
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of GdxFeyOz films using an atomic layer deposition-type approach†
Pengmei
Yu
 a,
Sebastian M. J.
Beer
b,
Anjana
Devi
b and
Mariona
Coll
*a
a,
Sebastian M. J.
Beer
b,
Anjana
Devi
b and
Mariona
Coll
*a
aInstitut de Ciéncia de Materials de Barcelona (ICMAB-CSIC), Campus de la UAB, 08193 Bellaterra, Barcelona, Spain. E-mail: mcoll@icmab.es; Tel: +34 935801853
bInorganic Materials Chemistry, Ruhr University Bochum, Universitätsstraβe 150, Bochum 44801, Germany
First published on 7th December 2020
Abstract
The growth of complex oxide thin films with atomic precision offers bright prospects to study improved properties and novel functionalities. Here we tackle the fabrication of gadolinium iron oxide thin films by an atomic layer deposition-type approach in which iron and gadolinium tailor-made metalorganic precursors (bis(N-isopropyl ketoiminate)iron(II), [Fe(ipki)2] and tris(N,N′-diisopropyl-2-dimethylamido-guanidinato)gadolinium(III), [Gd(DPDMG)3]) are alternately reacted with ozone and deposited on silicon substrates at 250 °C. The structure, chemical composition and magnetic properties of the resulting films are compared with those obtained from a commercially available ferrocene precursor [Fe(Cp)2] and [Gd(DPDMG)3]. All films resulted in cation ratio close to nominal stoichiometry with negligible amount of organic species. The tailor-made metalorganic precursors, designed to provide similar thermal behavior, result in the formation of polycrystalline Gd3Fe5O12 films coexisting with GdFeO3, Gd2O3 and Fe2O3 whereas the combination of [Fe(Cp)2] and [Gd(DPDMG)3] mainly favors the formation of Gd3Fe5O12 films coexisting with traces of Gd2O3. This study demonstrates that this is a viable route to prepare complex GdxFeyOz films and could be used for the design of complex oxide films with improved properties upon rigorous study of the compatibility of metalorganic precursors.
1 Introduction
GdFeO3 and Gd3Fe5O12 are two ternary oxide systems sharing fascinating magnetic and magneto-optical properties1,2 that can open new directions in the field of spintronics and magnonics.3–7 The ability to prepare stable GdxFeyOz phases with the appropriate stoichiometry,8,9 often as coatings of high aspect ratio structures, is becoming relevant to study new physics emerging from the films and at their interfaces.10,11 The best layer uniformity with atomic scale control is effectively achieved by atomic layer deposition (ALD).12,13 This is a versatile thin film deposition technique that relies on alternate pulsing of chemical precursors that react with the surface species in a self-limiting manner. Hence, the ALD process consists of a repetition of a basic sequence of reactant pulses and purges, i.e. a cyclic process. These characteristics offer exceptional control in film composition and thickness and excellent step coverage over complex structures. This is a well-established technique to deposit binary compounds14–16 and its unique deposition characteristics motivates the continuous research to develop deposition process for compositions with increased complexity.17 Unfortunately, the ALD synthesis of a ternary oxide is, in most of the cases, not a simple addition of the corresponding binary oxide processes.18,19 The compatibility of different chemical precursors confined under similar deposition conditions together with a varying surface species after every precursor dose increase considerably the level of intricacy.17,20,21 Indeed, LaFeO3,18 La1−xSrxFeO3 (ref. 18) and ErFeO3 (ref. 22) are some of the few examples reported on ALD rare-earth ferrites deposited by alternate pulsing of one reactant per component of the complex oxide. More recently, epitaxial GdFeO3 films have been grown by alternate dosing of a single heterobimetallic precursor, already containing the stoichiometric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cation ratio, and ozone.23
:1 cation ratio, and ozone.23
Clearly, the development of GdxFeyOz phases by ALD will require knowledge on the synthetic path which strongly depends on the chemistry of the metalorganic precursors. In this study we are interested in exploring the opportunities that the chemistry of homometallic Gd and Fe compounds can offer to grow Gd–Fe-oxide thin films by an ALD-type approach. To this end, a special focus has to be laid to the compatibility of Fe and Gd precursors in terms of reactivity, volatility and thermal stability24,25 as the commonly used precursors for these elements do not have matching properties. While there is a rich variety of suitable iron precursors based on cyclopentadienyl, halide, β-diketonate and acetylacetonate ligands, see Table S1,† there are limited sources for Gd mainly involving silylamide, β-diketonate and cyclopentadienyl-based ligand systems, see Table S2.† Among these precursors, we investigate the use of the cost-efficient and commercially available ferrocene precursor, [Fe(Cp)2], in which the metal is bonded to two η5-cyclopentadienyl rings exhibiting a high thermal stability, versus the use of bis(N-isopropyl ketoiminate)iron(II), [Fe(ipki)2],26 which possesses a ketoiminate ligand with mixed nitrogen and oxygen donors providing reactivity and stability, respectively. Their reactivity is evaluated against tris(N,N′-diisopropyl-2-dimethylamido-guanidinato)gadolinium(III), [Gd(DPDMG)3]27 which is a fully nitrogen coordinated complex and possesses an enhanced reactivity due to the rare-earth nitrogen bonds, see Scheme 1.
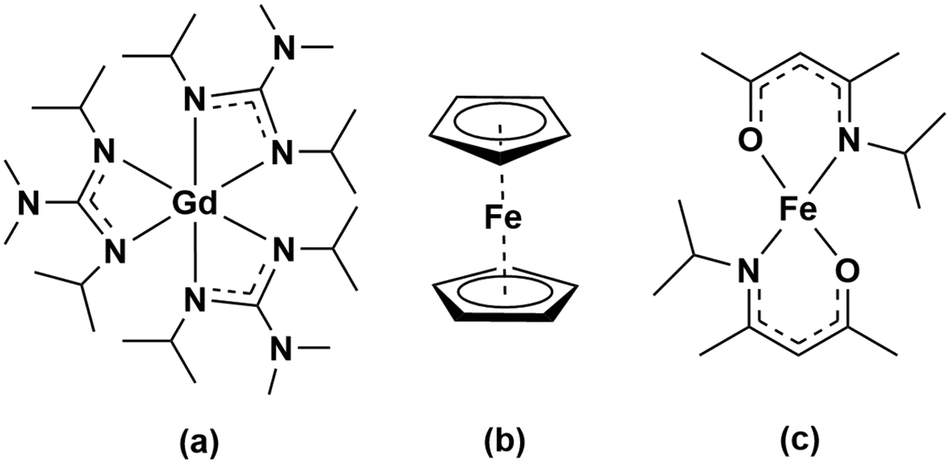 | ||
| Scheme 1 Molecular structure of the Gd and Fe precursors studied in this work: (a) [Gd(DPDMG)3], (b) [Fe(Cp)2], (c) [Fe(ipki)2]. | ||
First, we have studied the reactivity of each homo-metallic precursor with H2O and O3 to grow the corresponding binary oxides and identify the compatible deposition conditions for the subsequent ternary oxide system. By means of X-ray diffraction (XRD), atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS) and Rutherford backscattering spectrometry/nuclear reaction analysis (RBS/NRA), the phase purity, crystallinity, surface morphology and chemical composition of the grown complex compounds have been examined. Finally, the magnetic properties have been evaluated by superconducting quantum interference device (SQUID) magnetometry.
2 Experimental
2.1 Thin film deposition
The GdxFeyOz thin films deposition was carried out in a Savannah 100 ALD system from Cambridge NanoTech Inc. P-type boron-doped Si(100) and SiO2/Si(100) substrates were purchased from Active Business Company GmbH. Before introduction into the ALD chamber, the substrates were cleaned with acetone and ethanol followed by nitrogen gas blow drying. Prior to film growth, all substrates were kept in a constant high purity nitrogen flow for 10 min and subsequently exposed to ozone pulses to form a hydroxyl-terminated surface. Precursor sources were pulsed into the reaction chamber separated in time and purged by high purity nitrogen with a gas flow between 40–70 sccm. Ozone (O3) and Milli-Q water (H2O) were considered as oxygen sources. O3 was produced and delivered upon use from an LG-7 Corona discharge ozone generator with a 5 wt% concentration and an output of 5 g h−1. The generator was modulated to work at 5 V direct current voltage with an adjusted flow of 0.18 L min−1. The metalorganic precursors, [Gd(DPDMG)3] and [Fe(ipki)2] were synthesised as reported27,26 and handled under inert conditions, while the commercial Fe precursor [Fe(Cp)2] or ferrocene, (bis(cyclopentadienyl)iron (98% purity)), was purchased from Sigma-Aldrich and utilized as received. In this study we utilized three different valving systems that are here identified as flow mode (F), pressure-boost (PB) and exposure mode (E), scheme shown in Fig. S1.† F consists in having a continuous flow of inert gas to carry the precursor to the reaction chamber. For low-vapor pressure precursors, PB is used. That is, before precursor pulses, the gas exit valve was closed and ALD valve opened to allow carrier gas filling up the precursor cylinder and resulting in an elevated pressure in it. Afterwards, the exit valve is opened and the chamber pressure recovered, followed by precursor pulsing along with the filled nitrogen into the chamber. Under certain circumstances, E was applied. In this case, the gas exit valve was closed, exposing the sample to the precursor prolonged time to promote surface reaction and saturation over larger areas. Post-deposition thermal treatments, were performed on selected samples in an oxidising atmosphere (oxygen, 0.3 L min−1 flow) in a tubular furnace at 450–900 °C for 30–90 minutes.2.2 Thin film characterization
Qualitative elemental film composition studies were performed by energy disperse X-ray spectroscopy (EDX) employing a FEI Quanta 200 FEG microscope. Each spectrum was collected with 15 keV working voltage, 3.0 spot size for an acquisition time of 100 s. Quantitative compositional studies were carried out using X-ray photoelectron spectroscopy (XPS) using a SPECS PHOIBOS 150 hemispherical X-ray photoelectron spectroscopy analyser (Al Kα source, from SPECS GmbH, Berlin, Germany). The samples were analyzed with a pass energy of 10 eV at a base pressure of 1 × 10−10 mbar, taking a step size of 0.05 eV and 1 eV for the high-resolution and survey spectra, respectively. The acquired spectra were interpreted with CasaXPS software for binding energy determination and element assignment after spectrum calibration with C 1s reference peak at 284.8 eV.Thin film phase purity and crystallinity were investigated by X-ray diffraction (XRD) in the range of 2θ = 20–80° by grazing-incidence X-ray diffraction (GIXRD) at the incident angle of 1 °C. Specific samples were analyzed by θ–2θ scan on a Bruker D8 Discover A25 diffractometer with Cu Kα radiation (Kα = 1.5406 Å). The film texture was further evaluated with GADDS (General Area Detector Diffraction System) Bruker AXS, Inc equipped with a 2D detector.
