in PSII. Based on these findings, we propose a mechanism of structural changes which is functional towards the progression of the catalytic cycle in PSII.
Introduction
The presence of oxygen in the atmosphere is required for the survival of all life forms that breathe, and it has been ultimately generated by photosynthetic water oxidation occurring in plants, algae and cyanobacteria.4–6 Elucidation of the chemical mechanism of water oxidation is crucial for improvement and development of solar fuel systems and artificial photosynthesis devices.7–10 The role of photosystem II (PSII), the water splitting complex embedded in the thylakoid membrane, is to obtain the electrons from water oxidation for subsequent CO2 fixation to produce sugars.11–16 Water oxidation splits water into protons and oxygen molecules, and it takes place in the water-oxidizing center (WOC) located on the luminal side of the D1 protein in PSII (Fig. 1A).17–20 | ||
| Fig. 1 The atomic structure of (A) amino acid ligands and (B) the hydrogen bond network around the Mn4Ca cluster in the XFEL structure at a resolution of 1.95 Å (PDB code: 4UB6) with the direction of three channels linking to the bulk reported previously1–3 (green arrows). The interacting amino acids and the water molecules are selectively shown as the QM region. | ||
Several theoretical and experimental studies have provided considerable details regarding the electron transfer routes and rates in PSII.5,21–27 The light-induced excited state of the reaction center of chlorophylls (i.e., the special pair of chlorophyll moieties, P680 and ChlD1) releases an electron to the electron acceptor side. P680+ that is generated extracts an electron from a redox-active tyrosine, YZ. The resulting  can oxidize the Mn4Ca cluster and convert back to the reduced form on a time scale that ranges from 20 μs to 2 ms.28 Finally, the Mn4Ca cluster oxidizes two water molecules, passing through five intermediate states (Si → Si+1, i = 0–4), which is referred to as the Kok–Joliot cycle.29,30
can oxidize the Mn4Ca cluster and convert back to the reduced form on a time scale that ranges from 20 μs to 2 ms.28 Finally, the Mn4Ca cluster oxidizes two water molecules, passing through five intermediate states (Si → Si+1, i = 0–4), which is referred to as the Kok–Joliot cycle.29,30
The redox reaction of a tyrosine side chain involves a proton-coupled electron transfer (PCET) reaction. A tyrosine side chain releases a phenolic proton upon its oxidation due to the extremely low pKa (the pKa value is approximately −2).31 When YZ is oxidized by P680+, it releases a proton from the phenolic group to a neighboring histidine residue, D1-His190 (Fig. 1B).32–35 The recently reported high-resolution X-ray structure of photosystem II reveals an extremely short-distance H-bond (∼2.5 Å) between the Nτ atom of D1-His190 and the phenolic oxygen of YZ.17–19 The QM/MM data also revealed that YZ and His190 form a low-barrier H-bond (LBHB).36 This LBHB greatly facilitates the rapid PCET reaction of YZ due to the lower barrier proton transfer from the phenolic group to D1-His190. This is a basic mechanistic feature of the redox reaction of YZ.
Another redox-active tyrosine, YD, located symmetrically in the D2 protein of PSII, is also able to donate an electron to P680+. YD functions as a secondary electron transfer pathway and shows competitive electron transfer with YZ. The redox reaction rate of YD is much slower than that of YZ, the YD radical form being stabilized by a different arrangement of the Tyr–His pair and the surrounding water molecules.33,34 The H-bond structure and the PCET mechanism of YD have been elucidated in recent years. A theoretical study by Saito et al. predicted that the phenolic proton of YD is released to the bulk via an H-bonding network upon YD oxidation.37 Proton release to the bulk upon YD oxidation has indeed been demonstrated by Fourier transform infrared (FTIR) spectroscopy.38 The difference in the proton transfer reactions of the two tyrosines, YZ and YD, underlines their functional difference. These two proton-release reactions are characterized by the H-bond directions caused by the different protonation states of their neighboring histidine residues. Although data regarding the difference in PCET mechanisms of the two tyrosines have been reported previously, the factors determining the H-bond properties, such as the strength and the number of H-bonds of the tyrosine side chains, are still a matter of debate.
Although, recently, the different steps of the Kok–Joliot cycle have attracted a lot of attention and are studied actively,39–49 the role of YZ in water oxidation is still one of the main issues that need to be elucidated for the mechanism of water oxidation. The X-ray structure of PSII also shows that YZ is able to interact with the Mn4Ca cluster via H-bond networks, comprising several water molecules, and that it is located in the H-bond network linking the cluster to the luminal surface.17–19 Electron paramagnetic resonance (EPR) has been used to examine the magnetic and electronic interactions between YZ and the Mn4Ca cluster by detecting the split signal arising from the interaction of the spin states of YZ and the cluster itself. These EPR studies have shown the weakness of this interaction.50–53 It has also been postulated that the positive charge of 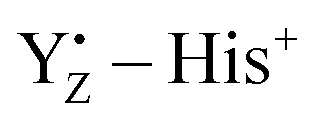 triggers the proton release reaction from substrate water through electrostatic interactions with the Mn4Ca cluster54–57 and that the proton of the phenolic oxygen of YZ is involved in the proton transfer from substrate water.58 However, a consensus has not yet been reached regarding the role of YZ in water oxidation.
triggers the proton release reaction from substrate water through electrostatic interactions with the Mn4Ca cluster54–57 and that the proton of the phenolic oxygen of YZ is involved in the proton transfer from substrate water.58 However, a consensus has not yet been reached regarding the role of YZ in water oxidation.
Our previous study also showed that the redox states of YZ control the equilibrium between the two isomers of the Mn4Ca cluster in the S2 → S3 transition.59 However, because the previous study focused on the role of the two isomers of the cluster in the S2 → S3 transition, the structural effects of the YZ oxidation on the cluster and the possible influence on the H-bond network around YZ were not analyzed in detail. In addition, in order to elucidate the mechanism of YZ involvement in water oxidation, a detailed study should include a systematic comparison between the H-bond properties of YZ and those of a basic tyrosine side chain.
So far, few studies have focused on the difference in the H-bond properties in each state of a free tyrosine molecule, although the structural and spectroscopic properties of each state have been well established.31,60–66 One of the reasons for this is the complexity of the system due to the presence of several H-bond patterns of a phenolic oxygen of a tyrosine side chain. A tyrosine can form an H-bond donor and/or acceptor, while a deprotonated tyrosine forms only an H-bond acceptor but with a different oxidation state. Thus, elucidating the H-bond properties of each state of a tyrosine is an essential and necessary step to understand the role of a key reactive tyrosine. This information can strongly contribute to clarifying the fundamental molecular mechanism of the protein functions as well as to predicting its function and structural changes from the structural information.
In the present study, we performed quantum mechanical-molecular dynamics (QM-MD) simulations of p-cresol in aqueous solution, which is a simple model of a tyrosine side chain, and quantum mechanics/molecular mechanics-molecular dynamics (QM/MM-MD) simulations of YZ and the Mn4Ca cluster in PSII. Since H-bonding is a dynamic process, the use of molecular dynamics simulations is crucial to pinpoint changes in the H-bond pattern, since shallow local minima can be easily overcome during the simulated time.
By comparing the results of the QM/MM simulations of YZ in PSII and the simple solvated p-cresol models, we are able to identify that the specificity of the H-bond properties of oxidized YZ is to form an anion state that promotes the formation of three H-bonds. The present simulations also show the S-state dependence of the H-bond structure of YZ and the rearrangements of the H-bond structure occurring around both YZ and the Mn4Ca cluster upon YZ oxidation. These rearrangements suggest that an arginine residue, CP43-Arg357, is involved in water oxidation by control of the H-bond structure around the O4 atom of the cluster. Based on the data of the structural effect upon YZ oxidation and the H-bond properties of YZ, we discuss the possible proton transfer mechanism from O5 in concert with YZ re-reduction with the rate-limiting electron transfer to 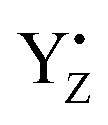 .
.
Methods
The computational setup for the QM-MD simulations of p-cresol was based on the classical MD simulation. The initial conditions for the classical MD simulations were created by TLEAP software in the AMBER program package. The p-cresol molecules were modeled using the GAFF force field67 and RESP charges calculated at the level of HF/6-31(d) by Gaussian09 software.68 We added 120 water molecules around the p-cresol moiety in a cubic boundary box. The full systems were treated at the quantum level.For the full QM-MD simulations of p-cresol in aqueous solution, we first performed individual classical MD simulations for each protonation or oxidation state (p-cresol, anionic p-cresol−, and oxidized p-cresol˙) to obtain the initial structures and set the boundary conditions using AMBER12 software.69 The MD simulations in the NPT ensemble were performed for 5.2 ns (2 fs per step) at 298 K using a Langevin heat bath after the NVT ensemble simulation for 100 ps. The equilibrated structures and the cubic boundary box were used for the QM-MD simulations, using CP2K ver.4.1 software.70,71 The QM-MD simulations in the NVT ensemble were performed for 80 ps (0.5 fs per step) at 298 K, using the PBE functional72 and the DZVP-MOLOPT-SR-GTH basis set73 with the GTH-PBE potential.74,75 The cutoff value for the plane-wave expansion in the QM calculation equaled 320 Rydberg.
