
Anion templated crystal engineering of halogen bonding tripodal tris(halopyridinium) compounds†
Émer M.
Foyle
and
Nicholas G.
White
 *
*
Research School of Chemistry, The Australian National University, Canberra, ACT, Australia. E-mail: nicholas.white@anu.edu.au; Web: www.nwhitegroup.com
First published on 24th March 2020
Abstract
In this work four new tripodal tris(halopyridinium) receptors containing potentially halogen bonding groups were prepared. The ability of the receptors to bind anions in competitive CD3CN/d6-DMSO was studied using 1H NMR titration experiments, which revealed that the receptors bind chloride anions more strongly than more basic acetate or other halide ions. The solid state self-assembly of the tripodal receptors with halide anions was investigated by X-ray crystallography. The nature of the structures was dependent on the choice of halide anion, as well as the crystallisation solvent. Halogen bond lengths as short as 80% of the sum of the van der Waals radii were observed, which is shorter than any halogen bonds involving halopyridinium receptors in the Cambridge Structural Database.
Introduction
In the last decade or so, there has been substantial development in the field of halogen bonding. Halogen bonds have received considerable attention in the fields of crystal engineering and also in solution phase supramolecular chemistry.1–5 Notably, halogen bond donor groups have been used to prepare strong and selective anion receptors that function in highly competitive media,6 including pure water.7–9 As well as their use in anion recognition, halogen bond donors10 have been used in anion binding catalysis11–13 and to template the self-assembly of a range of supramolecular architectures, including helices/helicates,14–17 interlocked molecules,18–21 and frameworks.22,23As well as these architectures, several halogen bonded capsules have been reported. In 2012, Aakeröy reported a capsule assembled by four I⋯N halogen bonds between fluoroiodobenzene and pyridine groups.24 The groups of Diederich and Rissanen have reported capsules based on resorcinarene cavitands assembled using halogen bonds. Typically these have used pyridine as the Lewis base, and either iodoalkynes or the iodonium cation as the Lewis acids,25–29 although Rissanen has reported an unusual example of an ammonium bearing resorcinarene which is assembled through R–NH3+⋯Cl−⋯I2⋯Cl−⋯+H3N–R interactions.30
Recently, Amendola, Mella and Metrangolo reported the tripodal tris(iodopyridinium) compounds 13+ and 23+ (Fig. 1) and showed that they formed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with halide, acetate and hydrogensulfate anions in CD3CN or 9:1 CD3CN:d6-DMSO solution.31 We were interested to see whether similar receptors containing solubilising alkoxy groups (i.e.33+–63+, Fig. 1) and halopyridinium motifs could be used to prepare halogen bonded capsules with anions if more than one equivalent of anion was used, i.e. if 1.5 equivalents of anion could be used to form receptor2·anion3 complexes.
:1 complexes with halide, acetate and hydrogensulfate anions in CD3CN or 9:1 CD3CN:d6-DMSO solution.31 We were interested to see whether similar receptors containing solubilising alkoxy groups (i.e.33+–63+, Fig. 1) and halopyridinium motifs could be used to prepare halogen bonded capsules with anions if more than one equivalent of anion was used, i.e. if 1.5 equivalents of anion could be used to form receptor2·anion3 complexes.