The film thickness was extracted from X-ray reflectivity (XRR) measurements using a SIEMENS D5000 diffractometer and fitted using ReMagX software.29 It was further validated with spectroscopic ellipsometry (SE) measurements using a GES5E Ellipsometer from SOPRA Optical Platform. The measurements were carried out in reflection mode with variant light incident angles (65–75°) at 30° analyzer angle for a spectral range of 200–900 nm. Thin film surface morphology was assessed from atomic force microscope topographic images, 5 μm × 5 μm in size using a Keysight 5100 microscope and further processed with Mountains®8 software.
The magnetic properties were probed by a Quantum Design MPMS-XL magnetometer based on a superconducting quantum interference device (SQUID). In-plane magnetization–temperature curve (M(T)) at 5 kOe and magnetic hysteresis loop (M(H)) at 50 K were explored.
Additional film composition and depth profiles were obtained from Rutherford backscattering spectrometry (RBS) coupled with nuclear reaction analysis (NRA). The experiments were carried out in the RUBION, the Central Unit for Ion Beams and Radionuclides at Ruhr University Bochum. For the measurements, a 2.0 MeV 4He+ beam with an intensity of 20–40 nA (tilt angle 7°) was used. The backscattered particles were detected at an angle of 160° by a Si detector with a resolution of 16 keV. NRA measurements were carried out with a 2H+ beam (1 MeV). The samples were tilted at an angle of 7°. The emitted protons were detected at an angle of 135° with respect to the beam axis. The detector covered a solid angle of 23 msrad and was shielded by a 6 μm Ni foil to eliminate elastically scattered deuterons. Typical beam currents on the samples were close to 40 nA in an area of ∼1 mm in diameter, whereas the collected beam charge for a sample was 12 μC. The RBS/NRA data were analyzed by means of the SIMNRA program suite.30 Since the detection limit of RBS is restricted especially for light atomic mass elements, NRA is used to detect C, N and O in thin film material. The calculation of the atomic percentages is achieved through the integration of the detected counts per area (in order of magnitudes of 1015 at cm−2). It must be considered that the total counts are logically lower for thin samples. Hence, not only the statistic error of the cross-section of the beam has an influence on the calculation of N and C, but also the subtraction of the background measurement (bare Si(100) substrate in our case) is required for the NRA analysis. Therefore an error margin of approximately 3 at% can be expected for C, N and O.
3 Results and discussion
Prior to study the deposition of ternary oxides, it is necessary to study the independent binary oxide processes, i.e. gadolinium oxide and iron oxide. The optimization procedure includes choice of oxidant co-reactants, precursor sublimation temperature, deposition temperature, carrier gas flow and valving system (flow, pressure boost, exposure described in the Experimental section), to identify the compatible conditions to finally assess the deposition of the multicomponent GdxFeyOz system.3.1 Gd2O3 films
The deposition of gadolinium oxide from [Gd(DPDMG)3] was studied in our reactor according to previous work.28 The viability of ozone versus water as co-reactant was investigated in the deposition range of 160–250 °C with 40 sccm N2 gas for 500 cycles under flow mode. The precursor sublimation temperature was optimized to 135 °C and the use of pressure boost to enhance its sublimation was evaluated. The use of water as co-reactant did not result in the deposition of gadolinium, regardless of the valving system. On the other hand, combining [Gd(DPDMG)3] with pressure boost, with ozone at 250 °C, Gd was detected in the films, Fig. S2.† The corresponding AFM topographic image illustrates high degree of film density, surface uniformity with a root mean square (RMS) roughness value below 1 nm, Fig. 1a.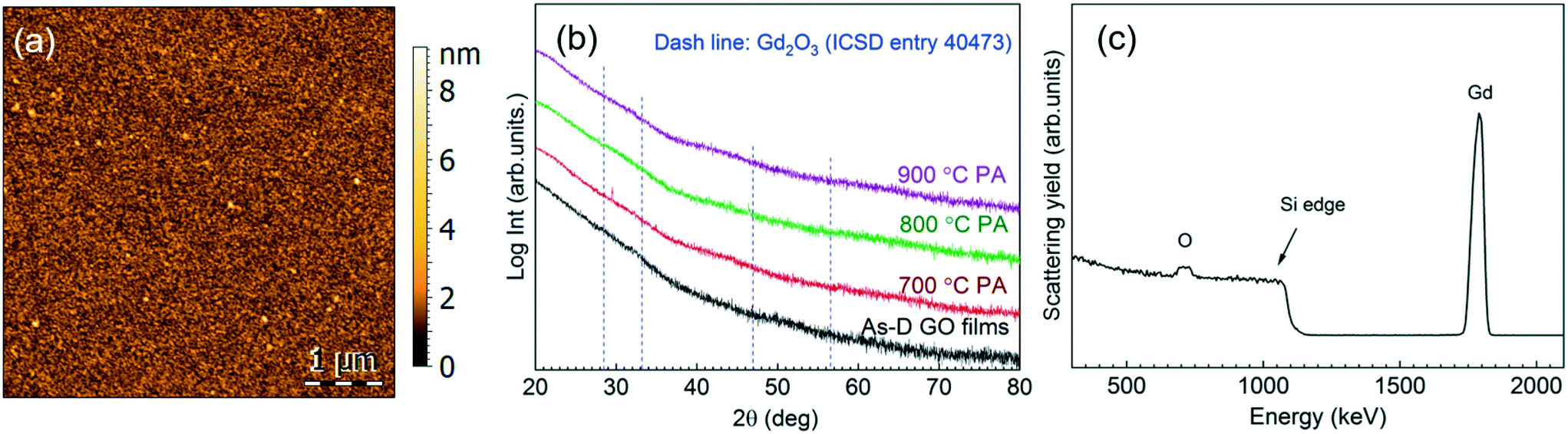 | ||
| Fig. 1 (a) AFM topographic image of an as-deposited sample from [Gd(DPDMG)3] and O3 at the deposition temperature of 250 °C. (b) GIXRD spectra of the as-deposited (As-D) and post-annealed (PA) films (700–900 °C). (c) RBS profile of the 800 °C post-annealed sample. | ||
Phase formation and degree of crystallinity of a 30 nm thick film was studied by GIXRD at the as-deposited stage and after different post annealing treatments (700–900 °C, 30 min in oxygen atmosphere), see Fig. 1b. The absence of Bragg reflections in the as-deposited step suggests the formation of amorphous films. The same patterns were obtained after post annealing treatments up to 900 °C, suggesting that the films remain amorphous. From RBS/NRA analysis the chemical composition of the film was studied in detail to rule out the presence of impurities trapped in the film that could hinder the crystallization process,31,32 see Fig. 1c. In the RBS profile it can be distinguished the Gd peak and the O peak superimposing the Si signal. Note that the tailing of the Si edge and the tailing of the Gd signal to lower energies indicate a diffusion of Gd and Si at the interface. This phenomenon can be explained with the tendency of rare-earth to form silicates and silicides at such interfaces,33 and could contribute to the poor crystallinity of the films in Fig. 1b. RBS/NBA quantification, Table S3,† reveals a low nitrogen and oxygen content of 0.4 at% and <3%, respectively. Note that the Gd/O ratio is 0.30 evidencing an oxygen overstoichiometry. Similar observations have been recently reported in ALD iron oxide films and attributed to the prolonged ozone exposure.34
To evaluate the deposition homogeneity across a 4′′ Si wafer using [Gd(DPDMG)3], the film thickness was analyzed at six different locations by means of XRR, see Fig. S3.† The areal thickness map was built in which the x axis corresponds to the gas flow pathway in the chamber, the y axis is the direction perpendicular to the flow, while z axis corresponds to the film thickness. As the wafer size is similar to the diameter of the ALD chamber, at x = 0 cm one can find the gas inlet whereas at x = 10 cm sits the gas outlet. For 40 sccm flow, Fig. 2a, the maximum thickness achieved is near the gas inlet and decreases along with the gas pathway inlet. This areal thickness gradient can result from insufficient gas flow, a leak upstream of the precursor or inadequate purge times.35 By increasing the gas flow to 70 sccm, the thickness homogeneity is improved although the areal gradient is still observed, Fig. 2b. Finally, longer precursor exposure to the reaction surface along with longer purging times (exposure mode) was explored but the amount of Gd deposited was below the EDX and XRR detection limit. This behavior suggests that prolonged exposure of [Gd(DPDMG)3] at 250 °C does not favor a saturation growth model either.