The calculation system for the QM/MM-MD simulations of WOC in PSII was based on previous studies.76,77 The initial structures were taken from the structure with PDB ID: 3ARC. Water molecules, ions and protons were added to the systems and equilibrated by means of classical MD simulations as previously described in ref. 76 and 77. The whole QM/MM system consisted of the D1, D2, and CP43 proteins, as well as the redox co-factor and water molecules located within these proteins. The protein environment topology and other cofactors were followed by the AMBER99SB78 and GAFF force fields, respectively, as previously described.79 The QM region of the Mn4Ca cluster in the S1, S2, and S3 states was constructed as described in our previous study.80 Such a region consists of 206 atoms, which includes the Mn4Ca cluster, the side chains of its ligands (D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Ala344, D1-Asp342, and CP43-Glu354), additional residues (D1-Asp61, D1-Tyr161, D1-His190, CP43-Arg357, D1-His337, D1-Ile60, D1-Ser169, and D1-Gly171), the Cl− ion and its ligand D1-Asn181, and a further 17 water molecules. The QM/MM-MD simulations of WOC in each oxidation state (S1, S2, S2YZ+, and S3) were performed as described previously,59,76,80,81 using the PBE+U functional with the Hubbard correction of 1.16 eV for Mn ions82 and the DZVP-MOLOPT-SR-GTH basis set with the GTH-PBE pseudopotential. A cubic cell of 28 Å on each side and a cutoff for the plane-wave expansion of 320 Rydberg were applied for the QM calculations. The QM/MM-MD simulations were performed in the NVT ensemble with a thermal bath at 298 K using a Nosè–Hoover thermostat83,84 for 20 ps (0.5 fs per step) after the equilibration, with the exception of S2YZ+. The MD simulation of the S2YZ+ model started from the equilibrated S2 structure and was performed for 8.5 ps (0.5 fs per step) with the same conditions as the other S states. The total spin of the Mn4Ca cluster was set to S = 0, 1/2, and 3 in the S1, S2, and S3 states, respectively. The spin configuration of the Mn4Ca cluster was simulated by means of broken-symmetry DFT, based on the results of previous works consistently with the high oxidation paradigm.26,46 Mulliken spin populations of Mn ions in QM/MM-MD simulations of each S state are reported in Table S1 (ESI†).
Results
Models of p-cresol, p-cresol−, and p-cresol˙ in aqueous solution
A tyrosine side chain can assume three different forms: i.e., neutral, high pH (anionic deprotonated), and oxidized (radical deprotonated). To estimate the H-bond properties of a tyrosine side chain, we used QM-MD simulations of solvated p-cresol, p-cresol−, and p-cresol˙ models, in aqueous solution. Fig. 2 shows the density maps of the solvation shell of the oxygen atoms of water around the phenolic oxygen for each p-cresol form. In the p-cresol model, two regions of oxygen atoms were observed in the H-bond donor and acceptor sides of the phenolic oxygen. Compared with the p-cresol, three regions of oxygen atom density were arranged around the phenolic oxygen in the p-cresol−, while two regions were observed in the p-cresol˙. The two density maps of the oxygen atoms of water molecules in the p-cresol and the p-cresol˙ overlap despite the different protonation states. The different densities for the two deprotonated states (i.e., the p-cresol− and the p-cresol˙) are to be expected: the anion form has a higher affinity for accepting an H-bond than the radical form due to the negative charge of the phenolic oxygen. It was noted that the two regions in the radical state were arranged perpendicularly with respect to the direction of the plane of the phenolic ring. This arrangement of the water density in the p-cresol˙ indicates that the tyrosyl radical behaves as an H-bond acceptor with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond in the phenolic group.
O double bond in the phenolic group.
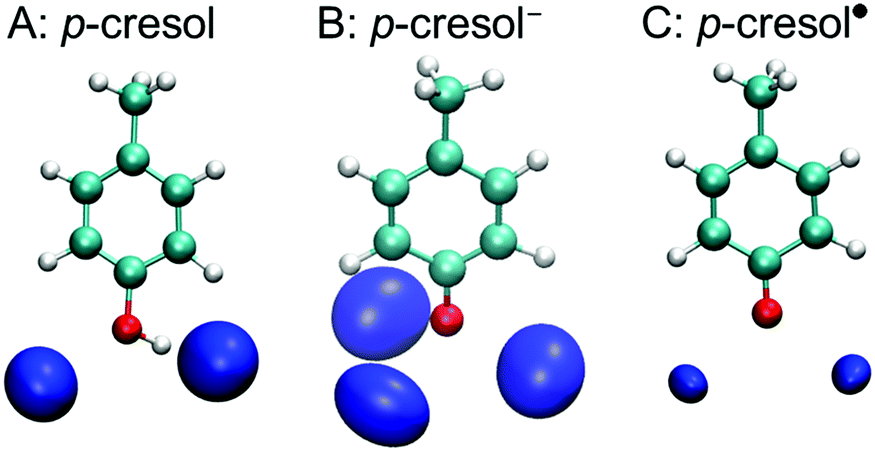 | ||
| Fig. 2 Density maps of the oxygen atoms of water molecules located within the H-bond distance (∼3.5 Å) in the neutral (A), the anion (B), and the radical (C) forms of p-cresol with the same isovalue of isosurface as determined by QM-MD simulations. | ||
Additionally, we report the calculated radial distribution functions of the oxygen and hydrogen atoms of water molecules around the phenolic oxygen in the ESI†, Fig. S1. The peak positions of the first hydration shell in the p-cresol− model are 2.60 Å and 1.50 Å in the Op-cresol–Owater and the Op-cresol–Hwater, respectively, which are shortened from those of the p-cresol (2.65 Å and 1.85 Å in the Op-cresol–Owater and Op-cresol–Hwater, respectively) and the p-cresol˙ (2.70 Å and 1.75 Å in the Op-cresol–Owater and Op-cresol–Hwater, respectively) models. This indicates, as expected, that the H-bonds are stronger in the anion than in the other forms. To quantitatively measure the structural properties around each model, we calculated the O–O and O–H coordination numbers, obtained by a definite integral of the radial distribution function up to its first minimum (Table 1). The coordination numbers of the Op-cresol–Owater in the first hydration shell in p-cresol− and p-cresol˙ were 3.00 and 1.96, respectively, which are consistent with the corresponding density maps. The coordination number of the Op-cresol–Owater of the p-cresol (2.33) instead was slightly larger than two, consistently with its oxygen density map. On the other hand, the coordination number of the Op-cresol–Hwater in p-cresol− was identical to that of the Op-cresol–Owater, whereas that in p-cresol˙ was slightly decreased. This indicates that the H-bonds of p-cresol− are more rigid than those of p-cresol˙. In the p-cresol case, there are several possible H-bond patterns, because p-cresol can become any of the H-bond donor, acceptor, and donor–acceptor forms. The difference in coordination numbers between Op-cresol–Owater and Op-cresol–Hwater (0.85) suggests the relatively stable H-bond on the donor side of p-cresol and the formation of an H-bond acceptor without donating an H-bond in ∼15% of the simulation. Interestingly, the shape of the Op-cresol–Owater in the second hydration shell in p-cresol˙ was significantly different from those of p-cresol and p-cresol−. The raising of the second hydration shell began from 3.15 Å to the long-distance region, which was a shorter distance than those of p-cresol (3.25 Å) and p-cresol− (3.20 Å). The peak position of the Op-cresol–Owater of the second hydration shell in p-cresol˙ (4.15 Å) was also a shorter distance than those of p-cresol (4.60 Å) and p-cresol− (4.35 Å).
 in the S1, S2, S2YZ+, and S3 states
in the S1, S2, S2YZ+, and S3 states
| Coordination number | Average of H-bond number | ||
|---|---|---|---|
| Ophenol–Owatera | Ophenol–Hwatera | ||
a The coordination numbers in the YZ and  cases include the nitrogen and the hydrogen atoms of D1-His190 H-bonding with the phenolic oxygen of YZ. cases include the nitrogen and the hydrogen atoms of D1-His190 H-bonding with the phenolic oxygen of YZ.
