
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein crystal occurrence domains in selective protein crystallisation for bio-separation
Xiaoyu
Li
a,
Wenqian
Chen
 a,
Huaiyu
Yang
a,
Zhongqiang
Yang
b and
Jerry Y. Y.
Heng
*a
a,
Huaiyu
Yang
a,
Zhongqiang
Yang
b and
Jerry Y. Y.
Heng
*a
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: jerry.heng@imperial.ac.uk
bDepartment of Chemistry, Tsinghua University, Beijing 100084, PR China
First published on 2nd June 2020
Abstract
Bio-separation is a key bottleneck in the manufacture of biopharmaceuticals. In this work, we report experimental evidence of direct selective protein crystallisation from a binary protein mixture solution. Lysozyme–thaumatin mixtures with a wide protein composition range (0–100 mg mL−1, respectively) were tested under the same crystallisation cocktail conditions using the hanging-drop vapour-diffusion (HDVD) crystallisation method. This work demonstrates the selectivity of crystallisation from a model binary protein mixture and four crystal occurrence domains were determined as the operation windows of selective crystallisation of the target protein: 1) an unsaturated region with no crystal formation, 2 & 3) target regions with only a single type of protein crystals (lysozyme crystals only or thaumatin crystals only) and 4) a mixture region which have a mixture of both types of protein. This study demonstrates that protein crystallisation is not only applicable to high-purity protein solutions and emphasizes the vital impacts of the presence of protein impurities in the process of target protein crystallisation. The study concludes that protein crystallisation is a feasible approach to separate a target protein from a complex mixture environment which can be achieved by manipulating the crystallisation operation conditions such as mixture composition, precipitant concentration, and operation time.
1. Introduction
Therapeutic proteins such as monoclonal antibodies have attracted major interest in the current pharmaceutical market. Progress in biotechnology in the past decades has resulted in the approval of 285 distinct active biopharmaceutical products which are predominated by monoclonal antibodies and the sales of these products continue to grow reaching a total revenue of $188 billion alone in 2017.1,2 Nevertheless, downstream separation processes have now become the bottleneck of cost-effective manufacturing of these protein-based products. Up to 80% of manufacturing costs are attributed to the downstream purification processes which mainly rely on multi-step chromatography technology.3 Meanwhile, advances in upstream processes such as cell technology have led to higher titres of secreted proteins which are now beyond 5 g L−1, creating a greater challenge for protein A chromatography as an effective purification step.4,5 Researchers are seeking alternative technologies to replace or partially substitute conventional chromatography steps with more cost effective, rapid, and robust downstream separation processes. Various alternative separation technologies have been investigated,6 including solvent extraction,7–10 membrane-based methods,11–14 precipitation15–17 and crystallisation.17,18 Crystallisation serves as a vital purification process for both inorganic and organic small molecule products in numerous conventional chemical industries. Moreover, crystalline proteins are believed to have higher purity and stability which can benefit formulation, storage, and drug delivery steps.19–22 Though most of the studies of protein crystallisation have focused on obtaining large single crystals for biomolecular structural studies, in the past few decades, researchers have demonstrated that protein crystallisation is also a feasible and scalable purification and isolation technology for downstream bio-separation. Judge et al. demonstrated the feasibility of ovalbumin recovery via bulk crystallisation in a 1 L stirred batch crystalliser.23 Jacobsen et al. were able to obtain microbial lipase crystals from clarified concentrated fermentation broths.24 Hekmat's group demonstrated that crystallisation is a scalable process by successfully transferring the crystallisation of antigen-binding fragment FabC225 from 10 μL vapour diffusion experiments to a 100 mL batch crystallisation process.25 A few continuous crystallisation platforms have also been developed on a lab-scale to demonstrate the potential of adapting crystallisation in a continuous manufacturing fashion, including stirred classified product removal tanks,26 tubular plug-flow crystallisers,27 continuous crystallisers with oscillatory flow,28,29 and meso-oscillatory flow crystallisers.30In most cases, the focus has been on the possibility of crystallisation from protein solution with high purity. Very limited cases of selective crystallisation were described. Ghatak and Ghatak reported selective crystallisation from a protein mixture which was realised by using specific additives (salts) accompanied with charged nano-patterned surfaces.31 A previous study by our group reported that mesoporous nucleants with a specific pore size, ordered structure, and narrow pore size distribution are able to promote selective protein crystallisation via controlling the nucleation process.32,33 Still, in all the cases mentioned above, the concentration range reported was at a relatively low level which may not be applicable for fast purification purposes. Judge et al. studied protein crystallisation in the presence of protein impurities,23,34 and preferential separation of lysozyme from lysozyme–ovalbumin mixtures was achieved by seeded batch crystallisation.35 Nonetheless, in the above cases, only a limited range of mixture composition was covered and only one of the proteins in the solution could be crystallised out under the solution conditions. Systematic knowledge of the crystallisation behaviour of the target protein from the mixture is still absent. Little information is available on introducing seeds/heterogeneous nucleants and future scale-up to proceed to selectively crystallise a target protein from a more complex solution. The reported preferential crystallisation cases only focused on specific scenarios, either with ultralow protein concentration or with a very limited range of protein composition of the mixture.
