
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructured zeolite with brain-coral morphology and tailored acidity: a self-organized hierarchical porous material with MFI topology†‡
Esun
Selvam§
a,
Rajesh K.
Parsapur
b,
Carlos E.
Hernandez-Tamargo
 bc,
Nora H.
de Leeuw
*cd and
Parasuraman
Selvam
*bef
bc,
Nora H.
de Leeuw
*cd and
Parasuraman
Selvam
*bef
aDepartment of Chemical Engineering, National Institute of Technology-Trichy, Tiruchirappalli 620 015, Tamil Nadu, India
bNational Centre for Catalysis Research and Department of Chemistry, Indian Institute of Technology-Madras, Chennai 600 036, Tamil Nadu, India. E-mail: selvam@iitm.ac.in
cSchool of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
dSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK
eSchool of Chemical Engineering and Analytical Science, The University of Manchester, Manchester M13 9PL, UK
fDepartment of Chemical and Process Engineering, University of Surrey, Guildford, Surrey GU2 7XH, UK
First published on 22nd September 2020
Abstract
We report here the synthesis of nano-zeolite, viz., ZSM-5 with MFI topology, having a unique brain-coral morphology, designated as n-ZSM-5. These nanoscopic structures (250–300 nm) are in turn formed by self-organization of uniform nanoparticles of zeolite nanospheres of sizes 10–40 nm. Such a remarkable crystal architecture endows the material with improved physico-chemical properties, viz., enhanced surface area, distributed acid sites, mesoporosity and reduced diffusion resistance, which make n-ZSM-5 a promising solid acid catalyst. For comparison, bulk or conventional ZSM-5, referred to as c-ZSM-5, was also synthesized and its performance for tertiary butylation of phenol was evaluated. Density functional theory modelling studies provide an adequate description of the variation of acid strength observed in temperature-programmed desorption of ammonia experiments.
Introduction
The remarkable success of zeolites as solid-acid catalysts is based on their distinctive ordered microporous structure and flexible aluminosilicate framework providing them with unique physicochemical characteristics.1 In particular, zeolites with MFI-topology, e.g., ZSM-5, are primarily important in petrochemical processing and fine chemical synthesis. Nevertheless, the presence of a well-organized network of micropores which is highly significant for spatial selectivity can also impose severe mass transfer constraints due to the sluggish diffusion inside the narrow pore channels.2 In this regard, the reduction of crystal size from bulk- to nano-dimensions can have a pronounced impact on the performance of these zeolitic structures owing to the significant change in the physico-chemical properties.3,4 However, regulating the growth of zeolites to nano dimensions is a challenging task owing to the highly alkaline synthesis conditions, which results in uncontrolled crystal growth and aggregation of crystals.5 Hence, attempts have been made to synthesize aluminium- and alkali-free siliceous structures owing to their framework simplicity and controlled crystal growth.3,6,7 On the other hand, numerous efforts were also devoted to the synthesis of nanocrystalline zeolites such as ZSM-5 by employing various strategies, viz., low-temperature synthesis from clear solutions, reflexing and hydrothermal treatments of alkali-free solutions and micro-emulsions, confined-space synthesis, and fluoride-assisted synthesis,8–13 but without much success. Although, fluoride media yield low-silanol groups with fewer framework defects,12,13 they also poses concerns with respect to safety and process economics. Therefore, the synthesis of nanosized zeolites, under alkaline synthesis conditions, with enhanced aluminium content, smaller crystal dimensions, and low framework defects is still a great challenge to the scientific community.Numerous efforts have also been made to alleviate the diffusion limitations by inducing mesoporosity in the zeolite matrix, however, these approaches use various expensive templating strategies and tedious post-synthetic modifications.14–19 In this regard, the development of novel nanostructured materials with unique morphology that exhibit inherent mesoporosity, high external surface area and enhanced diffusivity has gained significant interest owing to their exceptional performance in catalysis.20–22 However, tailoring the crystallization process to achieve the desired morphology and porosity without employing expensive mesoporogens is a major challenge. Therefore, in the present work, an attempt has been made to develop such novel nanostructured zeolite with inherent inter- and intra-crystalline porosity and smaller crystal dimensions to enhance the catalytic performance of the materials. Thus, we report here the synthesis of high-quality nano-ZSM-5 with a unique “brain-coral morphology”, designated as n-ZSM-5. These neuromorphic nanostructures are in turn formed by the self-organization of uniform nanosized ZSM-5 crystals.
Although alkali cations are known to increase particle aggregation,5,8 they also play a crucial role in increasing the aluminium content, and therefore the density of acid sites.23 Further, alkali cations along with templates such as tetrapropylammonium (TPA) cations are also well-known to influence the morphology of zeolites.24 Hence, we have adopted a careful synthesis procedure, and accordingly, a clear solution was prepared initially by slightly increasing the concentration of sodium cations. After the initial hydrolysis of silica and alumina precursors, water content was reduced by heating the precursor solution at 80 °C for 12 h to obtain a super-saturated gel. The resulting gel sterically stabilizes the sub-micron, pre-crystalline, colloidal nuclei and aids in the controlled growth and assembly of nanocrystalline zeolites. Furthermore, the rate of crystallization process was controlled by employing low-temperature polymerization and two-step variable hydrothermal treatment, i.e., an initial low temperature to promote nucleation followed by high-temperature treatment to complete the crystallization process.17,25,26 The resulting zeolite, i.e., n-ZSM-5, is consequently evaluated and tested for its catalytic activity in industrially important tertiary butylation reaction (Scheme S1‡).27 A preliminary account of the work is presented elsewhere.28
Experimental details
Starting materials
All the chemicals purchased were of analytical grade. The chemicals viz., tetraethyl orthosilicate (TEOS, 98%), aluminium isopropoxide (98%), sodium aluminate (40–45% Na2O; 50–56% Al2O3), tetrapropylammonium bromide (TPABr, 98%) and tetrapropylammonium hydroxide (TPAOH, 20 wt% in H2O) were obtained from Sigma Aldrich. The reactants (phenol and t-butyl alcohol) and sodium hydroxide were obtained from Merck.Nano- and bulk-zeolite synthesis
In our previous work, we have mainly employed the self-assembly mechanism to prepare hierarchical zeolites and ordered mesoporous aluminosilicates.17–20 In the present work, we report the successful synthesis of nano-zeolite, designated as n-ZSM-5, in the absence of mesoporogens under the designed synthesis conditions (Fig. 1).28 | ||
| Fig. 1 The formation of n-ZSM-5 with brain-coral type morphology (250−300 nm) and inherent mesoporosity organized by uniform nanospheres of 10−20 nm. Also shown in the figure is the reactor/apparatus used for the crystallization and calcination. | ||
In a typical synthesis of n-ZSM-5, 0.04 g NaOH was dissolved in 15.25 g TPAOH and 1.50 g H2O. To this mixture, 8.66 g of TEOS is added by stirring in an ice bath. The obtained mixture is stirred for 10 h before the addition of 0.44 g Al(O-iPr)3. Upon subsequent stirring for 14 h, a clear solution is obtained which is then dried at 80 °C for 24 h. The evaporation of water has led to the formation of a viscous gel which is hydrothermally treated in a stainless-steel autoclave (Fig. S1‡) at 100 °C for 24 h and 170 °C for 120 h. The resulting solution was washed thrice and centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The resulting solid product is dried at 100 °C overnight followed by calcination, in a tubular furnace (Fig. S2‡), at 550 °C in air for 6 h at a heating rate of 1 °C min−1.
