
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective polymorphism of α-glycine by acoustic levitation†
Adriana
Alieva
 a,
Matthew
Boyes
a,
Thomas
Vetter
b and
Cinzia
Casiraghi
*a
a,
Matthew
Boyes
a,
Thomas
Vetter
b and
Cinzia
Casiraghi
*a
aDepartment of Chemistry, University of Manchester, Manchester, UK. E-mail: cinzia.casiraghi@manchester.ac.uk
bDepartment of Chemical Engineering and Analytical Science, University of Manchester, Manchester, UK
First published on 8th October 2020
Abstract
In this work we investigate the crystallisation behaviour of glycine in water and in a binary solvent mixture in an acoustic levitator under controlled environmental conditions. High speed video microscopy was used to monitor the changes in the microdroplet volume upon evaporation of the solvent. The glycine crystals obtained from levitation form an agglomerate, whose exact morphology depends on the solvent system used. The agglomerates have been collected and precisely opened via laser cutting, allowing further investigation of the morphology and structure of the internal crystals. The crystals appear to grow from the external region towards the centre of the sphere, indicating the formation of a solid shell, whose formation depends on the solvent used. The polymorphic outcome was thoroughly investigated by Raman spectroscopy: all of the crystals measured, regardless of the region or the solvent used, were found to be exclusively of the α-form, despite the addition of IPA inducing changes in the induction time and morphology.
Introduction
Crystallisation from solution is at the heart of various phenomena occurring in nature1,2 and also the most widely applied approach for the isolation and purification of compounds in the chemical process industries.3 Although thermodynamic principles describing this phenomenon have been established more than a century ago,4 a full understanding of the underlying mechanism of nucleation is still not attained. This makes the production of organic crystals of desired size, shape and polymorph very challenging.It is well known that surfaces play an important role in crystallisation: the interaction of solute molecules with foreign surfaces, such as container walls, can change the energetics and kinetics of nucleation.5 A simple way to avoid the use of containers and other foreign elements affecting crystallisation is provided by levitation. A number of different levitation techniques are available, including magnetic,6 electric,7–9 optical,10,11 aerodynamic,12 and acoustic13–16 to mention a few. In particular, acoustic levitation is very attractive because it does not require the sample to have any specific properties, i.e. any material can be levitated. This versatility has made acoustic levitation a promising tool to investigate density,15,17 evaporation,18,19 and drying19,20 behaviour of droplets. Acoustic levitation has been also applied to study crystallisation, although only a very limited number of studies have been conducted up to now (see state-of-the-art table in ESI,† Section S1).19,21–30 To the best of our knowledge, there are no acoustic levitation studies conducted on glycine, the simplest amino acid, despite its wide use in crystallisation studies,31–35 due to its simple molecular structure and well-known polymorphism. Glycine crystallises in three distinct polymorphic forms at ambient conditions, denoted as α, β and γ, with relative stabilities: γ > α > β at room temperature.36 The α- and β-forms both have monoclinic structure with a space group symmetry of P21/n and P21, respectively.37,38 The most stable form, γ, has a trigonal crystal structure belonging to the P31 or P32 groups.39 Crystallisation from aqueous solutions typically yields the metastable α-form, as it is kinetically favoured.40 The least stable, β-form of glycine, is commonly obtained from cooling crystallisation by the addition of alcohol such as ethanol or methanol as an anti-solvent to aqueous solution and readily transforms to the α-form in the presence of water or upon heating;37,41 while the stable γ-form can be crystallised from acidic or basic solutions.39,42
Herein, we report an ex situ polymorph analysis of glycine crystals obtained from levitation experiments, in pure water and with isopropanol (IPA) as a co-solvent, by using Raman spectroscopy. Our results show that selective crystallisation of the α-form of glycine is obtained by acoustic levitation, regardless of the solvent investigated. The presence of the co-solvent in the crystallising solution influences the kinetics of the crystallisation of glycine and the morphology of the obtained crystals, but does not affect the polymorphic outcome from evaporative crystallisation. This polymorph outcome is very different from the one observed on evaporative droplets of glycine on a substrate, where both α- and β-forms are detected and the unstable β-form of glycine is exclusively located at the contact region of the droplet with the substrate, as a result of the higher supersaturation rate. Based on the same effect, one would expect to see the β-form of glycine on the surface of the crystallised droplet, while we find that all crystals are of the α-form. The selective crystallisation of the α-form of glycine under acoustic levitation could be attributed to several concomitant effects, such as geometric effects and the presence of the ultrasonic field, as selective crystallisation of the α-form over the β-form of glycine was also obtained in sono-crystallisation experiments.43–45 More experiments, such as a detailed comparison between crystallisation in an acoustic levitator and in a sonicator, under similar conditions, may help to elucidate the origin of the selectivity of the α-form observed in our work.