In this study, we provide the first direct experimental evidence that bio-separation is practical via crystallisation from a binary protein mixture where both proteins are supersaturated and crystallisable under identical crystallisation conditions.
2. Methodology
Materials
Lysozyme from chicken egg white (≥90%), thaumatin from Thaumatococcus daniellii, 1,4-piperazinediethanesulfonic acid (PIPES) (≥99%), potassium sodium tartrate tetrahydrate (99%), sodium chloride (≥ 99%), and anhydrous sodium acetate (≥ 99%) were purchased from Sigma-Aldrich (Dorset, UK). Sodium hydroxide (>98.5%) and hydrochloric acid (37% w/w) were purchased from VWR (Lutterworth, UK). Deionised water was obtained using a PURELAB Chorus 1 water purification system (ELGA LabWater). All chemicals were used as received without further purification.Protein crystallisation
The hanging-drop vapour-diffusion (HDVD) crystallisation method was used in this study. Sodium chloride (NaCl) precipitant solutions (5 mg mL−1 to 100 mg mL−1) were prepared by dissolving NaCl in 0.1 M sodium acetate (NaAc) buffer, pH 4.8. Potassium sodium tartrate tetrahydrate precipitant solutions (28 mg mL−1 to 560 mg mL−1) were prepared by dissolving potassium sodium tartrate tetrahydrate in 0.1 M PIPES buffer, pH 6.8. All precipitant solutions were filtered through 0.22 μm Millex-GS syringe filter units (Millipore) before crystallisation trials. A protein (lysozyme/thaumatin) solution was prepared by dissolving the protein powder into the buffer solution which was the same buffer used for precipitant preparation. The protein concentration in the solution was determined using a Nanodrop Onec microvolume UV-vis spectrophotometer (Thermo Scientific™) at 280 nm using a mass extinction coefficient (ε1%) of 26.4 L g−1 cm−1 for lysozyme and 12.7 L gm−1 cm−1 for thaumatin.36 The protein solution with an expected concentration higher than 100 mg mL−1 was diluted before measuring using a UV-vis spectrophotometer. Lysozyme–thaumatin mixtures were prepared by mixing the lysozyme solution and thaumatin solution with determined concentrations. All protein solutions were filtered through a 0.22 μm syringe filter (VWR) before crystallisation trials.HDVD crystallisation experiments were conducted in a 24-well VDX™ plate with sealant (Hampton Research). Each well was filled with 500 μL of precipitant solution as a reservoir solution. A 4 μL droplet with equal volumes of protein solution and precipitant solution (the same as the reservoir solution) was deposited on a borosilicate cover glass (VWR). The cover glass with the protein–precipitant droplet was carefully inverted and sealed onto the well filled with the reservoir solution. The crystallisation plates were then placed in an incubator (20 °C ± 0.5 °C). The plates were observed using a CX41 optical microscope (Olympus) regularly after they were set-up. Microscopy images were captured using a GXCAM HICHROME-MET camera (GT Vision).