000 rpm. The resulting solid product is dried at 100 °C overnight followed by calcination, in a tubular furnace (Fig. S2‡), at 550 °C in air for 6 h at a heating rate of 1 °C min−1.
In the final step, the calcined samples were ion-exchanged at 80 °C with 1 M NH4NO3 solution. The resulting materials were heated at 500 °C for 4 h to obtain the protonated forms of the zeolites. On the other hand, for comparison, we have also prepared and characterised conventional ZSM-5, referred to as c-ZSM-5, using the procedure reported elsewhere,17 with a typical gel composition of 1 Al2O3:10 TPABr:10 Na2O:38 SiO2:7200 H2O. As before, the as-synthesized samples were calcined, and the resulting calcined samples were ion-exchanged at 80 °C with 1 M NH4NO3 solution. The resulting materials were heated at 500 °C for 4 h to obtain the protonated forms of the zeolites.
Material characterization
Powder X-ray diffraction (XRD) measurements were carried out on a Bruker D8 Advance X-ray diffractometer with a Cu Kα (λ = 1.5418 Å) radiation source operating at 40 kV and 30 mA. The patterns were recorded with a divergent slit of 0.298° over the 2θ range of 1.5 to 7.5° and 5–50° with step size = 0.01° and a step time of 2 s. The diffraction patterns were refined by the Rietveld method using TOPAS software. Transmission electron microscopy (TEM) images and selected area electron diffraction (SAED) patterns were obtained with a 2100 JEOL microscope operated at 200 keV. Scanning electron microscopy (SEM) images were recorded using a QUANTA 400 instrument.Nitrogen sorption isotherms were measured at 77 K using a Micromeritics ASAP 2020 surface area analyzer, in which the catalysts were degassed at 300 °C for 8 h. Surface area measurements are carried out by employing the BET method for relative pressures (P/Po) between 0.1 to 0.3. Adsorption isotherms are used for measuring pore volumes. The micropore volume is obtained by using the t-plot. The micropore and mesopore sizes are calculated by using the Horvath–Kawazoe (HK) and Barrett–Joyner–Halenda (BJH) methods, respectively. Ammonia temperature programmed desorption of (NH3-TPD) was performed on a Micromeritics Autochem-II chemisorption analyzer. The samples were activated at 550 °C for 2 h in a helium flow, and later they were cooled and maintained at 120 °C before their exposure to NH3 vapour, followed by purging with helium gas for 30 min. Desorption of ammonia was performed by heating the reactor at a uniform rate of 10 °C min−1. The elemental compositions of the prepared zeolites were measured on a Rigaku Primini X-ray fluorescence (XRF) spectrometer using a palladium source operating at 40 kV. Dynamic light scattering (DLS) studies were performed using a Horiba Partica LA 950 instrument with a scattering angle of 173°, a 5 mW output power and a wavelength of 650 nm. Before the analysis, colloidal suspensions were prepared by dispersing the zeolites in water by ultrasonication.
Magic angle spinning-nuclear magnetic resonance (MAS-NMR) spectra of the samples were recorded on a Bruker DSX 400 spectrometer at a spinning frequency of 8.0 kHz at room temperature with a resonance frequency of 79.49 MHz, applying a pulse length of 1.38 μs and the recycle time was 1 s using a 5 mm diameter ZrO2 rotor. Pyridine in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed using a Bruker Tensor-27 FT-IR instrument in conjunction with a Harrick high vacuum cell in a praying mantis reaction chamber. The samples were outgassed at 400 °C for 4 h and then cooled to room temperature under dynamic vacuum. The background spectrum was measured before pyridine sorption followed by the sample spectra at various temperatures.