Results and discussion
The experimental set up of the acoustic levitator used in our studies is shown schematically in Fig. S2† (more details in Methods and Section S2). A conditioned gas is introduced with controlled gas flow rate, temperature and humidity around the droplet, allowing performing the crystallisation experiments at constant temperature (21 ± 0.5 °C) and dry conditions (RH = 0%). The setup consists of a 100 kHz ultrasonic droplet levitator, equipped with a CCD camera, backlight illumination and a controlled evaporator unit. The levitating droplets were monitored in situ during solvent evaporation using a CCD camera, the projected area of the droplet over the time is then analysed with a home-made automated images processing software. A representative video is included in the ESI,† Video S1. Full details of the levitator setup are provided in ref. 22.In a typical crystallisation experiment, a 2 μL droplet of 0.5 M undersaturated glycine solution, in either pure water or water/IPA (starting concentration, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v) mixture, was injected and levitated in one of the wave nodes of the acoustic levitator equipped with an environmentally controlled chamber. Note that this mixture ratio was selected as higher ratios of IPA result in the precipitation of glycine for the selected conditions.
:1 v:v) mixture, was injected and levitated in one of the wave nodes of the acoustic levitator equipped with an environmentally controlled chamber. Note that this mixture ratio was selected as higher ratios of IPA result in the precipitation of glycine for the selected conditions.
Fig. 1(a) shows the evaporation profiles of glycine microdroplets for different solvents. As expected, regardless of the type of the solvent used the droplet volume is found to decrease continuously till complete solvent removal. The evaporation rate of pure water shows a steady evolution with time. In the case of the binary solvent mixture, different stages can be seen: the initial slope of volume vs. time, Fig. 1(a), for the bi-component solvent system is higher compared to that of pure water. This is due to the preferential evaporation of the more volatile component (i.e., IPA) in the first stage of evaporation. The slope decreases over time indicating that the composition of the remaining liquid phase gets richer in water causing retardation in the evaporation rate.
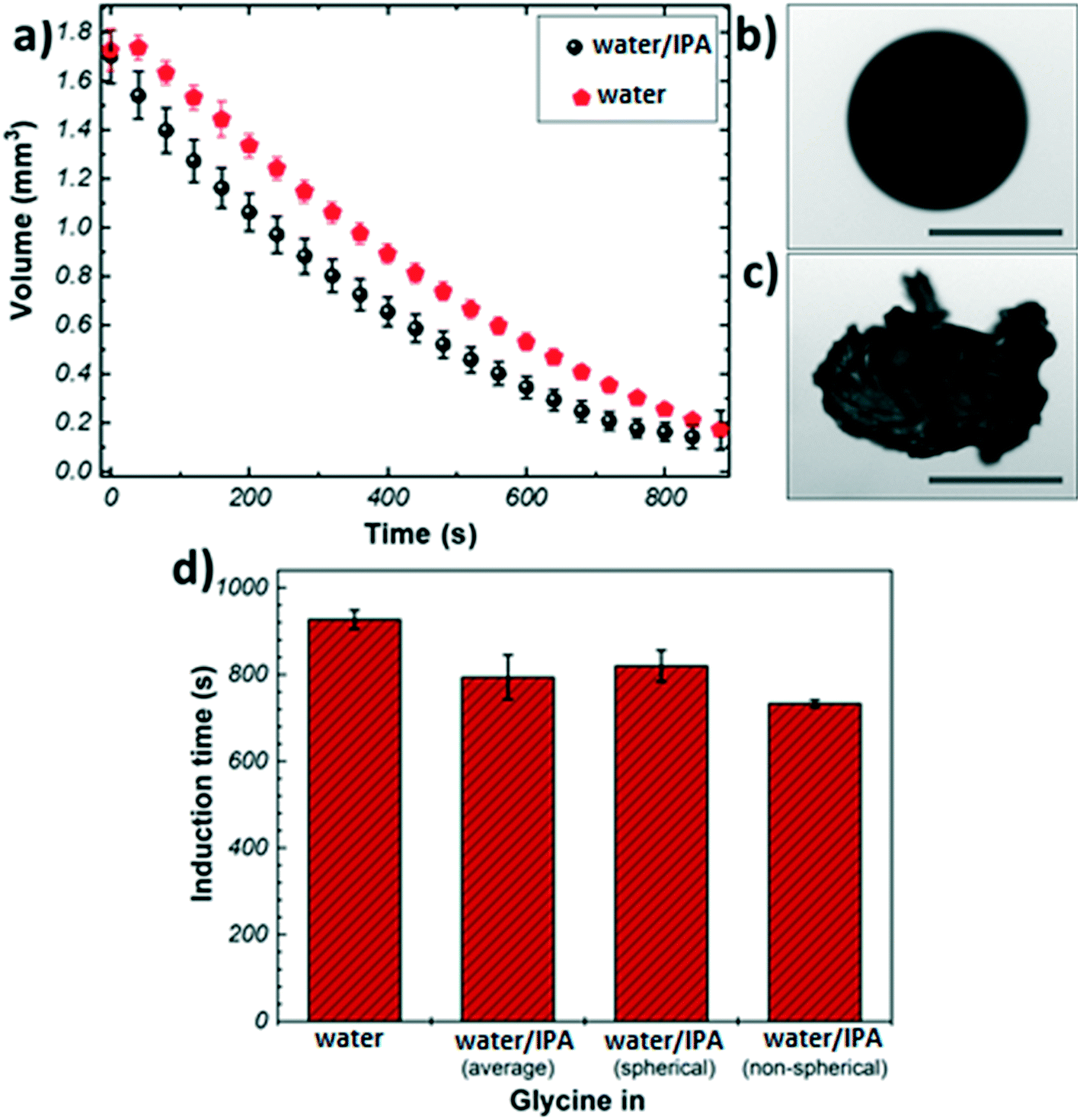 | ||
| Fig. 1 (a) Evaporation profiles of glycine microdroplets from different solvents (b) image of a spherical agglomerate of glycine crystals obtained from pure water; (c) image of a non-spherical agglomerate of glycine crystals obtained from water/IPA mixed solvent (d) induction time of glycine from different solvent systems. Scale bars ≈ 0.5 mm. | ||
Fig. 1(b) and (c) show representative images of crystals obtained from the levitation experiments. Typically, two distinct morphologies were observed: spherical agglomerates composed of small crystallites (Fig. 1(b)), and non-spherical agglomerates composed of large needle-like crystals (Fig. 1(c)). Only agglomerates with spherical morphology were obtained from pure water, whereas the addition of IPA resulted in a split in the morphology (7:3 split of spherical vs. non-spherical). This outcome could be attributed to the presence of IPA as an anti-solvent causing a widening of the metastable zone (MSZ) width in which crystal growth dominates over nucleation.46,47 Therefore, the probability of obtaining larger crystals increases in the presence of IPA. The evaporation profiles of the spherical and non-spherical agglomerates obtained from the binary solvent mixture exhibit a similar trend (Fig. S3†).