In this study, we determined the crystallisation results based on the observations of the droplets under the optical microscope. The droplets were categorised into (1) no crystal, (2) precipitation, (3) only lysozyme crystal(s), (4) only thaumatin crystal(s) and (5) both lysozyme crystal(s) and thaumatin crystal(s). Due to the limitation of the maximum amplification of the optical microscope, only crystals larger than about 5 μm can be observed and the shape of the crystal can be recognised, i.e. a result was marked as ‘(5) both types of crystals’ providing that at least one lysozyme crystal larger than 5 μm and at least one thaumatin crystal larger than 5 μm were observed in the droplet at the same time. Considering the inherently poor reproducibility of protein crystallisation and the numerous crystallisation conditions tested, it was not feasible to examine every single crystal using characterisation techniques such as single crystal X-ray diffraction which can be low-throughput and excessively time-consuming to provide representative analysis for the whole sample population. Usage of microscopy images of the droplets is a fast and robust way to screen and track the crystallisation conditions in a limited time and the results are real-time, relatively consistent and representative since all the droplets were examined rather than single crystals being sampled and tested off-line. To enhance the confidence level of our results, each condition was repeated at least 12 times to mitigate the inherently poor reproducibility of protein crystallisation due to the stochastic nucleation event. In the first run, experiments were repeated 12 times in 3 separate plates. If the same results were obtained from all the replicates, the condition was only repeated 12 times. In the second run, another 12 replicates of each condition were repeated. If 22 replicates have given the same results, providing about a 95% confidence level, the condition would be only repeated 24 times. For the remaining conditions, more replicates would be tested up to 144 times. In general, conditions with a protein concentration lower than 50 mg mL−1 were repeated for 48 to 144 droplets. Considering that the higher degree of supersaturation of the proteins would reduce the fluctuation and uncertainties of the crystallisation results, the conditions with higher protein concentration were repeated at least 12 to 24 times to reassure the accuracy of the results.
3. Results and discussion
Determination of protein crystallisation conditions in HDVD crystallisation experiments
The results in Table 1 show that lysozyme and thaumatin were able to be crystallised individually from their single-protein solutions under crystallisation conditions in which potassium sodium tartrate tetrahydrate was used as a precipitant. Yet, under the conditions investigated in this study, no thaumatin crystal was obtained by using sodium chloride as a precipitant. The droplets remained clear or only precipitation was observed in the period of observation.| Precipitant solution | |||
|---|---|---|---|
| Protein type | Temperature | Sodium chloride in 0.1 M sodium acetate buffer, pH 4.8 | Potassium sodium tartrate tetrahydrate in 0.1 M PIPES buffer, pH 6.8 |
| Lysozyme | 4 °C | Tetragonal crystal(s) | Tetragonal crystal(s) |
| 20 °C | Tetragonal crystal(s) | Tetragonal crystal(s) | |
| Thaumatin | 4 °C | Clear/precipitation | Bipyramidal crystal(s) |
| 20 °C | Clear/precipitation | Bipyramidal crystal(s) | |
Fig. 1 shows illustrative images of the crystallisation droplets crystallised using potassium sodium tartrate tetrahydrate as a precipitant. The shapes of the thaumatin crystals and lysozyme crystals grown using this precipitant were different. In the range of concentrations investigated in this study, tetragonal lysozyme crystals (Fig. 1A) were obtained from the lysozyme solution while bipyramidal thaumatin crystals (Fig. 1B) were obtained from the thaumatin solution. These two types of protein crystals can be distinguished under the optical microscope by their crystal shapes. Fig. 1C was captured after Izit Crystal Dye (Hampton Research, US) was added to the droplet. The dye test allows a quick test to see whether the crystals obtained were protein crystals or salt crystals. Protein crystals can be stained via dye molecule diffusion into their solvent channels since protein crystals are less packed compared to small molecule crystals like the salts in the buffer solutions. In Fig. 1C, both types of crystals were stained blue and thus they were protein crystals. The bipyramidal thaumatin crystals possess a deeper blue colour while the tetragonal lysozyme crystals possess a lighter blue colour which may due to the different crystal mosaicity. Fig. 1C reveals that both lysozyme crystals and thaumatin crystals can be crystallised out from a lysozyme–thaumatin mixture using the tartrate salt as the precipitant while still possessing distinct crystal shapes. The thaumatin crystals remained as bipyramidal in shape in the mixture. The lysozyme crystals were tetragonal crystals though defects might be detected under certain conditions. Therefore, further experiments, where preferential crystallisation from a lysozyme–thaumatin binary protein mixture was attempted, were conducted by using potassium sodium tartrate as the precipitant rather than sodium chloride.