|
|||
| p-Cresol | 2.33 | 1.48 | 2.29 |
| p-Cresol− | 3.00 | 3.00 | 2.99 |
| p-Cresol˙ | 1.96 | 1.77 | 1.66 |
| S1YZ | 2.92 | 1.71 | 2.71 |
| S2YZ | 3.00 | 1.99 | 2.98 |

|
2.99 | 2.15 | 2.06 |
| S3YZ | 3.00 | 2.00 | 2.95 |
H-bond network around YZ
For PSII, we performed QM/MM-MD simulations around the Mn4Ca cluster, including YZ and its H-bond network in the S1, S2, S2YZ+ and S3 states. Fig. 3 shows the evolution along the simulations of the two distances O–H and N–H in the H-bond between YZ and D1-His190 (d1 and d2 in Fig. 3) in each state, as well as the number of H-bonds of YZ. It should be noted that we collected the structural information in the S1, S2, and S3 states after an equilibration time, whereas the trajectories of S2YZ+ started from the equilibrated S2 structure. In other words, the initial structure (0 ps) of S2YZ+ corresponds to the last frame of the S2 dynamics.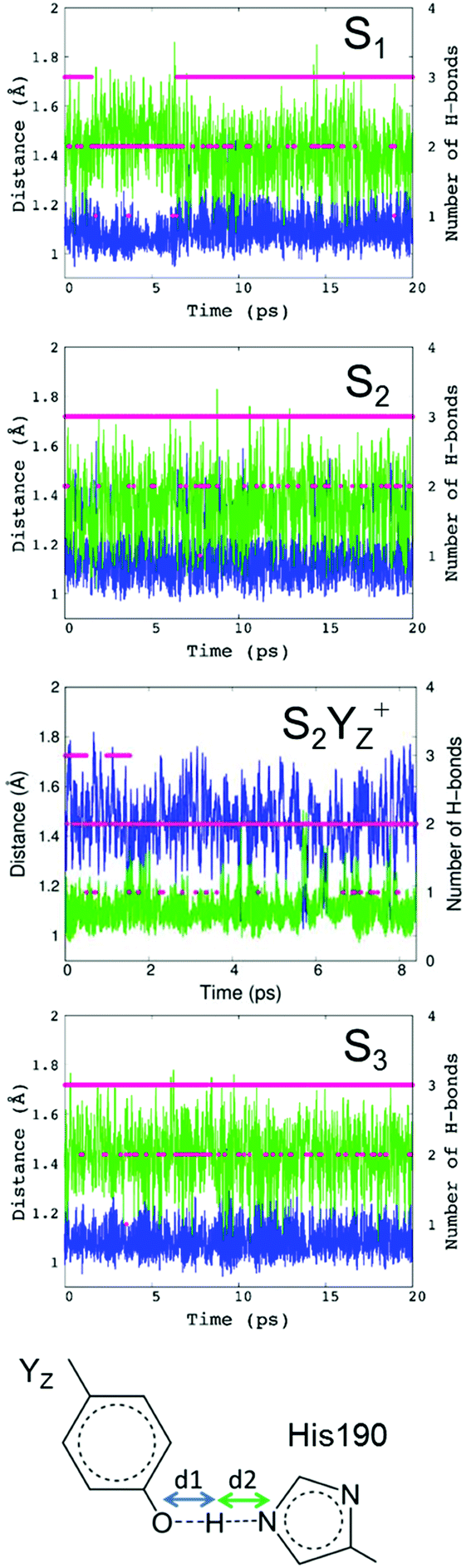 | ||
| Fig. 3 Time evolution of the number of H-bonds of the phenolic oxygen (red dots) of YZ and the distances of O–H (blue lines) and N–H (green lines) in S1, S2, S2YZ+, and S3, and the designation of the distance of O–H (shown at the bottom by a blue arrow) and N–H (shown at the bottom by a green arrow). | ||
In the simulation, YZ formed two or three H-bonds with D1-His190 and water molecules in the S1 state (Fig. 3; S1), whereas YZ in the S2 and S3 states tended to form three H-bonds (Fig. 3; S2 and S3). In order to compare such results with those of p-cresol, the coordination numbers of the first hydration shell of oxygen, nitrogen, and hydrogen atoms of water and D1-His190 from the phenolic oxygen of YZ and  were also calculated for each simulated S state and are shown in Table 1. The coordination numbers of the OYz–Hwater in the first hydration shell in YZ in the S2 and S3 states were 1.99 and 2.00, respectively, which indicated that the water molecules, W4 and W7, formed rigid H-bonds with YZ as H-bond donors. In contrast to the S2 and S3 states, the coordination number of the OYz–Hwater in the first hydration shell in YZ in the S1 state (1.71) was lower than those of the S2 and S3 states, which was consistent with the partial formation of two H-bonds in the S1 state (Fig. 3; S1).
were also calculated for each simulated S state and are shown in Table 1. The coordination numbers of the OYz–Hwater in the first hydration shell in YZ in the S2 and S3 states were 1.99 and 2.00, respectively, which indicated that the water molecules, W4 and W7, formed rigid H-bonds with YZ as H-bond donors. In contrast to the S2 and S3 states, the coordination number of the OYz–Hwater in the first hydration shell in YZ in the S1 state (1.71) was lower than those of the S2 and S3 states, which was consistent with the partial formation of two H-bonds in the S1 state (Fig. 3; S1).
In S2YZ+, the formation of two H-bonds was the most stable conformation (Fig. 3; S2YZ+), which is consistent with the coordination number of the OYz–Hwater(HHis190) in the first hydration shell in  in the S2YZ+ state (2.15) in Table 1. It has to be pointed out that the reason for the same coordination number of OYz–Owater(NHis190) shown by S2YZ and
in the S2YZ+ state (2.15) in Table 1. It has to be pointed out that the reason for the same coordination number of OYz–Owater(NHis190) shown by S2YZ and  is the presence of W4 within the H-bond distance from the phenolic oxygen even after breaking the H-bond between YZ and W4. So albeit similar coordination numbers, the two above mentioned states are characterized by a different number of H-bonds, as evident from Fig. 3.
is the presence of W4 within the H-bond distance from the phenolic oxygen even after breaking the H-bond between YZ and W4. So albeit similar coordination numbers, the two above mentioned states are characterized by a different number of H-bonds, as evident from Fig. 3.
Compared to S2, the O–H and N–H distances were fully switched around in S2YZ+ (Fig. 3; S2YZ+). Switching the two distances indicated that the PCET reaction of YZ was reproduced well in the S2YZ+ model. When YZ was oxidized, the proton of the phenolic group was released to D1-His190. The result for the number of H-bonds of  in S2YZ+ was in good agreement with the simulation of p-cresol in aqueous solution. It was noted that the distances of the O–H and N–H groups in the S1 state were dependent on the number of H-bonds. The difference between the two distances increased when the number of H-bonds decreased (Fig. 3; S2). This indicates that the strength of the H-bond between YZ and D1-His190 is dependent on the number of H-bonds of the phenolic oxygen of YZ.
in S2YZ+ was in good agreement with the simulation of p-cresol in aqueous solution. It was noted that the distances of the O–H and N–H groups in the S1 state were dependent on the number of H-bonds. The difference between the two distances increased when the number of H-bonds decreased (Fig. 3; S2). This indicates that the strength of the H-bond between YZ and D1-His190 is dependent on the number of H-bonds of the phenolic oxygen of YZ.
The distributions of the O–H and N–H distances for the H-bond between YZ and D1-His190 in the neutral YZ neutral state are also reported in the ESI†, Fig. S2. The distances between the two peaks of O–H and N–H in the S1 and S3 states were 0.36 Å and 0.37 Å, respectively, whereas for S2 the distance was 0.22 Å. This shorter distance in the S2 state indicates that the H-bond between YZ and D1-His190 is stronger. The overlapping region of the histograms of O–H and N–H in the S2 state was larger than that of the S1 and S3 states. The difference in the overlapping region indicates that switching of N–H and O–H bond distances is more frequent in the S2 state than in the other states. The differences in the number of H-bonds and the proton shuttle between YZ and D1-His190 indicate that the H-bond structure in YZ does depend on the S-state of the Mn4Ca cluster.
Structural effect of YZ oxidation on the Mn4Ca cluster, ligands and H-bond network
Decreasing the number of H-bonds of the phenolic oxygen from S2 to S2YZ+ leads to the structural effects of the H-bond structures not only around YZ but also around the Mn4Ca cluster. This is due to the presence of a water network linking between YZ and the Mn4Ca cluster. To determine the effect of the conformational changes of the H-bond network upon YZ oxidation, we compared the H-bonds and the ligand structure of the Mn4Ca cluster. Fig. 4 shows the distribution histograms of the distances of Ca–Mn and Ca–O in the WOC. There was a clear difference in the shape for the distance between Ca2+ and W4 H-bonded with YZ, in which the peak position increased from 2.33 to 2.43 Å upon YZ oxidation. Modifications of the ligand structure of the Ca2+ ion were also observed. The histograms of Ca–Glu189 and Ca–Asp170 in S2YZ+ were slightly shifted to the shorter distance region, while that of Ca–Ala344 was broader toward the region of longer distance. In the Ca-oxo distances, the Ca–O1 histogram became sharper and shifted slightly to the shorter distance region, while that of Ca–O5 became broader and its peak position was shifted from 2.57 to 2.63 Å in the S2YZ+ state. As in the case of the Ca2+ ion, the coordination structures of Mn1 and Mn4 were also partially changed upon YZ oxidation. The distance distribution histograms between Mn1 and its amino acid ligands in S2YZ+ were slightly downshifted to shorter distance although those between Mn1 and oxo-bridges were mostly the same as shown in the ESI†, Fig. S3. In the case of Mn4, the histogram of Mn4–Glu333 in S2YZ+ was broader toward the region of longer distance, whereas those of Mn4–O4 and Mn4–O5 were shifted to shorter distance by 0.02 and 0.04 Å, respectively (Fig. S4, ESI†). These modifications show that the ligand coordination of the Mn cluster is slightly modified by YZ oxidation. Notably, the histogram of the Ca–Mn4 distance was broadened and its peak position was shifted from 3.80 to 3.88 Å upon YZ oxidation (Fig. 4A). | ||
| Fig. 4 Distributions of the distance between the Ca2+ ion and Mn ions (A), Ca2+ and oxygen atoms of carboxylate ligands (B), Ca2+ and μ-oxo (C), and Ca2+ and water ligands (D) in the S2 (dotted line), and the S2YZ+ (solid line) state. | ||
The H-bond structure around the Mn4Ca cluster was also modified upon YZ oxidation. The conformational changes in the H-bond structure around the manganese cluster are shown in Fig. 5 and 7. The stable distances between O1 and Wb, which are indicated in Fig. 5C, were approximately 4.6 Å and 2.0 Å, respectively, in S2, whereas the distance was more than 8 Å in S2YZ+. This difference was a result of the conformational changes in the H-bond network around YZ due to W4 forming an H-bond with Wa instead of YZ in S2YZ+ (Fig. 7). Due to the formation of an H-bond between W4 and Wa, the O–O distance between W4 and D1-Glu189 decreased significantly from approximately 3.0 to approximately 2.7 Å. The H-bond pattern around CP43-Arg357 and D1-Asp61 also changed upon YZ oxidation (Fig. 6). We did not show the information for S2 in Fig. 6 because the structural properties in S2 were almost the same as those of S2YZ+ before the 5 ps point. In S2, Wc H-bonded with D1-Asp61 and O4 of the Mn4Ca cluster as an H-bond donor, while Wd H-bonded with Wc and CP43-Glu354 as an H-bond donor. The S2YZ+ simulation showed the rearrangement of this H-bond network upon YZ oxidation. In S2YZ+, Wd formed an H-bond with D1-Asp61 as soon as the H-bond between Wc and D1-Asp61 broke, and Wc then formed an H-bond with Wd as an H-bond donor (Fig. 7). At the same time, the H-bond structure of CP43-Arg357 was remarkably modified. Wd was located at the H-bond distance from a nitrogen of CP43-Arg357 in S2, whereas Wc was close to the nitrogen of CP43-Arg357 upon the conformational change at 5 ps in S2YZ+. Coupled with the conformational changes of the water network, CP43-Arg357 fully formed an H-bond with the C-terminus of D1-Ala344. The structural changes around CP43-Arg357 might not be directly related to the rearrangement of the H-bond network around YZ. The CP43-Arg357 electrostatic-mediated changes seemed to start at different times (5 ps) whereas the H-bond changes already happened (about 2 ps). In addition there is no direct link between the H-bond network and CP43-Arg357.
