
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymorphism and structural diversities of LiClO4–β-alanine ionic co-crystals†
Paulina H.
Marek
 *ab,
Grzegorz
Cichowicz
b,
Dorota M.
Osowicka
a,
Izabela D.
Madura
*a,
Łukasz
Dobrzycki
b,
Michał K.
Cyrański
b and
Arkadiusz
Ciesielski
b
*ab,
Grzegorz
Cichowicz
b,
Dorota M.
Osowicka
a,
Izabela D.
Madura
*a,
Łukasz
Dobrzycki
b,
Michał K.
Cyrański
b and
Arkadiusz
Ciesielski
b
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: pmarek@ch.pw.edu.pl; izabela@ch.pw.edu.pl
bUniversity of Warsaw, Faculty of Chemistry, Pasteura 1, 02-093 Warsaw, Poland
First published on 21st May 2020
Abstract
Three novel ionic co-crystals (ICCs) built from lithium perchlorate and β-alanine (LiClO4·βAla, LiClO4·2βAla-I, LiClO4·2βAla-II) were obtained and structurally characterized. Crystals with a twofold excess of amino acid, LiClO4·2βAla-I, were found to undergo a solvent-mediated phase transition, recrystallizing as the thermodynamically stable polymorph LiClO4·2βAla-II. The transition was characterized by a series of PXRD measurements, observations performed under a microscope with polarized light and DSC experiments. Both polymorphs were found to exhibit virtually the same square-grid topology of lithium–alanine coordination sheets, yet they differ in symmetry and geometrical parameters of the networks. In the LiClO4·βAla crystal structure, chain-like coordination polymers are formed. Responses to temperature change were determined for all three structures by performing a series of X-ray diffraction measurements in the 100–300 K range. Differences were elucidated with the help of a thermal tensor, which allowed us to identify the structural motifs most sensitive to temperature change.
Introduction
Ionic co-crystals (ICCs) are a class of multicomponent compounds formed by an organic, electrically neutral component and a salt.1 Since new strong ionic interactions are intentionally introduced into the system, species of new topology and thus properties can be formed. Structural modifications may alter properties such as thermal stability2 or dissolution rate, which is useful in many fields of the chemical industry.3 For example, urea-based ICCs were proven to be better soil fertilizers by improving the management of nitrogen and decreasing ammonia emission.4,5 One of the fastest developing fields of ionic co-crystal application is pharmacology. ICC formation is one of the methods that enable the enhancement of pharmaceutically important substance properties. The co-crystallization of APIs (active pharmaceutical ingredients) with salts, in general, leads to systems with boosted bioavailability6 or stability towards humidity.7 In some cases, the ionic component itself can be an approved API, as in the case of lithium cations, salts of which are used as mood stabilizers in psychiatry.8 The use of ICCs in such systems is aimed at improving the clinical performance of lithium therapeutics.9Moreover, lithium cations seem to be suitable four-coordinated nodes enabling the formation of topologically diverse ICCs, interesting for crystal engineering. Square, diamondoid, and zeolitic nets were reported when a non-equimolar ratio of components (lithium salt + amino acid) was applied.10 The various topologies formed by lithium-based ICCs were also attributed to specific properties of the investigated materials. Li+ based square-grid arrangements served as an example to illustrate the potential of hygroscopicity modification by ICC formation,11 whereas repeatable moieties in chain coordination polymers were found to be important in studies on spontaneous chiral resolution, observed in lithium halide–DL-amino acid (LiX·DL-aa) ionic co-crystals.12,13 Thus, understanding the tendencies in forming certain motifs and differences in Li-based ICC crystal structures might allow predicting the required crystal structure exhibiting the required properties.
In the Cambridge Structural Database14 (CSD ver. 5.40, February 2019), there are 36 lithium salt–amino acid ICC structures deposited. Our studies revealed that there are as many as 12 types of topologically distinguishable structural motifs driven by lithium nodes but they could be classified into 4 groups depending on the arrangement dimensionality (Fig. 1). Only one record contains an isolated 0-dimensional (0D) motif where a lithium cation is coordinated by two water and two alanine molecules.15 A 1D chain built from fused 6-membered rings can be found in 10 systems, and some of them have been extensively studied in terms of chiral recognition.12,13 In the structures deposited under refcodes ALUNEA (LiNO3·Gly)16 and HEFXEV (LiCl·GlyGly),17 a chain motif consisting of 8 and 4 membered rings is observed, while in the case of NEPWUC (LiBr·2Gly·H2O),18 a coordination polymer without ring motifs is formed. In this group, a 1D ribbon has been also found but it is represented by one crystal structure only (GARSUP, Li2SO4·βAla·H2O)19 where a combination of six-membered rings with additional interchain interactions through the anion can be distinguished (Fig. 1, in the middle). In 2D layers, 4 types of topological connections of ICCs are observed. The most commonly occurring is the square-grid topology (11 records), realized in both centrosymmetric (EVUWEY – LiNO3·2betaine, EVUWAU – LiNO3·2sarcosine)10 and non-centrosymmetric symmetry space groups (IFIVEB, IFIVIF, IFIVIF01 – lithium methoxybenzoate·L-Pro; IFIVOL, IFIVOL01, IFIVUR – lithium benzoate·L-Pro;11 MONDIE – lithium salicylate·L-Pro; MONDOK – lithium nicotinate·L-Pro;9 ROZTUW – LiNO3·2Gly20). However, three additional different connections are recognized in the ALGLYL – LiBr·L-AlaGly·2H2O (ref. 21) (five lithium nodes in a mesh), EVONAE22 and EVONAE01 (ref. 23) – Li2SO4·Gly (two nodes in a mesh) and UCIYOV – Li2CrO4·2Gly·H2O (ref. 24) (six nodes in a mesh) crystal structures. The 3D arrangements are represented by six records, where diamondoid and zeolite-like topologies consisting of 16, 24, or 32-membered rings can be distinguished.10 The diverse structural role of the anion should be noted, due to which the observed arrangements are electroneutral or positively charged. Unfortunately, too little data still prevent statistically justified correlation analysis taking into account the properties of anions, such as, for example, their basicity. Herein, we present novel ionic co-crystals composed of lithium perchlorate and β-alanine mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 molar ratios and scrutinize their topologies as well as the binding properties of the weakly basic perchlorate anion.
:1 and 1:2 molar ratios and scrutinize their topologies as well as the binding properties of the weakly basic perchlorate anion.