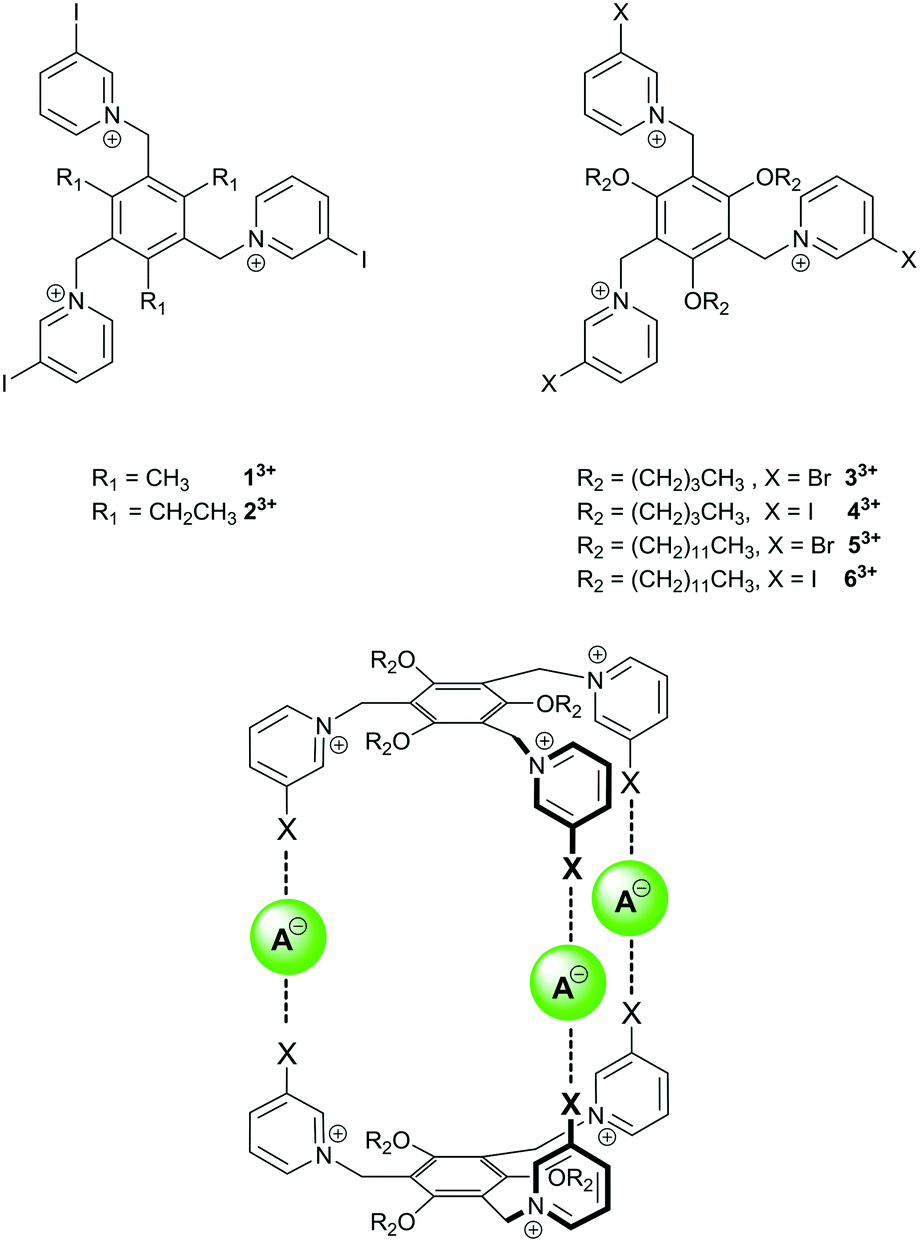 | ||
| Fig. 1 Previously-reported tris(iodopyridinium) anion receptors 13+ and 23+, compounds 33+–63+ used in this study, and targeted halogen-bonded capsules. | ||
We were also interested to see what effect varying the nature of the halogen bonding substituent and the crystallisation solvent has on the solid state structures formed. It has been demonstrated that solution phase halogen bonding is relatively insensitive to solvent,32 particularly when compared with hydrogen bonded systems.33 There have been few studies on the effect of crystallisation solvent on solid state halogen bonded structures. Notably, Perutz, Hunter and Brammer have shown that solvent could alter the balance of halogen and hydrogen bonding in a competitive co-crystallisation process by “turning off” hydrogen bonding in a competitive solvent, while the halogen bond was not affected.34 However, other authors have shown that solvent has a significant effect on halogen bonded product formation.16,35,36
Herein we report our investigation of the crystal engineering of complexes of halide anions and cationic tris(halopyridinium) receptors. While we were unable to isolate the targeted capsules, a range of interesting solid state supramolecular structures were realised.