 | ||
| Fig. 5 Time evolution of the distances between O1 and a hydrogen atom of Wb shown in C (blue arrow) in the S2 (A) and the S2YZ+ (B) state. Distributions of the distances between the oxygen atom of D1-Glu189 coordinated with the Ca2+ ion of the Mn4Ca cluster and that of W3 (green line in D and represented in E as a blue arrow) or W4 (red line in D and represented in E as a red arrow) in S2 (dotted line) and S2YZ+ (solid line). | ||
 | ||
| Fig. 6 Time evolution of the distances in the S2YZ+ state between the oxygen atom of D1-Asp61 H-bonded with W1 and hydrogen atoms of water molecules (A), between the nitrogen atom of CP43-Arg357 H-bonded with water molecules and oxygen atoms of water molecules (B), between the hydrogen atom of CP43-Arg357 H-bonded with carboxylate ligands and oxygen atoms of carboxylate ligands coordinated with the Ca2+ ion, D1-Ala344, and D1-Asp170 (C). The designation of each distance is shown on the right with arrows, in which the colors correspond to the lines on the left. | ||
 | ||
Fig. 7 The H-bond structure around YZ (top), CP43-Arg357 (middle) and O4 (bottom) of the Mn4Ca cluster in neutral YZ and the radical 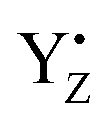 forms. forms. | ||
Discussion
In the present study, we first determined the H-bond patterns of p-cresol, a tyrosine side chain model, in aqueous solution. The QM-MD simulations of the three different protonation or oxidation states of p-cresol revealed the possible H-bond patterns in aqueous solution. The neutral p-cresol mostly formed two H-bonds (three partially formed H-bonds) with water molecules as an H-bond donor–acceptor (Fig. 2A). The H-bond structure was consistent with the vibrational insight of the p-cresol in H2O (D2O) solution.65 The conformation and the strength of the H-bonds in the radical form after the PCET reaction (Fig. 2C and Fig. S1, ESI†) were very similar to those of the neutral form due to the similarity of the charge distributions on the phenolic groups in the two states. Thus, in the case of the oxidation of a neutral tyrosine, a phenolic proton was released to an H-bond acceptor without major conformational changes. In contrast to the oxidation of the neutral form, there were significant differences in the H-bond properties of the oxidation of p-cresol− despite similarities in the atomic structure (Fig. 2B and Fig. S1, ESI†). The difference indicated that conformational changes occurred in the H-bond structure of a tyrosine anion upon its oxidation. The differences of the strength and the number of H bonds between the anion and the radical forms were mainly caused by those of the charge distribution of the phenolic group. Remarkably, the structural conformation of the H-bonded water molecules was determined by the phenolic C–O form, which was strongly influenced by the electronic structure of the phenolic ring. The anionic p-cresol− formed a single bond on the phenolic C–O group with three H-bonds arranged as the sp3 conformation (Fig. 2B), whereas the oxidized p-cresol˙ formed a partial double bond on the phenolic CO group, which could not form three H-bonds with the sp3 conformation (Fig. 2C). The difference of the C–O bond was consistent with the significant upshift of the CO stretching frequency upon the oxidation of anionic tyrosine and p-cresol molecules.85
The H-bond structure of YD was satisfactorily consistent with the model simulation of p-cresol. The H-bond structure of YD as revealed by the X-ray structure17–19 was very similar to that of neutral p-cresol in aqueous solution. For the neutral YD form, both proximal (∼2.8 Å from YD) and distal (∼4.3 Å from YD) positions were stable in the QM/MM simulations.37,86 A recent EPR study has indicated that, at equilibrium, a water molecule near YD can be located both proximally and distally.87 The H-bond pattern of YD and the proximal water corresponded to the results of the density map of neutral p-cresol (Fig. 2A). In the YD˙ state, the phenolic proton was released into the bulk upon its oxidation, and YD then became a neutral radical. The QM/MM simulations also predicted that the water would move to the distal site in YD˙ and that YD˙ would then only H-bond with D2-His189.37,86 The water movement was consistent with the decreasing H-bond affinity from neutral p-cresol to oxidized p-cresol˙, which was estimated by decreasing the coordination number and longer distance of the peak position of the radial distribution functions of the Op-cresol–Owater in the first hydration shell (Table 1). The proximal position was not, however, stable in YD and YD˙, although the density maps and the radial distribution function showed that neutral and radical forms of p-cresol were able to form two H-bonds. This may be due to D2-Arg180 being located in the H-bond distance from the distal water because the H-bond between the distal water and D2-Arg180 was more stable than that between YD˙ and the proximal water.