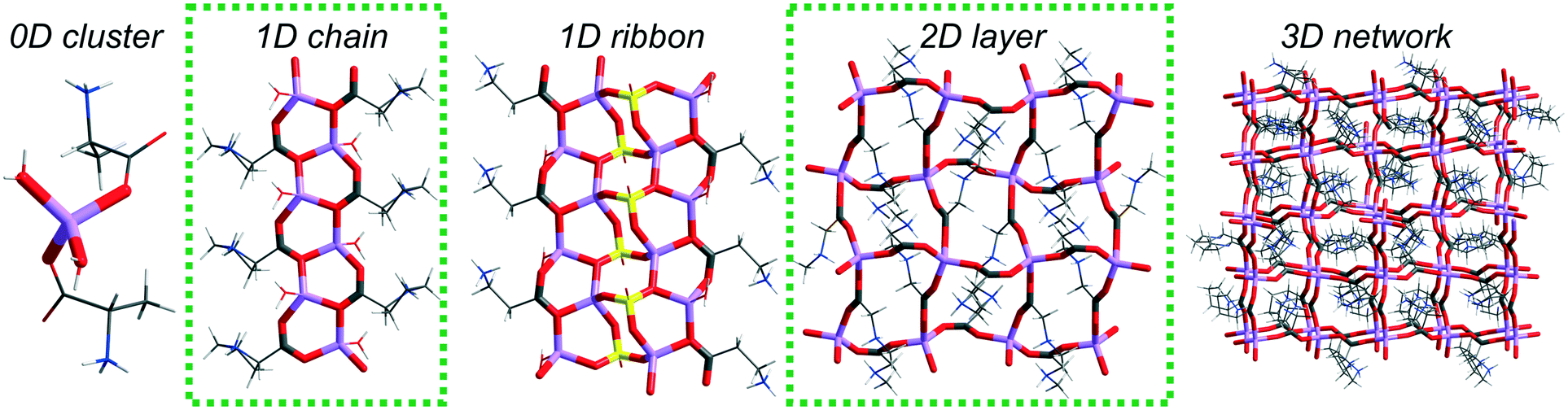 | ||
| Fig. 1 Diversity of different arrangements in LiX·aa ICCs. Examples from the CSD. Topologies of the newly obtained ICCs are marked with a green dotted frame. | ||
It is also important to note that the resulting chirality of the ICC can be related to the chirality of the amino acid, as in the cases described by Zaworotko and Lusi11 where the handedness of the amino acid was reflected in the chiral space group of the resulting ICC. However, in the case of ionic co-crystals based on achiral glycine, within search records, 3 crystal structures were centrosymmetric, 3 were polar (Pna21 space group), and one was in the Sohncke space group P212121. Since two non-centrosymmetric polymorphs of glycine have been reported under ambient conditions,25 affinity for formation of non-centrosymmetric crystal structures by this amino acid might be recognized. β-Alanine, which can be perceived as a homolog of glycine, crystallizes only in centrosymmetric crystal forms.26 Hence, we were interested in whether the formation of lithium ionic co-crystals could also, in this case, lead to non-centrosymmetric structures. The successfully obtained centrosymmetric and non-centrosymmetric crystals composed of lithium perchlorate and β-alanine are described in detail below, including the analysis of an unexpected, interesting solvent-mediated phase transition.
Experimental section
Co-crystal preparation
Lithium perchlorate trihydrate and β-alanine were mixed in 2:1, 1:1, 1:2, 1:3, and 1:4 molar ratios and dissolved in water, allowing for slow solvent evaporation. All the used reagents were provided by Alfa Aesar and used without purification. The results of all ICC crystallizations were confirmed by PXRD analyses and the corresponding diffractograms are presented in Fig. S1 in the ESI.†
Single crystal experiments
Single crystal diffraction experiments were carried out on a Bruker D8 Venture diffractometer equipped with a PhotonII CMOS area detector and using Mo Kα radiation (λ = 0.71073 Å) using an Oxford Cryosystems Cryostream cooling device. Temperature studies were performed in the 100–300 K temperature range with 25 K increments. In every case, data reduction was performed with APEX 3 software.27–30 All of the structures were solved and further refined using the SHELXT intrinsic phasing method31 and SHELXL least-squares minimization procedure,27 respectively, both implemented in the Olex2 suite.32 The structural and refinement parameters of the structures described herein (only measurements at 100 K) are gathered in Table 1. The unit cell dimensions obtained for all the phases during temperature measurements are presented in Tables S1–S3 in the ESI.† All ordered and main component disordered non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms bonded to carbon atoms were placed in geometrically idealized positions with Uiso (H) = 1.2 × Uiso (C). H-Atoms connected to nitrogen atoms were localized in the Fourier difference map and isotropically refined. In the case of the LiClO4·βAla and LiClO4·2βAla-II systems, the N–H distances were restrained, whereas in the LiClO4·2βAla-I phase, all the ammonium H atoms were free to refine. An ORTEP33 drawing depicting anisotropic displacement ellipsoids is available in the ESI† (Fig. S2). An absolute structure of LiClO4·2βAla-I was determined using anomalous scattering effects. The Flack parameter x (ref. 34) equal to 0.04(2) was determined using 1576 quotients. The Hooft parameter,35 calculated using 1661 Bijvoet pairs, equaled 0.03(2). In the structure of LiClO4·2βAla-I measured at 100 K, disorder of the perchlorate anion is observed. The anion is disordered over three positions with a refined occupancy ratio yielding 0.662(4):0.091(4):0.247(4) with common sites for O4 and Cl moieties, thereby simulating the rotation of the entire anion around the Cl–O4 bond. The minor occupancy atoms are localized on both sides of the main residue. The temperature studies of LiClO4·2βAla-I revealed that this disorder is dynamic as, at higher temperature, change of the occupancy of the moieties is visible. Indeed, at 300 K, the anion is almost equally distributed over two positions.
| LiClO 4 ·βAla | LiClO 4 ·2βAla-I | LiClO 4 ·2βAla-II | |
|---|---|---|---|
| Formula | C3H7ClLiNO6 | C6H14ClLiN2O8 | C6H14ClLiN2O8 |
| M x/g mol−1 | 195.49 | 284.58 | 284.58 |
| T/K | 100 | 100 | 100 |
| Space group | C2/c | P21 | Pbca |
| Unit cell dimensions | a = 24.143(3) Å | a = 4.9667(6) Å | a = 8.3651(6) Å |
| b = 5.0111(7) Å | b = 8.4410(11) Å | b = 9.9433(7) Å | |
| c = 14.528(2) Å | c = 13.9410(18) Å | c = 27.584(2) Å | |
| β = 125.825(3)° | β = 95.899(4)° | ||
| V/Å3, Z | 1425.1(3), 8 | 581.37(13), 2 | 2294.4(3), 8 |
| D x/g cm−3 | 1.822 | 1.626 | 1.648 |
| μ/mm−1 | 0.524 | 0.364 | 0.369 |
| F(000) | 800 | 296 | 1184 |
| Crystal size/mm3 | 0.3 × 0.15 × 0.13 | 0.29 × 0.15 × 0.13 | 0.22 × 0.18 × 0.15 |
| Radiation | MoKα | MoKα | MoKα |
| 2θmin, 2θmax | 4.162°, 61.12° | 5.66°, 61.12° | 5.696°, 60.996° |
| Completeness | 99.9% | 99.8% | 99.7% |
| Index ranges | −34 ≤ h ≤ 34, −7 ≤ k ≤ 7, −20 ≤ l ≤ 20 | −7 ≤ h ≤ 7, −12 ≤ k ≤ 12, −19 ≤ l ≤ 19 | −11 ≤ h ≤ 11, −14 ≤ k ≤ 14, −39 ≤ l ≤ 39 |
| Reflections collected/independent | 17655/2183 [Rint = 4.46%] |
17343/3541 [Rint = 4.59%] |
20002/3483 [Rint = 2.65%] |
| Data/restraints/parameters | 2183/3/122 | 3541/38/229 | 3483/6/187 |
| Gof on F2 | 1.109 | 1.081 | 1.178 |
| Final R indices [I ≥ 2σ(I)] | R 1 = 2.76%, wR2 = 7.06% | R 1 = 2.58%, wR2 = 6.44% | R 1 = 3.58%, wR2 = 8.35% |
| Final R indices [all data] | R 1 = 3.06%, wR2 = 7.27% | R 1 = 2.64%, wR2 = 6.48% | R 1 = 4.03%, wR2 = 8.60% |
| Δρmax, Δρmin/e Å−3 | 0.51, −0.47 | 0.24, −0.29 | 0.51, −0.56 |
| Flack parameter | — | 0.04(2) | — |
Optical microscopy