The crystallisation kinetics was determined by measuring the induction time of crystallisation (see details in Materials and methods), Fig. 1(d). It can be clearly seen than the addition of IPA to the crystallising solution has a promoting effect on the nucleation of glycine, as compared to the case of pure water: the presence of IPA increases the evaporation rate of the droplets generating higher supersaturation rate, hence inducing nucleation sooner. The induction times for the non-spherical glycine agglomerate obtained from the binary mixture (733 ± 8 s) were found to be slightly shorter than the spherical samples (820 ± 36 s)- this can be rationalised by considering the solubility curve of glycine and relative growth and nucleation rates. The solution begins well below the saturation concentration and it crosses the supersaturation threshold into glycine's MSZ with the evaporation of the solvent.5 In this region, crystal growth dominates over nucleation leading to the formation of larger crystals. If the solvent continues to evaporate, the system moves past the MSZ into high supersaturation, where nucleation events dominate over crystal growth.3
Further morphological characterisation was conducted on the spherical glycine agglomerates obtained from either water or the mixed solvent system by scanning electron microscopy (SEM). Fig. 2 shows SEM images of a broken glycine sphere crystallised from pure aqueous solution. These images show the morphology of the crystals in both the inner and outer parts of the sphere – the crystals seem to grow following the radial direction. Considering that the solvent evaporates from the surface of the droplet, one might infer that nucleation has been induced heterogeneously at the air–water interface due to the generation of higher supersaturation rate at the droplet surface. This is in agreement with previous studies, for example with mannitol22 and calcite crystals.26 This outcome can be explained by taking into account the mechanisms involved in drying of droplets containing solutes, where there are two types of drying processes happening: the first is driven by surface evaporation, and the second is driven by internal moisture migration.22,48,49 Initially, the solvent migrates towards the surface and the solute towards the centre due to solvent evaporation at the surface, which causes shrinking of the droplet diameter; at a critical point, the droplet stops shrinking, hence the molecules start diffusing on the surface, where supersaturation is reached first. Nucleation and crystallisation lead to a strong increase of the total solid fraction within the droplet, which ultimately produces a stable but permeable crust at the droplet surface.22 The exact morphology of the shell depends on the evaporation rate: a slow evaporative process will allow to produce a solid porous structure as molecules have enough time to diffuse.50 This description matches very well with the obtained morphology of the crystal agglomerate surface, as shown in Fig. 2, i.e. a rigid structure with holes of about 10–20 μm in size.