Catalytic activity studies
The vapour-phase tertiary butylation of phenol reaction was carried out in a fixed-bed downflow reactor (Fig. S3‡) using 500 mg of the catalyst.17–20,27 The reactor set-up was pre-heated to 350 °C in a flow of air for 2 h followed by cooling to the desired reaction temperature, i.e., 140 °C, in a nitrogen atmosphere. The ratio of reactants, weight hour space velocity, and Si/Al ratio of the catalysts has been varied to study the activity of the catalyst. Nitrogen was used as the carrier gas, and a liquid injection pump was used to feed the mixture of reactants. The products ortho-(2-t-BP), para-(4-t-BP) and 2,4-di-tert-butylphenols (2,4-di-t-BP) were analysed by using a Perkin-Elmer gas chromatograph with an HP-5 capillary column. The percentage conversion of the reactant and selectivity of products were calculated as follows:| Conversion = [(Total Area − Reactant Area)/Total area] × 100 |
| Selectivity = (Product Area/Total Area of Products) × 100 |
Computational details
The computer simulations were performed with the density functional theory (DFT) code VASP,29,30 using the generalized gradient approximation functional proposed by Perdew, Burke, and Ernzerhof (PBE).31 A plane-wave basis set with an energy cutoff of 400 eV was used to describe the Kohn–Sham orbitals of the valence electrons while describing their interaction with the inner part of the atom and the core electrons with the projected-augmented-wave method (PAW).32,33 The Grimme correction (D3) was employed to account for the dispersion interactions using the Becke–Johnson damping function.34,35 The convergence of the integral over the occupied bands and set of k points was improved by replacing the Dirac step function by a Gaussian-like function.36,37 Owing to the large size of the unit cell of a zeolite with the MFI framework type, only the Gamma k point was necessary to describe the first Brillouin zone of the system. The convergence thresholds for the electronic and ionic iterations were set at 10−5 eV and 0.03 eV Å−1, respectively.The unit cell of a pure silica zeolite with the MFI framework type was optimized using a set of fixed volume calculations.38 The optimized cell parameters were 20.296, 19.954 and 13.368 Å for the vectors a, b and c respectively, in close agreement with the experimentally reported values.39 The slab model was used to study the external surface of the zeolite (Fig. S4‡), with the vectors a and c parallel to the plane of the slab and the b vector along the normal direction to the slab surface.40 The cleaved Si–O bonds at the surface were saturated with OH groups, thus forming silanol groups. The Al atom replaced the Si atom at the 12 non-equivalent tetrahedral sites, at both the internal and external surfaces, allowing us to probe as many NH3 adsorption configurations as possible. The binding energy of NH3 to the different active sites was calculated by subtracting the energy of NH3 in the gas phase plus the bare zeolite structure from the system where the zeolite is loaded with NH3.
Results and discussion
Fig. 2 shows the Rietveld-refined XRD patterns of both nano- and bulk/conventional zeolite structures. It is clear from these diffraction patterns that the prepared materials show all the characteristic reflections typical of orthorhombic crystal symmetry (ICDD card No. 42-0023). At this juncture, it is noteworthy to point out here that the sample n-ZSM-5 (Fig. 2a), as expected, showed broad reflections to those of c-ZSM-5 (Fig. 2b) which can be ascribed to its smaller crystallite size. The computed unit cell parameters of both n-ZSM-5 (a = 20.15 Å; b = 19.99 Å; c = 13.43 Å) and c-ZSM-5 (a = 20.14 Å; = 19.93 Å; c = 13.40 Å) were obtained by profile fitting the patterns with the Pnma space group in the 2θ region 5–50° by the Le Bail method.41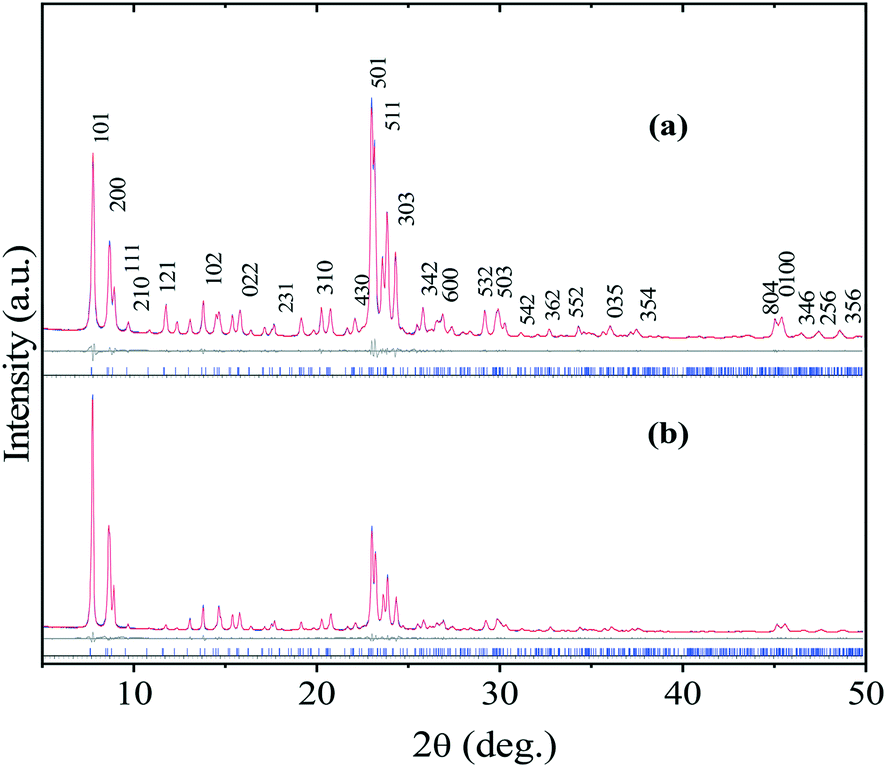 | ||
| Fig. 2 Rietveld-refined XRD patterns of: (a) n-ZSM-5 and (b) c-ZSM-5. The vertical lines indicate the Bragg positions of the different phases. | ||
Fig. 3 presents the N2 adsorption–desorption isotherms and pore-size distributions of both n-ZSM-5 and c-ZSM-5. It can be seen from this figure that the sample n-ZSM-5 (Fig. 3a) has a combination of both type-I and type-IV isotherms indicating the presence of zeolitic micropores of sizes ∼0.5 nm along with inter-crystalline mesopores formed by the characteristic self-assembly of uniform nanocrystals. This is further supported by the narrow micro- and mesopore-size distributions (see Fig. 3a and the inset) obtained by the Horvath–Kawazoe (HK) and Barrett–Joyner–Halenda (BJH) methods, respectively. Table 1 summarizes the relevant textural properties of the nanostructured and bulk ZSM-5.