A phase transition was visually observed using an optical microscope with a polarizing filter. A freshly prepared, wet sample of LiClO4·2βAla-I crystals was placed on a glass plate. Pictures of the proceeding phase transition were taken in two minute intervals during the first two hours, and then in sequences: every 10 minutes for 2 h, every 15 min for 4 h, every hour for 5 h and every 2 h for 36 h. Selected photographs are gathered in Fig. 7.Thermal tensor
The thermal expansion tensor was used to visualize changes in the analyzed structures with temperature. The tensor was fitted to the cell parameters recorded at 100–300 K.36 Parameters of the fitted functions, diagonalized thermal expansion tensor elements and the unit cell parameter changes are presented in the ESI† (Tables S4 and S5 and Fig. S3). All 3D representations presented in Fig. 6 correspond to the thermal expansion tensor at 100 K. Both fitting and visualization were performed with the Thermal Expansion Visualizing (TEV V1.0.1) program.37Periodic calculations
Molecular geometry and cell parameter optimizations for LiClO4·2βAla-I and LiClO4·2βAla-II were performed at the DFT(B3LYP)/TZVP38,39 level of theory with Grimme2 dispersion correction40,41 using the CRYSTAL09 (ref. 42 and 43) program for periodic calculations. To visualize minor changes, overlays of the structures from the crystals and calculations are presented in Fig. S4.†PXRD and DSC experiments
All PXRD patterns were recorded at room temperature on a Bruker D8 Advance diffractometer equipped with a LYNXEYE detector using Cu Kα radiation (λ = 1.5418 Å) in the Bragg–Brentano (θ/2θ) horizontal geometry (flat reflection mode) in continuous scan mode with 0.03° steps. The sample holder was rotated at an angular speed of 15 rpm. PXRD analysis of solution mediated phase transition was performed on the freshly prepared crystal sample of the first polymorphic form, not ground and not filtered from the crystallization solution. The sample was placed on a silicon antireflection plate. Data collection was narrowed to the 14–28° 2θ angle range to effectively gather sufficient data since, based on the visual observations, the polymorphic transition is relatively fast. Powder diffraction patterns were continuously gathered for three hours, yet no significant change was observed after 45 min (Fig. 8a). The same conditions were applied for acetone-dried crystals of the LiClO4·2βAla-I phase (Fig. S5†). Additionally, the dried sample was analyzed every three days for two weeks (Fig. 8b). DSC measurements were performed on a NETZSCH DSC 204 Phoenix apparatus, using ∼2 mg of dried crystalline samples. Experiments were carried out in a sealed aluminum crucible at a heating rate of 10 °C min−1 using N2 as a protective gas. The results for both LiClO4·2βAla polymorphs are shown in Fig. 9 while those for LiClO4·βAla are shown in Fig. S7.†Results and discussion
To investigate the role of the mutual ratio of salt and amino acid in the topology of formed coordination polymers, lithium perchlorate trihydrate and β-alanine were mixed in 2:1, 1:1, 1:2, 1:3, and 1:4 molar ratios. Novel ICC single crystals were obtained under equimolar (LiClO4·βAla) and twofold amino acid excess (LiClO4·2βAla-I and LiClO4·2βAla-II) conditions only. The use of a larger amount of organic compound resulted in separate crystallization of the salt and amino acid. Interestingly, the formation of the LiClO4·βAla phase was observed in the case of the 2:1 salt and amino acid molar ratio, as well. The results of all ICC crystallizations were confirmed by PXRD analyses and the corresponding diffractograms are presented in Fig. S1 in the ESI.†
Unexpectedly, formation of a second polymorphic form of the 1:2 stoichiometry (LiClO4·2βAla-II) was ascertained while observing the wet sample of form I (LiClO4·2βAla-I) under the microscope. The obtained crystals exhibited a distinctly different morphology (Fig. 2) which also enabled visual analysis of spontaneously occurring phase transition. Both phases were successfully structurally determined and compared in terms of thermal expansion. Herein, a detailed crystal structure analysis of the three newly obtained ICCs is presented, complemented by the characterization of the observed polymorphic transformation.
 | ||
| Fig. 2 Crystal morphology of (a) LiClO4·βAla, (b) LiClO4·2βAla-I and (c) LiClO4·2βAla-II. The scale was set the same for every microscopy image. | ||
Crystal structures of LiClO4 and βAla ICCs
In all three obtained structures, coordination polymers are formed: one-dimensional chains in the case of LiClO4·βAla and 2D layers in both polymorphs of LiClO4·2βAla. In the ICCs, β-alanine occurs exclusively in the zwitterionic form, which enables the deprotonated carboxylic group to coordinate lithium cations. LiClO4·βAla crystallizes in the centrosymmetric C2/c space group, with the main motif being the 1D infinite neutral chain built from six-membered rings (Fig. 3), virtually the same as that reported for glycine (refcode: HEFWUK),16,17 glycylglycylglycine (GLYLIB),21 proline (NOCXIO,44 NOCXIO01,45 YOXBET46) and histidine (AZIPIK, AZIPOQ, AZIPUW, AZIQUAD)12 ICCs.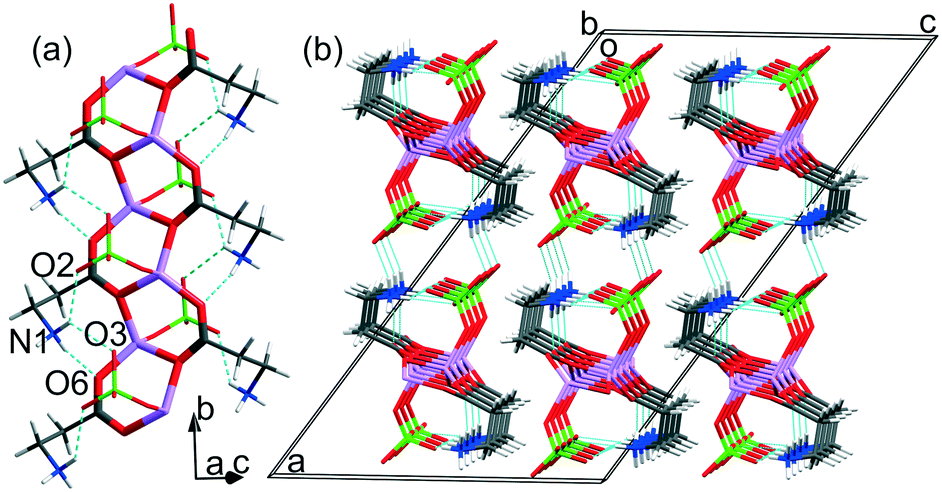 | ||
| Fig. 3 (a) Neutral chain formed by fused six-membered rings in the LiClO4·βAla crystal structure. (b) Crystal packing of chains viewed along the [010] direction. | ||
LiClO 4 ·2βAla-I and LiClO4·βAla-II crystalize in the Sohncke P21 and centrosymmetric Pbca space groups, respectively. Both ICCs form 2D layer structures exhibiting a square-grid-like topology (Fig. 4). Although the topology of the cationic layers is the same, their geometry differs significantly. In the firstly formed polymorph LiClO4·2βAla-I, one type of chiral mesh can be observed. In the second form, LiClO4·2βAla-II, the layer is built from two different meshes, both of which being centrosymmetric. The symmetry of the mesh building block (16-membered ring) is strictly connected to the placement of the side chain of β-alanine molecules. In polymorph I, when one side chain is facing forward, three others are facing backward, while in the case of form II, the amino acid side chain distribution is equal (two forward, two backward; Fig. 4b).