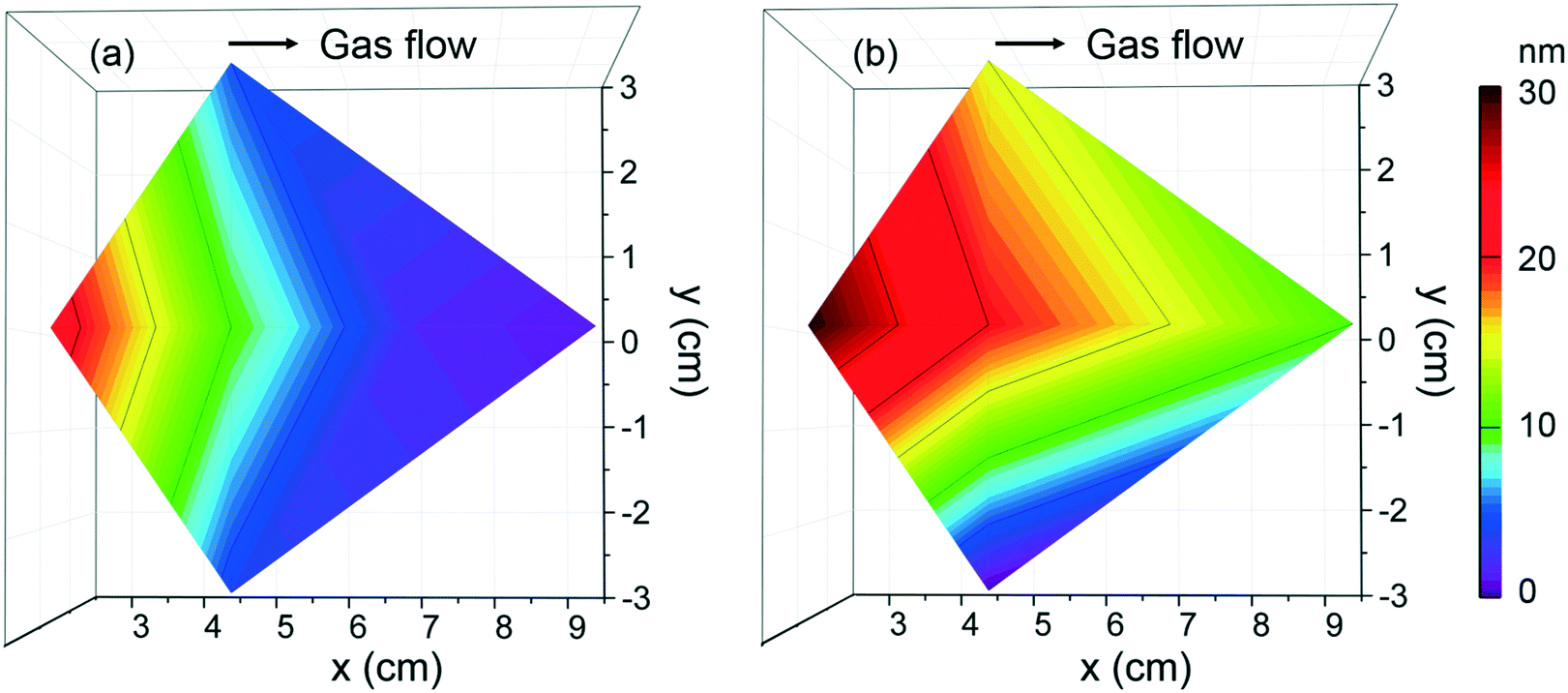 | ||
| Fig. 2 Thickness mappings of Gd2O3 films grown at (a) 40 sccm and (b) 70 sccm N2 flow located at different positions of a 4′′ Si wafer in the ALD chamber. Deposition conditions: Tdep = 250 °C with ozone using PB mode, 500 cycles. | ||
From deposition at 250 °C and 70 sccm gas flow, a growth per cycle (GPC) of ∼0.6 Å per cycle is obtained near the inlet. Similar growth rates are reported when combined Gd(thd)3/O3 (ref. 36) although higher growth rates have been reported when using N-bonded Gd precursors and water (1 Å per cycle).27 It is very likely that the use of O3 and N-containing ligands can form intermediate NOx and C–O species37 resulting in a more complex surface reaction mechanism affecting the growth rate.31
According to this study, the selected Gd2O3 deposition conditions for further studies were selected: TGd-subl = 135 °C and pressure boost, Tdep = 250 °C, 70 sccm N2 carrier gas flow and ozone as co-reactant. Note that experimental conditions here reported do not lead to a self-limiting surface reaction.
3.2 Fe2O3 films
There is a divergence of deposition conditions reported for the preparation of ALD Fe2O3 from ferrocene.34,35,38–40 One main purpose of our study is to investigate the feasibility of [Fe(Cp)2] to react with H2O or O3 to form Fe2O3 in our reactor and to evaluate its compatibility with the [Gd(DPDMG)3] precursor.The deposition of [Fe(Cp)2] was studied in the 160–250 °C temperature window in flow mode (70 sccm) repeating 2000 cycles, Fig. S4.† Fe was only detected when ozone was used as co-reactant for a temperature range of 200–250 °C. The 15 nm as-deposited films (250 °C) present a mesh-like surface morphology with a surface RMS roughness of 3 nm, Fig. 3a, and crystallize into the α-Fe2O3 phase (ICSD entry 24004), Fig. 3b. EDX spectrum of an as-deposited film is shown in Fig. 3c.
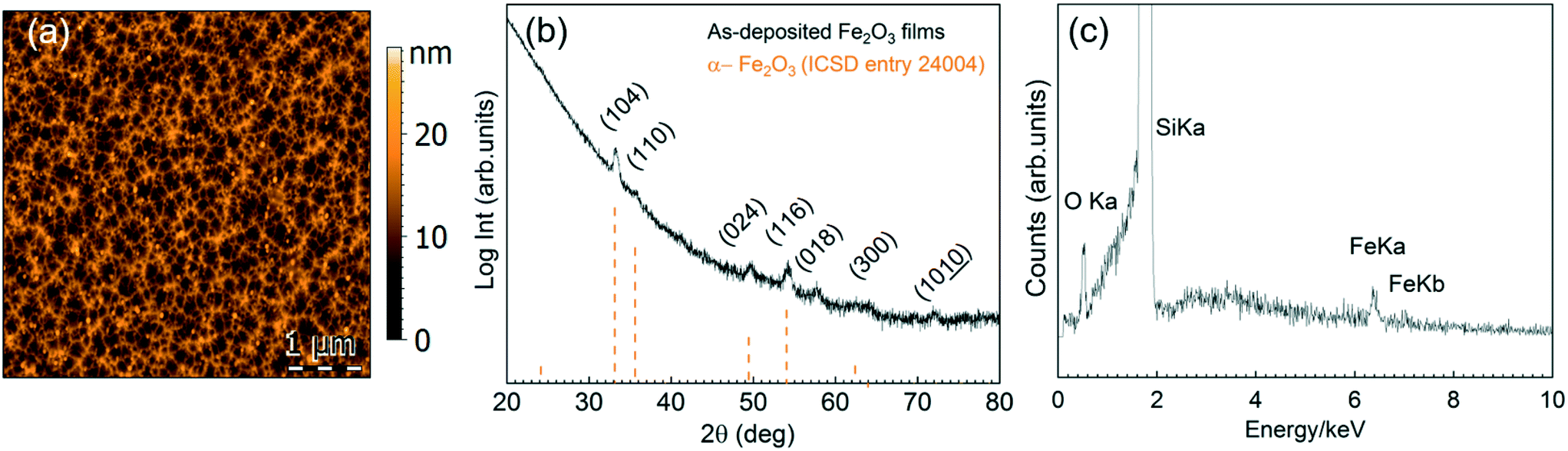 | ||
| Fig. 3 (a) 5 μm × 5 μm AFM topography image from the as-deposited 15 nm film prepared from [Fe(Cp)2] and ozone at Tdep = 250 °C, (b) corresponding GIXRD pattern. (c) EDX spectrum showing Fe in the deposited films. | ||
According to the deposition conditions identified above, the thickness homogeneity along a 4′′ silicon wafer was investigated using [Fe(Cp)2] and ozone at 250 °C at 70 sccm, Fig. 4a. The maximum thickness, 15 nm, is identified towards the gas outlet revealing an evident thickness gradient. To improve the Fe2O3 deposition homogeneity, exposure mode was adopted enabling ferrocene and ozone to stay longer period of time in the chamber and promote surface saturation. In this case, an average thickness of 20 nm was obtained across the whole Si wafer, Fig. 4b. The resulting GPC is ∼0.1 Å per cycle, significantly lower than the reported GPC values when combined [Fe(Cp)2] and O3 (0.6–1.4 Å per cycle).39,40 Schneider et al.34 recently reported that lower growth per cycles at 250 °C can be assigned to increased decomposition of ozone.