 | ||
| Fig. 3 N2 adsorption–desorption isotherms and pore size distribution (inset) of (a) n-ZSM-5 and (b) c-ZSM-5. | ||
Fig. 4(A–C) and S5(A–D)‡ show the SEM images of n-ZSM-5. For comparison, the SEM images of c-ZSM-5 are given in Fig. 4D and S5(E and F).‡ As expected, c-ZSM-5 prepared by the given method exhibits a spherical morphology with an average particle size of about 1500–2000 nm. On the other hand, it can be seen from Fig. 4(A–C) that n-ZSM-5 shows a unique brain-coral like nanostructure (250–300 nm) formed by the self-organization of uniform nanospheres of size around 10–20 nm. Furthermore, it is also clear from the images that these nanostructured zeolites, which mimic the human brain and/or underwater coral morphology, are self-organized compactly to form intercrystalline bimodal mesopores. Materials with such multiple levels of porosity are known to increase the diffusion by few orders of magnitude.
 | ||
| Fig. 4 SEM images of (A–C) n-ZSM-5 and (D) c-ZSM-5. | ||
Fig. 5 and S6(A–D)‡ show the TEM images of n-ZSM-5, which clearly show the occurrence of small zeolite crystals with dimensions around 10–20 nm, indicating the nanocrystalline nature of the material. Furthermore, the presence of lattice fringes (Fig. 5B, inset) of size 1.1 nm corresponding to the (111) plane of the ZSM-5 lattice indicates the crystalline nature of the sample. This is well-supported by the SAED patterns which show characteristic orthorhombic crystal symmetry distinctive of MFI-type zeolites. For comparison, the TEM images of c-ZSM-5 are shown in Fig. S6(E and F).‡ Further, DLS analysis (Fig. 6) presents a uniform and well-dispersed hydrodynamic diameters of both nano- and c-ZSM-5 particles with an average size of about 291 and 1640 nm, respectively, which is consistent with the TEM data.
 | ||
| Fig. 5 TEM images of n-ZSM-5; inset – SAED. (A) Low magnification and (B) high-magnification. | ||
 | ||
| Fig. 6 DLS particle size distribution: (a) n-ZSM-5 and (b) c-ZSM-5. | ||
Fig. 7 shows the 29Si and 27Al MAS-NMR spectra of both n-ZSM-5 and c-ZSM-5. In the case of both nano- and bulk-ZSM-5 (Fig. 7A), the resonances above −110 ppm can be attributed to the silicon in tetrahedral coordination (SiT). Remarkably, the 29Si NMR spectrum of n-ZSM-5 exhibits many satellite resonances around −114 ppm indicating short-range atomic ordering in agreement with NMR studies performed at ultra-high magnetic field.42 The observed satellite resonances can be attributed to the 12 crystallographically distinct tetrahedral sites originating from the orthorhombic crystal lattice of ZSM-5.43,44 On the other hand, the 27Al NMR spectra (Fig. 7B) of both zeolites have shown a single peak around ‘56 ppm’ indicating tetrahedral aluminium coordination in the framework structure (AlT). However, unlike c-ZSM-5 which exhibits a sharp signal, n-ZSM-5 shows a broad NMR resonance. This could be attributed to the dependence of the resonance signal width on the crystallinity of the sample.9,45,46 Nonetheless, the broadness of the chemical shift around ‘35 ppm’ indicates the presence of small amounts of penta-coordinated aluminium (AlP) as structural Lewis acid sites.19
 | ||
| Fig. 7 (A) 29Si MAS-NMR and (B) 27Al MAS-NMR spectra of (a) n-ZSM-5 and (b) c-ZSM-5. | ||
Crystallization process
Based on the characterization results, we have attempted to explain the plausible mechanism for the formation of such self-assembled zeolite structures (see Fig. 1) with complex NMR spectra and modified acidity. Unlike bulk/conventional ZSM-5 synthesis, here, we have initially used a low concentration of alkali to avoid rapid crystallization. At this stage, the mild alkalinity and high concentration of TPAOH induce the hydrolysis of the silica precursor. Subsequently, the formed oligomers encapsulate TPA+ cations to form colloidal particles resulting in a clear solution. In the next step, the aluminium precursor is added, and the temperature of the mixture is increased to 80 °C. The evaporation of water increases the alkalinity of the solution resulting in the formation of viscous aluminosilicate gel/glossy solid. At this stage, the supersaturated ultra-fine uniform colloidal aluminosilicate particles start to crystallize as zeolitic structures, and agglomerate such ultra-small particles to form large aggregates of the zeolitic material. On further crystallization under hydrothermal conditions, the primary particle aggregates to form nanocrystalline spherical particles of size 10–20 nm with intercrystalline mesopores formed by the assembly of aggregates of size 250–300 nm, in agreement with the literature.47A series of conditions, viz., high concentration of TPA+ and low concentration of water and alkali cations, may restrict the movement of aluminosilicate species resulting in the formation of small islands of silicalite-I species which is evident from the 29Si NMR spectrum, which shows small indentations due to the presence of the orthorhombic MFI framework typical of silicalite-I type structure. Besides, the broad resonance in 27Al NMR spectra indicates that the tetracoordinate aluminium is mostly present in the periphery of the nanocrystals and present as type-i and type-ii acid sites. This is further evident from the NH3 TPD and FT-IR studies, wherein the major density of acid sites as type ii and type-iii corresponds to vibrational bands at 3646 cm−1 and 3475 cm−1.