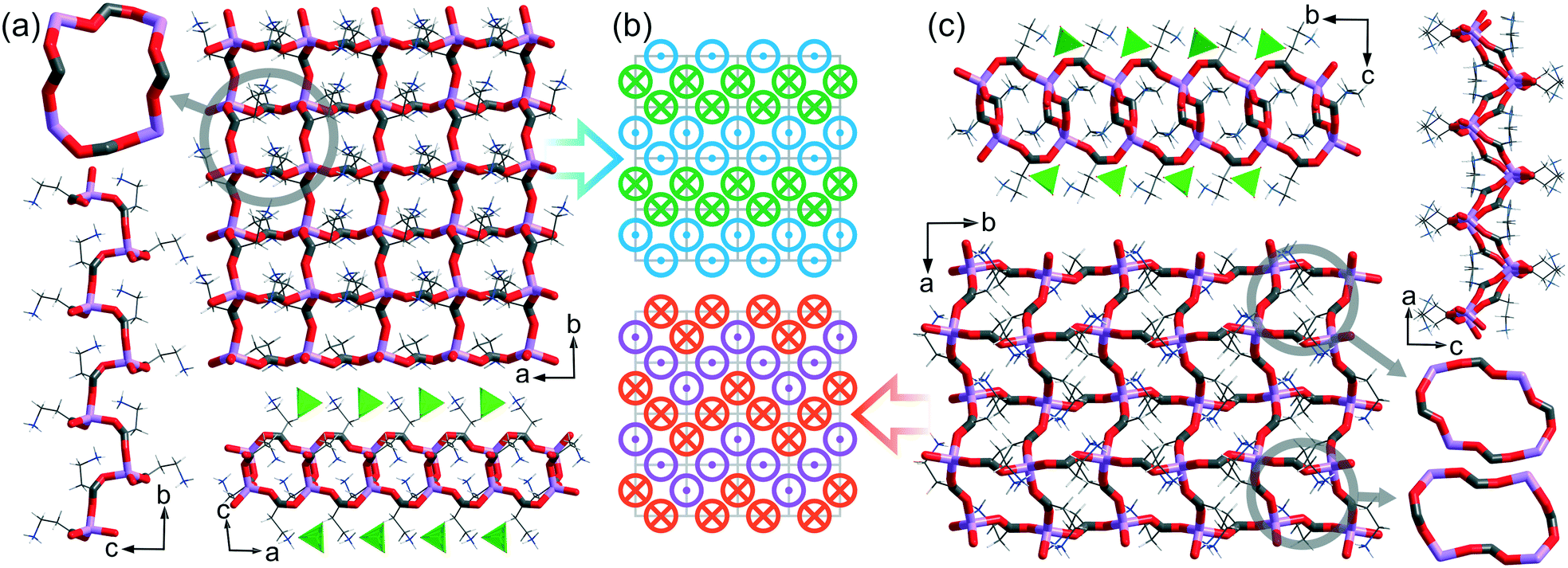 | ||
| Fig. 4 (a) and (c) Square-grid layers in LiClO4·2βAla-I and LiClO4·2βAla-II, respectively, with the geometry of the highlighted 16-membered ring building block and side views of the layer (green tetrahedra represent perchlorate counterions); (b) β-alanine arrangements in the polymorph I (top) and II (bottom) topologies; the amino acid side chains facing forward and backward are marked with a dot and a cross, respectively. Low occupancy disordered ClO4− anions in LiClO4·2βAla-I omitted for clarity. | ||
Furthermore, the structures differ significantly in terms of the undulation of the cationic surface. The same square-grid topology of cationic coordination polymers was also observed in three other similar structures revealed by the CSD search, namely: LiNO3·2Gly (ROZTUW), LiNO3·2Bet (Bet – betaine, EVUWEY) and LiCl·2Sar (Sar – sarcosine, EVUWAU). However, when the spatial arrangement of the side chain of the amino acid is considered, one more grid topology can be distinguished (Fig. S6†). Further insight into the 2D layer geometry discloses that in the centrosymmetric structures (LiClO4·2βAla-II, EVUWEY, and EVUWAU), two geometrically distinguishable meshes can be noticed, while in non-centrosymmetric ROZTUW (Pca21) and LiClO4·2βAla-I, only one kind of 16-membered ring is formed. The mean distance of atoms from the mean plane of the ring motif can play the role of a local folding indicator. Both rings in the centrosymmetric structures seem to be rather flat, with a mean deviation between 0.210 and 0.497 Å, when comparing to the rings of ROZTUW or LiClO4·2βAla-I (0.839 and 0.695 Å, respectively). All values for individual rings, and the mean values are gathered in Table S6.† It might be concluded that for centrosymmetric structures, the layer undulation is realized by differences in the geometry of two individual rings, rather than by folding of one basic unit, as in the case of non-centrosymmetric structures. In chain structures with the most common 6-membered fused ring topology, the building blocks are less distorted with mean deviations in the range 0.106–0.251 for analyzed structures.
In all the reported structures, as well as in the quoted ones, lithium is four-coordinated solely by oxygen atoms. Analysis of the distortion from an ideal tetrahedron of the Li+ coordination sphere was performed with the τ4′ parameter47 (Table S6†). Values close to 1 indicate small distortions from an ideal tetrahedron. No correlation was found between this parameter and rings folding or symmetry.
Herein, in the analyzed ICC crystal structures of different stoichiometry, the perchlorate anion plays a different role in the crystal structure stabilization. In the LiClO4·βAla crystal structure, perchlorate anions are bonded with the polymeric chain and thus complete the lithium coordination sphere. It is somehow surprising as in the majority of reported ICC structures, the fourth coordination site on Li+ is filled by a water molecule (except for GLYLIB). Yet, in the case of LiClO4·βAla, regardless of the temperature conditions of the crystallization process, the anhydrous structure was formed. However, the analysis of the CSD shows that in lithium perchlorate compounds, where Li+ is the only metal and is surrounded exclusively by oxygen atoms, the ClO4− anion is engaged in the lithium coordination sphere in 7 out of 18 structures. However, in the LiClO4·2βAla-I and LiClO4·2βAla-II crystal structures, the perchlorate units interact with cationic (Li-2βAla)nn+ layers through a network of charge assisted hydrogen bonds (Fig. 5 and Table 2).
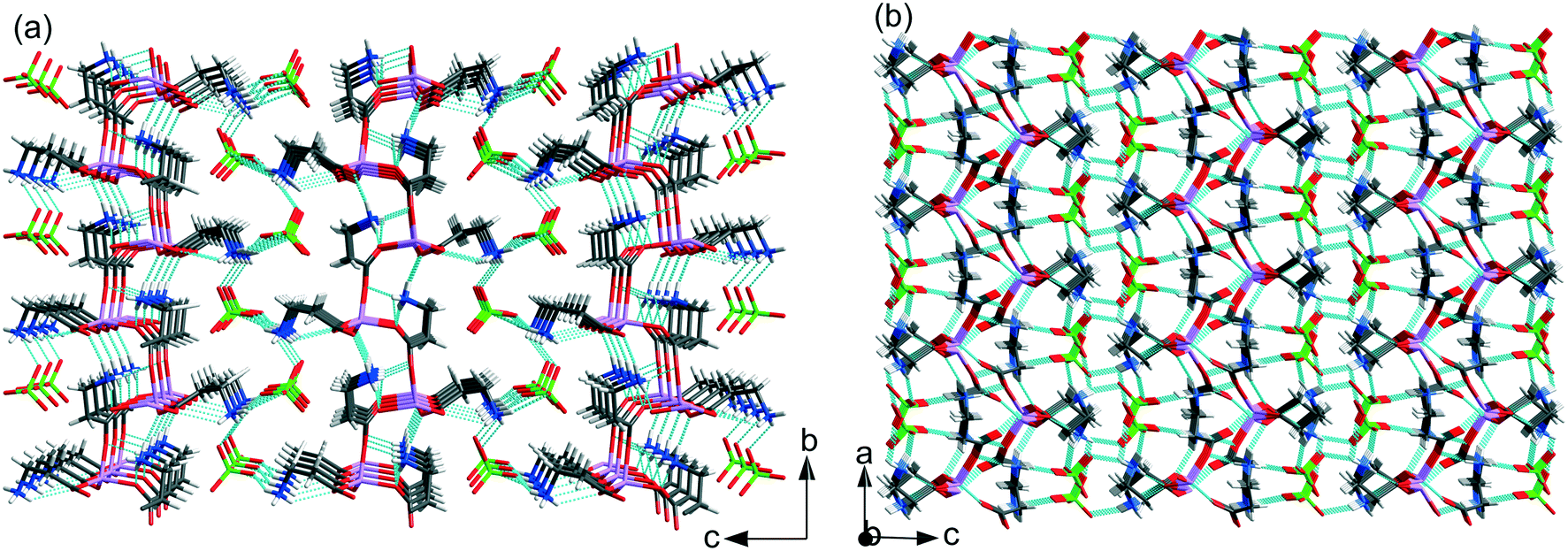 | ||
| Fig. 5 Hydrogen bond networks in (a) LiClO4·2βAla-I and (b) LiClO4·2βAla-II. Low occupancy disordered ClO4− anions in LiClO4·2βAla-I omitted for clarity. | ||
| Structure | DH⋯O | D–H/Å | H⋯O/Å | D⋯A/Å | DHA/° | |
|---|---|---|---|---|---|---|
| 13/2 − x, −1/2 + y, 1/2 − z; 2x, 1 + y, z; 31/2 + x, 3/2 − y, 1/2 + z; 43/2 − x, 3/2 − y, 1 − z; 5−1 + x, y, z; 61 − x, −1/2 + y, 2 − z; 7−x, 1/2 + y, 1 − z; 8x, −1 + y, z; 9−1/2 + x, 3/2 − y, 1 − z; 101/2 − x, −1/2 + y, z; 111 − x, 1 − y, 1 − z; 123/2 − x, 1/2 + y, z; 13−1/2 + x, y, 3/2 − z.a In the case of LiClO4·2βAla-I, the hydrogen bonds listed are for the main component ClO4− residue only. | ||||||
| LiClO 4 ·βAla | Intra | N1H1A⋯O21 | 0.900(15) | 2.364(15) | 2.9555(16) | 123.4(12) |
| N1H1A⋯O41 | 0.900(15) | 2.159(17) | 2.9100(16) | 140.5(11) | ||
| N1H1C⋯O52 | 0.901(17) | 1.902(18) | 2.7936(16) | 170.1(16) | ||
| N1H1B⋯O33 | 0.901(16) | 2.008(15) | 2.8849(17) | 164.2(14) | ||
| C3H3A⋯O34 | 0.99 | 2.54 | 3.4298(15) | 149 | ||
| LiClO 4 ·2βAla-I | Intra | N1H1A⋯O6 | 0.85(3) | 2.32(3) | 2.872(2) | 123(3) |
| N11H11A⋯O165 | 0.91(3) | 1.97(3) | 2.870(2) | 169(2) | ||
| N11H11B⋯O16 | 0.88(3) | 2.51(3) | 2.999(2) | 115(2) | ||
| N11H11B⋯O156 | 0.88(3) | 2.02(3) | 2.892(2) | 168(3)′ | ||
| N1H1B⋯O3 | 0.90(3) | 1.99(3) | 2.810(5) | 151(3) | ||
| N1H1B⋯O45 | 0.90(3) | 2.55(3) | 3.024(2) | 113(2) | ||
| N1H1C⋯O27 | 0.83(3) | 2.20(3) | 2.923(7) | 146(3) | ||
| N11H11C⋯O58 | 0.86(3) | 1.93(3) | 2.773(2) | 167(3) | ||
| LiClO 4 ·2βAla-II | Intra | N1H1B⋯O59 | 0.901(13) | 2.55(2) | 3.1253(16) | 122.1(15) |
| N1H1B⋯O69 | 0.901(13) | 1.877(14) | 2.7775(16) | 177(2) | ||
| N1H1C⋯O159 | 0.900(13) | 1.986(14) | 2.8571(15) | 162.6(18) | ||
| N11H11C⋯O1610 | 0.899(16) | 1.847(16) | 2.7401(15) | 172.2(16) | ||
| N1H1A⋯O111 | 0.900(14) | 2.071(14) | 2.9613(16) | 170.2(13) | ||
| N11H11A⋯O312 | 0.899(15) | 2.106(15) | 2.9617(16) | 158.6(15) | ||
| N11H11B⋯O4 | 0.901(15) | 2.429(17) | 2.9208(17) | 114.6(12) | ||
| N11H11B⋯O213 | 0.901(15) | 2.053(15) | 2.9310(16) | 164.4(15) | ||
In LiClO4·2βAla-I, one of the amino groups (N11H3) of β-alanine forms N–H⋯O hydrogen bonds only within the layer, while the second one (N1H3) interacts with one carboxyl atom in the layer and two oxygen atoms from separate perchlorate anions. In the LiClO4·2βAla-II polymorph, one amino group (N1H3) is H-bonded to the carboxyl O6 and O15 atoms in the layer and one oxygen atom from ClO4−, while the second β-alanine side chain forms H-bonds with two perchlorate anions and one within the layer. This diversity in the hydrogen bond array can be connected to the different folding of layers and the changed placement of amino acid side chains. It is worth noting that subsequent layers interact with each other only through the H-bonded anions.
The H-bond network in the LiClO4·βAla crystal structure is dominated by intramolecular N–H⋯O hydrogen bonds since the side chains of β-alanine are bent in such a way that the interactions between the terminal NH3 group and oxygen atoms from perchlorate anions are facilitated. The neutral chain is further linked with two others by the formation of another set of N–H⋯O interactions involving the remaining atom of the perchlorate unit. The resulting layers of H-bonded jointed chains extend parallel to the (10−1) plane and interact with each other through residual C3–H⋯O3 contacts (Fig. 3b). The geometrical parameters of hydrogen bonds present in all three structures are collected in Table 2.
Temperature studies and the thermal expansion tensor
All three systems have been tested for response to temperature changes to determine how the polymorphic forms differ from each other and the LiClO4·βAla structure exhibiting a different stoichiometry and topology. Changes of the cell parameters for all three structures are within the 1.5% range, without any discontinuity in the cell parameters, suggesting the stability of the crystal structures in the analyzed temperature range. Gathered X-ray data allowed for determining thermal expansion tensors, the anisotropy of which may indicate the direction of changes propagating in the structures with temperature (Fig. 6). It was stated that thermal structural deformation is more pronounced in the directions where weak interactions dominate.48 Therefore, it is not surprising that in the case of LiClO4·βAla, the biggest changes appear along the [001] crystallographic direction, where only weak interchain C–H⋯O contacts are responsible for the H-bonded layer packing. This can be easily seen with thermal expansion tensor projections (Fig. 6a), in which the shape indicates the biggest distortion in this exact direction. Surprisingly, in the case of both LiClO4·2βAla polymorphs, the thermal expansion tensor indicates that the most significant change takes place parallel to the cationic layer, exactly along the [100] crystallographic direction. This suggests that the most sensitive to thermal changes are the Li–O coordination bonds in the 16-membered rings forming a grid, and not, as expected, the hydrogen bonds between layers. This observation also indicates the high lability and thus the ease of deformation of the ring. In addition, it might be presumed that the overall energy of H-bonds between the layers and anions is a result of the synergy effect of many individual interactions and also dispersed charge in the cationic layer.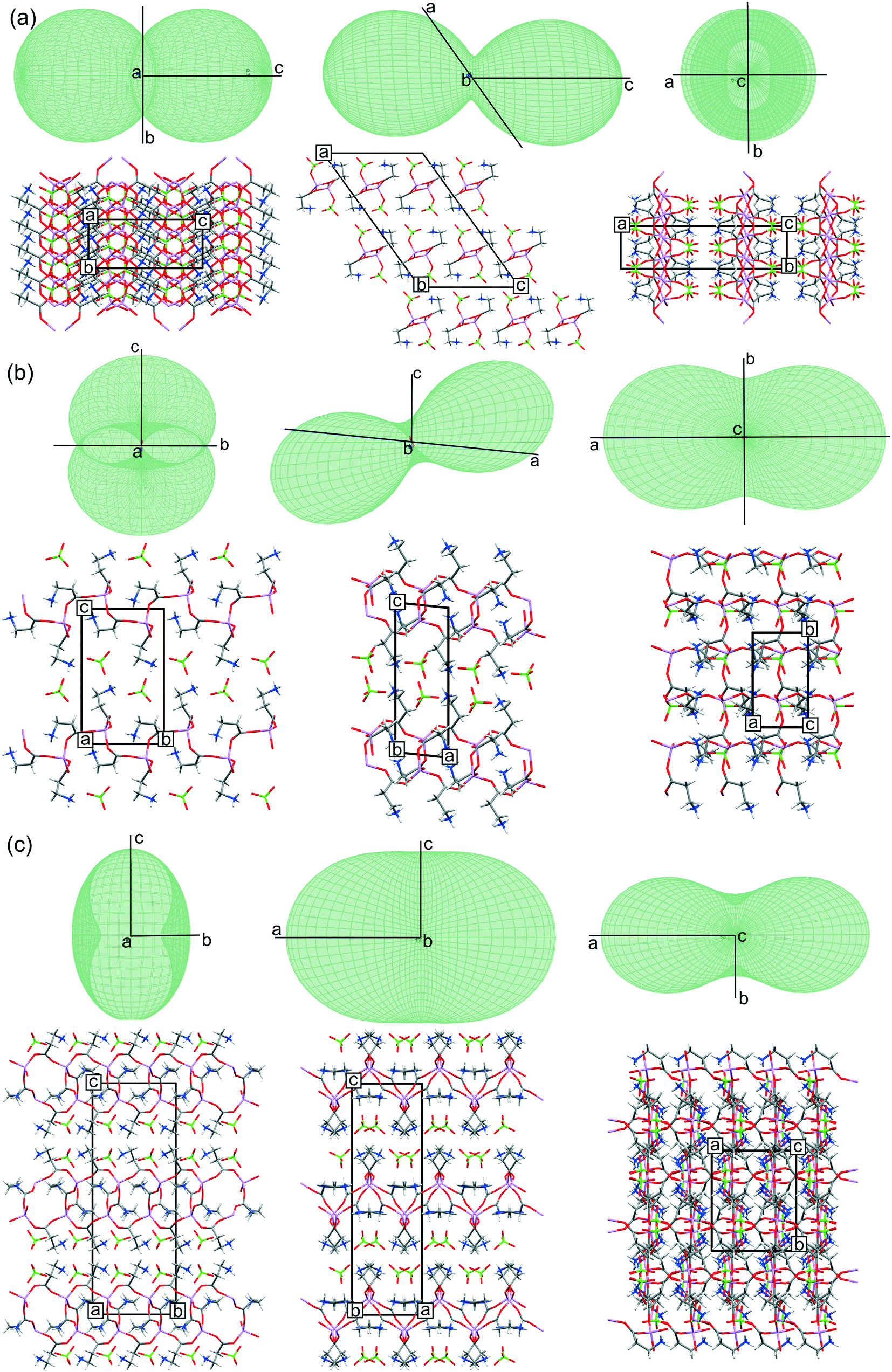 | ||
| Fig. 6 Projections of the thermal expansion tensor along all three crystallographic directions in accordance to the molecular packing of (a) LiClO4·βAla, (b) LiClO4·2βAla-I and (c) LiClO4·2βAla-II. Low occupancy disordered ClO4− anions in LiClO4·2βAla-I omitted for clarity. | ||
In the LiClO4·2βAla-I crystal structure, dynamic disorder of perchlorate anions was detected. Analysis of the placement of ClO4− anions in the structure shows that in this polymorph, the space occupied by the anions is about 14.1% of the unit cell volume, while in LiClO4·2βAla-II, it is 13.7%. It may be concluded that additional space gives the anions the required space to rotate when the temperature rises. Moreover, the looser structure of LiClO4·2βAla-I may be a reason why the structure undergoes the phase transition. In the resulting thermodynamically preferable second polymorph, the perchlorate anions are intertwined into the structure to a higher degree, and maybe this is why they occupy specific positions while strengthening the H-bond network between cationic layers.
Polymorphic transition
It was noticed that when LiClO4·2βAla-I plate crystals were left in the crystallization solution in a closed vial, crystals exhibiting a bulk morphology appeared (Fig. 2), replacing the primarily obtained crystallites. Based on single-crystal X-ray diffraction experiments, a new phase, LiClO4·2βAla-II, was confirmed (Table 1). Since the morphology differences between both forms are significant, optical observations of the undergoing phase transition were performed (Fig. 7). Visual changes in the sample were noticed only when LiClO4·2βAla-I single crystals were submerged in the crystallization solution. Bulk, single crystals of the new phase can be visualized already after four minutes. However, even after two days, crystals of the first phase remain present. This is most likely due to the conditions of the experiments, where the saturated crystallization solution was limited and exposed to drying, unlike that in a closed vial, where vapor–liquid equilibrium is achieved, and full transformation proceeds. | ||
| Fig. 7 Polarization microscopy images showing the growth of the LiClO4·2βAla-II polymorph on LiClO4·2βAla-I crystals. Results for wet crystals. | ||
Growth of the LiClO4·2βAla-II phase can be also observed with X-ray powder diffraction experiments. PXRD patterns of both phases were measured and compared with simulated patterns from single-crystal data (Fig. S1†). As optical observations showed, polymorphic transition in the wet sample is rather fast, and thus only a selected range of 2θ angle was measured to enable fast data acquisition. Based on the preliminary PXRD pattern analysis, a 14–28° angular range has been selected for further monitoring as the most varied in both polymorphs. A series of powder patterns taken in 3 minute intervals allows us to observe the formation of the LiClO4·2βAla-II phase; see the reflections appearing at the 19.5 and 24.8° 2θ angles (Fig. 8a). It should be noted that the reflection sets from the LiClO4·2βAla-I phase do not disappear, which can be connected to the sample drying during data acquisition, similar to that noticed in the microscopy observations. An analogous PXRD experiment was carried out on dry, ground crystals of polymorph I, and no change in the diffraction patterns was observed (Fig. S5 in the ESI†). The collection of the powder patterns was also repeated after 3, 5 and 14 days to rule out the pace of the transition in the dried crystals. Similarly, no change was spotted (Fig. 8b). The results may suggest the nature of the phase transition to be mediated by the liquid state, similar to the case of the PhCOONa·PhCOOH ionic co-crystal reported by Butterhof et al.49 To verify the lack of single-crystal to single-crystal solid-state transition, differential scanning calorimetry experiments were carried out. The DSC profile of LiClO4·2βAla-I shows only a melting peak at 164.6 °C (Fig. 9), which proves that phase transition does not occur in the dry sample. In addition, the lower melting temperature of phase I compared to that of II is consistent with the observed polymorph stability.
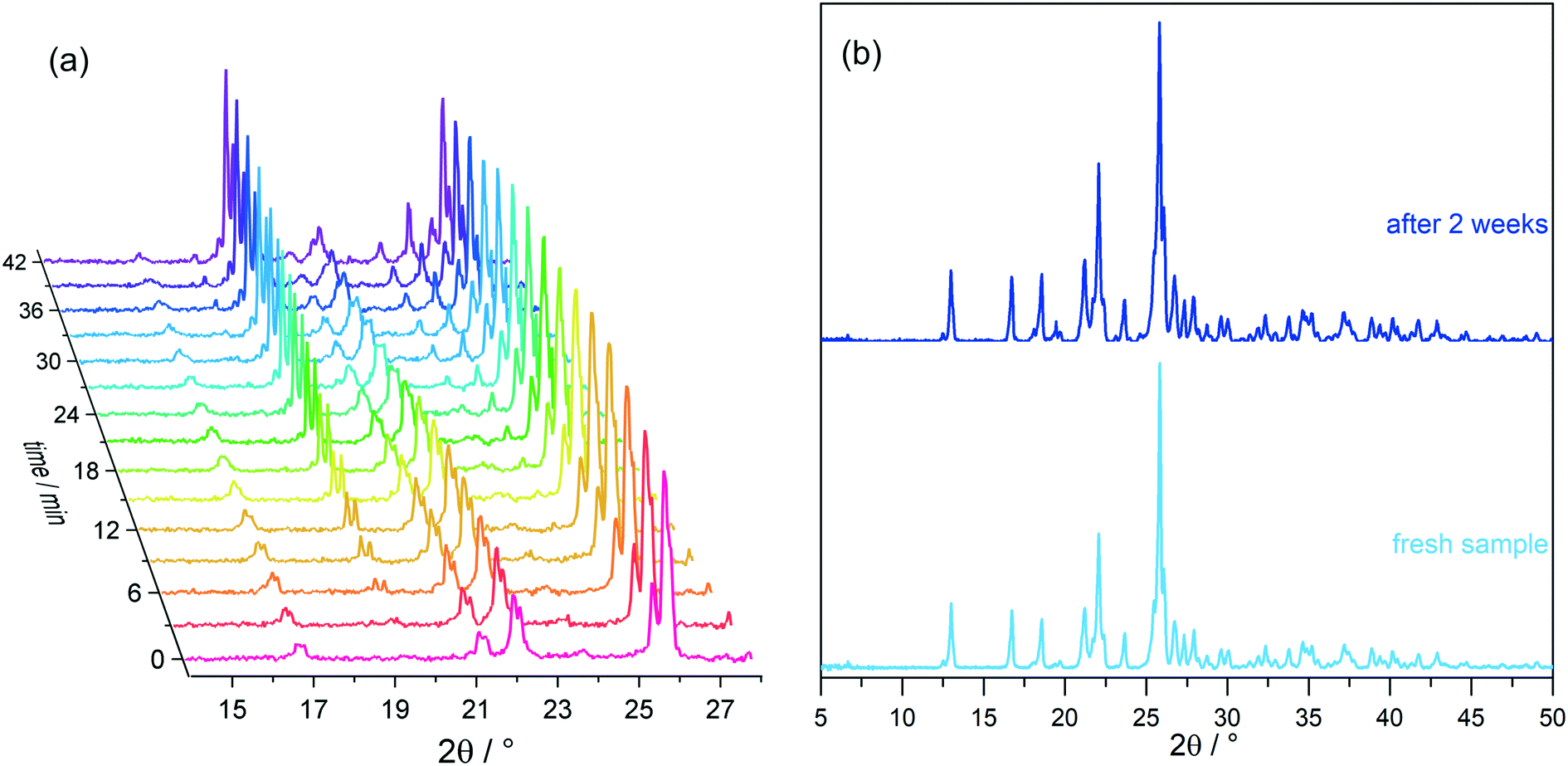 | ||
| Fig. 8 LiClO 4 ·2βAla-I powder diffraction patterns of (a) a wet sample taken in 3 minute intervals and (b) dried crystals, freshly prepared and after two weeks. | ||
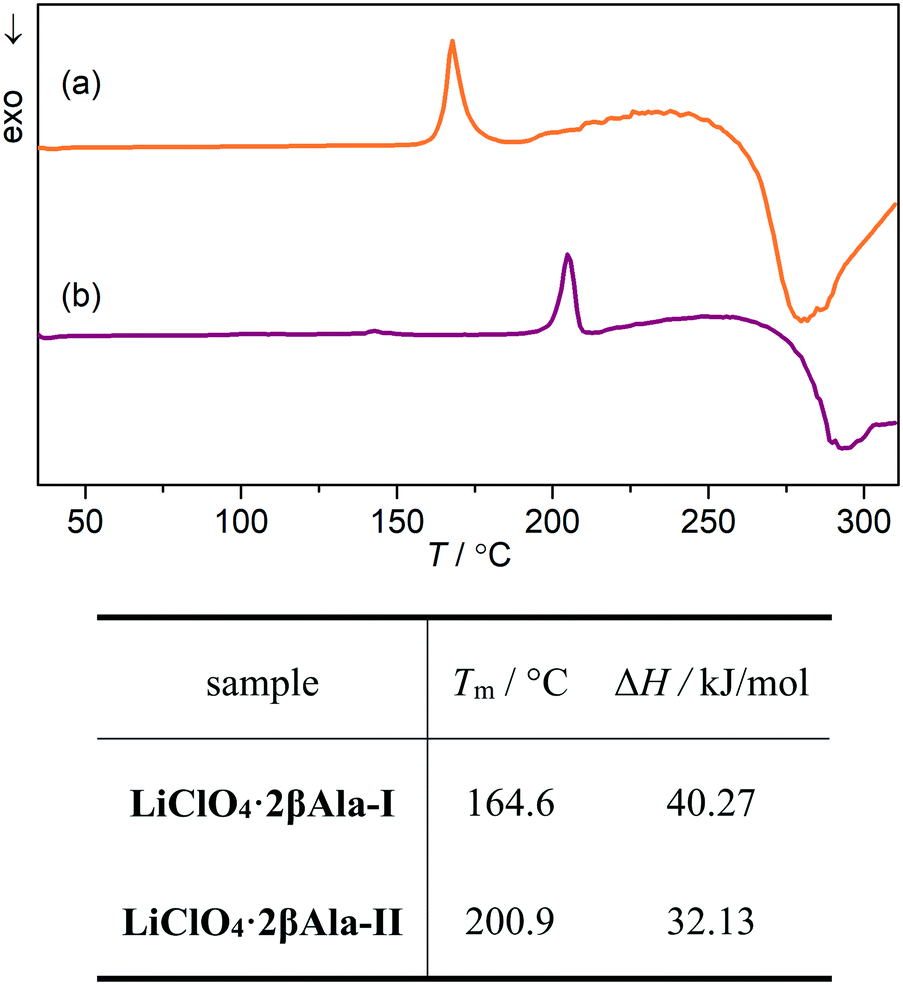 | ||
| Fig. 9 DSC profiles of (a) LiClO4·2βAla-I and (b) LiClO4·2βAla-II along with the melting points (Tm) and enthalpies of fusion (ΔH) derived from DSC measurements. | ||
The performed periodic calculations enabled us to determine the difference in total energies between the two polymorphic forms, which equaled 15.9 kJ mol−1, confirming that LiClO4·2βAla-II exhibits a lower energy value. Hence, phase II can be regarded as the more stable, thermodynamic phase, while LiClO4·2βAla-I is the less energetically preferable, firstly formed kinetic form. Structural differences between the two described polymorphs concern the differences in layer geometry – β-alanine side chain spatial orientations and hydrogen-bond networks. Therefore, due to the need for significant structural changes, a phase transition in the solid-state (single-crystal to single-crystal) is highly energetically unfavorable and hence not observed. However, in concentrated water solution (only a small amount of water is needed to start the process), it might be assumed that the coordination layers are sustained but more flexible, so the energy barrier might lower enough to enable the formation of the second polymorph. Full understanding of the phase transition nature remains impossible to grasp, yet presumably, the disorder of perchlorate anions existing in the kinetic phase might influence the weaker binding of two adjacent polycationic layers, consequently facilitating their easier separation at the first step of dissolution. Simultaneously, water molecules themselves may support the amino acid side chain spatial rearrangement and modifications in the hydrogen-bond network.
Conclusions
Herein, we report three novel ICCs built from lithium perchlorate and β-alanine and their crystal structures, both centrosymmetric and non-centrosymmetric. β-Alanine seems to be a building block as versatile as its predecessor in the homologous series, glycine, even though centrosymmetric forms of pure β-alanine are only known. The structure with a 1:1 molar ratio crystalizes in a centrosymmetric space group in a manner typical for 1:1 systems, creating neutral chain coordination polymers built from fused six-membered rings. Two polymorphic structures of the ionic co-crystal with a 2:1 ratio are formed, non-centrosymmetric LiClO4·2βAla-I and centrosymmetric LiClO4·2βAla-II, both having the same topology, but different geometries and packing. The observed phase transition was investigated with PXRD measurements and optical observations under the microscope, proving the crucial role of the solvent in mediating the formation of the thermodynamically preferable polymorph. An energy gain of about 15.9 kJ mol−1 according to periodic calculations was found. The single-crystal to single-crystal nature of the transformation was excluded since no change in the diffraction patterns of the dried sample even after a two week aging period and no phase transition peak in the DSC plots were detected. Taking into consideration one of the differences between the polymorphic forms, which is the placement of the β-alanine side-chain, it can be explained why a solid-state phase transition is not observed. The flipping of the amino acid side-chain to the other side of the mesh is probably too energetically expensive, and thus, the formation of the LiClO4·2βAla-II phase takes place only in a saturated solution. Our observations may also suggest that the coordination polymer square-grid topology might be preserved after the dissolution of LiClO4·2βAla-I, and loosening the flexible 16-membered mesh may allow for the side-chain flipping and formation of the novel phase. The presumed transition state mechanism may also be supported by the observed diversity in layered ICC structures found in the CSD (Fig. S6†). It might be even postulated that polymorphism in this class of ICCs may be connected to the ease of mesh deformation with a possible energy landscape of many local minima. Additionally, the thermal tensor analysis allowed us to determine which bonds in every structure were the easiest to be modified with temperature. In the case of the 1D chain-like structure of LiClO4·βAla, thermal expansion was associated with elongation of the weakest C–H⋯O contacts. In LiClO4·2βAla-I and LiClO4·2βAla-II, the most significant change was observed in the plane of the coordination polymer. This additionally underlines the ease of layer deformations and high flexibility supporting our assumptions on the probable mechanism of this polymorphic transition.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financed by a grant from the National Science Centre (DEC-2018/29/N/ST4/00451). P. H. M. would like to thank the Wrocław Centre for Networking and Supercomputing for providing computational facilities enabling periodic calculations (grant no. WCSS#27198765). This work was implemented as a part of the Operational Project Knowledge Education Development 2014-2020 co-financed by the European Social Fund. Single crystal X-ray diffraction and DSC experiments were performed at the Czochralski Laboratory of Advanced Crystal Engineering (Faculty of Chemistry, University of Warsaw) while PXRD measurements were carried out at the Faculty of Chemistry, Warsaw University of Technology.References
- S. P. Kelley, A. Narita, J. D. Holbrey, K. D. Green, W. M. Reichert and R. D. Rogers, Cryst. Growth Des., 2013, 13, 965–975 CrossRef CAS.
- D. Braga, F. Grepioni, L. Maini, S. Prosperi, R. Gobetto and M. R. Chierotti, Chem. Commun., 2010, 46, 7715–7717 RSC.
- D. Braga, F. Grepioni, G. I. Lampronti, L. Maini and A. Turrina, Cryst. Growth Des., 2011, 11, 5621–5627 CrossRef CAS.
- L. Casali, L. Mazzei, O. Shemchuk, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, Chem. Commun., 2018, 54, 7637–7640 RSC.
- K. Honer, E. Kalfaoglu, C. Pico, J. McCann and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2017, 5, 8546–8550 CrossRef CAS.
- M. L. Cheney, D. R. Weyna, N. Shan, M. Hanna, L. Wojtas and M. J. Zaworotko, J. Pharm. Sci., 2011, 100, 2172–2181 CrossRef CAS PubMed.
- N. K. Duggirala, A. J. Smith, Ł. Wojtas, R. D. Shytle and M. J. Zaworotko, Cryst. Growth Des., 2014, 14, 6135–6142 CrossRef CAS.
- R. S. B. Williams, L. Cheng, A. W. Mudge and A. J. Harwood, Nature, 2002, 417, 292–295 CrossRef CAS PubMed.
- A. J. Smith, S.-H. Kim, N. K. Duggirala, J. Jin, L. Wojtas, J. Ehrhart, B. Giunta, J. Tan, M. J. Zaworotko and R. D. Shytle, Mol. Pharmaceutics, 2013, 10, 4728–4738 CrossRef CAS PubMed.
- T. T. Ong, P. Kavuru, T. Nguyen, R. Cantwell, Ł. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 9224–9227 CrossRef CAS PubMed.
- M. Lestari, M. Lusi, A. O'Leary, D. O'Nolan and M. J. Zaworotko, CrystEngComm, 2018, 20, 5940–5944 RSC.
- D. Braga, L. Degli Esposti, K. Rubini, O. Shemchuk and F. Grepioni, Cryst. Growth Des., 2016, 16, 7263–7270 CrossRef CAS.
- O. Shemchuk, L. Degli Esposti, F. Grepioni and D. Braga, CrystEngComm, 2017, 19, 6267–6273 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A. I. Ilin, CSD Communication, 2017 DOI:10.5517/ccdc.csd.cc1pz2g6.
- J. Baran, Pol. J. Chem., 2003, 1561–1577 CAS.
- G. Müller, G.-M. Maier and M. Lutz, Inorg. Chim. Acta, 1994, 218, 121–131 CrossRef.
- T. Balakrishnan, K. Ramamurthi, J. Jeyakanthan and S. Thamotharan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m60–m61 CrossRef CAS PubMed.
- M. D. Sweetlin, S. M. Eapen, S. Perumal and S. Ramalingom, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m206–m207 CrossRef CAS PubMed.
- J. Baran, M. Drozd, H. Ratajczak and A. Pietraszko, J. Mol. Struct., 2009, 927, 43–53 CrossRef CAS.
- J. P. Declercq, R. Meulemans, P. Piret and M. Van Meerssche, Acta Crystallogr., Sect. B: Struct. Sci., 1971, 27, 539–544 CrossRef CAS.
- M. Fleck and L. Bohatý, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m291–m295 CrossRef PubMed.
- M. R. Hudson, D. G. Allis, W. Ouellette, P. M. Hakey and B. S. Hudson, J. Mol. Struct., 2009, 934, 138–144 CrossRef CAS.
- M. Fleck, K. Schwendtner and A. Hensler, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m122–m125 CrossRef PubMed.
- G. L. Perlovich, L. K. Hansen and A. Bauer-Brandl, J. Therm. Anal. Calorim., 2001, 66, 699–715 CrossRef CAS.
- B. A. Zakharov, N. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2015, 17, 2074–2079 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- SAINT, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- APEX3, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect. Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249–259 CrossRef CAS PubMed.
- R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2010, 43, 665–668 CrossRef CAS.
- P. Paufler and T. Weber, Eur. J. Mineral., 1999, 11, 721–730 CrossRef CAS.
- T. Langreiter and V. Kahlenberg, Crystals, 2015, 5, 143–153 CrossRef.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Dovesi, Z. Kristallogr., 2005, 220, 571 CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, Ph. D’Arco and M. Llunell, CRYSTAL09 User’s Manual, University of Torino, Torino, 2009, vol. 220, p. 571 Search PubMed.
- M. Shkir, S. Alfaify, M. A. Khan, E. Dieguez and J. Perles, J. Cryst. Growth, 2014, 391, 104–110 CrossRef CAS.
- S. Sathiskumar, T. Balakrishnan, K. Ramamurthi and S. Thamotharan, Spectrochim. Acta, Part A, 2015, 138, 187–194 CrossRef CAS.
- T. U. Devi, N. Lawrence, R. Ramesh Babu, S. Selvanayagam, H. Stoeckli-Evans and K. Ramamurthi, Cryst. Growth Des., 2009, 9, 1370–1374 CrossRef CAS.
- D. Rosiak, A. Okuniewski and J. Chojnacki, Polyhedron, 2018, 146, 35–41 CrossRef CAS.
- K. Durka, A. A. Hoser, R. Kamiński, S. Luliński, J. Serwatowski, W. Koźmiński and K. Woźniak, Cryst. Growth Des., 2011, 11, 1835–1845 CrossRef CAS.
- C. Butterhof, K. Bärwinkel, J. Senker and J. Breu, CrystEngComm, 2012, 14, 6744–6749 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7 show additional powder diffraction patterns, ORTEP drawings of asymmetric units, cell parameter changes with temperature, overlay of DFT optimized and experimental packing diagrams for both polymorphs, possible arrangements of amino acid side chains in layered structures, and DSC profile for the compound with 1:1 stoichiometry. Tables S1–S7 contain unit cell parameter changes with temperature, data for thermal expansion tensor calculations and analysis, geometrical parameters for the ring description and selected bond lengths in the analyzed crystal structures. Additionally, atom coordinates for DFT optimized structures are included (PDF). CCDC 1994864–1994866 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00592d |
| This journal is © The Royal Society of Chemistry 2020 |