In contrast to YD, a complete agreement regarding the H-bond structure between YZ and the neutral p-cresol model in the QM-MD simulation was not obtained. D1-His190 and two water molecules were located within the H-bond distance from the phenolic oxygen of YZ in the X-ray structure.17–19 To understand its specificity and the respective contribution to water oxidation, we performed QM/MM-MD simulations based on our previous models (see Methods). Neutral YZ formed three H-bonds for a long time in the simulation (Fig. 3), which was not the same tendency of the neutral p-cresol tending to form two H-bonds in aqueous solution. The coordination numbers of both of the OYz–Hwater and OYz–Owater in the first hydration shell in YZ are also higher than that of p-cresol in aqueous solution (Table 1). The comparison between the neutral solvated p-cresol and the neutral YZ demonstrates that the protein environment plays a crucial role in triggering the conformational changes upon oxidation. Since the protein hydrogen bond network of neutral YZ is not optimal, as found by solvated tyrosine simulations, the structural changes upon oxidation are amplified. One of the keys for the elucidation of the specificity of the number of H-bonds in neutral YZ was the formation of the LBHB between YZ and D1-His190. Our simulation showed the shuttling of a proton via this short H-bond (Fig. S2, ESI†). This proton shuttling suggests that YZ momently formed an anion state due to transient deprotonation. The partial anion strongly contributed to stabilizing three H-bonds with W4, W7, and D1-His190 in the neutral YZ state because the H-bond distance between YZ and D1-His190 depended on the number of H-bonds (Fig. 3; S1). The influence of the H-bond of W4 on the LBHB between YZ and D1-His190 was consistent with the results of the QM/MM study by Saito et al.36
The present simulation in S2YZ+ reproduced not only the proton transfer from YZ to D1-His190 upon YZ oxidation but also breaking of the H-bond between  and W4 (Fig. 7), which was suggested by the QM/MM simulation reported by Noguchi and co-workers.58 This H-bond pattern of
and W4 (Fig. 7), which was suggested by the QM/MM simulation reported by Noguchi and co-workers.58 This H-bond pattern of  corresponds to the H-bond property of oxidized p-cresol˙ in aqueous solution. The changing H-bond pattern upon YZ oxidation induced direct connection of the water chain from the Ca2+ ion of the Mn4Ca cluster to the bulk surface via the “large channel system”1–3 (shown in Fig. 1B as channel A) due to the formation of the new H-bond of W4 with Wa (Fig. 7). This causes the breaking of H-bond between Wb and O1 in the Mn4Ca cluster. A recent study by Sakashita et al.88 based on MD simulations showed that this channel has an elevated degree of water accessibility due to its large radius. Thus, the direct connection of the Ca2+ ion and this channel upon YZ oxidation leads to higher accessibility of bulk water to the Mn4Ca cluster. Moreover, the structural changes in the H-bond network around YZ cause W4 to come in the proximity of D1-Glu189 at the same time that the new H-bond network forms. The changes are indicative of the proton transfer pathway from the O5 site of the Mn4Ca cluster to the bulk via D1-Glu189. Our previous study showed that D1-Glu189 can be involved in the proton transfer from the O5 site of the Mn4Ca cluster in the S3 → S4 transition.89 Our data strongly support the notion that the proton located at the O5 site can be released via D1-Glu189 and W4 coupled with YZ reduction (Fig. 8). This means that this mechanism is used in the S0 → S1 transition in particular because it promoted the proton release from the O5 site. Since
corresponds to the H-bond property of oxidized p-cresol˙ in aqueous solution. The changing H-bond pattern upon YZ oxidation induced direct connection of the water chain from the Ca2+ ion of the Mn4Ca cluster to the bulk surface via the “large channel system”1–3 (shown in Fig. 1B as channel A) due to the formation of the new H-bond of W4 with Wa (Fig. 7). This causes the breaking of H-bond between Wb and O1 in the Mn4Ca cluster. A recent study by Sakashita et al.88 based on MD simulations showed that this channel has an elevated degree of water accessibility due to its large radius. Thus, the direct connection of the Ca2+ ion and this channel upon YZ oxidation leads to higher accessibility of bulk water to the Mn4Ca cluster. Moreover, the structural changes in the H-bond network around YZ cause W4 to come in the proximity of D1-Glu189 at the same time that the new H-bond network forms. The changes are indicative of the proton transfer pathway from the O5 site of the Mn4Ca cluster to the bulk via D1-Glu189. Our previous study showed that D1-Glu189 can be involved in the proton transfer from the O5 site of the Mn4Ca cluster in the S3 → S4 transition.89 Our data strongly support the notion that the proton located at the O5 site can be released via D1-Glu189 and W4 coupled with YZ reduction (Fig. 8). This means that this mechanism is used in the S0 → S1 transition in particular because it promoted the proton release from the O5 site. Since 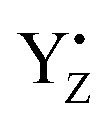 cannot form three H-bonds, the proton release into the bulk has to be coupled with the electron transfer to
cannot form three H-bonds, the proton release into the bulk has to be coupled with the electron transfer to 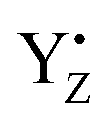 . In particular, the recent time-resolved infrared measurement showed a fast single phase with a small kinetic isotope effect (H/D) in the S0 → S1 transition, which strongly suggested the PCET with electron transfer as the rate-limiting step.90 Thus, we propose that the proton release mechanism via D1-Glu189 from the O5 site is effective in such a PCET reaction, and therefore cannot precede the oxidation of the Mn4Ca cluster by
. In particular, the recent time-resolved infrared measurement showed a fast single phase with a small kinetic isotope effect (H/D) in the S0 → S1 transition, which strongly suggested the PCET with electron transfer as the rate-limiting step.90 Thus, we propose that the proton release mechanism via D1-Glu189 from the O5 site is effective in such a PCET reaction, and therefore cannot precede the oxidation of the Mn4Ca cluster by  .
.
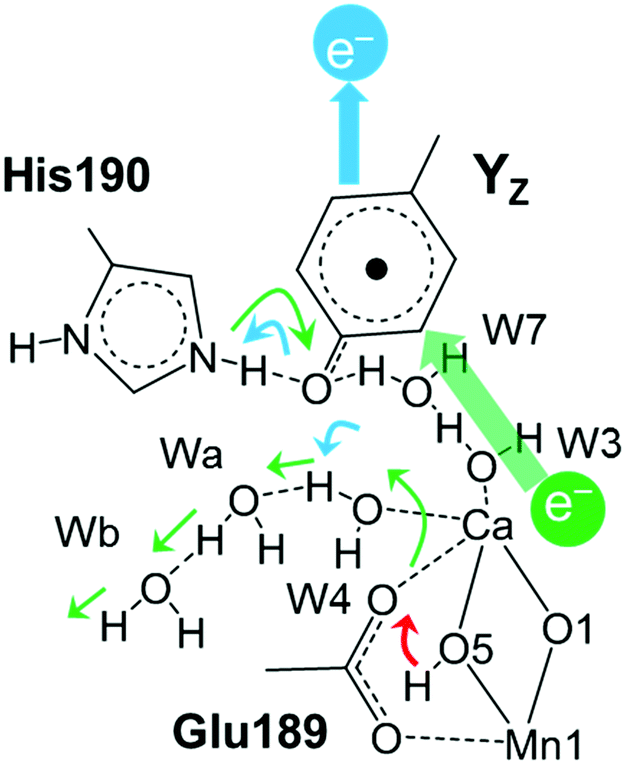 | ||
Fig. 8 Possible proton transfer mechanism from the site of O5 of the Mn4Ca cluster to the bulk. In the first step, YZ is oxidized by P680+ with structural changes in the H-bond network around YZ (blue arrows). After this, the proton of O5 is oriented toward the oxygen atom of D1-Glu189 (red arrow). In the electron transfer step from Mn to  , a proton between D1-Glu189 and O5 moves to W4 and the proton forms an H-bond with YZ with a concerted proton transfer relay analogous to the Grotthuss mechanism from W4 into the bulk via the large channel system (green). , a proton between D1-Glu189 and O5 moves to W4 and the proton forms an H-bond with YZ with a concerted proton transfer relay analogous to the Grotthuss mechanism from W4 into the bulk via the large channel system (green). | ||
The comparison between the dynamics of different S-states reports that the H-bond structure of YZ is also dependent on the S-state of the Mn4Ca cluster (Fig. S2, ESI†). In particular, the H-bond between YZ and W4 was not rigid in the S1 state, while the proton shuttling on the H-bond between YZ and D1-His190 was more frequent in the S2 state than in the other states. This was due to the different charge distribution on the Mn4Ca cluster by different oxidation states of Mn ions because the topologies of the S1 and S2 models were the same. Changing the H-bond interaction of phenolic oxygen upon Mn oxidation was consistent with the FTIR study, which detected the structural coupling between YZ and the Mn4Ca cluster in the S1 and S2 states using isotope labeling.91 The Mn4Ca cluster in the S2 and S3 states had excess positive charge since the proton release pattern was 0:1:2:1 for the S1 → S2, S2 → S3, S3 → S0, and S0 → S1 transitions, respectively.92–94 The partial anion of neutral YZ in the S2 state was more stable than that in the S1 state due to the Coulombic interaction between YZ and the excess positive charge on the Mn4Ca cluster. Although the Mn4Ca cluster in the S3 state also had excess positive charge, the H-bond structure of YZ in the S3 state was slightly different from that in the S2 state. This may have been caused by the structural and/or screening effect due to binding an additional hydroxide (i.e. O6) in the S3 state. The stabilization of the LBHB and the three-H-bond state of YZ contributed to the rapid conformational changes of the re-reduction of YZ in the S1 → S2 and S2 → S3 transitions. Thus, the excess positive charge on the Mn4Ca cluster may play a key role in controlling the H-bond interaction of the phenolic oxygen of YZ.
The H-bond network around O4 is a part of the “narrow channel”1–3 shown in Fig. 1B as channel B although this network and that around YZ are divided. In addition, the hydrogen bond distances among the water cluster between YZ and the Mn4Ca cluster and its interatomic distance are virtually unchanged (Tables S1 and S2, ESI†). As a remarkable difference between the S2 and S2YZ+ states, the  moiety has a remaining positive charge by its oxidation. Thus, it is reasonable that the hydrogen bond rearrangements around O4 arose from the charge repulsion of the positive charges of CP43-Arg357 and
moiety has a remaining positive charge by its oxidation. Thus, it is reasonable that the hydrogen bond rearrangements around O4 arose from the charge repulsion of the positive charges of CP43-Arg357 and  . This interaction mechanism strongly suggests that CP43-Arg357 modified the H-bond structure of the water network around O4, coupled with the redox reaction of YZ. The role of CP43-Arg357 is a key issue for the elucidation of the water oxidation mechanism. The CP43-Arg357Lys point mutation has very low oxygen-evolving activity (∼18% of wild-type).95 These results mean that CP43-Arg357 not only functioned as a positive charge around the Mn4Ca cluster but that it also stabilized the structure around the Mn4Ca cluster through the guanidine group. Our previous study showed that the water network around D1-Asp61 was involved in the delivery of the substrate water in the S2 → S3 transition.96 The QM/MM study by Saito et al. suggested that the water network around O4 serves as the proton transfer pathway in the S0 → S1 transition.97 Thus, we propose that CP43-Arg357 plays a key role in controlling the H-bond structure around O4 to facilitate the proton transfer and/or the water movement in the S-state transitions. However, further careful studies are needed to obtain a more detailed understanding of the function of CP43-Arg357 in water oxidation.