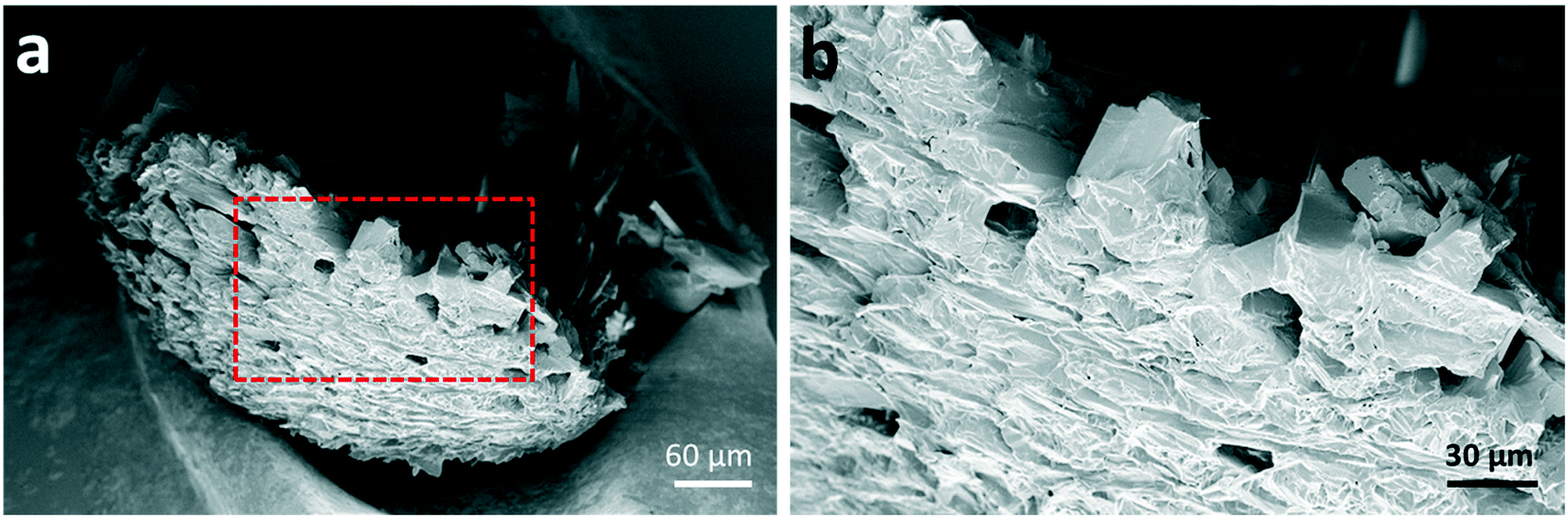 | ||
| Fig. 2 (a) SEM image of a broken spherical agglomerate of glycine crystals, revealing the morphologies of both inner and outer parts of the sample (b) magnified view of the area marked with a red-dashed rectangle in (a). | ||
The shell is expected to form at a critical point, where the solid particles are unable to move sufficiently relative to each other.22 Since the induction time is measured by looking at the changes in shape of the droplet due to solid formation and the images only show the droplet surface, in first approximation one can assume the induction time as the time at which the critical solid fraction is reached first on the surface. Hence, the shell formation is expected at around 900 s for pure water, and at around 733 s and 820 s for the mixed solvent system, depending on the final morphology of the crystals aggregate. The time of shell formation reduces when IPA is used as co-solvent because the higher evaporation rate results in faster and higher enrichment at the droplet boundary. It is interesting to note the difference in induction times (∼90 s) between the two morphologies obtained in the mixed solvent: this seems to indicate that the addition of IPA may lead to the formation of regions within the droplet with very high initial solute concentration, which in turn will produce particles with larger size (i.e., non-spherical agglomerates), making shell formation more difficult to achieve.
Let us now move to the polymorph characterisation. Raman spectroscopy is a simple and fast technique that allows taking individual measurements on crystals with spatial resolution of ∼300–500 nm. In contrast to powder X-ray diffraction (XRD), where the samples have to be ground and fair amount of material is required, little or no sample preparation and a small amount of material is sufficient for Raman spectroscopy measurements. In addition, it has been shown that Raman spectroscopy is able to easily identify glycine polymorphs: each crystal structure show Raman peaks in distinct positions due to the variations in the intermolecular hydrogen bonding interactions.51 In this work, the characteristic C–H stretching modes, representing the symmetric (lower shift) and asymmetric (higher shift) stretches of the C–H bonds of glycine were used for the identification of glycine polymorphs.32 The positions of these modes were found to be at 2972 and 3007 cm−1 for α-form; at 2953 and 3008 cm−1 for β-form, and at 2962 and 3000 cm−1 for γ-form (see representative Raman spectra of the three polymorphs in Fig. S5†).
Raman measurements have been taken first from the agglomerates obtained directly after levitation, i.e. the measurements are performed on the outer part. The samples were then cut in half with high precision using a laser cutter (Materials and methods). This allowed us to get access also to the inner region of the samples in a controlled way. Raman measurements were collected from at least 50 crystals for each sample in order to obtain statistically significant results.
Fig. 3 shows representative Raman spectra obtained from different regions of glycine samples and optical images of the spherical agglomerates after laser cutting. The crosses in Fig. 3(b) and (c) indicate the points where measurements were taken. Fig. 3(a) (see also Section S6†) shows that the Raman spectra of glycine crystals do not show any significant variation amongst different crystals. Furthermore, C–H stretching mode positions are found at ∼2972 and ∼3008 cm−1, indicating that the crystals are of α-form. Hence, our results show that the α-form is found for all crystals, regardless of the region or the solvent used.