 | ||
| Fig. 1 Representative images of lysozyme crystals and thaumatin crystals crystallised with a 141 mg mL−1 potassium sodium tartrate tetrahydrate precipitant solution. A: 50 mg mL−1 lysozyme; only tetragonal lysozyme crystals in the droplet. B: 50 mg mL−1 thaumatin; only bipyramidal thaumatin crystals in the droplet. C: 50 mg mL−1 thaumatin + 50 mg mL−1 lysozyme; both lysozyme and thaumatin crystals in the droplet and this photo was captured after Izit Crystal Dye (Hampton Research, US) was added to the droplet. All the transparent crystals absorbed dye molecules and turned blue afterward. The bipyramidal thaumatin crystals were darker compared to the lysozyme crystals. | ||
Protein crystallisation from a binary-protein mixture
The HDVD crystallisation method was used in this stage of crystallisation condition screening to investigate the operating window for crystallisation from the model mixture. An initial precipitant concentration of 141 mg mL−1 potassium sodium tartrate tetrahydrate in the buffer solution and a temperature of 20 °C were kept the same for the whole set of experiments. The crystallisation plates were observed regularly and for simplicity of the diagram, the results listed only included 6 hours, 24 hours, and 48 hours after the plates were set up.Four crystal occurrence domains were observed from the crystallisation droplets as shown in Fig. 2: 1) a clear domain with no crystal formation in which no crystal >5 μm was detected (crystals smaller than 5 μm were not detectable using the optical microscope used in this study), 2 & 3) target domains with only one type of protein crystals (lysozyme crystals or thaumatin crystals only) and 4) a mixture domain with a mixture of both types of protein crystals. Fig. 2 shows the evolution of the crystallisation domains over time. The results also indicate that one type of protein would be crystallised out first from the mixture, followed by the other protein later, and the sequence was dependent on the mixture composition. This suggests that within a certain period, harvesting a single type of protein crystals from the mixture for separation purposes is possible. Comparing the crystallisation results from the single protein solution and the results from the protein mixture, Fig. 2 also reveals that the presence of another protein in the mixture would normally hinder the crystallisation process of the target protein. For instance, after 6 hours, crystals were observed in the droplet containing 50 mg mL−1 lysozyme, but it remained clear after the same period of time when more than 20 mg mL−1 of thaumatin existed on top of the lysozyme in the droplet.
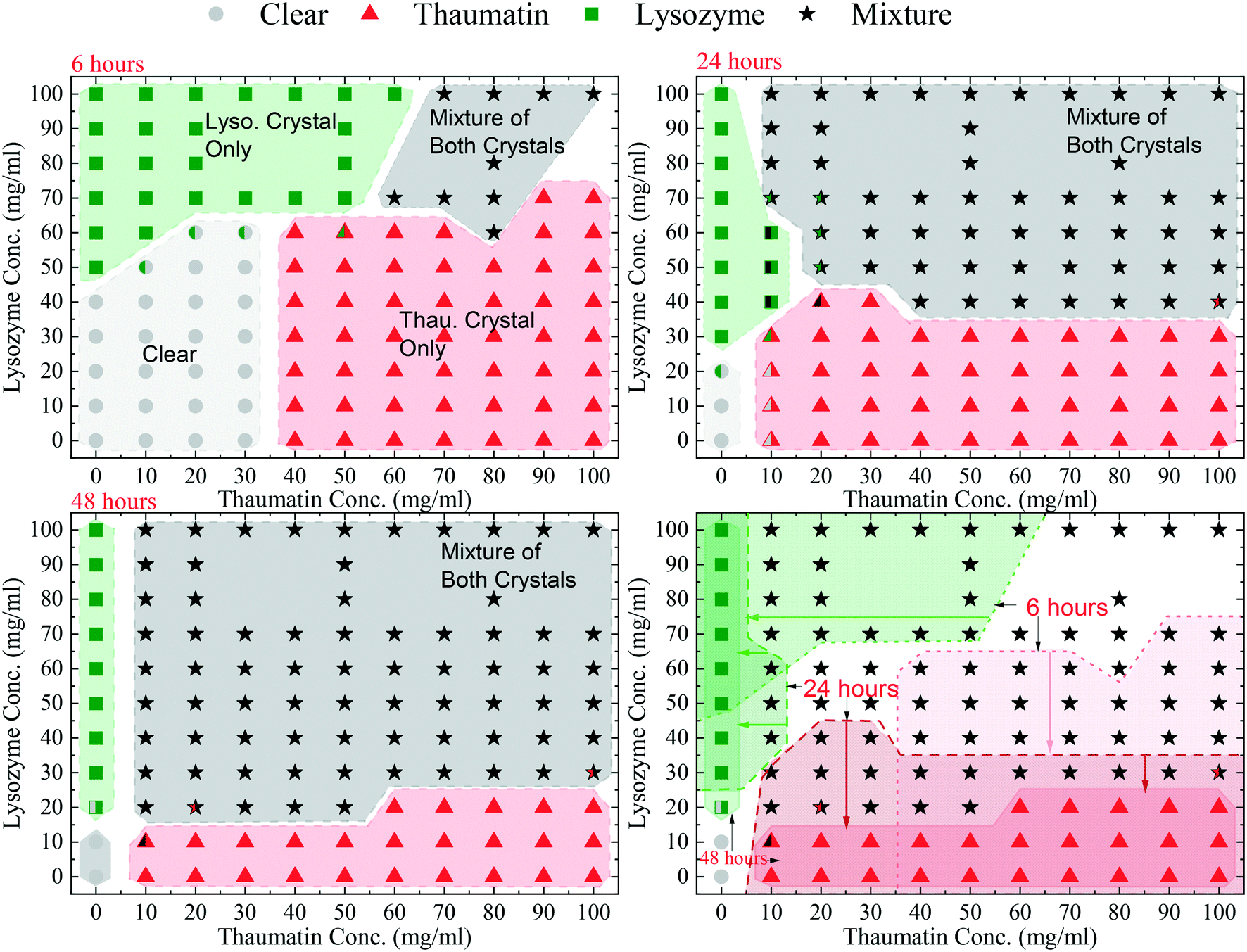 | ||