. This interaction mechanism strongly suggests that CP43-Arg357 modified the H-bond structure of the water network around O4, coupled with the redox reaction of YZ. The role of CP43-Arg357 is a key issue for the elucidation of the water oxidation mechanism. The CP43-Arg357Lys point mutation has very low oxygen-evolving activity (∼18% of wild-type).95 These results mean that CP43-Arg357 not only functioned as a positive charge around the Mn4Ca cluster but that it also stabilized the structure around the Mn4Ca cluster through the guanidine group. Our previous study showed that the water network around D1-Asp61 was involved in the delivery of the substrate water in the S2 → S3 transition.96 The QM/MM study by Saito et al. suggested that the water network around O4 serves as the proton transfer pathway in the S0 → S1 transition.97 Thus, we propose that CP43-Arg357 plays a key role in controlling the H-bond structure around O4 to facilitate the proton transfer and/or the water movement in the S-state transitions. However, further careful studies are needed to obtain a more detailed understanding of the function of CP43-Arg357 in water oxidation.
Recent X-ray studies gave more insight into the intermediate structures in the water oxidation cycle.98,99 The simulations here presented reproduced well the metal–metal distances in the Mn4Ca cluster in the stable S1, S2, and S3 states (Table S2, ESI†). In contrast, the Mn1–Mn4 distances obtained by the simulation of the YZ-oxidized S2 state showed different behavior from the transient state at 150 μs of the S2 → S3 transition.99 The simulation of the S2YZ+ state, that preceded the insertion of a water molecule, provided a virtually identical Mn1–Mn4 distance to that in the S2 state. On the other hand, the X-ray transient structure showed a longer Mn1–Mn4 distance by ∼0.2 Å than that of the structure after single illumination. This longer distance is maintained in the stable structure after the second flash illumination. This indicates that the significant structural changes in the Mn4Ca cluster to insert a new water molecule (O6) between Mn1 and Mn4 occur before 150 μs in the S2 → S3 transition. This mismatch arose from the different time scales between the X-ray measurement (>100 μs) and the dynamics simulation (∼10 ps).
The hydrogen bond distances among the water molecules around the Mn4Ca cluster in the recent X-ray structures17,19,98,99 were also satisfactorily reproduced by the present simulations in the stable S states (Table S3, ESI†). In terms of the S2 → S3 transition, the X-ray structure after 150 μs from the second illumination provided a longer O–O distance between Wb and O1 (3.68 Å and 4.00 Å),99 which indicated the absence of the hydrogen bond. This appears in good agreement with our simulations for the S2YZ+ state. In contrast with the hydrogen bond between Wb and O1, the hydrogen bond between W4 and Wa in the transient structure (3.45 Å and 3.40 Å) is longer than that in the S2YZ+ simulation (2.95 Å). This difference suggests that further hydrogen bond rearrangement takes place around YZ on the time scale of microseconds.
Previous theoretical studies proposed the formation of Mn1-oxidized form (i.e. closed cubane structure) of the Mn4Ca cluster in the S2 → S3 transition.59,76,96,100,101 In this proposed model, the equilibrium between Mn4- and Mn1-oxidized forms in the S2 states is inverted upon YZ oxidation, favoring the Mn1-oxidized structure in a closed cubane fashion. The present study showed that YZ oxidation induced modifications of the coordination and the geometry of the Mn4Ca cluster. The coordinate bonds between Mn1 and amino acid ligands strengthened upon YZ oxidation (Fig. S3, ESI†), which can contribute to a stabilization of the Mn1-oxidized S2 state. On the other hand, the increased distance between Ca2+ and Mn4 (Fig. 4 with a broadening of the distribution shown upon YZ oxidation in the S2 state), which indicates the respective weakening of the bond Ca–Mn4 with higher flexibility, may promote the reduction of Mn4, also triggered by the presence of the positive 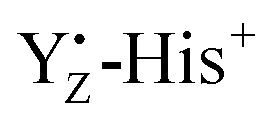 moiety. Remarkably, the Mn4–O5 distance distribution is slightly broadened upon YZ oxidation, despite shortening the average value. The observation is consistent with our proposed S2 → S3 transition model. Thus, these structural changes upon YZ oxidation suggest that radical
moiety. Remarkably, the Mn4–O5 distance distribution is slightly broadened upon YZ oxidation, despite shortening the average value. The observation is consistent with our proposed S2 → S3 transition model. Thus, these structural changes upon YZ oxidation suggest that radical 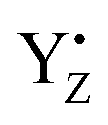 may act not only as a mere electron acceptor, but can induce conformational changes in the cluster promoting the transition between isomers and can have a functional role in the progress of the catalytic cycle. The formation of the closed cubane structure was not revealed as an intermediate by the recent X-ray study.99 Also in the present simulations we did not observe spontaneous shifts of the O5 position from the open to the closed cubane conformation in the S2YZ+ state. This is not surprising since the energy barrier (∼10 kcal mol−1) present between the two conformers cannot be overcome in the simulated time.59 Thus, the present results do not exclude our previous hypothesis, also corroborated by the evidence that direct water insertion between Mn1 and O5 provides a much higher barrier (∼20 kcal mol−1) than the W2 insertion between Mn4 and O5 (∼8 kcal mol−1).96
may act not only as a mere electron acceptor, but can induce conformational changes in the cluster promoting the transition between isomers and can have a functional role in the progress of the catalytic cycle. The formation of the closed cubane structure was not revealed as an intermediate by the recent X-ray study.99 Also in the present simulations we did not observe spontaneous shifts of the O5 position from the open to the closed cubane conformation in the S2YZ+ state. This is not surprising since the energy barrier (∼10 kcal mol−1) present between the two conformers cannot be overcome in the simulated time.59 Thus, the present results do not exclude our previous hypothesis, also corroborated by the evidence that direct water insertion between Mn1 and O5 provides a much higher barrier (∼20 kcal mol−1) than the W2 insertion between Mn4 and O5 (∼8 kcal mol−1).96
It has been proposed that the role of YZ and D1-His190 in water oxidation involves the release of a proton from substrate water, especially in the S2 → S3 transition.34,54,56 In particular, an FTIR study has indicated that the proton between  and protonated D1-His190 is involved in the proton transfer of water oxidation due to the low NH stretching frequency as an extremely broad band.58 The possibility of proton transfer via YZ in water oxidation cannot be fully excluded because our simulation also reproduced a strong H-bond between
and protonated D1-His190 is involved in the proton transfer of water oxidation due to the low NH stretching frequency as an extremely broad band.58 The possibility of proton transfer via YZ in water oxidation cannot be fully excluded because our simulation also reproduced a strong H-bond between  and protonated D1-His190 with proton shuttling. Furthermore, a recent theoretical study by Saito et al. proposed the oxidized and deprotonated D1-His190 instead of YZ, with the rotation of D1-Asn298.102 D1-Asn298 is involved in the large channel system and the H-bond network from YZ to PsbV-Lys129.17 All of the mutations of D1-Asn298 that have been tested exhibited very low O2 evolution activity,103 while an FTIR study using the D1-Asn298Ala mutation has shown that the efficiency of the S2 → S3 transition was lowered and the S3 → S0 transition was inhibited by this mutation.104 Thus, it seems clear that D1-Asn298 is a key amino acid involved in the mechanism of water oxidation. Therefore, further dynamic simulations with a larger QM region than that of the present study are necessary to elucidate the involvement of YZ and its H-bond network.
and protonated D1-His190 with proton shuttling. Furthermore, a recent theoretical study by Saito et al. proposed the oxidized and deprotonated D1-His190 instead of YZ, with the rotation of D1-Asn298.102 D1-Asn298 is involved in the large channel system and the H-bond network from YZ to PsbV-Lys129.17 All of the mutations of D1-Asn298 that have been tested exhibited very low O2 evolution activity,103 while an FTIR study using the D1-Asn298Ala mutation has shown that the efficiency of the S2 → S3 transition was lowered and the S3 → S0 transition was inhibited by this mutation.104 Thus, it seems clear that D1-Asn298 is a key amino acid involved in the mechanism of water oxidation. Therefore, further dynamic simulations with a larger QM region than that of the present study are necessary to elucidate the involvement of YZ and its H-bond network.
The data in the present study add further structural information regarding the rearrangement of the H-bond network around the Mn4Ca cluster upon YZ oxidation. YZ is able to interact with not only the large channel system but also the narrow channel (Fig. 6). The interaction mechanism of the two channels with YZ is, however, completely different. The large channel system directly interacts with YZvia the H-bond network, whereas the narrow channel interacts with YZvia CP43-Arg357 by Coulombic interactions. Even though the length of our ab initio molecular dynamics is restricted to the ps time scale we have clearly observed both the initial movements that are triggered by YZ oxidation in the large and narrow water channels.