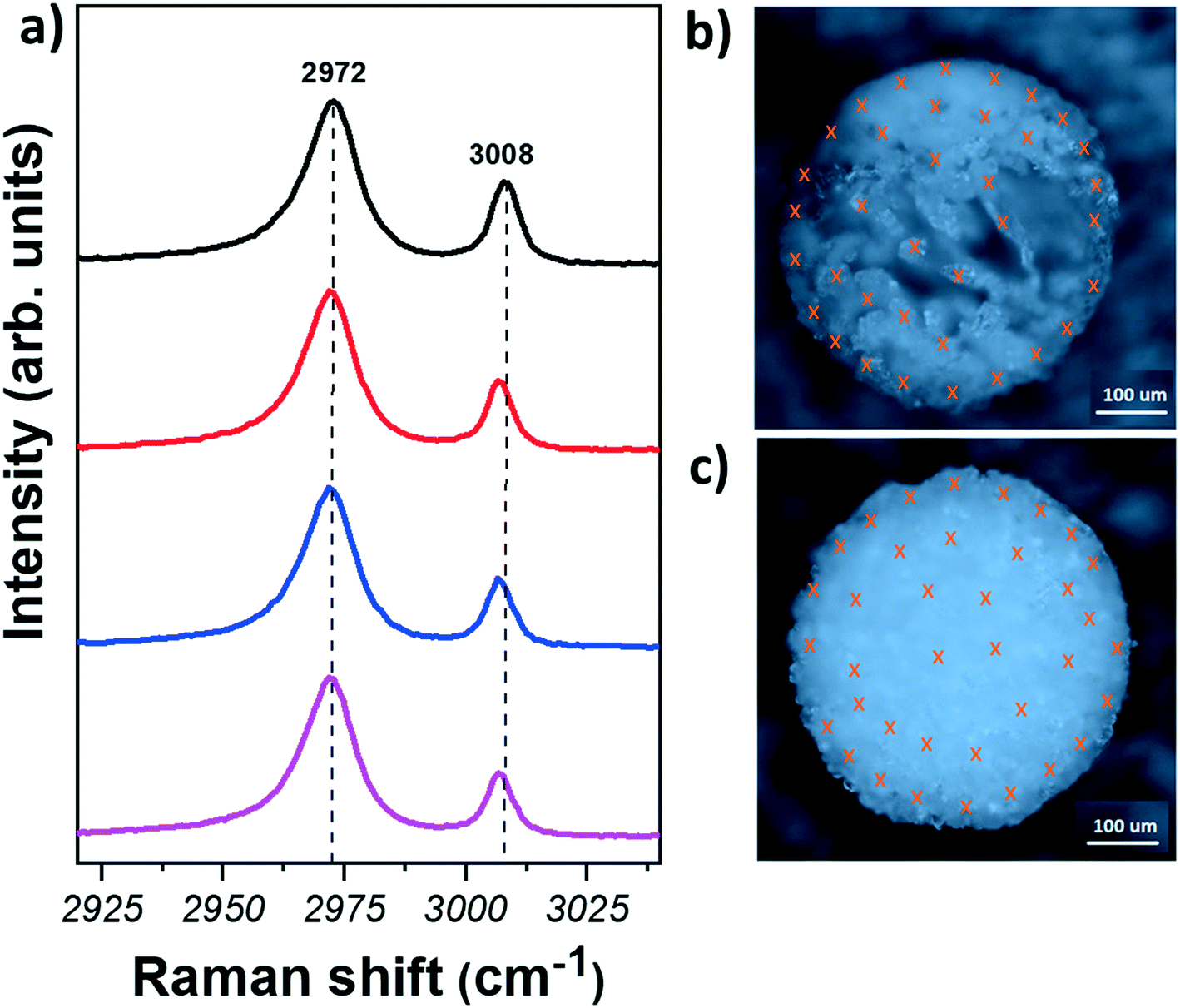 | ||
| Fig. 3 (a) Raman spectra of the CH region measured on glycine crystals from the spherical agglomerates, obtained from different solvent systems (from top to bottom: outer region, solvent = pure water; inner region, solvent = pure water; outer region, solvent = water/IPA; inner region, solvent = water/IPA). Optical microscopy images of glycine spherical agglomerates after laser cutting (b) crystallised from pure water (c) crystallised from water/IPA solvent system. Crosses represent different points where the Raman measurements were taken. | ||
It is interesting to compare our results with those obtained from evaporating droplets of glycine placed on a substrate. In this case, concomitant polymorph formation was observed: both α- and β-form have been identified, with the β-form detected only at the contact region of the droplet with the substrate and air, due to the higher supersaturation rate generated in this region.31,35,51–53 In contrast, under acoustic levitation, the α-form is observed even in the region of higher supersaturation. Similarly, polymorph selectivity of caffeine under acoustic levitation has been also demonstrated: both α- and β-forms were obtained when caffeine was crystallised on different substrates while only pure α-form was obtained from the levitated droplets.27 There are several explanations to elucidate our results. First, one could assume that the β-form is indeed formed on the outer surface and it transforms to the α-form once the droplet is removed from the levitator and exposed to humid air. This would be especially relevant for the mixed solvent system, as the addition of alcohol as an anti-solvent to aqueous solution has been observed to yield the unstable β-form, which transforms to the α-form in the presence of water or upon heating.37,41 However, we did not observe any difference in the polymorphic outcome between the crystals obtained from water and the mixed solvent. Furthermore, the β-crystals obtained from crystallisation of microdroplets on a surface have shown to be very stable in ambient conditions, hence we tend to rule out the possibility of polymorph transformation. The second explanation is related to a pure geometric effect: the droplet has a different shape when levitated and when deposited on a substrate. In surface-assisted crystallisation, the contact angle is known to determine the energy barrier of nucleation.5 In the case of a levitating droplet, from a geometric effect, the crystallisation is expected to be homogeneous, so the energy barrier of nucleation is the highest. It is however unclear how this would reflect on the polymorph outcome. Therefore, an interesting comparison to elucidate the difference in the polymorph outcome can be drawn by determining the nucleation rates.54 Although levitation is considered as a method to achieve homogenous nucleation, currently there are discussion on the effect of mass transport and rotation induced by the acoustic pressure on crystallisation, hence the theory developed for homogeneous nucleation may not be directly applicable to crystallisation in an acoustic levitator.55–57 Finally, exclusive nucleation of the α-form in water or mixed solvent has been also observed in sono-crystallisation experiments using bath or tip sonication:43–45 it has been shown that ultrasound promotes the α-form by inhibiting the formation of the β-form. The presence of ultrasonic waves also narrows down the size distribution and enhances the growth rate of the crystals. Hence, the exclusive formation of the α-form in a levitated droplet of glycine could be attributed to the effect of the acoustic waves, which results in the formation of the metastable α-form, even in the regions of higher supersaturation. As the processes of levitation and sono-crystallisation are not exactly the same, some differences, for example in size distribution of the crystals, may arise between the two methods, but this would require further investigation, which is beyond the scope of this work.