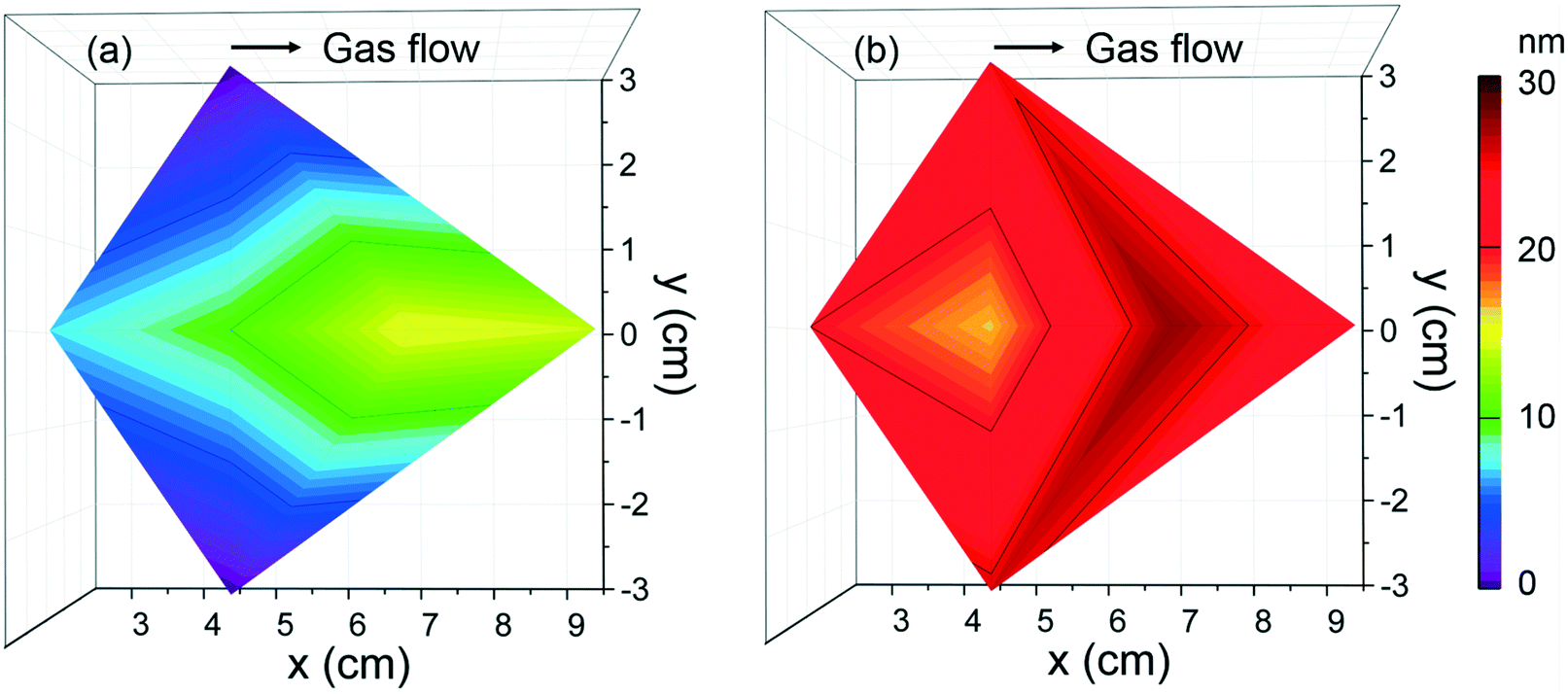 | ||
| Fig. 4 Thickness mappings of Fe2O3 films deposited from [Fe(Cp)2] and O3 at Tdep = 250 °C, 2000 cycles at (a) flow mode and (b) exposure mode with 70 sccm N2 gas. | ||
From this study the suitable processing conditions identified for Fe2O3 using ferrocene are TFe-subl = 90 °C, Tdep = 250 °C with ozone as co-reactant under 70 sccm N2 gas and exposure mode, which could be compatible with those previously found for the [Gd(DPDMG)3] precursor.
The suitability of the ketoiminate iron precursor [Fe(ipki)2],26 to deposit Fe2O3 films was evaluated and compared to the commercial [Fe(Cp)2] and to [Gd(DPDMG)3].
Based on the previous studies on [Fe(ipki)2],26 the iron precursor was sublimed at 100 °C (with and with no pressure boost) in the temperature window of 150–250 °C using H2O or O3 as co-reactant under 70 sccm N2 gas flow for 2000 cycles. Based on EDX studies, no iron was detected at any of these processing conditions, see Fig. S5a.† Therefore, the sublimation temperature of [Fe(ipki)2] was increased up to 130 °C and the processing conditions were re-evaluated. Depositing Fe2O3 using [Fe(ipki)2] and O3 at 250 °C resulted in a 65 nm film, Fig. S5b and c.† AFM analysis on as-deposited films reveals a dense and homogeneous surface with very low surface roughness (RMS ∼ 1 nm), Fig. 5a. The crystallinity of 65 nm Fe2O3 films was investigated by GIXRD. In the as-deposited stage, the films are amorphous. Subsequent post thermal treatments for 30 min in oxygen at 500 °C and at 800 °C did not favor the crystallization of Fe2O3. However, typical peaks of α-Fe2O3 were observed at 900 °C, Fig. 5b. It can be seen from the RBS analysis, Fig. 5c, that distribution of Fe and O is homogeneous throughout the film thickness with a ratio of Fe to O of 0.52 and C and N abundance below <0.8% indicating a clean surface reaction and desorption of byproducts during the growth of Fe2O3 (see Table S3†). Fig. 6 shows the film thickness homogeneity along the 4′′ silicon wafer. In this case, a thickness gradient from 65 nm to 45 nm was identified, showing a preferred deposition near the gas inlet, similar to the Gd2O3. Accordingly, the preferred conditions for Fe2O3 deposition from [Fe(ipki)2] using an ALD-type approach is: TFe-subl = 130 °C, Tdep = 250 °C, ozone as co-reactant at 70 sccm N2 flow.
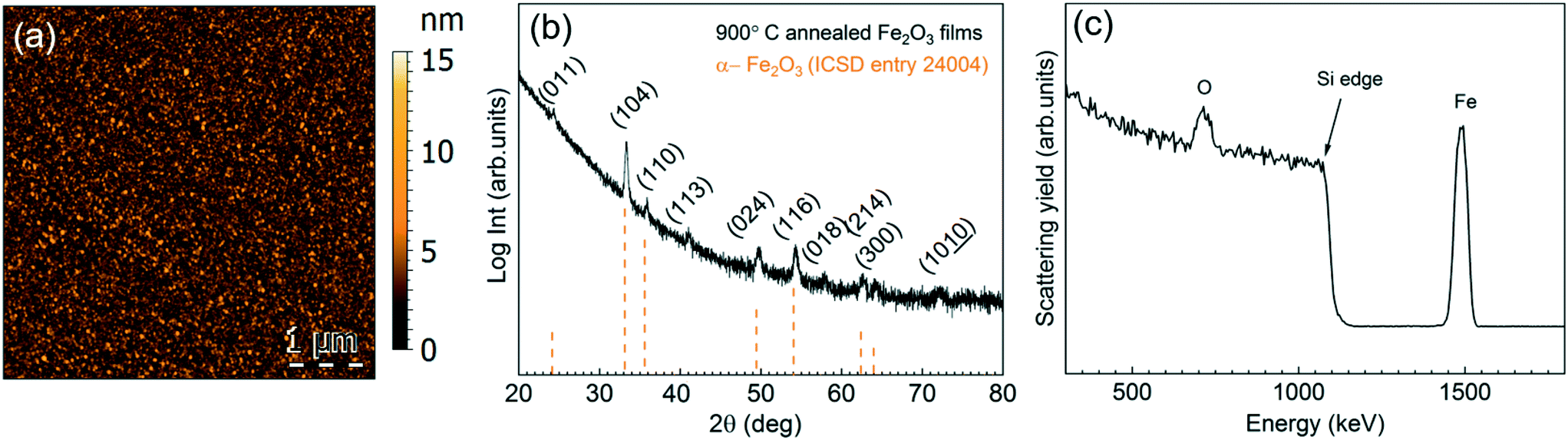 | ||
| Fig. 5 Characteristics of the Fe2O3 films prepared by alternate pulsing of [Fe(ipki)2] (in pressure boost mode) and ozone at 250 °C. (a) AFM topographic image of the as-deposited Fe2O3 films, (b) GIXRD spectrum of a 900 °C annealed sample, (c) RBS spectrum of the Fe2O3 annealed at 800 °C. | ||
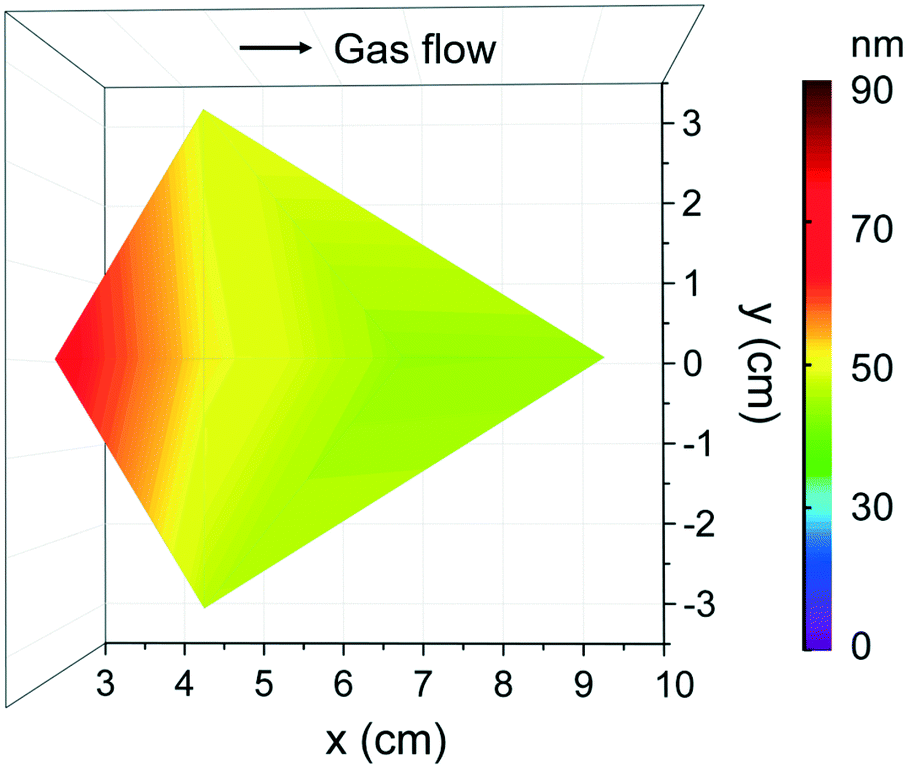 | ||
| Fig. 6 Thickness homogeneity map of Fe2O3 films that are located at various chamber positions on a 4′′ Si wafer from [Fe(ipki)2] and O3 under 70 sccm flow at Tdep = 250 °C with 2000 cycles. | ||
The resultant growth rate of iron oxide films is ∼0.4 Å per cycle, which is more than four times higher than that from [Fe(Cp)2] (0.1 Å per cycle). The higher GPC of [Fe(ipki)2] could be attributed to the enhanced reactivity from the mixed N–O-ligand chemistry26,41 compared to the stable Fe–metallocene bonding in [Fe(Cp)2].39 The steric hindrance of the ligands will also play a key role. The low reactivity of Cp can certainly slow the reaction mechanism when compared to the ketoiminate ligands.42–44 This difference in precursor chemistry also affects the surface morphology of the final film noting that [Fe(ipki)2] leads to smooth and dense films whereas [Fe(Cp)2] favors higher degree of porosity (Fig. 3a).