| Fig. 2 A schematic illustration of the protein crystal domains of crystallisation from lysozyme–thaumatin mixtures in this study. The time denotes the period from when the crystallisation plates were set up. The closed symbols with only one colour represent 100% of the droplets in the study having the stated results. The symbols with two colours followed the results with the highest possibility. The circle symbols (grey) represent the experimental results where the droplets remained clear with no crystals observed; the square symbols (green) represent the experimental results where only lysozyme crystals were observed in the droplet; the triangle symbols (red) represent the results where only thaumatin crystals were observed in the droplet; the star symbols (black) represent the results where both lysozyme crystals and thaumatin crystals were observed in the same droplet. The shaded regions in the figure only serve as a visual guide to highlight the domains where different results were observed. | ||
After 6 hours, a clear droplet domain existed in the low protein concentration range with a lysozyme concentration ≤40 mg mL−1 and thaumatin concentration ≤30 mg mL−1. In this region, the degree of supersaturation was relatively low and thus the crystallisation process was slow. However, after 2 weeks, crystals were observed in this region. Additionally, this clear region reached the range of lysozyme concentration of 50–60 mg mL−1 with a thaumatin concentration ≤30 mg mL−1. In this extended part, though lysozyme crystals were observed from a pure lysozyme solution with the same initial lysozyme concentration, no lysozyme crystallised out as thaumatin was present in the mixture. Furthermore, when the lysozyme concentration was 50–60 mg mL−1 with a thaumatin concentration >30 mg mL−1, there was still no lysozyme crystals observed. Lysozyme crystallisation was inhibited within 6 hours due to thaumatin in the mixture. A similar tendency was observed when the initial concentration of thaumatin was 40–60 mg mL−1; when a higher amount of lysozyme is present in the mixture, no thaumatin crystals were observed after 6 hours while thaumatin crystals were observed in droplets with the same initial thaumatin concentrations. Thaumatin crystallisation was inhibited due to high amount of lysozyme in the solution. With a higher initial lysozyme concentration (>70 mg mL−1), lysozyme always crystallised out regardless of the thaumatin concentration in the mixture in the range studied in this work. In the mixture containing both high concentrations of lysozyme and thaumatin, a mixture of both types of protein crystals was observed.
After 24 hours, the mixture domain expanded, and protein crystals were observed in the regions with lower initial protein concentrations. The thaumatin crystal only domain still existed for all mixture droplets with an initial lysozyme concentration no more than 30 mg mL−1. However, the domain with only lysozyme crystals almost retreated to the region where no thaumatin was added from the beginning, i.e. a pure lysozyme solution.