Summary and conclusions
An acoustic levitator has been used to study the evaporation of microdroplets of glycine in pure water and in water/IPA solutions. The resulting crystals were analysed by scanning electron microscopy and Raman spectroscopy. The changes in the shape of the droplet and the formation of solid on the surface were monitored with a CCD camera during evaporation. Our results show that selective crystallisation of the α-form of glycine is achieved by crystallisation under acoustic levitation, even in the presence of an anti-solvent. The morphology of the spherical agglomerate obtained from water suggests the formation of small and high density crystals growing from the outer part of the droplet towards the centre, due to the higher supersaturation rate generated in the region in contact with air. Despite the higher supersaturation rate, the crystals at the outer region have been found to be of the α-form, in contrast to crystallisation of glycine on substrates, where the β-form is formed at the highest supersaturation region. The selective crystallisation of the α-form of glycine under acoustic levitation could be attributed to several concomitant effects, such as geometric effects and the presence of an ultrasonic field.Materials and methods
Materials
Glycine powder (Reagent Plus ≥99%) and isopropyl alcohol (IPA, ≥99.5%) were purchased from Sigma-Aldrich. Deionised (DI) water (Millipore SIMPAK® 1, 18.2 MΩ cm) was used for all experiments requiring water.Acoustic levitation
The experimental set up of the acoustic levitator used in our studies is shown schematically in Fig. S2.† In a typical crystallisation experiment, 2 μL of a 0.5 M undersaturated glycine solution (either in pure water or water/IPA (3:1 v:v) mixture) was injected in an acoustic levitator (tec5 AG, Oberursel, Germany). The levitator operated at a frequency of 100 kHz generating several nodes and antinodes between the emitter and the reflector. For each levitation experiment, the microdroplet was injected under the same node and the distance between the emitter and the reflector was adjusted to stabilise the droplet throughout the evaporation. Each set was repeated minimum 6 times for the generation of the average evaporation profiles of the solvents. A Manta G-505 CCD camera (Allied Division, Stadtroda, Germany) was used to monitor the levitation process with a backlight illumination of the droplet. MATLAB© was used for video processing.
In order to make sure that experiments performed during different days are reproducible, a 2 μL droplet of pure water was introduced first in the levitator and its evaporation monitored. Although the evaporation rate was not exactly the same in all experiments, the deviations were typically within ±6.9%.The samples obtained from the levitation experiments were carefully collected using a fine mesh net after the solvent evaporation was completed.
Volumes of the droplets were calculated using MATLAB© by determining the width, a, and the height, b, of the ellipsoid droplet and then applying the formula V = 4π/3a2b to find the volume of an ellipsoid.
Induction times were extracted by analysing the collected images and determined by the change in the shape of the droplets when a solid is present.