The optimal deposition conditions for each binary oxide are summarized in Table 1. Along with the difference in GPC of [Fe(Cp)2] compared to [Gd(DPDMG)3] and [Fe(ipki)2] already mentioned above, distinct thickness homogeneity along the silicon wafer was detected (Fig. 2b, 4b and 6). [Fe(ipki)2] and [Gd(DPDMG)3] showed a clear trend to be deposited near the gas inlet, whereas [Fe(Cp)2] resulted in a highly homogeneous distribution. This discrepancy could also arise from the pronounced reactivity of the [Gd(DPDMG)3] and [Fe(ipki)2] precursors containing N– and O–metal bonds which are prone to be depleted at the reaction chamber inlet.28
| T dep (°C) | T subl (°C) | Flow (sccm) | Co-reactant | Mode | Growth rate (Å per cycle) | |
|---|---|---|---|---|---|---|
| Gd–O from [Gd(DPDMG)3] | 250 | 135 pressure-boost | 70 | O3 | Flow | 0.6 |
| Fe–O from [Fe(Cp)2] | 250 | 90 | 70 | O3 | Exposure | 0.1 |
| Fe–O from [Fe(ipki)2] | 250 | 130 pressure-boost | 70 | O3 | Flow | 0.4 |
It is important to consider the differences observed between the three metal oxides to elaborate the deposition process of the ternary systems. Importantly, when combining the different reactants, i.e. Fe-precursor, Gd-precursor and O3, the active surface sites will differ from the studied binary system and therefore differences in the reactivity, composition and film crystallinity might be expected.20
3.3 GdxFeyOz system
The deposition of the GdxFeyOz system was first assessed by studying the combination of [Gd(DPDMG)3] and [Fe(Cp)2] with ozone at 250 °C. The deposition process was designed by alternating nanolaminates of the respective oxides and the influence of subcycle ratio and sequence were evaluated on both film stoichiometry and spatial distribution. Subsequently, the impact of post thermal treatments on phase crystallization were also studied. Table 2 lists the investigated ALD subcycle ratio and sequence along with the experimental cation ratios found by EDX analysis. According to the GPC identified in the binary oxides study (0.6 Å per cycle for Gd–O and 0.1 Å per cycle for Fe–O), a process with larger proportion of Fe–O subcycles over Gd–O subcycles was designed. Nonetheless, from this approach the amount of Gd found in the film largely exceeds that of Fe (GFO-Cp-1). In an attempt to increase the Fe content in the film while minimizing larger bilayer periods, ferrocene pulse and purge length were increased to 5 s/10 s and the superlattice ratio decreased to 10:10 (GFO-Cp-2) which results in the intended increase of Fe content (28%). The number of Cp and DPDMG ligands that react at the Gd–O and Fe–O terminated surfaces, respectively, is very likely to differ from the binary oxide system and therefore the nucleation scenario changes. Decrease in the growth rate of the mixture18,45 or even etching of the metal–O surface by the precursor ligands may occur.46 Then, by keeping a theoretical 10:10 supercycle ratio while changing the sequence from Gd–O/Fe–O to Fe–O/Gd–O (sample GFO-Cp-3), the experimental cation ratio went to 49:51 revealing that the sequence of nanolaminates deposition has also a critical role on cation incorporation. It is likely that Si–OH surface are preferred nucleation sites for ferrocene compared to Gd–O terminated surface to activate the growth.
The Gd and Fe distribution along a 4′′ silicon wafer was investigated by EDX repeating the recipe GFO-Cp-3. The compositional map reveals that Gd is mostly located near the gas inlet (left side of the map), Fig. 7a, following the same trend observed for the binary Gd2O3 process (Fig. 2). Taking advantage of this spatial Gd/Fe distribution, samples located at three different regions of the chamber corresponding to Gd/Fe ratio of 11.5, 0.6 and 1 were investigated by GIXRD analysis, Fig. 7b. In the as-deposited stage, all films turned out to be amorphous (not shown). Then, these three samples were exposed to a post annealing process at 800 °C under 0.3 L min−1 oxygen flow for 30 min. Gd-Rich films (Gd/Fe = 11.5) show the characteristic Bragg reflections corresponding to the cubic Gd2O3 phase (ICSD entry 40473). For the Gd/Fe = 0.6 sample, the formation of crystalline Gd3Fe5O12 (garnet) film is obtained (ICSD entry 27127) with minor traces of Gd2O3. These films present an homogeneous surface morphology defined by fine grain size (50 nm) and very smooth surface showing rms of 0.7 nm ± 0.1 nm, see Fig. S6.† Finally, for the Gd/Fe = 1 sample, the film remains amorphous. Therefore, gadolinium orthoferrite has not been stabilized under these conditions.
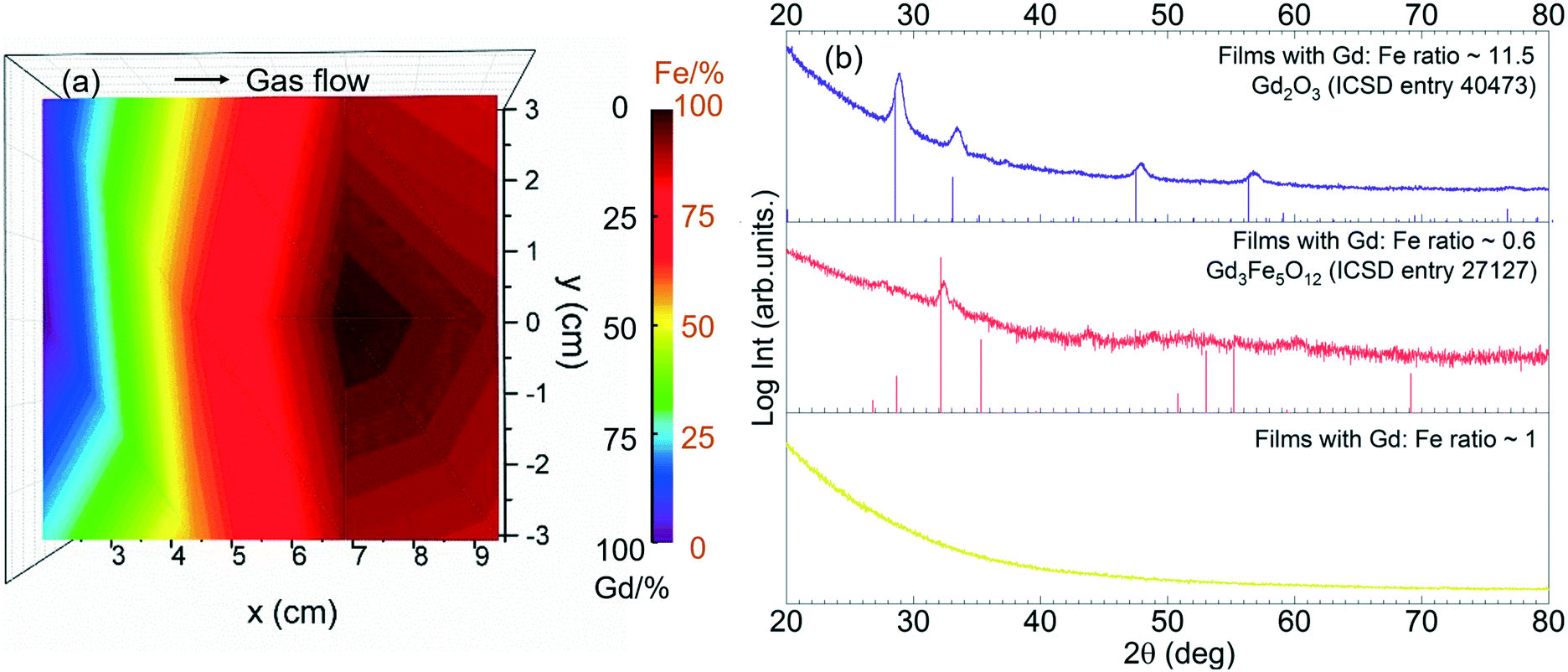 | ||
| Fig. 7 (a) Elemental distribution map of GdxFeyOz films under GFO-Cp-3 conditions. The ALD process was carried out using [Gd(DPDMG)3], [Fe(Cp)2] and O3 at the deposition temperature of 250 °C. (b) GIXRD spectra of post-annealed Gd–Fe–O samples from [Fe(Cp)2] and [Gd(DPDMG)3] which are Gd:Fe = 11.5, Gd:Fe = 0.6 and Gd:Fe = 1. Reference XRD patterns from Gd2O3, and Gd3Fe5O12 are displayed under each panel. | ||