After 48 hours, the clear region shrank dramatically to the region where the initial lysozyme concentration was not greater than 10 mg mL−1. So did the lysozyme crystal only domain and thaumatin crystal only domain. There was no domain with only lysozyme crystals in all the mixture compositions tested. Additionally, the domain with a mixture of both protein crystals expanded further. Apart from the experiments with 20 mg mL−1 lysozyme initially and a thaumatin concentration higher than 50 mg mL−1, in which only thaumatin crystals were observed after 48 hours, all droplets with a lysozyme concentration higher than 10 mg mL−1 had a mixture of both types of protein crystals.
Fig. 3 shows a set of representative microscopy photos of the crystallisation droplets 24 hours after the experiments started. Both the crystal size and crystal number of thaumatin crystals were reduced dramatically as the lysozyme concentration increased. Nevertheless, when the initial lysozyme concentration was higher than 80 mg mL−1, the number of thaumatin crystals increased. This increase may due to that lysozyme crystallisation was faster resulting from the high degree of supersaturation. Consequently, free lysozyme in the solution decreased and thus thaumatin crystallisation was less affected by lysozyme in the mixture. Another assumption is based on the nature of protein crystals, that is, protein crystals retain a relatively high solvent content compared to small molecule crystals.37 Therefore, as more protein crystals formed from the mixture, less solvent was left in the mixture and the thaumatin concentration might have increased accordingly.
 | ||
| Fig. 3 Representative images of lysozyme crystals and thaumatin crystals obtained under the crystallisation conditions after 24 hours, starting with the same thaumatin concentration (20 mg mL−1) but with different lysozyme concentrations (A: 0 mg mL−1; B: 10 mg mL−1; C: 30 mg mL−1; D: 50 mg mL−1; E: 70 mg mL−1; F: 90 mg mL−1). A–C only thaumatin crystallised. D–F a mixture of lysozyme and thaumatin crystallised which were circled out in D and E. | ||
Furthermore, as shown in Fig. 2, when the initial thaumatin concentration was 10–20 mg mL−1 with a moderate lysozyme concentration of 30–70 mg mL−1, the reproducibility of the HDVD crystallisation results was the worst. The success chance of crystallisation of each type of protein was not 100% and thus led to an overlap of the clear, target, and mixture domains. As shown in Fig. 3(D and E), in this composition region, due to the presence of lysozyme in the solution, both the crystal size and crystal number of thaumatin crystals were reduced significantly. Lysozyme crystallisation dominated in this region and there were chances that thaumatin did not crystallise when the initial thaumatin concentration was low.
In general, as shown in Fig. 2, there was always one type of protein crystallised out from the solution first, followed by the other protein crystals, and the sequence was decided by the composition of the mixture, i.e. degrees of supersaturation of the proteins. When enough time was provided, both lysozyme and thaumatin would crystallise out from the mixture. This suggests that when the operation time was controlled properly, bio-separation can be achieved via preferential protein crystallisation even if protein impurities in the mixture are supersaturated and able to be crystallised out under certain crystallisation conditions.
We also suggest that the presence of another protein, acting as an impurity in the solution, will slow down the crystallisation process of both the target proteins and the impurity protein itself. Still, the crystallisation process was not inhibited completely. Additionally, in the model system studied in this work, we did not find evidence that the existing lysozyme protein crystal could stimulate thaumatin crystallisation or vice versa. A strategy using previously obtained target protein crystals as seeds can facilitate protein crystallisation from mixtures for bioseparation.
4. Conclusion
In this study, we successfully demonstrated preferential protein crystallisation using a lysozyme–thaumatin binary mixture as the model. Four domains were identified: 1) a clear domain, 2 & 3) target domains with only one type of protein crystals (lysozyme crystals or thaumatin crystals only) and 4) a mixture domain with a mixture of both types of protein crystals. The size of these domains depended on the mixture composition and shifted with time. There was no direct evidence in this study that protein solubility is changed due to the existence of protein impurities. In the model binary protein mixture, the presence of another protein impurity can slow down the crystallisation process of the target protein. This implies that crystallisation kinetics plays a key role for selective crystallisation from the mixture. Furthermore, as the kinetics of crystallisation can also be manipulated by the presence of protein impurities, this highlights the importance of further investigation of the effect of protein impurities in a more complex crystallisation system. This work demonstrates that protein crystallisation is not only applicable to high-purity protein solutions but also a feasible approach to separate a target protein from a more complex mixture environment, even for protein mixtures with both proteins at the supersaturated state. This work also provides a working model system with essential foundation knowledge for future work on protein crystallisation for bioseparation, such as scale-up crystallisation processes, seeding, and crystallisation facilitated by heterogeneous nucleants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Seeding and Continuous Biopharmaceutical Crystallisation (SCoBiC) Project (EP/N015916/1) funded by the EPSRC.References
- G. Walsh, Biopharmaceutical benchmarks 2018, Nat. Biotechnol., 2018, 36(12), 1136–1145 CrossRef CAS PubMed.