Laser cutting
The spherical glycine samples were cut in half using a laser based sample preparation system (microPREP) equipped with a 532 nm laser. In order to avoid any heating effects generated by the laser the power was kept at 0.1 W. 30 line cuts with a distance of 2.5 μm from each other were performed for each cut.Raman spectroscopy
A Renishaw inVia Raman spectrometer equipped with a 514.5 nm laser was employed to acquire all spectra for the polymorph assignment of glycine crystals. All measurements were taken using a 100× (NA = 0.85) objective lens, 2400 l mm−1 grating and laser power less than 1.3 mW. 50 measurements were taken from each sample prior to laser cutting in half for the identification of the crystals grown outside of the spheres. After cutting, the crystals grown in the centre of the spheres were also extensively characterised by taking 50 point measurements from different crystals. Minimum 2 spherical samples of glycine crystals grown from each solvent system were characterised.Scanning electron microscopy (SEM)
ZEISS Sigma field emission SEM with an acceleration voltage of 5 kV was used to obtain images from glycine agglomerate coated with ∼15 nm Pt layer.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No 648417. T. V. thanks the Royal Academy of Engineering for the support through an Engineering for Development research fellowship (Grant No. RF1516/15/22).References
- A. Sigel, H. Sigel and R. K. O. Sigel, Biomineralization: from nature to application, 2008 Search PubMed.
- S. K. Haldar and J. Tišljar, Introduction to Mineralogy and Petrology, Elsevier Inc., 2013 Search PubMed.
- R. Davey and J. Garside, From molecules to crystallizers, Oxford University Press, New York, 2000 Search PubMed.
- J. W. Gibbs, Am. J. Sci., 1878, s3–16, 441–458 CrossRef.
- J. W. Mullin, Crystallization, Butterworth-Heinemann, Oxford, Boston, 4th edn, 2001 Search PubMed.
- M. V. Berry and A. K. Geim, Of flying frogs and levitrons, Eur. J. Phys, 1997, 18, 307 CrossRef.
- M. D. Barnes, K. C. Ng, W. B. Whitten and J. M. Ramsey, Anal. Chem., 1993, 65, 2360–2365 CrossRef CAS.
- W. B. Whitten, J. Michael Ramsey, S. Arnold and B. V. Bronk, Anal. Chem., 1991, 63, 1027–1031 CrossRef CAS.
- K. C. Ng, W. B. Whitten, S. Arnold and J. M. Ramsey, Anal. Chem., 1992, 64, 2914–2919 CrossRef CAS.
- G. G. Hoffmann, E. Lentz and B. Schrader, Rev. Sci. Instrum., 1993, 64, 823–824 CrossRef CAS.
- A. Ashkin, Phys. Rev. Lett., 1970, 24, 156–159 CrossRef CAS.
- E. G. Lierke, Forsch. Ingenieurwes., 1995, 61, 201–216 CrossRef CAS.
- A. R. Hanson, E. G. Domich and H. S. Adams, Rev. Sci. Instrum., 1964, 35, 1031–1034 CrossRef.
- E. H. Trinh, Rev. Sci. Instrum., 1985, 56, 2059–2065 CrossRef CAS.
- C. J. Hsu, J. Acoust. Soc. Am., 1986, 79, 1335–1338 CrossRef.
- E. Welter and B. Neidhart, Fresenius' J. Anal. Chem., 1997, 357, 345–350 CrossRef CAS.
- E. H. Trinh and C. J. Hsu, J. Acoust. Soc. Am., 1986, 80, 1757–1761 CrossRef CAS.
- E. H. Trinh, P. L. Marston and J. L. Robey, J. Colloid Interface Sci., 1988, 124, 95–103 CrossRef CAS.
- Y. Maruyama and K. Hasegawa, RSC Adv., 2020, 10, 1870–1877 RSC.
- A. L. Yarin, G. Brenn, O. Kastner and C. Tropea, Phys. Fluids, 2002, 14, 2289–2298 CrossRef CAS.
- H. Schiffter and G. Lee, J. Pharm. Sci., 2007, 96, 2284–2295 CrossRef CAS.
- H. Abdullahi, C. L. Burcham and T. Vetter, Chem. Eng. Sci., 2020, 224, 115713 CrossRef CAS.
- H.-L. Cao, D.-C. Yin, Y.-Z. Guo, X.-L. Ma, J. He, W.-H. Guo, X.-Z. Xie and B.-R. Zhou, J. Acoust. Soc. Am., 2012, 131, 3164–3172 CrossRef.
- J. Leiterer, W. Leitenberger, F. Emmerling, A. F. Thünemann and U. Panne, J. Appl. Crystallogr., 2006, 39, 771–773 CrossRef CAS.
- J. Leiterer, F. Delißen, F. Emmerling, A. F. Thünemann and U. Panne, Anal. Bioanal. Chem., 2008, 391, 1221–1228 CrossRef CAS.
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342–12347 CrossRef CAS.
- J. Leiterer, F. Emmerling, U. Panne, W. Christen and K. Rademann, Langmuir, 2008, 24, 7970–7978 CrossRef CAS.