To further investigate the chemical composition and cation valence state of the post annealed Gd3Fe5O12 sample, RBS and XPS studies were performed. From the RBS spectrum of the Gd3Fe5O12 film post annealed at 800 °C, O, Gd and Fe are present, superimposed to the signal from the silicon substrate, see Fig. 8. The elemental composition of Gd, Fe and O has been determined from RBS/NRA and listed in Table S3.† The Gd/Fe/O amount is 12.4/17.6/67.6 in at% close to the ideal Gd3Fe5O12 stoichiometry. Also, C and N content remain below 2% revealing once more that the reactivity of the precursor towards ozone is sufficient to ensure a clean reaction of the surface species with desorption of the organics. On the other hand, from XPS analysis (survey spectrum shown in Fig. S7†) of the Gd 3d and Fe 2p core level spectra were investigated to elucidate the oxidation state, Fig. 9a and b respectively. For Gd 3d core level spectra, main peaks of 3d3/2 and 3d5/2 were found at 1221.1 eV and 1189.1 eV, respectively. These values are slightly shifted to higher binding energy than the ones reported, yet the spin–orbit splitting energy difference ΔEGd 3d3/2–Gd 3d5/2 is 32.0 eV, in good agreement with the average value 32.1 ± 0.3 eV for Gd3+ containing compounds47 therefore confirming the Gd3+ oxidation number, see Table S4.† For Fe 2p, the peaks appearing at ∼725.0 eV and 711.4 eV coincide with 2p1/2 and 2p3/2 doublets. The Fe 2p3/2 peak position and ΔE2p1/2–2p3/2 being 13.6 eV indicate the +3 oxidation state of iron.48,49 The shake-up energy separation of the 2p3/2 satellite peak was found to be 7.8 eV from the main peak, in accordance with literature for Fe3+ compounds (see Table S4†), further corroborating the Fe3+ oxidation state.50,51
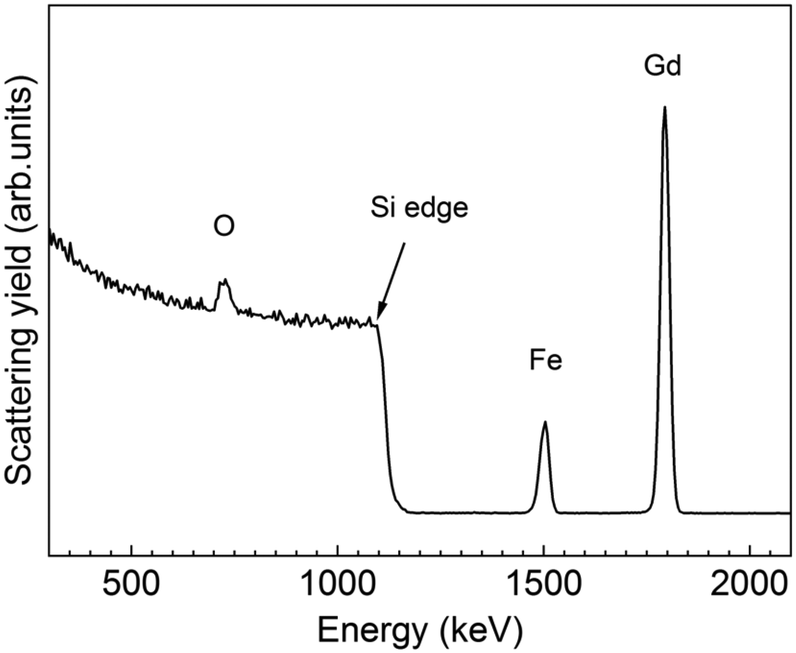 | ||
| Fig. 8 RBS profile spectrum of a 30 nm thick Gd3Fe5O12 film annealed at 800 °C for 30 min in oxygen. | ||
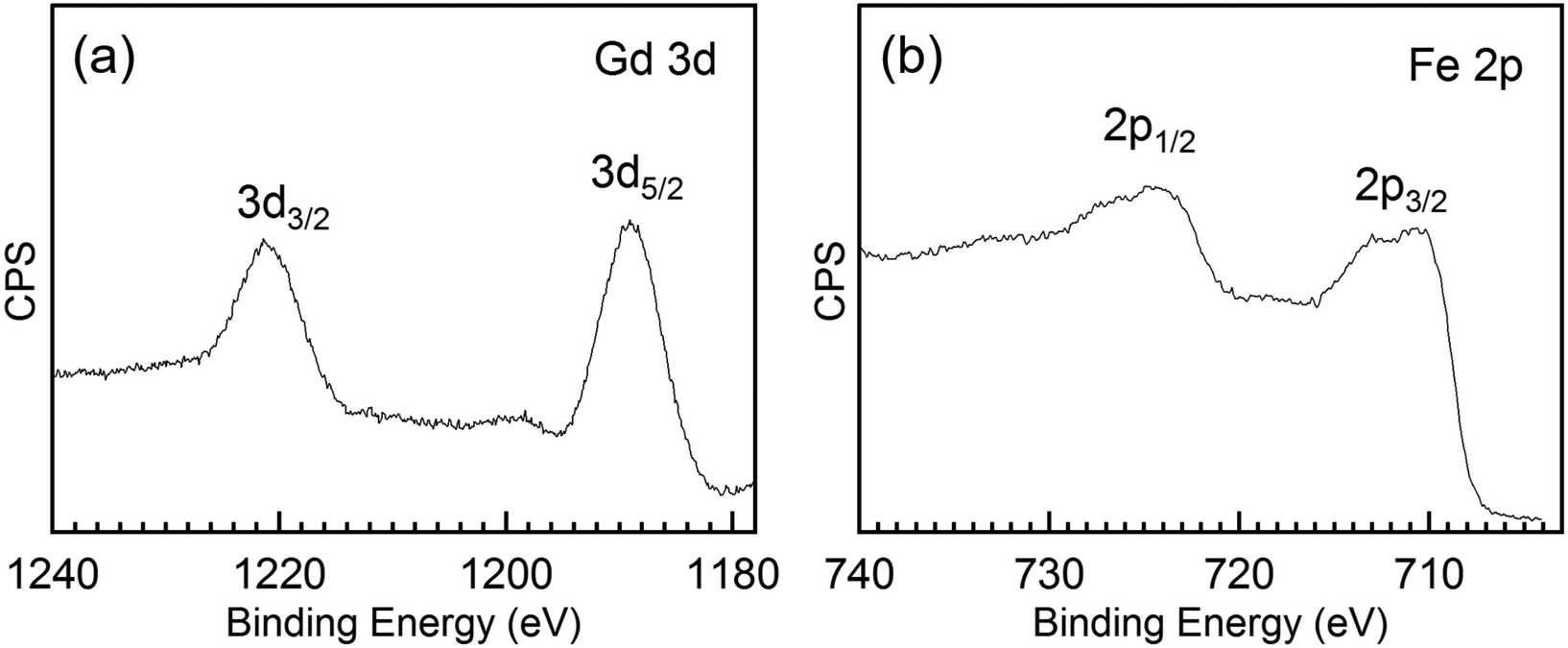 | ||
| Fig. 9 (a) Gd 3d core level and (b) Fe 2p core level spectra of a 30 nm thick Gd3Fe5O12 film annealed at 800 °C for 30 min in oxygen. | ||
Then the magnetic properties were further explored by SQUID magnetometry on a 30 nm thick Gd3Fe5O12 film. Fig. S8a† presents the field dependent magnetic hysteresis loop (M–H) of the sample at 50 K with the magnetic field applied along the in-plane direction. From the hysteresis loop, a saturation magnetization (Ms) of 8.5 emu g−1, remnant magnetization Mr of 2.86 emu g−1 and coercive field (Hc) of 130 Oe can be extracted. Compared to ∼500 nm thick sol–gel Gd3Fe5O12 films,51 our samples show significantly lower Ms and larger Hc values. We attribute this difference to the slight off-stoichiometry identified by RBS and the presence of secondary phases identified by XRD.52
The temperature dependent magnetization behavior (M–T) was investigated in the range of 10–350 K at 5 kOe magnetic field under zero field cooling (ZFC) and field cooling conditions (FC). A rapid decrease of magnetization was observed below 50 K with temperature increase, then this trend diminished in 50–350 K with magnetization approaching zero. Gd3Fe5O12 bulk material has a typical compensation point at the vicinity of ∼300 K where Gd and Fe moments reverse directions and result in the total magnetization crosses zero. In our investigation, no compensation temperature (Tcomp) was observed, see Fig. S8b.† Shifts in Tcomp from the bulk value has been attributed to film off-stoichiometry but also to a thickness/strain effect. Indeed, ultra-thin films can result in a significant rising of Tcomp.52–54
Finally, the synthesis of GdxFeyOz films from [Gd(DPDMG)3] and [Fe(ipki)2] precursors is here investigated. The starting recipes are shown in Table 3.
| Sample | Gd–O subcycles | Fe–O subcycles | Theoretical ratio | Subcycle sequence | Gd (%) | Fe (%) |
|---|---|---|---|---|---|---|
| GFO-Ki-1 | 2 | 1 | 2:1 |
Gd:Fe |
50 | 50 |
| GFO-Ki-2 | 2 | 1 | 2:1 |
Fe:Gd |
50 | 50 |
In this case, as the respective binary oxide GPC are very similar ([Fe(ipki)2] 0.4 Å per cycle vs. [Gd(DPDMG)3] 0.6 Å per cycle), simple alternate subcycles was performed (no nanolaminates). Note that alternating the subcycles sequence the final composition of the film is not affected (being 50:50), which is probably due to the resemblance of the precursor chemistry.
Analogous to GdxFeyOz study from [Gd(DPDMG)3] and [Fe(Cp)2] sources, the samples were attached in different positions in the chamber to study the elemental distribution. Based on the EDX spectra (Fig. S9†), a Gd/Fe ratio map is generated and shown in Fig. 10a. Samples near the gas inlet are identified with ∼1 Gd/Fe cation ratio, the rest of the area is Gd-deficient which is consistent with the high reactivity of [Gd(DPDMG)3] and its susceptibility to be depleted near the inlet. In addition, the pulse sequence does not affect the stoichiometry of the films which could be ascribed to the chemistry resemblance between [Gd(DPDMG)3] and [Fe(ipki)2].