Results and discussion
Synthesis
The new halopyridinium receptors were synthesized starting from the known triethers 7 (ref. 37) and 8,38 which were themselves prepared by alkylating commercially-available phloroglucinol with the appropriate 1-bromoalkane (Scheme 1). The triethers were then bromomethylated using paraformaldehyde and HBr in acetic acid, as described by Pittelkow,39 to give the new trifunctional scaffolds 9 and 10. Alkylation with either 3-bromopyridine or 3-iodopyridine gave the halopyridinium compounds 33+–63+ as bromide salts. Anion exchange using ammonium hexafluorophosphate gave the corresponding PF6− salts [3·(PF6)3–6·(PF6)3]. The yields for the alkylation reactions were relatively modest, and while we attempted to increase the yields by increasing the equivalents of halopyridine added or by prolonging the reaction time, neither of these significantly increased reaction yield.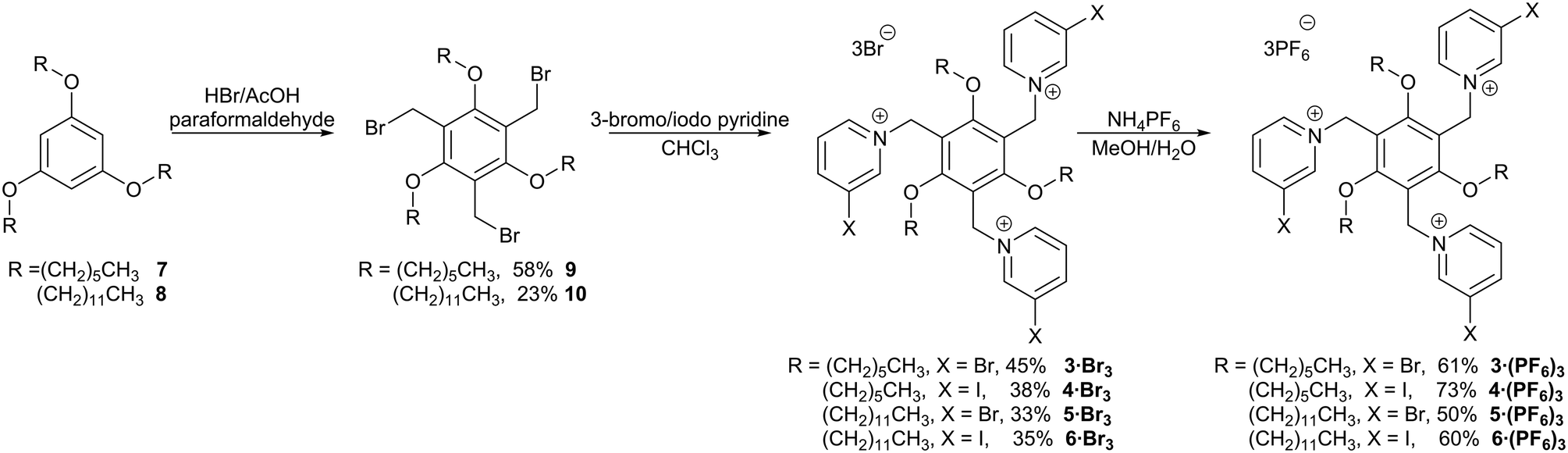 | ||
| Scheme 1 Synthesis of potentially halogen bonding tectons 33+–63+. | ||
Solution binding studies
In order to gain information about the strength of the halogen bonding interactions in solution, we conducted 1H NMR anion titration experiments using the dodecyl substituted receptors 5·(PF6)3 and 6·(PF6)3. Quantitative experiments were not conducted with hexyl containing receptors 3·(PF6)3 and 4·(PF6)3 as these compounds tended to precipitate from solution upon the addition of halide anions. Anions were added as their tetrabutylammonium (TBA) salts and titrations were conducted in the competitive solvent system of 9:1 CD3CN:d6-DMSO, to allow for comparison with the work of Amendola, Mella and Metrangolo, who reported solution phase binding data for the similar compounds 1·(PF6)3 and 2·(PF6)3 in this solvent mixture.31