The effect of 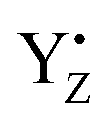 on the H-bond network around the Mn4Ca cluster may be crucial for water oxidation because the proton transfer and the water movements are coupled with or are triggered by the YZ redox reaction, especially in the S2 → S3 and S3 → S0 transitions.54–57 In the S2 → S3 transition, the rearrangement of the H-bond network, including the water movement, occurs before the concerted reduction of YZ and the proton transfer.105 The present data and the mechanism of water movement in the S2 → S3 transition, which we previously reported,96 strongly support the notion that the rearrangements and the water movement in the S2 → S3 transition are triggered by the H-bond rearrangement upon YZ oxidation. As with the S2 → S3 transition, the first proton transfer in the S3 → S0 transition takes place before the reduction of YZ.54,106,107 We previously proposed a proton transfer mechanism whereby D1-Glu189 mediates transfer of a proton from O5–O6 of the Mn4Ca cluster in the S3 → S4 transition.89 The present study supports this mechanism because the ligand structure of the Ca2+ ion of the Mn4Ca cluster is modified slightly upon YZ oxidation.
on the H-bond network around the Mn4Ca cluster may be crucial for water oxidation because the proton transfer and the water movements are coupled with or are triggered by the YZ redox reaction, especially in the S2 → S3 and S3 → S0 transitions.54–57 In the S2 → S3 transition, the rearrangement of the H-bond network, including the water movement, occurs before the concerted reduction of YZ and the proton transfer.105 The present data and the mechanism of water movement in the S2 → S3 transition, which we previously reported,96 strongly support the notion that the rearrangements and the water movement in the S2 → S3 transition are triggered by the H-bond rearrangement upon YZ oxidation. As with the S2 → S3 transition, the first proton transfer in the S3 → S0 transition takes place before the reduction of YZ.54,106,107 We previously proposed a proton transfer mechanism whereby D1-Glu189 mediates transfer of a proton from O5–O6 of the Mn4Ca cluster in the S3 → S4 transition.89 The present study supports this mechanism because the ligand structure of the Ca2+ ion of the Mn4Ca cluster is modified slightly upon YZ oxidation.
Finally it has to be pointed out that the QM/MM-MD simulation of the S2YZ+ state reported here captured only beginning of the structural changes upon YZ oxidation due to its reduced length (∼10 ps). Nonetheless, such conformational changes may be a trigger for further changes (i.e. proton transfer and water movements), which require longer times (i.e. ns to μs).
Conclusions
In the present study, we performed QM MD simulations of p-cresol as a model of a tyrosine side chain in aqueous solution in different oxidation/protonation states and QM/MM MD simulations of YZ in its protein environment for different states of the Kok–Joliot cycle. From the systematic comparison between simulated systems, we assessed the specificity of the H-bond properties of YZ in PSII with respect to the tyrosine side chain model. We found that oxidized formed two H-bonds, whereas neutral YZ tended to form three H-bonds. The structure of YZ with three H-bonds was not in agreement with the model simulations using p-cresol, whereas the H-bond conformation was almost the same as that of the anionic p-cresol− model. This difference was due to the transient formation of the anion state as a result of the LBHB between YZ and D1-His190. Furthermore, the H-bond structure of YZ was slightly changed by the S state transitions. In particular, the Mn4Ca cluster of the S2 state clearly lowered the barrier in the LBHB. The YZ simulation also showed that the redox reaction of YZ affected the two main channels linking to the bulk and the Mn4Ca cluster. Our results clearly indicated that YZ is actively involved in the water movement in the S2 → S3 transition and in the proton transfer along the S0 → S1 transition. Last but not least, this study revealed that the proton transfer mechanism can occur in a concerted manner with the
formed two H-bonds, whereas neutral YZ tended to form three H-bonds. The structure of YZ with three H-bonds was not in agreement with the model simulations using p-cresol, whereas the H-bond conformation was almost the same as that of the anionic p-cresol− model. This difference was due to the transient formation of the anion state as a result of the LBHB between YZ and D1-His190. Furthermore, the H-bond structure of YZ was slightly changed by the S state transitions. In particular, the Mn4Ca cluster of the S2 state clearly lowered the barrier in the LBHB. The YZ simulation also showed that the redox reaction of YZ affected the two main channels linking to the bulk and the Mn4Ca cluster. Our results clearly indicated that YZ is actively involved in the water movement in the S2 → S3 transition and in the proton transfer along the S0 → S1 transition. Last but not least, this study revealed that the proton transfer mechanism can occur in a concerted manner with the 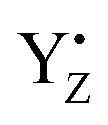 re-reduction, only after the radicalization of YZ and the respective change in the H-bond network. Based on these results, we proposed a model of structural changes involving H-bond network rearrangements around the Mn4Ca cluster and the coordination sphere of YZ, occurring upon the tyrosine oxidation, and functional towards the progression of the catalytic cycle. Our study corroborates the hypothesis that YZ plays a key role in the Kok–Joliot cycle not only as a mere electron acceptor, but also by triggering structural changes required for the progress of the cycle itself.
re-reduction, only after the radicalization of YZ and the respective change in the H-bond network. Based on these results, we proposed a model of structural changes involving H-bond network rearrangements around the Mn4Ca cluster and the coordination sphere of YZ, occurring upon the tyrosine oxidation, and functional towards the progression of the catalytic cycle. Our study corroborates the hypothesis that YZ plays a key role in the Kok–Joliot cycle not only as a mere electron acceptor, but also by triggering structural changes required for the progress of the cycle itself.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge PRACE infrastructure (project id: Pra16-3574) for computational time. SN was supported by a JSPS Overseas Research Fellowships.Notes and references
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, 2012, 1817, 1671–1678 CrossRef CAS PubMed.
- F. M. Ho and S. Styring, Biochim. Biophys. Acta, 2008, 1777, 140–153 CrossRef CAS PubMed.
- K. Linke and F. M. Ho, Biochim. Biophys. Acta, 2014, 1837, 14–32 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- J. Barber, Q. Rev. Biophys., 2016, 49, e14 CrossRef PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- J. Barber, Sustainable Energy Fuels, 2018, 2, 927–935 RSC.
- R. J. Debus, Biochim. Biophys. Acta, 1992, 1102, 269–352 CrossRef CAS.
- W. Hillier and J. Messinger, in Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, ed. T. J. Wydrzynski and K. Satoh, Springer, Dordrecht, The Netherlands, 2005, pp. 567–608 Search PubMed.
- J. Messinger, T. Noguchi and J. Yano, in Molecular solar fuels, ed. T. J. Wydrzynski and W. Hillier, Royal Society of Chemistry, Cambridge, UK, 2011, ch. 7, pp. 163–207 Search PubMed.
- G. Renger, in Photosynthesis: Plastid Biology, Energy Conversion and Carbon Assimilation, ed. J. J. Eaton-Rye, B. C. Tripathy and T. D. Sharkey, Springer, Dordrecht, The Netherlands, 2012, pp. 359–414 Search PubMed.
- A. Grundmeier and H. Dau, Biochim. Biophys. Acta, 2012, 1817, 88–105 CrossRef CAS PubMed.
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- A. Tanaka, Y. Fukushima and N. Kamiya, J. Am. Chem. Soc., 2017, 139, 1718–1721 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- I. D. Young, M. Ibrahim, R. Chatterjee, S. Gul, F. Fuller, S. Koroidov, A. S. Brewster, R. Tran, R. Alonso-Mori, T. Kroll, T. Michels-Clark, H. Laksmono, R. G. Sierra, C. A. Stan, R. Hussein, M. Zhang, L. Douthit, M. Kubin, C. de Lichtenberg, P. Long Vo, H. Nilsson, M. H. Cheah, D. Shevela, C. Saracini, M. A. Bean, I. Seuffert, D. Sokaras, T. C. Weng, E. Pastor, C. Weninger, T. Fransson, L. Lassalle, P. Brauer, P. Aller, P. T. Docker, B. Andi, A. M. Orville, J. M. Glownia, S. Nelson, M. Sikorski, D. Zhu, M. S. Hunter, T. J. Lane, A. Aquila, J. E. Koglin, J. Robinson, M. Liang, S. Boutet, A. Y. Lyubimov, M. Uervirojnangkoorn, N. W. Moriarty, D. Liebschner, P. V. Afonine, D. G. Waterman, G. Evans, P. Wernet, H. Dobbek, W. I. Weis, A. T. Brunger, P. H. Zwart, P. D. Adams, A. Zouni, J. Messinger, U. Bergmann, N. K. Sauter, J. Kern, V. K. Yachandra and J. Yano, Nature, 2016, 540, 453–457 Search PubMed.
- J. Kern and G. Renger, Photosynth. Res., 2007, 94, 183–202 CrossRef CAS PubMed.
- R. J. Debus, Biochim. Biophys. Acta, 2015, 1847, 19–34 CrossRef CAS PubMed.
- M. R. Blomberg, T. Borowski, F. Himo, R. Z. Liao and P. E. Siegbahn, Chem. Rev., 2014, 114, 3601–3658 CrossRef CAS PubMed.
- J. Barber, Q. Rev. Biophys., 2003, 36, 71–89 CrossRef CAS PubMed.
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6(20), 4764–4771 RSC.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- A. Guskov, A. Gabdulkhakov, M. Broser, C. Glockner, J. Hellmich, J. Kern, J. Frank, F. Muh, W. Saenger and A. Zouni, ChemPhysChem, 2010, 11, 1160–1171 CrossRef CAS PubMed.
- G. T. Babcock, B. A. Barry, R. J. Debus, C. W. Hoganson, M. Atamian, L. McIntosh, I. Sithole and C. F. Yocum, Biochemistry, 1989, 28, 9557–9565 CrossRef CAS PubMed.
- P. Joliot, G. Barbieri and R. Chabaud, Photochem. Photobiol., 1969, 10, 309–329 CrossRef CAS.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- W. T. Dixon and D. Murphy, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1221–1230 RSC.
- G. Renger, Biochim. Biophys. Acta, 2012, 1817, 1164–1176 CrossRef CAS PubMed.