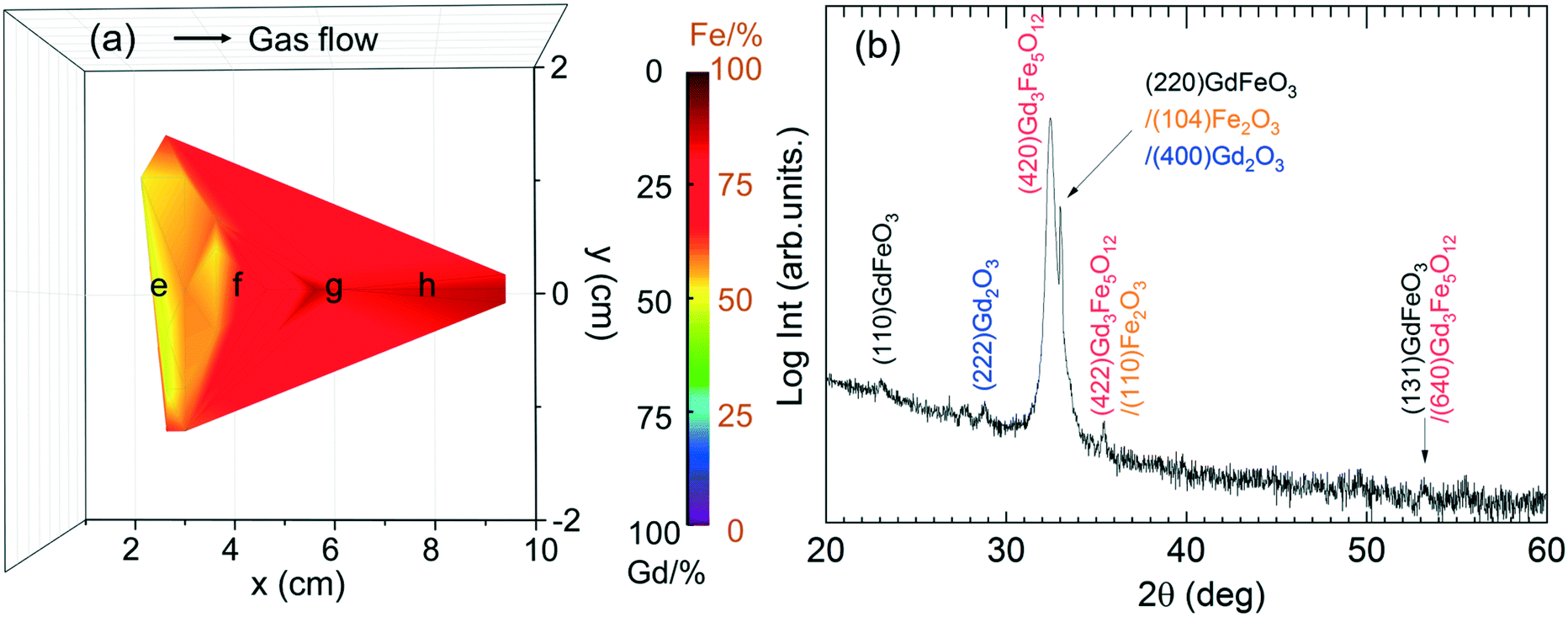 | ||
| Fig. 10 (a) Gd/Fe map of GdxFeyOz films from 2Gd:1Fe deposition process combining [Gd(DPDMG)3] and [Fe(ipki)2]. (b) XRD θ–2θ scan of a 800 °C annealed 120 nm thick Gd–Fe–O thin films with Gd/Fe ratio ∼1 prepared from [Gd(DPDMG)3] and [Fe(ipki)2] at 250 °C. | ||
In order to evaluate the film crystallinity and phase formation of the Gd/Fe ∼ 1 samples, XRD θ–2θ scan was carried out. In the as-deposited stage the films are amorphous. Upon 800 °C annealing, two intense reflections are observed at 32–33°, see Fig. 10b. The most intense reflection at 32.5° agrees well with the main reflection of Gd3Fe5O12 phase. Also, weak peaks at 2θ of 28.5°, 36° and 54° would further support the formation of Gd3Fe5O12. The Bragg reflection that appears as a shoulder at 33° could be attributed to GdFeO3, Fe2O3 and Gd2O3 which hinders an unambiguously identification of this peak. Therefore, the coexistence of Gd3Fe5O12 with GdFeO3, Fe2O3 and Gd2O3 is in well agreement with the reported difficulty to stabilize pure phase GdFeO3.9 These films show an homogeneous and smooth surface morphology with surface roughness values of 1 nm and average grain size of 70 nm, see Fig. S6b.†
From RBS/NRA, the elemental composition was estimated allowing a comparison of the Fe/Gd and M/O ratio exhibiting values of 0.96 and 0.5, respectively, see Table S3,† consistent with the EDX data presented in Table 3. The level of C and N contamination was found to be low with 0.9 at% and 0.3 at%, respectively. This indicates that the growth mechanisms of the subcycles are not influenced by the different active surface sites. The thin film thickness of 120 nm was calculated from RBS and it is in good agreement with XRR. However, from the RBS graph it is visible that a gradient of Gd and Fe is formed at the interface. This phenomenon could be in part explained because of the initial deposition of Gd2O3 cycles acting as seeding layer for the garnet. Consequently, the concentration of Gd at the Si interface is higher, whereas the Fe signal decreases at the low energy edge indicating a decreased Fe concentration in this area. Nonetheless, it is well reported that gadolinium oxide tend to precipitate during the growth of garnet resulting in a gradient depletion along the garnet matrix,54 which could also contribute to the compositional gradient identified from the RBS profile spectrum.
Magnetic properties of the 1Gd:1Fe Gd–Fe–O sample were explored using SQUID magnetometry with the magnetic field applied in the in-plane direction. From the magnetic hysteresis loop at 50 K, a Ms value of 26.7 emu g−1, Mr of 13.7 emu g−1, and coercive field (Hc) of 0.036 kOe were observed, Fig. S10a.† The magnetization as a function of temperature was investigated at 5 kOe from 5–300 K, Fig. S10b.† The curve shows no phase transitions in this temperature range. Compared to the previous sample prepared from [Gd(DPDMG)3] and [Fe(Cp)2], here it is observed a slightly stronger Ms although no Tcomp is observed. We attribute this different behavior to the existence of Gd gradient composition identified by RBS, Fig. 11, along with the coexistence of GdFeO3, Gd2O3 and Fe2O3 both being already identified in Fig. 10b as key factors on the magnetic behavior of garnet films (Fig. 11).52
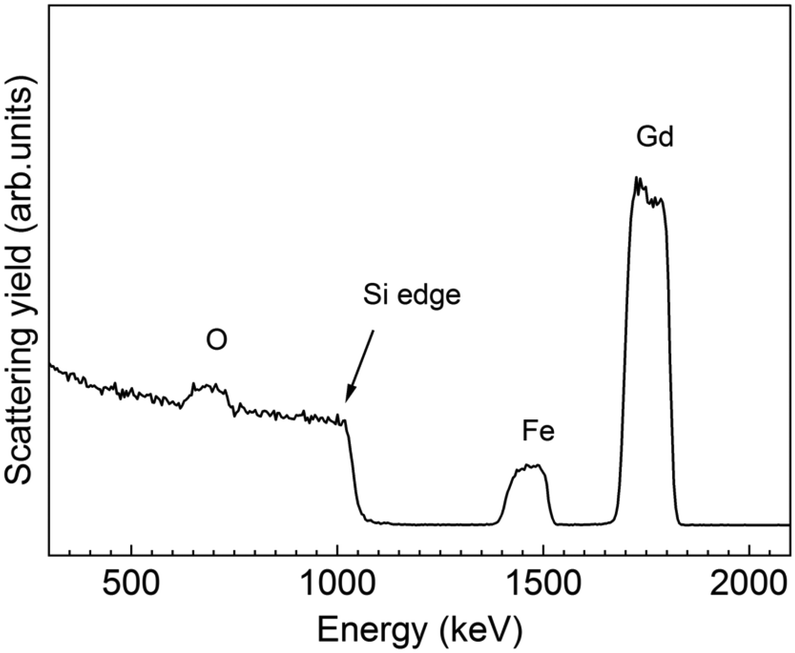 | ||
| Fig. 11 RBS profile spectrum of a 120 nm thick GdxFeyOz film annealed at 800 °C in oxygen. | ||
4 Conclusions
We have evaluated the chemical compatibility of tailor-made [Gd(DPDMG)3] and [Fe(ipki)2] and the traditional [Fe(Cp)2] precursors to be deposited by an ALD-type approach. Compatible deposition conditions at 250 °C using ozone as co-reactant and 70 sccm N2 carrier gas flow have been identified and subsequently tested to prepare a series of GdxFeyOz thin films. RBS/NRA analysis helped identifying the metal to oxygen ratio while confirming that ozone ensured a clean reaction of the surface species with complete desorption of the organics. The combination of [Fe(Cp)2] with [Gd(DPDMG)3] resulted in the formation of garnet Gd3Fe5O12 as the main phase while the combination of [Fe(ipki)2] and [Gd(DPDMG)3], led to a mixture of crystalline Gd3Fe5O12, GdFeO3, and Gd2O3 and traces of Fe2O3, both showing weak magnetization response. Therefore, an ALD-type approach has been successfully used to stabilize garnet thin films mixed with minor oxide secondary phases offering new opportunities to advance in the synthesis of complex compounds, difficult to obtain by traditional deposition techniques. Importantly, thorough understanding of the reaction mechanism during the film growth enabled by the use of in situ characterization tools will be key factors to fully unravel the synthesis of pure phase garnet films.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported through the “Severo Ochoa” Programme for Centres of Excellence in R & D (CEX2019-000917-S), and MAT2017-83169-R from the Spanish Ministry of Economy and Competitiveness. This article is based upon work from COST Action MP1402 “Hooking together European research in atomic layer deposition (HERALD)”, supported by COST (European Cooperation in Science and Technology). The authors gratefully thank the Scientific Services at ICMAB and G. Sauthier from ICN2 core support facilities for XPS analysis. This work has been done in the framework of the doctorate in Materials Science of the Autonomous University of Barcelona. We also acknowledge financial support from China Scholarship Council fellowship to P. Yu (201606920073). The authors from RUB thank the DFG-DE-790-11-1 project for partly supporting this project. M. C. acknowledges Beca Leonardo from Fundación BBVA.Notes and references
- S. Geller, J. Chem. Phys., 1956, 24, 1236–1239 CrossRef CAS.