As shown in Fig. 2, the resonances corresponding to pyridinium protons H1 and H4 moved downfield from their initial position, while the other peaks did not show significant shifts.40 Peak H4 moved the most and therefore was used to calculate an association constant. The peak movement was fitted to a 1:1 binding isotherm using Bindfit.41 We also attempted to fit the titration to data to 1:2 or 2:1 binding isotherms, but these did not give sensible fits.42 Interestingly, a value of 2316(87) M−1 was obtained, which is smaller than the chloride association constants reported for 1·(PF6)3 and 2·(PF6)3 in the same solvent (5000–11000 M−1).31
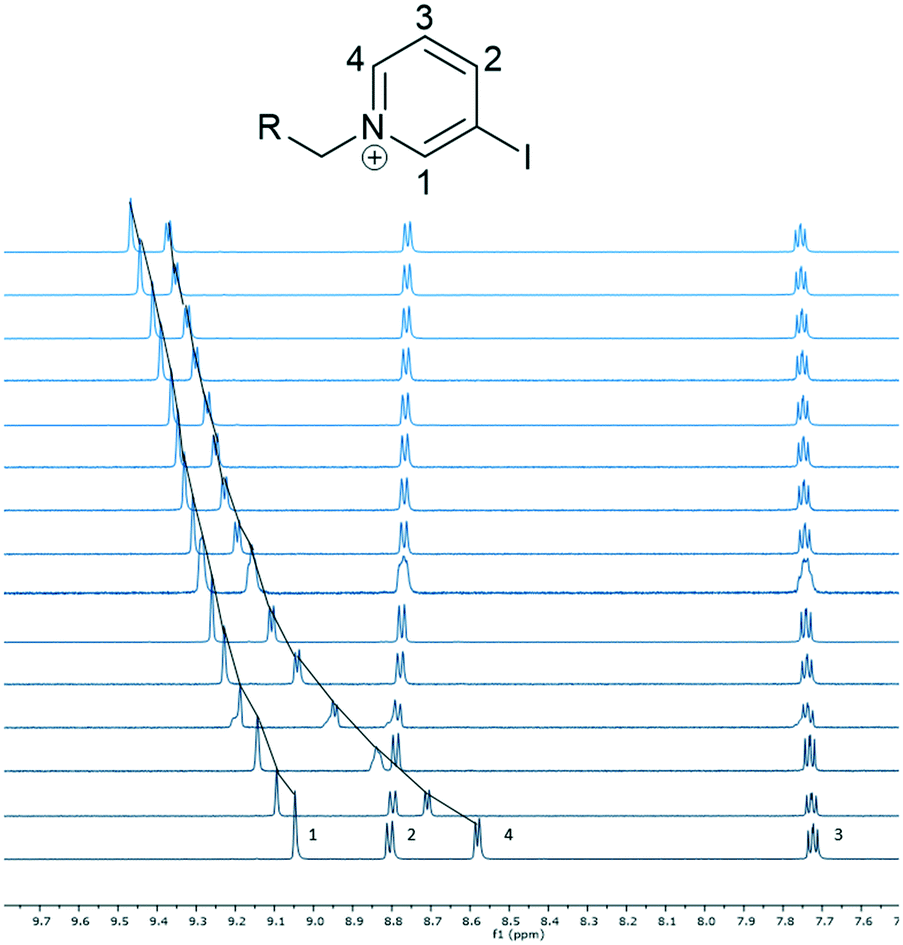 | ||
| Fig. 2 Truncated 1H-NMR spectra of 6·(PF6)3 upon addition of TBA·Cl (9:1 CD3CN:d6-DMSO, 600 MHz, 298 K).40 | ||
The solution phase anion binding of tris(bromopyridinium) compound 5·(PF6)3 was also investigated. Similar to 6·(PF6)3, H1 and H4 moved downfield during the titration experiments, with H4 again moving the most. Therefore, the chemical shifts of H4 were used to calculate 1:1 binding constants using Bindfit.41 We also attempted to fit the titration data to 1:2 or 2:1 binding isotherms, but these did not give sensible fits.42
The association constant for bromopyridinium receptor 53+ with chloride was found to be 2608 (± 231) M−1, which is within error of that obtained for iodopyridinium receptor 63+. This is surprising given previous studies that have shown that iodo-halogen bond donors tend to give stronger binding than their bromo counterparts.43–45 The binding constants for bromide and iodide were also determined with their respective values being 850 ± 41 M−1 and 544 ± 42 M−1 (Table 1 and Fig. 3). Both of these values are lower than the association constant obtained for chloride, which is expected as chloride is a more basic anion then bromide or iodide.