- B. A. Diner and R. D. Britt, in Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, ed. T. J. Wydrzynski and K. Satoh, Springer, Dordrecht, The Netherlands, 2005, pp. 207–233 Search PubMed.
- S. Styring, J. Sjöholm and F. Mamedov, Biochim. Biophys. Acta, 2012, 1817, 76–87 CrossRef CAS PubMed.
- F. Rappaport, A. Boussac, D. A. Force, J. Peloquin, M. Brynda, M. Sugiura, S. Un, R. D. Britt and B. A. Diner, J. Am. Chem. Soc., 2009, 131, 4425–4433 CrossRef CAS PubMed.
- K. Saito, J. R. Shen, T. Ishida and H. Ishikita, Biochemistry, 2011, 50, 9836–9844 CrossRef CAS PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7690–7695 CrossRef CAS PubMed.
- S. Nakamura and T. Noguchi, Biochemistry, 2015, 54, 5045–5053 CrossRef CAS PubMed.
- S. Luber, I. Rivalta, Y. Umena, K. Kawakami, J. R. Shen, N. Kamiya, G. W. Brudvig and V. S. Batista, Biochemistry, 2011, 50, 6308–6311 CrossRef CAS PubMed.
- M. Askerka, D. J. Vinyard, J. Wang, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 1713–1716 CrossRef CAS PubMed.
- J. Wang, M. Askerka, G. W. Brudvig and V. S. Batista, ACS Energy Lett., 2017, 2, 2299–2306 CrossRef CAS PubMed.
- S. Petrie, R. Stranger and R. J. Pace, Phys. Chem. Chem. Phys., 2017, 19, 27682–27693 RSC.
- S. Petrie, R. J. Pace and R. Stranger, Angew. Chem., Int. Ed., 2015, 54, 7120–7124 CrossRef CAS PubMed.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J. R. Shen and K. Yamaguchi, Dalton Trans., 2012, 41, 13727–13740 RSC.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- M. Askerka, J. Wang, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2016, 55, 981–984 CrossRef CAS PubMed.
- M. Shoji, H. Isobe, T. Nakajima, Y. Shigeta, M. Suga, F. Akita, J. R. Shen and K. Yamaguchi, Faraday Discuss., 2017, 198, 83–106 RSC.
- H. Isobe, M. Shoji, J. R. Shen and K. Yamaguchi, Inorg. Chem., 2016, 55, 502–511 CrossRef CAS PubMed.
- A. Haddy, Photosynth. Res., 2007, 92, 357–368 CrossRef CAS PubMed.
- K. G. V. Havelius, J. Sjöholm, F. M. Ho, F. Mamedov and S. Styring, Appl. Magn. Reson., 2009, 37, 151–176 CrossRef CAS.
- M. Chrysina, G. Zahariou, N. Ioannidis and V. Petrouleas, Biochim. Biophys. Acta, 2010, 1797, 487–493 CrossRef CAS PubMed.
- J. Sjoholm, S. Styring, K. G. Havelius and F. M. Ho, Biochemistry, 2012, 51, 2054–2064 CrossRef PubMed.
- A. Klauss, M. Haumann and H. Dau, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16035–16040 CrossRef CAS PubMed.
- A. Klauss, T. Sikora, B. Suss and H. Dau, Biochim. Biophys. Acta, 2012, 1817, 1196–1207 CrossRef CAS PubMed.
- F. Rappaport, M. Blanchard-Desce and J. Lavergne, Biochim. Biophys. Acta, 1994, 1184, 178–192 CrossRef CAS.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2012, 14, 4849–4856 RSC.
- S. Nakamura, R. Nagao, R. Takahashi and T. Noguchi, Biochemistry, 2014, 53, 3131–3144 CrossRef CAS PubMed.
- D. Narzi, D. Bovi and L. Guidoni, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8723–8728 CrossRef CAS PubMed.
- A. Hinchliffe, Chem. Phys. Lett., 1974, 27, 454–456 CrossRef CAS.
- P. J. O'Malley, Chem. Phys. Lett., 2000, 325, 69–72 CrossRef.
- J. Spanget-Larsen, M. Gil, A. Gorski, D. M. Blake, J. Waluk and J. G. Radziszewski, J. Am. Chem. Soc., 2001, 123, 11253–11261 CrossRef CAS PubMed.
- P. J. O'Malley, Biochim. Biophys. Acta, 2002, 1553, 212–217 CrossRef.
- R. Ramaekers, J. Pajak, M. Rospenk and G. Maes, Spectrochim. Acta, Part A, 2005, 61, 1347–1356 CrossRef PubMed.
- R. Takahashi and T. Noguchi, J. Phys. Chem. B, 2007, 111, 13833–13844 CrossRef CAS PubMed.
- W. J. McDonald and O. Einarsdottir, J. Phys. Chem. A, 2008, 112, 11400–11413 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., Wallingford CT, 2009.
- D. Case, T. Darden, T. Cheatham III, C. Simmerling, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang and K. Merz, AMBER12, University of California, San Francisco, USA, 2012.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. Laino, F. Mohamed, A. Laio and M. Parrinello, J. Chem. Theory Comput., 2005, 1, 1176–1184 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., Int. Ed., 2013, 52, 11744–11749 CrossRef CAS PubMed.
- D. Narzi, G. Mattioli, D. Bovi and L. Guidoni, Chemistry, 2017, 23, 6969–6973 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- D. Narzi, E. Coccia, M. Manzoli and L. Guidoni, Biophys. Chem., 2017, 229, 93–98 CrossRef CAS PubMed.
- M. Capone, D. Bovi, D. Narzi and L. Guidoni, Biochemistry, 2015, 54, 6439–6442 CrossRef CAS PubMed.
- D. Bovi, M. Capone, D. Narzi and L. Guidoni, Biochim. Biophys. Acta, 2016, 1857, 1669–1677 CrossRef CAS PubMed.
- D. Bovi, D. Narzi and L. Guidoni, New J. Phys., 2014, 16, 015020 CrossRef.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- C. Berthomieu, C. Boullais, J.-M. Neumann and A. Boussac, Biochim. Biophys. Acta, 1998, 1365, 112–116 CrossRef CAS.
- A. Sirohiwal, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2019, 141, 3217–3231 CrossRef CAS PubMed.
- N. Ahmadova, F. M. Ho, S. Styring and F. Mamedov, Biochim. Biophys. Acta, 2017, 1858, 407–417 CrossRef CAS PubMed.
- N. Sakashita, H. C. Watanabe, T. Ikeda, K. Saito and H. Ishikita, Biochemistry, 2017, 56, 3049–3057 CrossRef CAS PubMed.
- D. Narzi, M. Capone, D. Bovi and L. Guidoni, Chemistry, 2018, 24, 10820–10828 CrossRef CAS PubMed.
- T. Shimizu, M. Sugiura and T. Noguchi, J. Phys. Chem. B, 2018, 122, 9460–9470 CrossRef CAS PubMed.
- T. Noguchi, Y. Inoue and X. S. Tang, Biochemistry, 1997, 36, 14705–14711 CrossRef CAS PubMed.
- C. F. Fowler, Biochim. Biophys. Acta, 1977, 462, 414–421 CrossRef CAS.
- E. Schlodder and H. T. Witt, J. Biol. Chem., 1999, 274, 30387–30392 CrossRef CAS.
- H. Suzuki, M. Sugiura and T. Noguchi, J. Am. Chem. Soc., 2009, 131, 7849–7857 CrossRef CAS PubMed.
- H. J. Hwang, P. Dilbeck, R. J. Debus and R. L. Burnap, Biochemistry, 2007, 46, 11987–11997 CrossRef CAS PubMed.
- M. Capone, D. Narzi, D. Bovi and L. Guidoni, J. Phys. Chem. Lett., 2016, 7, 592–596 CrossRef CAS PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L. J. Yu, T. Sakamoto, T. Motomura, J. H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J. R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- M. Shoji, H. Isobe and K. Yamaguchi, Chem. Phys. Lett., 2015, 636, 172–179 CrossRef CAS.
- I. Ugur, A. W. Rutherford and V. R. Kaila, Biochim. Biophys. Acta, 2016, 1857, 740–748 CrossRef CAS PubMed.
- K. Kawashima, K. Saito and H. Ishikita, Biochemistry, 2018, 57, 4997–5004 CrossRef CAS PubMed.
- H. Kuroda, N. Kodama, X. Y. Sun, S. Ozawa and Y. Takahashi, Plant Cell Physiol., 2014, 55, 1266–1275 CrossRef CAS PubMed.
- R. Nagao, H. Ueoka-Nakanishi and T. Noguchi, J. Biol. Chem., 2017, 292, 20046–20057 CrossRef CAS.
- H. Sakamoto, T. Shimizu, R. Nagao and T. Noguchi, J. Am. Chem. Soc., 2017, 139, 2022–2029 CrossRef CAS PubMed.
- T. Noguchi, H. Suzuki, M. Tsuno, M. Sugiura and C. Kato, Biochemistry, 2012, 51, 3205–3214 CrossRef CAS PubMed.
- A. Klauss, M. Haumann and H. Dau, J. Phys. Chem. B, 2015, 119, 2677–2689 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04605d |
| This journal is © the Owner Societies 2020 |