Surface acidity
Many experimental methods have been developed to determine the acidity in zeolites, viz., types of acid sites, their strength, and concentrations. Ammonia temperature-programmed desorption (NH3-TPD) is typically employed to determine the amounts and strength of the acid sites while infrared spectroscopy is widely used to study the types of hydroxyl groups. Fig. 8A shows the NH3-TPD profiles of various zeolites which are deconvoluted into various peaks based on the distribution of acid sites, and the obtained values are listed in Table 2. The acid sites type (i) and type (ii) can be attributed to surface silanols and weak acid sites, respectively, Meanwhile, the acid sites, type (iii) and type (iv) can be assigned to moderate and strong Brønsted acid sites, respectively, originating from the framework aluminium in distinct tetrahedral coordination at the core of the framework. On the other hand, type (v) sites can be attributed to the structural Lewis acid sites originating from the framework defects on the surface. | ||
| Fig. 8 NH3-TPD profiles (A) and pyridine-DRIFT spectra (B) of (a) n-ZSM-5 and (b) c-ZSM-5. | ||
| Catalyst | Acidityc (mmol g−1) | Conv. (%) | Selectivity (%) | TOFd (h−1) | ||||
|---|---|---|---|---|---|---|---|---|
| Total | Type (iii) | Type (iv) | 2-t-BP | 4-t-BP | 2,4-Di-t-BP | |||
| a Si/Al ratio = 20. b Reaction Conditions: T = 140 °C; WHSV = 7 h−1; nt-BA/nPhenol = 4; TOS = 24 h. c Determined by NH3-TPD. d n phenol/(nAl × t). | ||||||||
| n-ZSM-5 | 0.55 | 0.17 | 0.13 | 18.5 | 25.8 | 48.2 | 25.9 | 18.5 |
| c-ZSM-5 | 0.83 | 0.10 | 0.45 | 7.2 | 14.6 | 64.5 | 20.8 | 2.5 |
On the contrary, n-ZSM-5 has shown distinct acidities owing to the unique arrangement of nanocrystalline structure. Unlike c-ZSM-5, which shows characteristic low and high-temperature profile peaks, n-ZSM-5 has shown a broad second peak from 250 to 500 °C. This distinct feature can be attributed to the wide distribution of acid sites (types ii, iii, iv, and v) owing to the smaller crystallite size, high external surface area and low framework density, and is in good agreement with 27Al MAS-NMR data.48 Such a wide distribution of discrete acid sites of various strengths can result in the unusual selectivity in a catalytic reaction.
Fig. 8B shows the pyridine-DRIFTS spectra of various zeolites. The high-intense peak at wavelength 1545 cm−1 can be attributed to the Brønsted acid sites originating from the aluminium ions in perfect tetrahedral coordination in the framework. On the other hand, the peak around 1600 cm−1 can be ascribed to the physically adsorbed pyridine on various acid sites. Meanwhile, the peaks around 1450 and 1445 cm−1 could be attributed to the geometrical defects of the framework and due to the penta-coordinated aluminium species, which is in good agreement with 27Al MAS-NMR spectra of n-ZSM-5.
Surface hydroxyl groups
The surface acidity of zeolites is an important property and responsible for the catalytic activity in many acid catalysed reactions. IR spectroscopy is a direct and routinely used method to study various types of acidity or hydroxyl (silanol) groups.49Fig. 9 shows the typical room temperature IR spectra of both n-ZSM-5 and c-ZSM-5. The spectra show different types of hydroxyl groups associated with terminal (isolated; 3745 cm−1), bridging/Brønsted acid (Si–O(H)–Al; 3646 cm−1), silanol nest (3475 cm−1), and hydration (3247 cm−1) in agreement with most zeolites.50–53 It can clearly be seen from this figure that the narrow ν(OH) band at 3745 cm−1 indicates the existence of isolated silanol groups (Si–OH) in high surface area n-ZSM-5 (Fig. 9a and Table 1) leading to a higher concentration of isolated silanol which is in line with the particle size-dependent characteristics, i.e., samples with a large particle size show a relatively low intensity of the Si–OH groups on the external surface and vice versa. As expected, a very weak or nearly no signal is noticed for c-ZSM-5 (Fig. 9b) owing to the larger particle size/low surface area of the sample indicating that, in general, conventional zeolites are free of isolated silanol groups and therefore expected to have very small or no weak acid sites. In terms of qualitative estimation, it can be seen from Fig. S7‡ that the ratio of bridged/terminal hydroxyl groups in n-ZSM-5 is lower than that in ZSM-5. Indeed, such structural defects (weakly acidic) greatly influence the catalytic activity, e.g., Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam over silicalite-1 having MFI-type structure.54 | ||
| Fig. 9 FT-IR spectra of (a) n-ZSM-5 and (b) c-ZSM-5. | ||
The spectral region of 3600–3100 cm−1 normally shows hydroxyl stretch vibrational modes, an intense band (3646 cm−1) and a weak OH stretch mode (3247 cm−1). The former is often ascribed to silanol nests that consist of several hydroxyl groups interacting through extended hydrogen bonding and is generally attributed to weakly bonded hydrous species while the latter is mainly attributed to strongly hydrogen-bonded hydrous species.50,53 Further, the signal intensity of the 3247 cm−1 band is relatively weaker (Fig. 9a) for n-ZSM-5; this might be due to the filling of internal pores, while an intense band is observed for c-ZSM-5 (Fig. 9b) which could be attributed to the growth of the uniform layer of strongly-bonded water on the ordered surface.55
Modelling studies
Previous reports indicate that Al substitution in the MFI framework is not random, but it takes place following an order of preference among the tetrahedral sites, which depends on the Si/Al ratio and synthesis method.56,57 However, the varying interaction strength between probe molecules and zeolite active sites, which is commonly explained by weak, medium and strong acidity,58,59 cannot be solely associated with Al substitutions at different T sites that create BA of different acid strength.60 Our preliminary DFT calculations show that the adsorption energy of NH3 at each of the twelve non-equivalent BA sites in the MFI framework varies within a very narrow range, thus not justifying the reasoning of BA sites with weak, medium and strong acid strength. Therefore, a wider set of interactions, including differences in the adsorption environment, has to be invoked to provide a suitable explanation to the experimental observations.Pinto et al.61 calculated the heat of ammonia adsorption for weak acids that ranges between 50 and 65 kJ mol−1 based on TPD measurements; these weak acid sites have been previously assigned to physisorbed NH3 on external silanol groups.62 Our computer simulations (Fig. 10) show that silanol groups display the weakest affinity for the NH3 molecules among the sites analyzed here, with a DFT-calculated average binding energy of −55 ± 25 kJ mol−1, satisfactorily agreeing with the reported values.61 Therefore, we relate the acid site (i) to silanol groups located at the external surface of the zeolite.
 | ||
| Fig. 10 Representation of the different site where an NH3 molecule is adsorbed on the internal and external surfaces of the ZSM-5 zeolite. (a) Silanol group, (b) NH4 ion at an external Brønsted acid site, (c) NH4 ion at an internal Brønsted acid site, (d) external Brønsted acid site, (e) internal Brønsted acid site and (f) Lewis acid site. Color code: Si (orange), Al (grey), O (red), N (blue) and H (white). | ||
Lónyi et al.63 have reported that desorption of NH3 from NH4+ is responsible for the existence of peaks in the low temperature region of the TPD profile (<400 °C). Consequently, we consider that the interaction between a second NH3 molecule and an NH4+ ion results in the peaks (iia) and (iib) in the deconvolution of the TPD profile. Our calculated adsorption energies for the NH3⋯NH4+ interaction are approximately 20 kJ mol−1 higher at the interior of the pore compared to the values computed at the external surface, −94 ± 10 kJ mol−1 against −76 ± 16 kJ mol−1; this is a result of stronger dispersion interactions with the wall of the pores, which together with higher diffusion constraints at the interior of the material, acceptably explains the position of the peaks (iia) and (iib) in the NH3-TPD.