:1 CD3CN:d6-DMSO
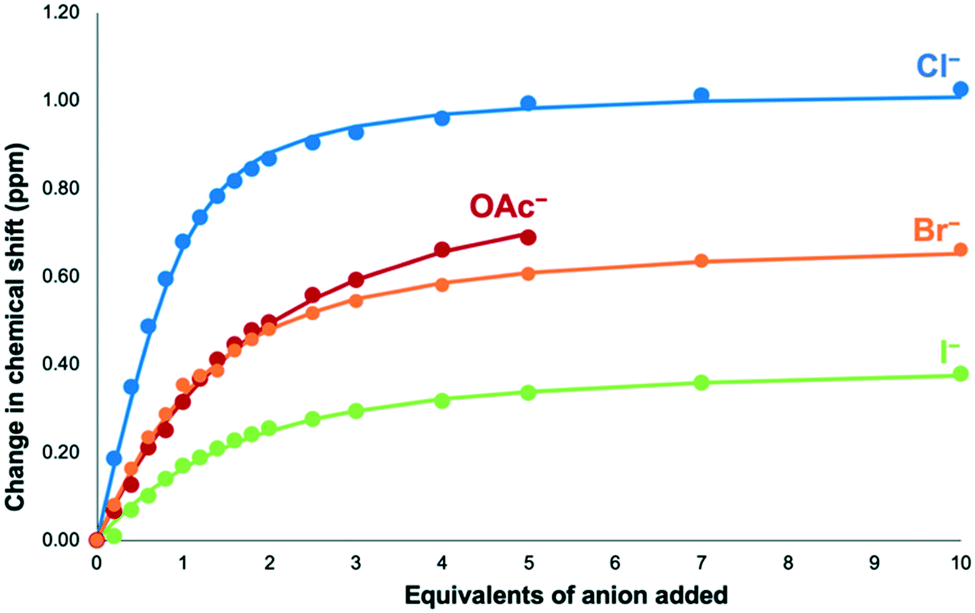 | ||
| Fig. 3
1H NMR binding curve for addition of TBA·Cl, TBA·Br, TBA·I and TBA·OAc to 5·(PF6)3, showing the change in chemical shift of H4 (9:1 CD3CN:d6-DMSO, 298 K). Dots represent the observed points and the line represents 1:1 binding isotherms fitted using Bindfit.41 Precipitation was observed after addition of five equivalents of TBA·OAc. | ||
NMR titrations with acetate were also completed to see if a non-spherical anion affects the binding ability of 5·(PF6)3. In a similar fashion to the other anion titrations H1 and H4 both shift downfield with H4 shifting the most. The data were fitted to a 1:1 stoichiometry with the binding constant found to be 419 ± 17 M−1. This value is lower than that calculated for the halide anions despite acetate being more basic than these anions. This may result from an innate preference for softer halide anions over harder oxoanions,10 or may be due to the differing size requirements of the spherical halide anions compared to larger acetate.
Solid state structures
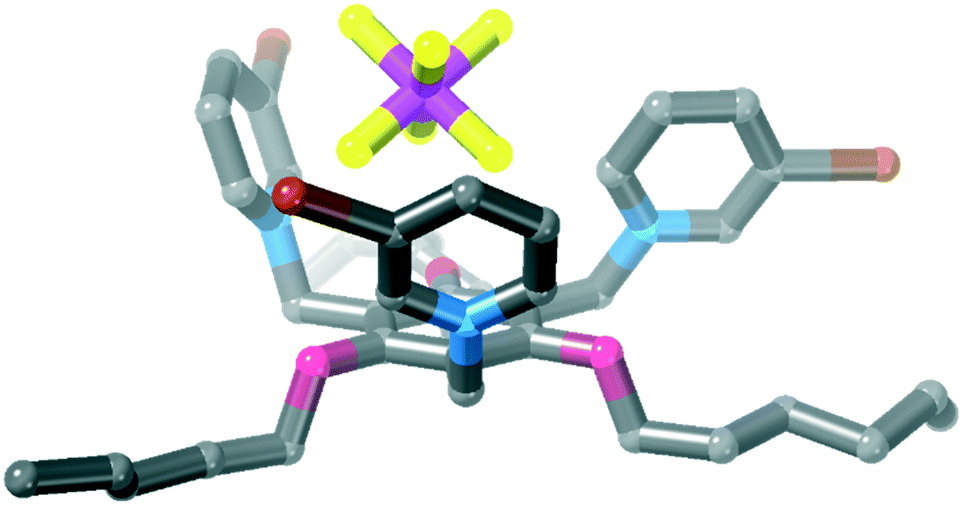 | ||
| Fig. 4 Crystal structure of 3·(PF6)3. Hydrogen atoms, disorder of one bromopyridinium ring and counter ions omitted for clarity. | ||
Structures crystallised from aprotic solvents
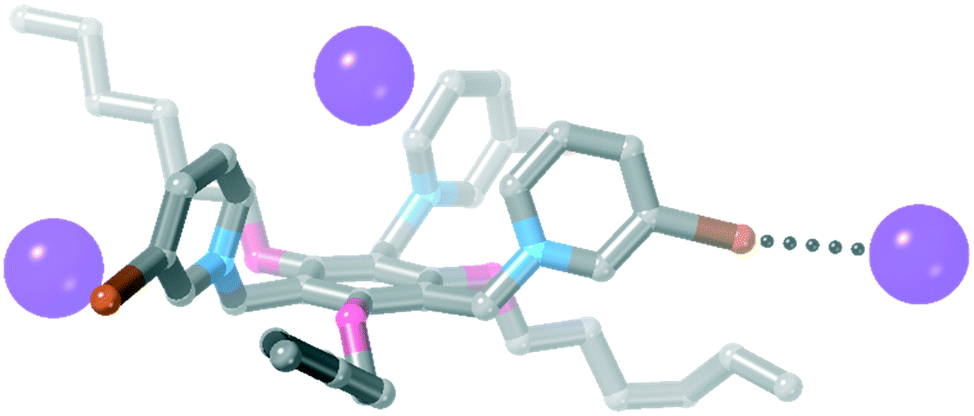 | ||
| Fig. 5 The asymmetric unit of 3·I3. Hydrogen atoms have been omitted for clarity. | ||
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) and the asymmetric unit contains 1/3 of the receptor, one iodide anion and 1/3 of an acetonitrile molecule. Each iodopyridinium group forms a C–I⋯I− halogen bond [Fig. 6, I⋯I = 3.5955(4) Å, 88% ∑rvdW].47
and the asymmetric unit contains 1/3 of the receptor, one iodide anion and 1/3 of an acetonitrile molecule. Each iodopyridinium group forms a C–I⋯I− halogen bond [Fig. 6, I⋯I = 3.5955(4) Å, 88% ∑rvdW].47
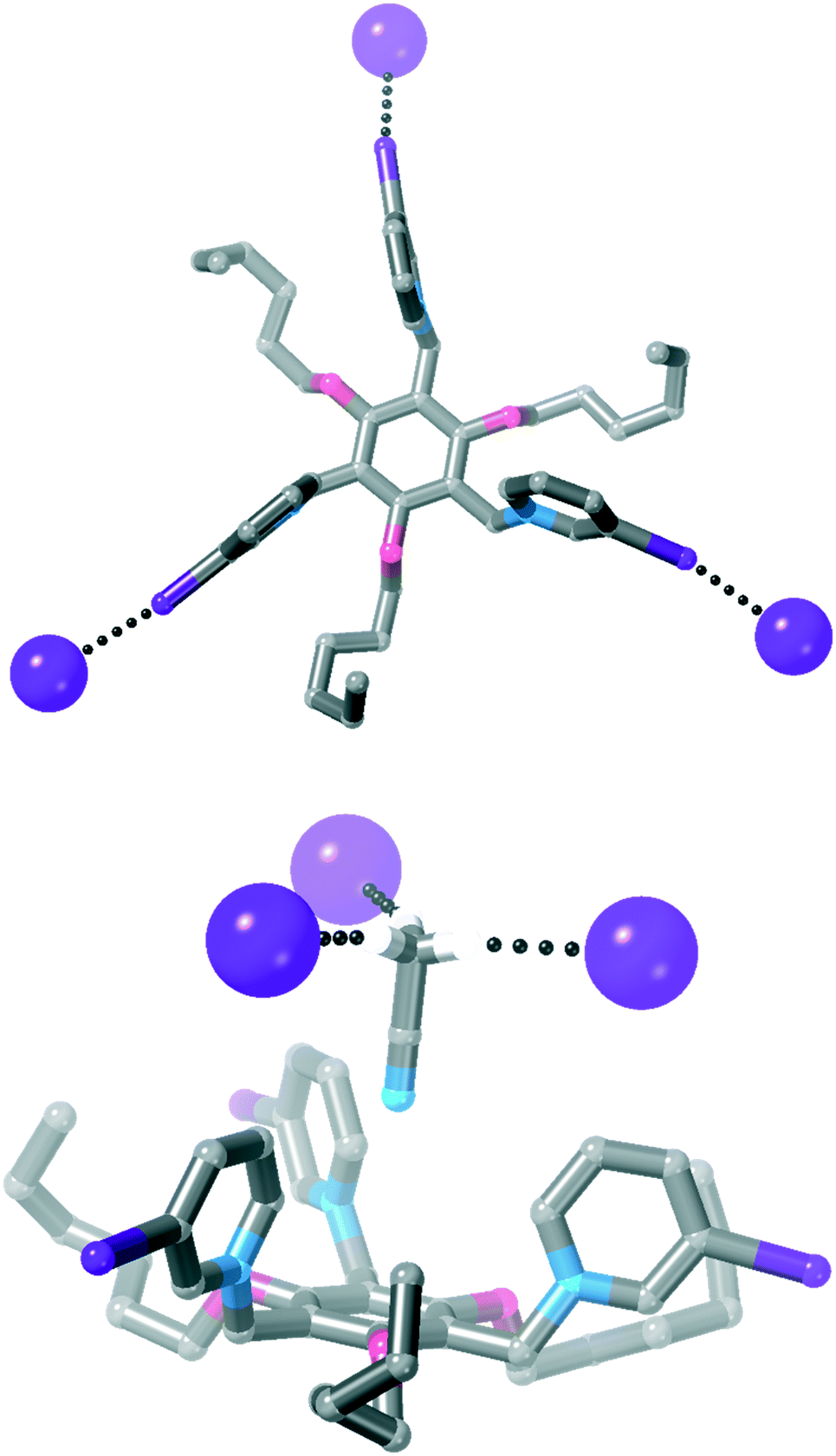 | ||
| Fig. 6 Two views of the crystal structure of 4·I3 (most hydrogen atoms are omitted for clarity, as is the acetonitrile solvent molecule in the top view of the structure). | ||
An acetonitrile solvent molecule sits in the centre of the tripodal receptor, forming relatively long C–H⋯N hydrogen bonds with iodopyridinium C–H groups [H⋯N = 2.59 Å, 94% ∑rvdW].47 The methyl hydrogen atoms on this solvent molecule then form further hydrogen bonds to iodide anions [H⋯I− = 3.16 Å, 98% ∑rvdW],47 which assemble the 3D crystal structure.
space group and are isostructural with 4·I3 (see Fig. S37 and S38† for structures). The halogen bond length in iodopyridinium containing 4·Br3 [I⋯Br−: 3.4685(6) Å, 89% ∑rvdW]47 is notably shorter than that in bromopyridinium containing 3·Br3 [Br⋯Br−: 3.640537(16) Å, 93% ∑rvdW].47
space group with a molecule of acetonitrile in the centre of the tripodal receptor. Interestingly, when 3·Cl3 was crystallised by vapour diffusion of diethyl ether into an acetonitrile solution of 3·(PF6)3 and three equivalents of TBA·Cl, the crystal structure was different. The asymmetric unit contains one receptor, three chloride anions and two water molecules. One of the bromopyridinium rings is rotationally disordered (see ESI† for full crystallographic details).
The two water molecules hydrogen bond to two of the chloride anions (H⋯Cl− = 2.28 and 2.29 Å, 75 and 76% ∑rvdW).47 The other chloride anion sits inside the cavity of the tripodal receptor forming hydrogen bonds to C–H groups of the pyridinium ring (H⋯Cl− = 2.46–2.83 Å, 81–94% ∑rvdW).47 This halide anion also forms a halogen bond to an adjacent receptor [Br⋯Cl− = 3.129(6) Å, 85% ∑rvdW],47 and this halogen bond assembles the receptors into a 1D polymeric structure (Fig. 7). We note that a somewhat similar structure of 1·Br·(PF6)2 was obtained by Amendola, Mella, Metrangolo and co-workers when a halogen bond between one receptor and a bromide anion sitting in the cavity of the next receptor gave a polymeric structure.31
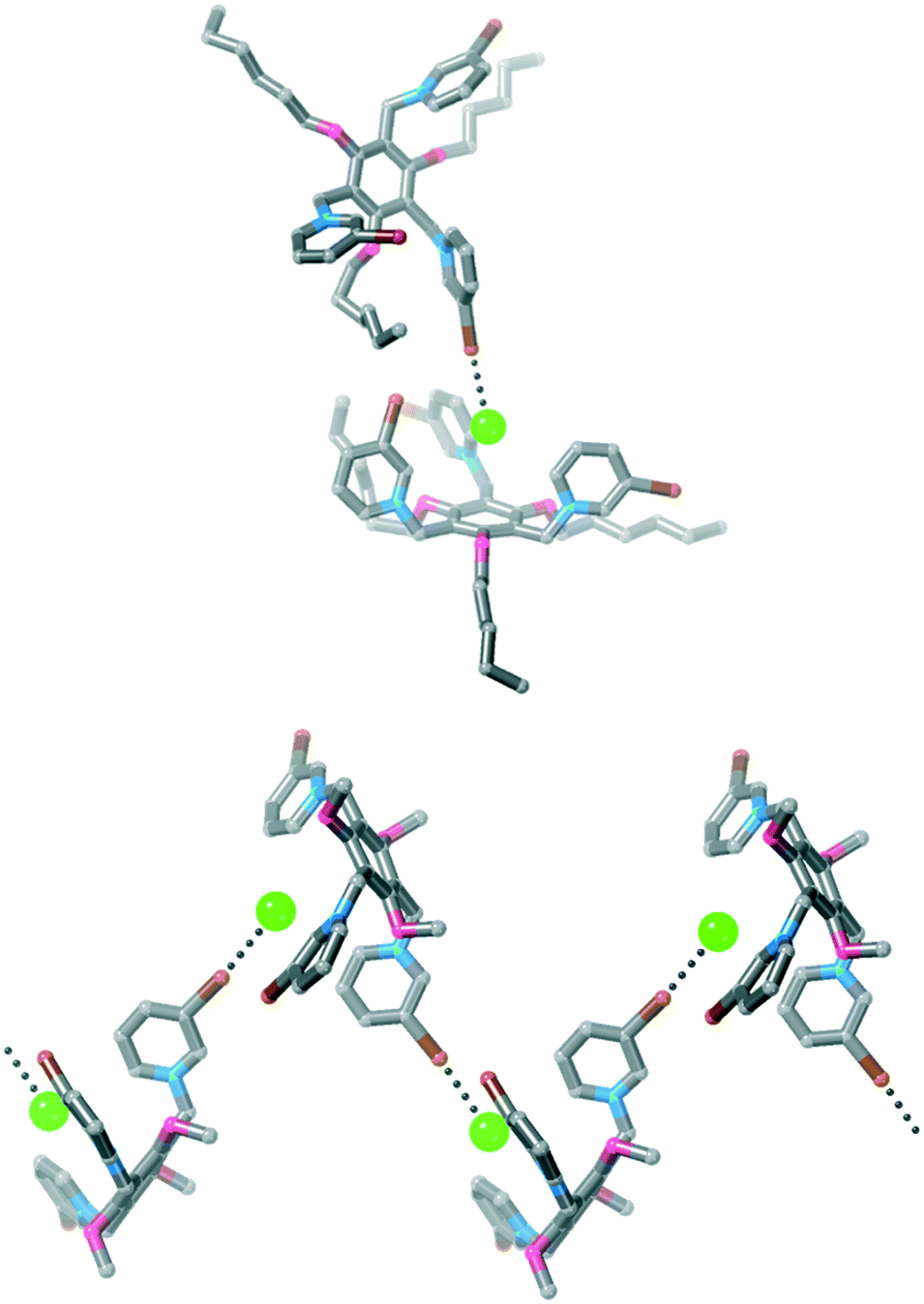 | ||
| Fig. 7 Two views of the structure of 3·Cl3 showing the 1D polymeric structure. Hydrogen atoms, disorder, solvent molecules and some chloride anions have been omitted for clarity. | ||