- L. M. B. Intelligence, http://www.lamerie.com.
- J. A. V. Costa, H. Treichel, L. O. Santos and V. G. Martins, Chapter 16 - Solid-State Fermentation for the Production of Biosurfactants and Their Applications, in Current Developments in Biotechnology and Bioengineering, ed. A. Pandey, C. Larroche and C. R. Soccol, Elsevier, 2018, pp. 357–372 Search PubMed.
- V. Natarajan and A. L. Zydney, Protein A Chromatography at High Titers, Biotechnol. Bioeng., 2013, 110(9), 2445–2451 CrossRef CAS PubMed.
- A. A. Shukla and J. Thommes, Recent advances in large-scale production of monoclonal antibodies and related proteins, Trends Biotechnol., 2010, 28(5), 253–261 CrossRef CAS PubMed.
- J. Thommes and M. Etzel, Alternatives to chromatographic separations, Biotechnol. Prog., 2007, 23(1), 42–45 CrossRef PubMed.
- G. A. Krei and H. Hustedt, Extraction of enzymes by reverse micelles, Chem. Eng. Sci., 1992, 47(1), 99–111 CrossRef CAS.
- R. Hu, X. J. Feng, P. Chen, M. Fu, H. Chen, L. Guo and B. F. Liu, Rapid, highly efficient extraction and purification of membrane proteins using a microfluidic continuous-flow based aqueous two-phase system, J. Chromatogr. A, 2011, 1218(1), 171–177 CrossRef CAS PubMed.
- S. Y. Lee, I. Khoiroh, C. W. Ooi, T. C. Ling and P. L. Show, Recent Advances in Protein Extraction Using Ionic Liquid-based Aqueous Two-phase Systems, Sep. Purif. Rev., 2017, 46(4), 291–304 CrossRef CAS.
- P. A. J. Rosa, I. F. Ferreira, A. M. Azevedo and M. R. Aires-Barros, Aqueous two-phase systems: A viable platform in the manufacturing of biopharmaceuticals, J. Chromatogr. A, 2010, 1217(16), 2296–2305 CrossRef CAS PubMed.
- A. Saxena, B. P. Tripathi, M. Kumar and V. K. Shahi, Membrane-based techniques for the separation and purification of proteins: An overview, Adv. Colloid Interface Sci., 2009, 145(1–2), 1–22 CrossRef CAS PubMed.
- C.-C. Ho, Chapter 7 - Membranes for Bioseparations, in Bioprocessing for Value-Added Products from Renewable Resources, ed. S.-T. Yang, Elsevier, Amsterdam, 2007, pp. 163–183 Search PubMed.
- A. L. Zydney and R. van Reis, Bioseparations—Membrane Processes☆, in Reference Module in Biomedical Sciences, Elsevier, 2016 Search PubMed.
- V. Orr, L. Zhong, M. Moo-Young and C. P. Chou, Recent advances in bioprocessing application of membrane chromatography, Biotechnol. Adv., 2013, 31(4), 450–465 CrossRef CAS.
- F. W. F. Wong, A. B. Ariff and D. C. Stuckey, Downstream protein separation by surfactant precipitation: a review, Crit. Rev. Biotechnol., 2018, 38(1), 31–46 CrossRef CAS PubMed.
- F. Hilbrig and R. Freitag, Protein purification by affinity precipitation, J. Chromatogr., B, 2003, 790(1–2), 79–90 CrossRef CAS.
- R. dos Santos, A. L. Carvalho and A. C. A. Roque, Renaissance of protein crystallization and precipitation in biopharmaceuticals purification, Biotechnol. Adv., 2017, 35(1), 41–50 CrossRef CAS PubMed.
- D. Hekmat, Large-scale crystallization of proteins for purification and formulation, Bioprocess Biosyst. Eng., 2015, 38(7), 1209–1231 CrossRef CAS PubMed.
- S. K. Basu, C. P. Govardhan, C. W. Jung and A. L. Margolin, Protein crystals for the delivery of biopharmaceuticals, Expert Opin. Biol. Ther., 2004, 4(3), 301–317 CrossRef CAS.