As to the direct interaction of NH3 with the BA sites, we did not observe significant differences between the adsorption of NH3 at the internal and external surfaces, with mean values of −147 ± 9 and −145 ± 15 kJ mol−1, respectively. However, re-adsorption events in the microporous system should make the desorption temperature from internal Brønsted acid sites higher compared to the sites at the external surface.64,65 We thus assign the peaks (iii) and (iv) in the NH3-TPD to the external and internal acid sites, respectively, after considering their position and intensity in the TPD profiles. Regarding the peak at the highest measured temperature, we can observe that its intensity is considerably lower than the signals (iii) and (iv), and increases for n-ZSM5. We can therefore tentatively assign this peak to the adsorption of ammonia on Lewis acid sites, considering that the desorption temperature related to this interaction has been reported to appear in the high-temperature region of the TPD profile.63 Three-coordinated Lewis acid sites can be formed as a result of the dehydration of terminal Al atoms located at intra-framework positions at the external surface of the zeolite.38,66,67 In the present calculations, the adsorption of NH3 on these Lewis acid sites is especially strong as a consequence of the simultaneous interaction of the N atom with Al and the formation of hydrogen-bonds NH⋯OH with nearby silanol groups, generating binding energies as large as −214 kJ mol−1. This agrees with the assignment given to the acid site type (v) in the NH3-TPD. Table 3 lists the full set of adsorption energies and their TPD assignment, with Fig. 10 showing a representation of each adsorption site.
| Acid site | Adsorption energy (kJ mol−1) | TPD signal | |
|---|---|---|---|
| Bulk | Surface | ||
| Silanol group | — | −55 ± 25 | (i) |
| NH4 ion at external BA site | — | −76 ± 16 | (iia) |
| NH4 ion at internal BA site | −94 ± 10 | — | (iib) |
| External BA site | — | −145 ± 15 | (iii) |
| Internal BA site | −147 ± 9 | — | (iv) |
| Lewis site | — | −191 ± 22 | (v) |
Reaction studies
The catalytic properties of the synthesized zeolites have been evaluated for vapour phase tertiary butylation of phenol, and the obtained values are listed in Table 3. The strong correlation between the nature of acid sites and selectivity of alkylated isomers in the tert-butylation reaction was well-established in the literature.67–69 While weak Lewis/Brønsted acid sites (type-ii and type-v) are known to direct ortho-alkylations (2-t-BP), the moderate acid sites (type-iii) preferably yield 4-t-butylphenol (4-t-BP) (see Scheme S1‡). Further, the strong Brønsted acid sites (type-iv) can result in dialkylations to yield 2,4-di-t-butylphenol (2,4-di-t-BP).17,18In this context, a strong Brønsted acid catalyst such as ZSM-5 was expected to give high conversion and enhanced selectivity for 2,4-di-t-BP. However, the narrow micropores (d ∼ 0.5 nm) of the MFI-type frameworks will restrict the facile diffusion of the reactant and product molecules (kinetic diameter >0.5 nm) to decrease the effective catalytic activities of the catalyst. Such limitation is reflected in the poor selectivity of di-alkylated 2,4-di-t-BP, whose kinetic dimensions are larger than the micropore size of c-ZSM-5. On the other hand, nanocrystalline sample n-ZSM-5 has shown improved catalytic activity and a better selectivity for 2,4-di-t-BP when compared to c-ZSM-5 (see Table 2 and Fig. 10). Such activity can be attributed to the high external surface and intercrystalline mesoporosity of n-ZSM-5, which can promote di-alkylations.
More importantly, n-ZSM-5 has shown better selectivity towards the ortho-alkylated isomer, 2-t-BP. Such ortho-alkylations are unusual for zeolite catalysts with a high density of strong Brønsted acid sites. As mentioned before, ortho-alkylations are directed by weak/moderate acid sites present in the mesopores. Therefore, the increased selectivity of n-ZSM-5 towards 2-t-BP can be attributed to the presence of weak Brønsted acid sites. The origin of such weak acid sites can be explained by considering the synthesis conditions of n-ZSM-5. Unlike the traditional strategies of synthesis of c-ZSM-5, the n-ZSM-5 is prepared from viscous gels at low alkali concentration and less amount of water. Such conditions may restrict the movement of aluminosilicate oligomers and hence result in the formation of numerous geometric defects (type-v; weak Lewis acid sites). It is evident from the FT-IR spectra of n-ZSM-5, which show less-intense vibrational modes at 3245 cm−1 (cf.Fig. 9) corresponding to core lattice vibrations of weak Lewis acid sites (Fig. 8). Further, it is also observed in the TPD profile of n-ZSM-5, which shows an increased amount of type (ii) and type (v) acid sites (Fig. 7A). Based on these characterization studies, it can be perceived that the distinct acid sites of n-ZSM-5 play a key role in ortho-alkylations apart from para-alkylations to form the final product, 2,4-di-t-BP.
Fig. 11 shows the catalytic lifetime of n-ZSM-5, which shows better activity for a longer period of time when compared to the bulk sample owing to the facile diffusion of the reactant molecules through the intercrystallite mesopores and also due to reduced diffusion path lengths of the zeolite nanocrystals. The sample has shown unaltered activity and excellent selectivity for a reaction time of 24 h. More importantly, the selectivity towards 2-t-BP is not momentary and is maintained in the time-on-stream studies of 24 h.
 | ||
| Fig. 11 Time-on-stream studies of n-ZSM-5 (filled symbols) and c-ZSM-5 (open symbols). | ||
Conclusions
In summary, we report here the synthesis of nanostructured zeolite aggregates, designated as n-ZSM-5, consisting of well-defined ultra-small nanospheres of MFI-type topology having a unique brain-coral morphology. The remarkable self-organization of such nanocrystals endows the zeolite with intercrystalline mesoporosity, enhanced surface area, remarkable acidity and reduced diffusion path length, making n-ZSM-5 a promising solid-acid catalyst. The improved physico-chemical and textural characteristics of the nanocrystalline zeolite are reflected in terms of increased catalytic activity and high selectivity towards the desired products of the tertiary butylation reaction of phenol, viz., 2-t-BP and 2,4-di-t-BP. The assignment of the signals in the TPD experiments is further supported by DFT calculations. The calculated adsorption energies of NH3 on different sites follow the order of strength: silanol groups (external surface) < NH4+ (external surface) < NH4+ (internal surface) < Brønsted acid sites (external surface) < Brønsted acid sites (internal surface) < Lewis acid sites (external surface), with a satisfactory agreement of the number and position of the TPD signals in the n-ZSM-5 profile, and the variations observed with c-ZSM-5.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank DST, New Delhi, for funding NCCR, IIT-Madras; Prof. B. Viswanathan for encouragement and support; Mr. T. V. R. Mohan for the help with TPD data. NHdL thanks EPSRC, grant number EP/K009567, for funding. Information on the data underpinning the results presented here, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2020.0115255376.Notes and references
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS.
- J. Grand, S. N. Talapaneni, A. Vicente, C. Fernandez, E. Dib, H. A. Aleksandrov, G. N. Vayssilov, R. Retoux, P. Boullay, J.-P. Gilson, V. Valtchev and S. Mintova, Nat. Mater., 2017, 16, 1010 CrossRef CAS.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246 CrossRef CAS.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494 CrossRef CAS.
- S. Mintova and V. Valtchev, Microporous Mesoporous Mater., 2002, 55, 171 CrossRef CAS.
- C. M. Lew, Z. Li, S. Li, S.-J. Hwang, Y. Liu, D. I. Medina, M. Sun, J. Wang, M. E. Davis and Y. Yan, Adv. Funct. Mater., 2008, 18, 3454 CrossRef CAS.
- R. Van Grieken, J. L. Sotelo, J. M. Menéndez and J. A. Melero, Microporous Mesoporous Mater., 2000, 39, 135 CrossRef CAS.
- W. Song, R. E. Justice, C. A. Jones, V. H. Grassian and S. C. Larsen, Langmuir, 2004, 20, 8301 CrossRef CAS.
- P. Morales-Pacheco, J. M. Domínguez, L. Bucio, F. Alvarez, U. Sedran and M. Falco, Catal. Today, 2011, 166, 25 CrossRef CAS.
- S. S. Kim, J. Shah and T. J. Pinnavaia, Chem. Mater., 2003, 15, 1664 CrossRef CAS.
- B. Louis and L. Kiwi-Minsker, Microporous Mesoporous Mater., 2004, 74, 171 CrossRef CAS.
- Z. Qin, L. Lakiss, L. Tosheva, J.-P. Gilson, A. Vicente, C. Fernandez and V. Valtchev, Adv. Funct. Mater., 2014, 24, 257 CrossRef CAS.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Science, 2011, 333, 328 CrossRef CAS.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718 CrossRef CAS.
- D. Xu, Y. Ma, Z. Jing, L. Han, B. Singh, J. Feng, X. Shen, F. Cao, P. Oleynikov, H. Sun, O. Terasaki and S. Che, Nat. Commun., 2014, 5, 4262 CrossRef CAS.
- R. K. Parsapur and P. Selvam, ChemCatChem, 2018, 10, 3978 CrossRef CAS.
- R. K. Parsapur and P. Selvam, Sci. Rep., 2017, 23, 16291 Search PubMed.
- N. V. Krishna and P. Selvam, Chem. – Eur. J., 2017, 23, 1604 ( Dalton Trans. , 2017 , 46 , 770 ) CrossRef CAS.
- A. Inayat, I. Knoke, E. Spiecker and W. Schwieger, Angew. Chem., Int. Ed., 2012, 51, 1962 CrossRef CAS.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684 CrossRef CAS.
- K. Möller, B. Yilmaz, R. M. Jacubinas, U. Müller and T. Bein, J. Am. Chem. Soc., 2011, 133, 5284 CrossRef.
- Z. Gabelica, N. Blom and E. G. Derouane, Appl. Catal., 1983, 5, 227 CrossRef CAS.
- A. Nastro and L. B. Sand, Zeolites, 1983, 3, 57 CrossRef CAS.
- Q. Li, D. Creaser and J. Sterte, Chem. Mater., 2002, 14, 1319 CrossRef CAS.
- K. Shen, W. Qian, N. Wang, C. Su and F. Wei, J. Mater. Chem. A, 2014, 2, 19797–19808 RSC.
- P. Selvam, N. V. Krishna and A. Sakthivel, Adv. Porous Mater., 2013, 1, 239 CrossRef CAS.
- E. Selvam, R. K. Parsapur and P. Selvam, 23rd Natl. Symp. Catal., Bengaluru, 17-19 January 2018, p. 148 Search PubMed; Faraday Discussion Meeting, London, 16-18 May 2018 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS ; 1994, 49, 14251.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 ( Phys. Rev. B: Condens. Matter Mater. Phys. , 1996 , 54 , 11169 ) CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS.
- K. M. Ho, C. L. Fu, B. N. Harmon, W. Weber and D. R. Hamann, Phys. Rev. Lett., 1982, 49, 673 CrossRef CAS.
- C. L. Fu and K. M. Ho, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 5480 CrossRef CAS.
- C. E. Hernandez-Tamargo, A. Roldan and N. H. de Leeuw, Mol. Catal., 2017, 433, 334 CrossRef CAS.
- S. Quartieri, R. Arletti, G. Vezzalini, F. Di Renzo and V. Dmitriev, J. Solid State Chem., 2012, 191, 201 CrossRef CAS.