- M. A. Gilleo, J. Chem. Phys., 1956, 24, 1239–1243 CrossRef CAS.
- Y. Tokunaga, N. Furukawa, H. Sakai, Y. Taguchi, T. H. Arima and Y. Tokura, Nat. Mater., 2009, 8, 558–562 CrossRef CAS PubMed.
- A. Panchwanee, V. Raghavendra Reddy, A. Gupta, R. Choudhary, D. Phase and V. Ganesan, Thin Solid Films, 2019, 669, 301–305 CrossRef CAS.
- E. E. Anderson, J. R. Cunningham and G. E. McDuffie, Phys. Rev., 1959, 116, 624–625 CrossRef CAS.
- Y. Tokunaga, Y. Taguchi, T.-h. Arima and Y. Tokura, Nat. Phys., 2012, 8, 838–844 Search PubMed.
- A. A. Serga, A. V. Chumak and B. Hillebrands, J. Phys. D: Appl. Phys., 2010, 43, 264002 CrossRef.
- C. Suchomski, C. Reitz, C. T. Sousa, J. P. Araujo and T. Brezesinski, Chem. Mater., 2013, 25, 2527–2537 CrossRef CAS.
- N. Kimizuka, A. Yamamoto, H. Ohashi, T. Sugihara and T. Sekine, J. Solid State Chem., 1983, 49, 65–76 CrossRef CAS.
- M. Bibes, J. E. Villegas and A. Barthélémy, Adv. Phys., 2011, 60, 5–84 CrossRef CAS.
- M. Coll, J. Fontcuberta and M. Althammer, et al. , Appl. Surf. Sci., 2019, 482, 1–93 CrossRef CAS.
- J. Schmitz, Surf. Coat. Technol., 2018, 343, 83–88 CrossRef CAS.
- H. H. Sønsteby, E. Skaar, Ø. S. Fjellvåg, J. E. Bratvold, H. Fjellvåg and O. Nilsen, Nat. Commun., 2020, 11, 1–7 Search PubMed.
- M. Ritala and M. Leskelä, in Handbook of Thin Films, Elsevier, 2002, pp. 103–159 Search PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- H. Knoops, S. Potts, A. Bol and W. Kessels, in Handbook of Crystal Growth, Elsevier, 2nd edn, 2015, vol. 3, pp. 1101–1134 Search PubMed.
- M. Coll and M. Napari, APL Mater., 2019, 7, 110901 CrossRef.
- M. Lie, O. Nilsen, H. Fjellvåg and A. Kjekshus, Dalton Trans., 2009, 481–489 RSC.
- A. J. M. Mackus, J. R. Schneider, C. MacIsaac, J. G. Baker and S. F. Bent, Chem. Mater., 2019, 31, 1142–1183 CrossRef CAS.
- J.-H. Kim, J.-Y. Kim and S.-W. Kang, J. Appl. Phys., 2005, 97, 093505 CrossRef.
- H. H. Sønsteby, H. Fjellvåg and O. Nilsen, Adv. Mater. Interfaces, 2017, 4, 1–15 Search PubMed.
- S. Vangelista, A. Lamperti, C. Wiemer, M. Fanciulli and R. Mantovan, Thin Solid Films, 2016, 604, 18–22 CrossRef CAS.
- C. Bohr, P. Yu, M. Scigaj, C. Hegemann, T. Fischer, M. Coll and S. Mathur, Thin Solid Films, 2020, 698, 137848 CrossRef CAS.
- R. G. Gordon, in ALD Precursors and Reaction Mechanisms, ed. C. S. Hwang, Springer, Boston, MA, 2014, pp. 15–46 Search PubMed.
- T. Hatanpää, M. Ritala and M. Leskelä, Coord. Chem. Rev., 2013, 257, 3297–3322 CrossRef.
- D. Peeters, A. Sadlo, K. Lowjaga, O. Mendoza Reyes, L. Wang, L. Mai, M. Gebhard, D. Rogalla, H.-W. Becker, I. Giner, G. Grundmeier, D. Mitoraj, M. Grafen, A. Ostendorf, R. Beranek and A. Devi, Adv. Mater. Interfaces, 2017, 4, 1700155 CrossRef.
- A. P. Milanov, R. A. Fischer and A. Devi, Inorg. Chem., 2008, 47, 11405–11416 CrossRef CAS PubMed.
- A. P. Milanov, K. Xu, A. Laha, E. Bugiel, R. Ranjith, D. Schwendt, H. J. Osten, H. Parala, R. A. Fischer and A. Devi, J. Am. Chem. Soc., 2010, 132, 36–37 CrossRef CAS PubMed.
- S. Macke, A. Radi, J. E. Hamann-Borrero, A. Verna, M. Bluschke, S. Brück, E. Goering, R. Sutarto, F. He, G. Cristiani, M. Wu, E. Benckiser, H.-U. Habermeier, G. Logvenov, N. Gauquelin, G. A. Botton, A. P. Kajdos, S. Stemmer, G. A. Sawatzky, M. W. Haverkort, B. Keimer and V. Hinkov, Adv. Mater., 2014, 26, 6554–6559 CrossRef CAS PubMed.
- M. Mayer, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 332, 176–180 CrossRef CAS.
- Y. Wang, M. Dai, M.-T. Ho, L. S. Wielunski and Y. J. Chabal, Appl. Phys. Lett., 2007, 90, 022906 CrossRef.
- F. Sandiumenge, A. Cavallaro, J. Gàzquez, T. Puig, X. Obradors, J. Arbiol and H. C. Freyhardt, Nanotechnology, 2005, 16, 1809–1813 CrossRef CAS.
- S. Seppälä, J. Niinistö, M. Mattinen, K. Mizohata, J. Räisänen, W. Noh, M. Ritala and M. Leskelä, Thin Solid Films, 2018, 660, 199–206 CrossRef.
- J. R. Schneider, J. G. Baker and S. F. Bent, Adv. Mater. Interfaces, 2020, 7, 2000318 CrossRef CAS.
- H. H. Sønsteby, A. Yanguas-Gil and J. W. Elam, J. Vac. Sci. Technol., A, 2020, 38, 020804 CrossRef.
- J. Niinistö, N. Petrova, M. Putkonen, L. Niinistö, K. Arstila and T. Sajavaara, J. Cryst. Growth, 2005, 285, 191–200 CrossRef.
- J. H. Han, A. Delabie, A. Franquet, T. Conard, S. Van Elshocht and C. Adelmann, Chem. Vap. Deposition, 2015, 21, 352–359 CrossRef CAS.
- B. M. Klahr, A. B. Martinson and T. W. Hamann, Langmuir, 2011, 27, 461–468 CrossRef CAS PubMed.
- A. B. F. Martinson, M. J. DeVries, J. A. Libera, S. T. Christensen, J. T. Hupp, M. J. Pellin and J. W. Elam, J. Phys. Chem. C, 2011, 115, 4333–4339 CrossRef CAS.
- X. Li, N. C. Fan and H. J. Fan, Chem. Vap. Deposition, 2013, 19, 104–110 CrossRef.
- A. Devi, Coord. Chem. Rev., 2013, 257, 3332–3384 CrossRef CAS.
- M. Leskelä and M. Ritala, Angew. Chem., Int. Ed., 2003, 42, 5548–5554 CrossRef PubMed.
- A. Zydor, V. G. Kessler and S. D. Elliott, Phys. Chem. Chem. Phys., 2012, 14, 7954–7964 RSC.
- G. Dey and S. D. Elliott, J. Phys. Chem. C, 2015, 119, 5914–5927 CrossRef CAS.
- K. L. Pickrahn, A. Garg and S. F. Bent, ACS Catal., 2015, 5, 1609–1616 CrossRef CAS.
- J. W. Elam and S. M. George, Chem. Mater., 2003, 15, 1020–1028 CrossRef CAS.
- D. Raiser and J. Deville, J. Electron Spectrosc. Relat. Phenom., 1991, 57, 91–97 CrossRef CAS.
- E. Paparazzo, Appl. Surf. Sci., 1986, 25, 1–12 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- P. C. J. Graat and M. A. J. Somers, Appl. Surf. Sci., 1996, 100–101, 36–40 CrossRef.
- F. Söderlind, L. Selegård, P. Nordblad, K. Uvdal and P.-O. Käll, J. Sol-Gel Sci. Technol., 2009, 49, 253–259 CrossRef.
- E. Sawatzky and E. Kay, J. Appl. Phys., 1971, 42, 367–375 CrossRef CAS.
- E. Sawatzky and E. Kay, J. Appl. Phys., 1968, 39, 4700–4706 CrossRef CAS.
- M. Oron, I. Barlow and W. Traber, J. Mater. Sci., 1969, 4, 271–281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: List of Fe and Gd precursors tested by ALD to prepare Fe2O3 and Gd2O3, respectively. EDX spectra of as-deposited films, summary of the composition and thickness of Gd2O3, Fe2O3 and GdxFeyOz. XPS Survey spectra of Gd3Fe5O12 films and the magnetic measurements (M(H) and M(T)) for the GdxFeyOz films. See DOI: 10.1039/d0ce01252a |
| This journal is © The Royal Society of Chemistry 2021 |