- M. Klimakow, J. Leiterer, J. Kneipp, E. Rössler, U. Panne, K. Rademann and F. Emmerling, Langmuir, 2010, 26, 11233–11237 CrossRef CAS.
- T. Gnutzmann, Y. Nguyen Thi, K. Rademann and F. Emmerling, Cryst. Growth Des., 2014, 14, 6445–6450 CrossRef CAS.
- Y. Nguyen Thi, K. Rademann and F. Emmerling, CrystEngComm, 2015, 17, 9029–9036 RSC.
- A. Y. Lee, I. S. Lee, S. S. Dette, J. Boerner and A. S. Myerson, J. Am. Chem. Soc., 2005, 127, 14982–14983 CrossRef CAS.
- K. Kim, A. Centrone, T. A. Hatton and A. S. Myerson, CrystEngComm, 2011, 13, 1127–1131 RSC.
- B. D. Hamilton, M. A. Hillmyer and M. D. Ward, Cryst. Growth Des., 2008, 8, 3368–3375 CrossRef CAS.
- X. Yang and A. S. Myerson, CrystEngComm, 2015, 17, 723–728 RSC.
- A. B. M. Buanz and S. Gaisford, Cryst. Growth Des., 2017, 17, 1245–1250 CrossRef CAS.
- G. L. Perlovich, L. K. Hansen and A. Bauer-Brandl, J. Therm. Anal. Calorim., 2001, 66, 699–715 CrossRef CAS.
- Y. Iitaka and IUCr, Acta Crystallogr., 1960, 13, 35–45 CrossRef CAS.
- G. Albrecht and R. B. Corey, J. Am. Chem. Soc., 1939, 61, 1087–1103 CrossRef CAS.
- Y. Iitaka, Acta Crystallogr., 1961, 14, 1–10 CrossRef CAS.
- G. Albrecht and R. B. Corey, The Crystal Structure of Glycine, J. Am. Chem. Soc., 1939, 61, 1087 CrossRef CAS.
- I. Weissbuch, V. Y. Torbeev, L. Leiserowitz and M. Lahav, Angew. Chem., Int. Ed., 2005, 44, 3226–3229 CrossRef CAS.
- W. Tang, H. Mo, M. Zhang, J. Gong, J. Wang and T. Li, Cryst. Growth Des., 2017, 17, 5028–5033 CrossRef CAS.
- K. Renuka Devi, A. Raja and K. Srinivasan, Ultrason. Sonochem., 2015, 24, 107–113 CrossRef CAS.
- M. Louhi-Kultanen, M. Karjalainen, J. Rantanen, M. Huhtanen and J. Kallas, Int. J. Pharm., 2006, 320, 23–29 CrossRef CAS.
- H. U. Rodríguez Vera, F. Baillon, F. Espitalier, P. Accart and O. Louisnard, Ultrason. Sonochem., 2019, 58, 104671 CrossRef.
- V. Bhamidi, S. H. Lee, G. He, P. S. Chow, R. B. H. Tan, C. F. Zukoski and P. J. A. Kenis, Cryst. Growth Des., 2015, 15, 3299–3306 CrossRef CAS.
- W. El Bazi, C. Porte, I. Mabille and J. L. Havet, J. Cryst. Growth, 2017, 475, 232–238 CrossRef CAS.
- W. E. Ranz and W. Marshall, Chem. Eng. Prog., 1952, 48, 141 CAS.
- D. H. Charlesworth and W. R. Marshall, AIChE J., 1960, 6, 9–23 CrossRef CAS.
- D. E. Walton, Drying Technol., 2000, 18, 1943–1986 CrossRef CAS.
- A. Y. Lee, I. S. Lee and A. S. Myerson, Chem. Eng. Technol., 2006, 29, 281–285 CrossRef CAS.
- S. K. P. Poornachary, J. V. Parambil, P. S. Chow, R. B. H. Tan and J. Y. Y. Heng, Cryst. Growth Des., 2013, 13, 1180–1186 CrossRef CAS.
- M. Boyes, A. Alieva, J. Tong, V. Nagyte, M. Melle-Franco, T. Vetter and C. Casiraghi, ACS Nano, 2020, 14, 10394–10401 CrossRef CAS.
- M. Bienfait and R. Kern, Bull. Soc. Fr. Mineral. Cristallogr., 1964, 87, 604 CAS.
- K. Hasegawa, Y. Abe, A. Fujiwara, Y. Yamamoto and K. Aoki, Microgravity Sci. Technol., 2008, 20, 261–264 CrossRef.
- Y. Yamamoto, Y. Abe, A. Fujiwara, K. Hasegawa and K. Aoki, Microgravity Sci. Technol., 2008, 20, 277–280 CrossRef.
- K. Hasegawa, Y. Abe and A. Goda, npj Microgravity, 2016, 2, 1–5 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce00856g |
| This journal is © The Royal Society of Chemistry 2020 |