- C. E. Hernandez-Tamargo, A. Roldan and N. H. De Leeuw, J. Solid State Chem., 2016, 237, 192 CrossRef CAS.
- A. Le Bail, Powder Diffr., 2005, 20, 316 CrossRef CAS.
- D. H. Brouwer and G. D. Enright, J. Am. Chem. Soc., 2008, 130, 3095 CrossRef CAS.
- I. Kabalan, L. Michelin, S. Rigolet, C. Marichal, T. J. Daou, B. Lebeau and J. L. Paillaud, Solid State Sci., 2016, 58, 111 CrossRef CAS.
- H. van Koningsveld, J. C. Jansen and H. van Bekkum, Zeolites, 1990, 10, 235 CrossRef CAS.
- W. Zhang, X. Bao, X. Guo and X. Wang, Catal. Lett., 1999, 60, 89 CrossRef CAS.
- R. W. Joyner, A. D. Smith, M. Stockenhuber and M. W. E. van der Berg, Phys. Chem. Chem. Phys., 2004, 6, 5435 RSC.
- Q. Zhang, A. Mayoral, O. Terasaki, Q. Zhang, B. Ma, C. Zhao, G. Yang and J. Yu, J. Am. Chem. Soc., 2019, 141, 3772 CrossRef CAS.
- J. Liu, G. Jiang, Y. Liu, J. Di, Y. Wang, Z. Zhao, Q. Sun, C. Xu, J. Gao, A. Duan, J. Liu, Y. Wei, Y. Zhao and L. Jiang, Sci. Rep., 2015, 4, 7276 CrossRef.
- In zeolites, the silanol groups are typically characterised by absorptions (vibration modes/bands)50–52 in the hydroxyl stretching frequency region of 4000–3000 cm−1. Owing to the existence of silanol groups in different environments, four relevant features are typically observed and they are classified as: (i) terminal (isolated – external) and (vicinal/geminal – internal) silanol groups (3750–3740 cm−1); (ii) bridging (Brønsted acid) silanol groups (3680–3550 cm−1) which act as proton donor; (iii) silanol nests (associated) that consist of a number of silanol groups (3680–3550 cm−1) interacting through extended hydrogen bonding; (iv) adsorbed water (3300–3200 cm−1), i.e., hydrogen bonding interaction between silanol groups and water molecules.
- V. Bolis, C. Busco, S. Bordiga, P. Ugliengo, C. Lamberti and A. Zecchina, Appl. Surf. Sci., 2002, 196, 56 CrossRef CAS.
- J.-P. Gallas, J.-M. Goupil, A. Vimont, J.-C. Lavalley, B. Gil, J.-P. Gilson and O. Miserque, Langmuir, 2009, 25, 5825 CrossRef CAS.
- K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, J. Catal., 2011, 280, 196 CrossRef CAS.
- K. Sillar and P. Burk, Chem. Phys. Lett., 2004, 393, 285 CrossRef CAS.
- V. R. R. Marthala, Y. Jiang, J. Huang, W. Wang, R. Gläser and M. Hunger, J. Am. Chem. Soc., 2006, 128, 14812 CrossRef CAS.
- D. Ngo, H. Liu, Z. Chen, H. Kaya, T. J. Zimudzi, S. Gin, T. Mahadevan, J. Du and S. H. Kim, npj Mater. Degrad., 2020, 4, 1 CrossRef.
- C. W. Kim, N. H. Heo and K. Seff, J. Phys. Chem. C, 2011, 115, 24823 CrossRef CAS.
- V. Pashkova, S. Sklenak, P. Klein, M. Urbanova and J. Dedecek, Chem. – Eur. J., 2016, 22, 3937 CrossRef CAS.
- Q. Zhao, W.-H. Chen, S.-J. Huang, Y.-C. Wu, H.-K. Lee and S.-B. Liu, J. Phys. Chem. B, 2002, 106, 4462 CrossRef CAS.
- Y. Seo, K. Cho, Y. Jung and R. Ryoo, ACS Catal., 2013, 3, 713 CrossRef CAS.
- C. E. Hernandez-Tamargo, A. Roldan and N. H. de Leeuw, J. Phys. Chem. C, 2016, 120, 19097 CrossRef CAS.
- R. R. Pinto, P. Borges, M. A. N. D. A. Lemos, F. Lemos, J. C. Védrine, E. G. Derouane and F. R. Ribeiro, Appl. Catal., A, 2005, 284, 39 CrossRef CAS.
- N.-Y. Topsøe, K. Pedersen and E. G. Derouane, J. Catal., 1981, 70, 41 CrossRef.
- F. Lónyi and J. Valyon, Microporous Mesoporous Mater., 2001, 47, 293 CrossRef.
- T. Omojola, N. Cherkasov, A. I. McNab, D. B. Lukyanov, J. A. Anderson, E. V. Rebrov and A. C. van Veen, Catal. Lett., 2018, 148, 474 CrossRef CAS.
- B. E. Handyla, A. Jacobo, M. G. Cárdenas Galindo, M. González, M. E. LLanos, M. de Lourdes Gúzman and F. Hernández, Top. Catal., 2002, 19, 249 CrossRef CAS.
- C. E. Hernandez-Tamargo, A. Roldan and N. H. de Leeuw, J. Phys. Chem. C, 2018, 123, 7604 CrossRef.
- A. Sakthivel, S. E. Dapurkar, N. M. Gupta, S. K. Kulshreshtha and P. Selvam, Microporous Mesoporous Mater., 2003, 65, 177 CrossRef CAS.
- A. Sakthivel, S. K. Badamali and P. Selvam, Microporous Mesoporous Mater., 2000, 39, 457 CrossRef CAS.
- Y. Kubota, S. Inagaki, Y. Nishita, K. Itabashi, Y. Tsuboi, T. Syahylah and T. Okubo, Catal. Today, 2015, 243, 85 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Akira Miyamoto on the occasion of his 73rd birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental and computational details along with additional characterisation data, and reaction scheme and models. See DOI: 10.1039/d0ce00989j |
| § Present Address: Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716–3110, United States of America. |
| This journal is © The Royal Society of Chemistry 2020 |