- R. G. Harrison, P. Todd, S. R. Rudge and D. P. Petrides, Bioseparations Science and Engineering, Oxford University Press, 2nd edn, 2015 Search PubMed.
- M. X. Yang, B. Shenoy, M. Disttler, R. Patel, M. McGrath, S. Pechenov and A. L. Margolin, Crystalline monoclonal antibodies for subcutaneous delivery, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(12), 6934–6939 CrossRef CAS.
- S. Stolnik and K. Shakesheff, Formulations for delivery of therapeutic proteins, Biotechnol. Lett., 2009, 31(1), 1–11 CrossRef CAS PubMed.
- R. A. Judge, M. R. Johns and E. T. White, Protein purification by bulk crystallization: The recovery of ovalbumin, Biotechnol. Bioeng., 1995, 48(4), 316–323 CrossRef CAS PubMed.
- C. Jacobsen, J. Garside and M. Hoare, Nucleation and growth of microbial lipase crystals from clarified concentrated fermentation broths, Biotechnol. Bioeng., 1998, 57(6), 666–675 CrossRef CAS.
- D. Hebel, S. Huber, B. Stanislawski and D. Hekmat, Stirred batch crystallization of a therapeutic antibody fragment, J. Biotechnol., 2013, 166(4), 206–211 CrossRef CAS PubMed.
- D. Hekmat, M. Huber, C. Lohse, N. von den Eichen and D. Weuster-Botz, Continuous Crystallization of Proteins in a Stirred Classified Product Removal Tank with a Tubular Reactor in Bypass, Cryst. Growth Des., 2017, 17(8), 4162–4169 CrossRef CAS.
- P. Neugebauer and J. G. Khinast, Continuous Crystallization of Proteins in a Tubular Plug-Flow Crystallizer, Cryst. Growth Des., 2015, 15(3), 1089–1095 CrossRef CAS PubMed.
- H. Y. Yang, W. Q. Chen, P. Peczulis and J. Y. Y. Heng, Development and Workflow of a Continuous Protein Crystallization Process: A Case of Lysozyme, Cryst. Growth Des., 2019, 19(2), 983–991 CrossRef CAS.
- H. Y. Yang, P. Peczulis, P. Inguva, X. Y. Li and J. Y. Y. Heng, Continuous protein crystallisation platform and process: Case of lysozyme, Chem. Eng. Res. Des., 2018, 136, 529–535 CrossRef CAS.
- F. Castro, A. Ferreira, J. A. Teixeira and F. Rocha, Influence of Mixing Intensity on Lysozyme Crystallization in a Meso Oscillatory Flow Reactor, Cryst. Growth Des., 2018, 18, 5940–5946 CrossRef CAS.
- A. S. Ghatak, G. Rawal and A. Ghatak, Precipitant-Free Crystallization of Protein Molecules Induced by Incision on Substrate, Crystals, 2017, 7(8), 245 CrossRef CAS.
- U. V. Shah, D. R. Williams and J. Y. Y. Heng, Selective Crystallization of Proteins Using Engineered Nanonucleants, Cryst. Growth Des., 2012, 12(3), 1362–1369 CrossRef CAS.
- U. V. Shah, N. H. Jahn, S. Huang, Z. Yang, D. R. Williams and J. Y. Y. Heng, Crystallisation via novel 3D nanotemplates as a tool for protein purification and bio-separation, J. Cryst. Growth, 2017, 469, 42–47 CrossRef CAS.
- R. A. Judge, E. L. Forsythe and M. L. Pusey, The effect of protein impurities on lysozyme crystal growth, Biotechnol. Bioeng., 1998, 59(6), 776–785 CrossRef CAS.
- S. Maosoongnern, C. Flood, A. E. Flood and J. Ulrich, Crystallization of lysozyme from lysozyme – ovalbumin mixtures: Separation potential and crystal growth kinetics, J. Cryst. Growth, 2017, 469(Supplement C), 2–7 CrossRef CAS.
- N. Asherie, C. Ginsberg, S. Blass, A. Greenbaum and S. Knafo, Solubility of Thaumatin, Cryst. Growth Des., 2008, 8(6), 1815–1817 CrossRef CAS.
- B. W. Matthews, Solvent content of protein crystals, J. Mol. Biol., 1968, 33(2), 491–497 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |