
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A three-in-one crystal of mixed sized cucurbit[n]uril homologues†
Oksana
Danylyuk
 * and
Volodymyr
Sashuk
* and
Volodymyr
Sashuk
Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: odanylyuk@ichf.edu.pl
First published on 2nd April 2020
Abstract
Crystallization-driven self-assembly of three macrocyclic members of the cucurbit[n]uril family is reported. The largest cucurbit[10]uril serves as a host for the smallest cucurbit[5]uril forming an inclusion complex of the macrocycle-within-macrocycle type. This supramolecular ensemble undergoes cocrystallization with cucurbit[7]uril. Such unusual cocrystallization is explained by the strong tendency of all cucurbit[n]urils to associate via multiple C–H⋯O hydrogen bonds.
Crystallization is a peculiar example of self-assembly.1 The propensity of a given molecule to assemble on its own and/or with certain partners may result in one or different crystalline forms ranging from simple solvates, through salts, polymorphs, pseudopolymorphs, cocrystals, solid-state host–guest complexes, to advanced framework architectures.2 Crystallization from multi-component mixtures often yields the resolution of the individual chemical entities due to differences in their solubility. This simple and powerful separation method is well-known as fractional crystallization. Failures of fractional crystallization3 usually imply the formation of stoichiometric solid-state compounds (cocrystals)4 or solid solutions.5 Formation of cocrystals arise from non-covalent intermolecular interactions such as hydrogen bonds, halogen bonds, π–π interactions, etc. between two or more chemical species.6 Crystal engineering uses knowledge on these non-covalent interactions of supramolecular synthons to design systems for which fractional crystallization will fail predictably, due to strong specific attraction between molecular components.7
Fractional crystallization is often used to isolate individual macrocyclic homologues from reaction mixtures. Indeed, cucurbit[n]uril (CBn) synthesis yields a mixture of different homologues with n = 5–10, with CB6 being the major component, with traces of CB5, CB7, CB8, CB10 and other oligomers.8 Isolation of each component is based on their solubility differences in water, water/methanol and hydrochloric acid solutions. Due to rather small differences in solubility, crystallization accompanied with careful examination of each fraction by NMR spectroscopy should be repeated multiple times (10–30) to achieve the separation of the individual CBn homologues. Even then, some of the fractions are contaminated (contain mixture of CBn) and may be further added to the next batch of mixed CBn to be separated. We accidentally left one of such contaminated fractions to evaporate very slowly under ambient conditions for several months and as a result, we discovered a small amount of crystals in the forgotten vial. Single crystal X-ray analysis revealed surprising presence of three different CBn macrocycles in the crystal lattice. The asymmetric unit consists of CB5 included into the CB10 macrocycle and CB7 partnering them (Fig. 1). Although the formation of the ternary complex was not ‘engineered’, we decided to report it as it is a really unusual example of the failure of fractional crystallization. We explain the formation of a ‘three-in-one’ cocrystal by strong tendency of all CBn to associate via multiple C–H⋯O hydrogen bonds.
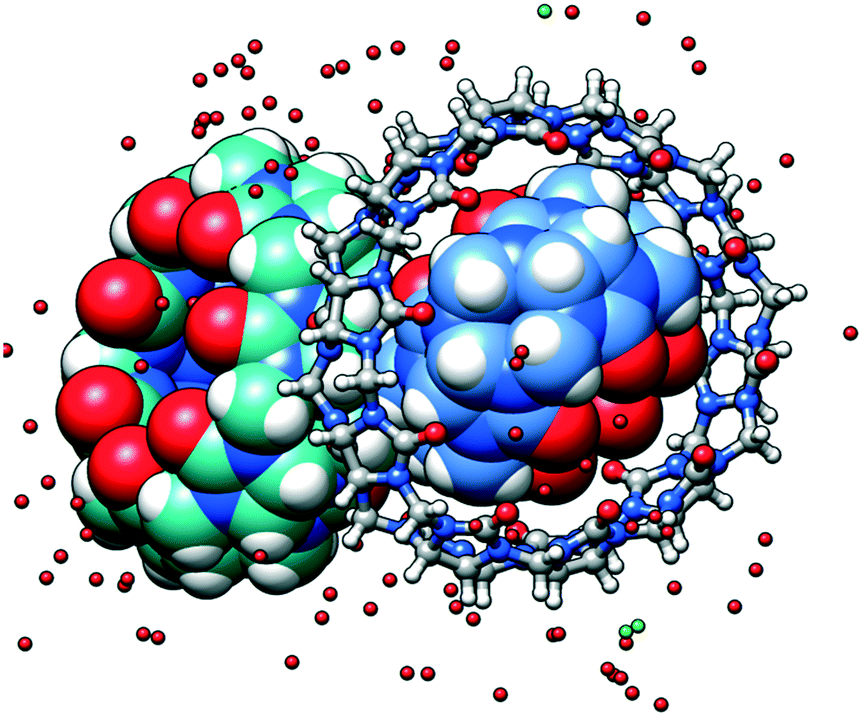 | ||
| Fig. 1 The asymmetric unit of the multi-component crystal comprising CB5 (in blue) included into the cavity of CB10 and CB7 (in sea green). The solid-state complex is highly hydrated. | ||
There are several levels of the inclusion happening in the multi-component crystal. The cavity of the smallest CB5 is filled with one water molecule disordered over two positions. Each of the CB5 portals holds additional water molecules with O⋯O distances between carbonyl oxygen atoms and central water molecule typical for hydrogen bonding (Fig. 2a). The H2O@CB5(H2O)2 assembly is surrounded by the cavity of large CB10 (Fig. 2c and d). The centroids of the CB10 and CB5 are practically overlapped thus, the macrocycle-within-macrocycle assembly is concentric. The inclusion complex CB5@CB10 was isolated and described for the first time by A. Day and co-workers.9 This beautiful concentric fitting of a small macrocycle within a large one has been known in the supramolecular chemistry as a cucurbituril-based gyroscane. Structural studies on this supramolecular ensemble are quite scarce, and besides the initial report, only two crystallographic works on the CB5@CB10 complex have appeared concerning the rigidifying role of potassium10 and neodymium11 on this structure.
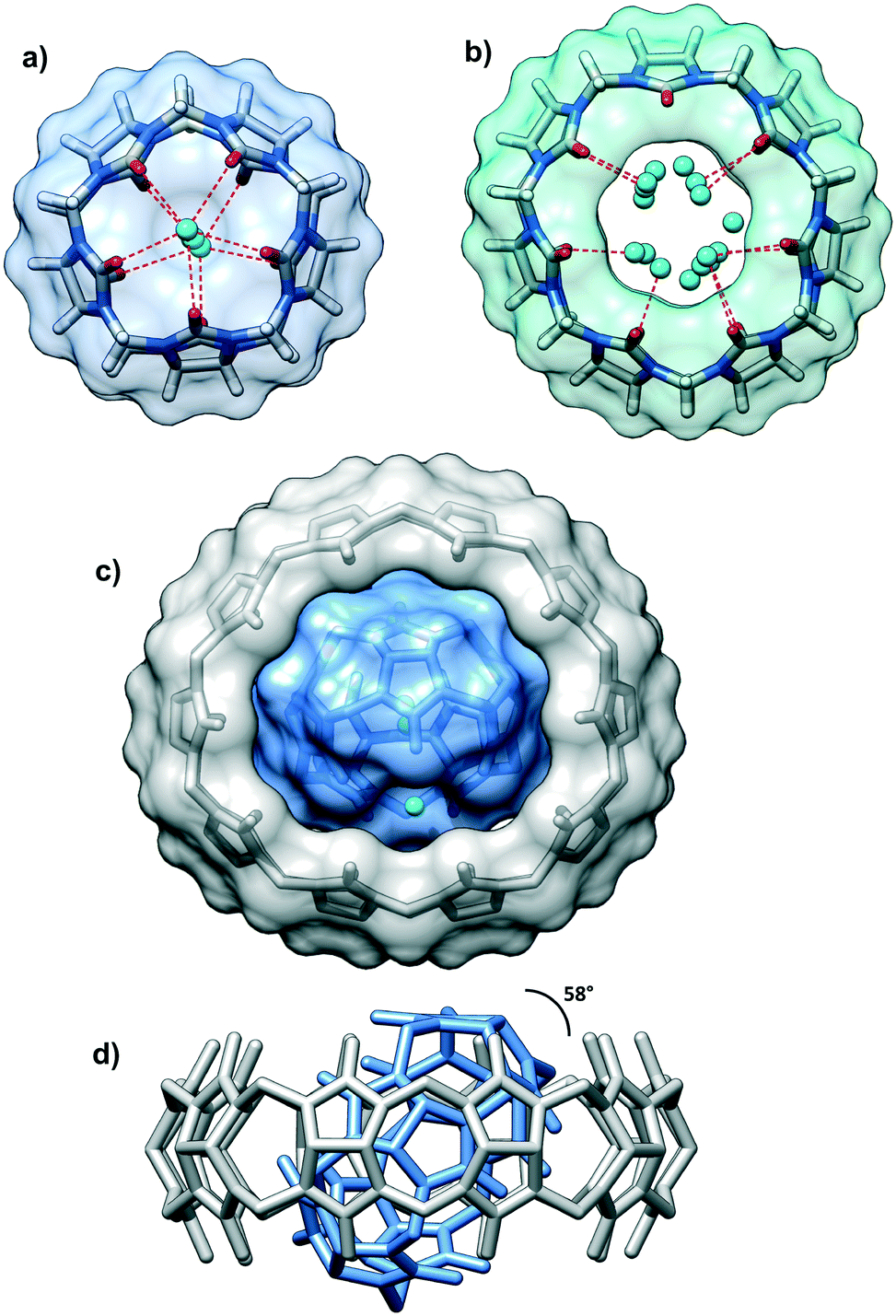 | ||
| Fig. 2 (a) The CB5 cavity contains disordered water molecules and is capped by two water molecules, and the O⋯O distances between carbonyl oxygen atoms and water molecules are in the range of 2.77–2.99 Å; (b) the CB7 cavity contains disordered water molecules (over 16 positions), and the O⋯O distances between carbonyl oxygen atoms and water molecules are in the range of 2.59–2.96 Å; (c) the inclusion complex CB5@CB10 (top view) and d) its side view. The dihedral angle between the mean planes of the CB5 and CB10 is 58°; two ring systems are concentric. | ||
Taking into account the persistent nature of the CB5@CB10 assembly, it was quite astonishing to find out that it is able to cocrystallize with another member of the CBn family. Indeed, individual CB5@CB10 units are well separated in the crystal lattice by CB7 molecules (Fig. 3a). A closer look at the CB7 (Fig. 2b) reveals that it is filled with multiple water molecules, which were modelled as disordered over 16 positions. The water molecules reside in the inner cavity and in the proximity of the portals. The outer surface of the CB7 is richly engaged in multiple C–H⋯O hydrogen bonds with the CB5@CB10 entities acting both as a donor (Fig. 3b) and acceptor (Fig. 3c). As a result, each CB5@CB10 complex is surrounded by four CB7 molecules in the crystal lattice and vice versa. Such an alternating packing mode in the favorable ‘cross’ arrangement provides more CB–CB interaction possibilities than that in the native CB5@CB10 assembly.9 It is well known in the cucurbit[n]uril solid state chemistry that multiple C–H⋯O hydrogen bonds are responsible for the peculiar properties of the homologues, such as solubility, thermal stability, self-assembly and crystal-to-crystal transformations.12 We postulate that also the unusual cocrystallization of three different homologues can be explained by the cumulative effect of multiple C–H⋯O hydrogen bonds that are effectively realized between CB5@CB10 and CB7. Surprisingly, despite the intensive CB–CB interactions, the complex is highly hydrated in the solid state, and we have been able to identify at least 79 water molecules per three CB molecules. For comparison, there are only 25 water molecules per CB5@CB10 solid state supramolecular assembly.9 The careful examination of the crystal packing reveals that the efficient CB–CB interactions are achieved only in two dimensions within the tightly associated layers composed of CB5@CB10 and CB7 (Fig. 4). There are no CB–CB interactions between the layers. The spaces between the layers are richly loaded with water molecules that render the crystals vulnerable when out from the mother solution due to rapid solvent loss. Besides multiple water molecules, the aqueous layers contain disordered molecules of hydrochloric acid. The hydrochloric acid solution was used upon fractional crystallization in the attempt to separate individual CBn homologues.
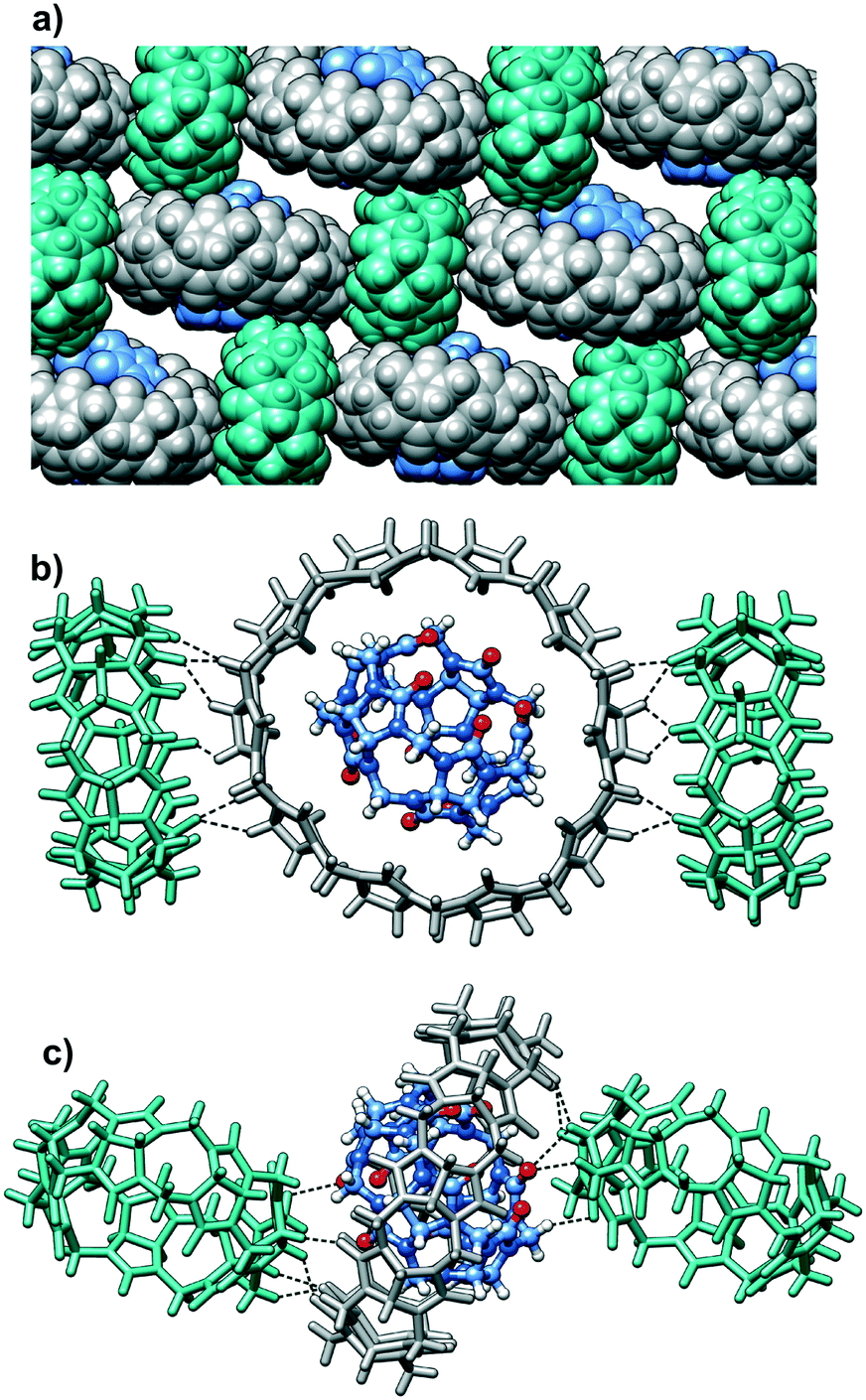 | ||
| Fig. 3 (a) Part of the supramolecular assembly of CB5@CB10 and CB7. Note that each CB5@CB10 ensemble is surrounded by four CB7 molecules in the ‘cross’ arrangement and vice versa; (b) the methine and methylene groups of CB10 donate multiple C⋯H⋯O hydrogen bonds to the carbonyl oxygen atoms of CB7; (c) the methine and methylene groups of CB7 form C–H⋯O hydrogen bonds toward CB5 and CB10 portals. | ||
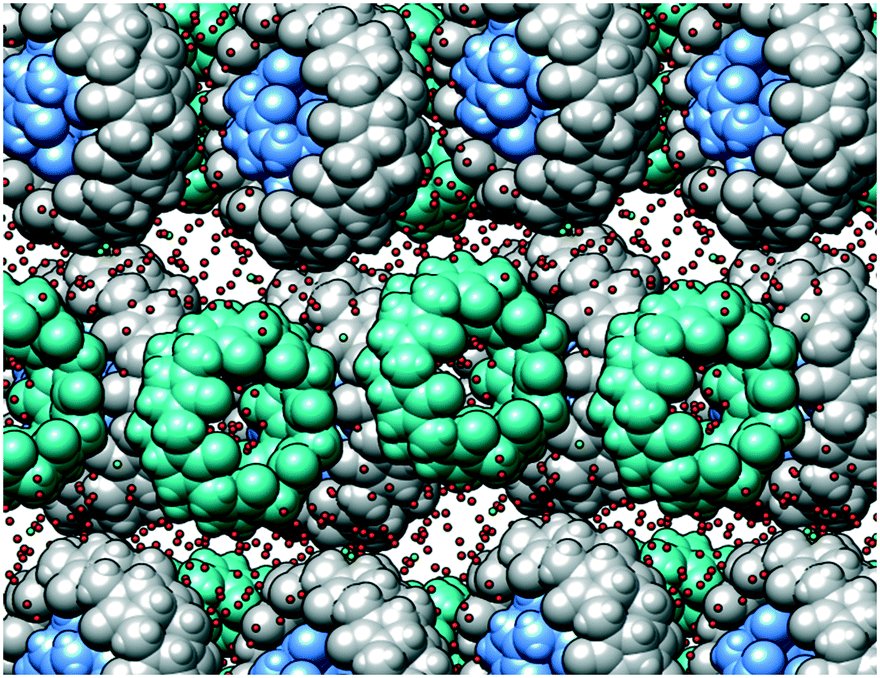 | ||
| Fig. 4 The crystal packing viewed along the a direction. The CB layers are separated by rich aqueous regions. CB7 is colored sea green, CB10 is grey and CB5 is cornflower blue. There are no CB–CB interactions between the layers. | ||
In conclusion, crystallization driven self-assembly between cucurbit[5]uril, cucurbit[7]uril and cucurbit[10]uril is described. The cocrystal comprises the CB5@CB10 inclusion complex interacting with CB7via outer surface interactions. The small differences in solubility of the cucurbit[n]uril homologues, the rigid geometry of the macrocycles and the cumulative effect of the multiple C–H⋯O hydrogen bonds contribute to this peculiar ‘failure’ of fractional crystallization. The example demonstrates that besides the significant advances in the understanding, prediction and control of molecular solids,13 the crystallization outcome is sometimes rather unpredictable. The cocrystallization of different cucurbit[n]uril homologues is not a mere crystallographic curiosity.14 We believe that this phenomenon can shed some light on the nucleation/crystallization from multi-component mixtures15 and supramolecular alignment of different macrocycles in space without the help of a linker.16 Since CB7 and CB5@CB10 interact with each other in the specific direction (‘cross’ geometry) and the overlap between them is partial, such an assembly mode potentially enables future utilization of the cavity features of the heteromacrocyclic system towards even more complex geometries.
Experimental
Cucurbit[n]urils were synthesized according to the literature procedure.8b The cocrystals of mixed sized cucurbit[n]urils were obtained serendipitously upon very slow evaporation of the ‘contaminated’ fraction containing differently sized cucurbit[n]urils upon fractional crystallization from the reaction mixture.Crystal data: (C30H30N20O10)@(C60H60N40O20)·(C42H42N28O14)·2HCl·79(H2O), Mr = 5169.6, colourless, monoclinic, space group P21/c, a = 20.3464(1), b = 37.9267(3), c = 30.4684(2) Å, β = 97.040(1)°, V = 23334.3(3) Å3, Z = 4, ρcalc = 1.47 g cm−3, SuperNova Agilent diffractometer, Cu-Kα radiation, T = 100.0(1) K, μ(CuKα) = 1.38 mm−1, θmax = 65.1°, 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 788 reflections measured, 39678 unique, 3495 parameters, R = 0.102, wR = 0.291 (R = 0.119, wR = 0.314 for all data), GooF = 1.08. CCDC 1986639.
788 reflections measured, 39678 unique, 3495 parameters, R = 0.102, wR = 0.291 (R = 0.119, wR = 0.314 for all data), GooF = 1.08. CCDC 1986639.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. A. Grzybowski, C. E. Wilmer, J. Kim, K. P. Browne and K. J. M. Bishop, Soft Matter, 2009, 5, 1110 RSC.
- G. Coquerel, Chem. Soc. Rev., 2014, 43, 2286 RSC; Z. Gao, S. Rohani, J. Gong and J. Wang, Engineering, 2017, 3, 343 CrossRef CAS.
- S. P. Kelley, L. Fábián and C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 79 CrossRef CAS PubMed.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. RodriguezHornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147 CrossRef CAS.
- M. Lusi, CrystEngComm, 2018, 20, 7042 RSC.
- C. Aakeröy, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 387–391 CrossRef CAS PubMed; N. K. Duggirala, M. L. Perry, O. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640 RSC; L. Sun, Y. Wang, F. Yang, X. Zhang and W. Hu, Adv. Mater., 2019, 31, 1902328 CrossRef PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952 CrossRef CAS PubMed.
- (a) J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS; (b) A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094 CrossRef CAS PubMed; (c) A. Chakraborty, A. X. Xu, D. Witt, J. Lagona, J. C. Fettinger and L. Isaacs, J. Am. Chem. Soc., 2002, 124, 8297 CrossRef CAS PubMed; (d) H. Cong, X. L. Ni, X. Xiao, Y. Huang, Q.-J. Zhu, S.-F. Xue, Z. Tao, L. F. Lindoy and G. Wei, Org. Biomol. Chem., 2016, 14, 4335 RSC.
- A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and Ian Dance, Angew. Chem., Int. Ed., 2002, 41, 275 CrossRef CAS.
- J.-X. Liu, R.-L. Lin, L.-S. Long, R.-B. Huang and L.-S. Zheng, Inorg. Chem. Commun., 2008, 11, 1085 CrossRef CAS.
- L. X. Chen, J. L. Kan, H. Cong, T. J. Prior, Z. Tao, X. Xiao and C. Redshaw, Molecules, 2017, 22, 1147 CrossRef PubMed.
- D. Bardelang, K. Udachin, D. M. Leek, J. M. Margeson, G. Chan, C. I. Ratcliffe and J. A. Ripmeester, Cryst. Growth Des., 2011, 11, 5598 CrossRef CAS.
- J. W. Steed, Chem. Commun., 2018, 54, 13175 RSC.
- W. Clegg, Acta Crystallogr., Sect. C: Struct. Chem., 2019, 75, 833 CrossRef CAS PubMed.
- S. Cherukuvada, R. Kaur and T. N. G. Row, CrystEngComm, 2016, 18, 8528 RSC.
- A. Dawn, A. Eisenhart, M. Mirzamani, T. L. Beck and H. Kumari, Chem. Commun., 2018, 54, 7131 RSC.
Footnote |
| † CCDC 1986639. X-ray crystallographic file in CIF format. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00296h |
| This journal is © The Royal Society of Chemistry 2020 |