Despite numerous attempts, we were unable to obtain single crystals of 4·Cl3 from aprotic solvents. It appears that this complex is significantly less soluble that the other halide-containing complexes, which complicates growing crystals of it. We were however able to crystallise 43+ and chloride from methanol (see next section).
Structures crystallised from protic solvents
When 3·(PF6)3 was crystallised from ethanol/methanol in the presence of TBA·Cl, no chloride was incorporated into the structure (see previous). | ||
| Fig. 8 Structure of 4·Cl·(PF6)2. Hydrogen atoms and one of the hexafluorophosphate anions are omitted for clarity. | ||
Interestingly, in this structure the 1,3,5-trialkoxybenzene scaffolding48 of the host does not occur and rather than the six substituents alternating up-down around the central benzene ring, two substituents located para to one another are one face of the ring, while the other four are co-facial (Fig. 9). A hexafluorophosphate anion is located in the centre of the tripodal host, and two iodopyridinium rings form halogen bonds to bromide anions. One halide anion is located on a two-fold rotation axis and forms two crystallographically-equivalent halogen bonds, linking two receptors into a halogen bonded dimer [I⋯Br− = 3.3124(11) Å, 85% ∑rvdW].47 The other halide anion forms a halogen bond to another iodopyridinium ring [I⋯Br− = 3.2163(9) Å, 82% ∑rvdW].47 It is notable that these halogen bonds are significantly longer (as % ∑rvdW) than those in 4·Cl·(PF6)2, which contains a halogen bonded square incorporating a methanol solvate.
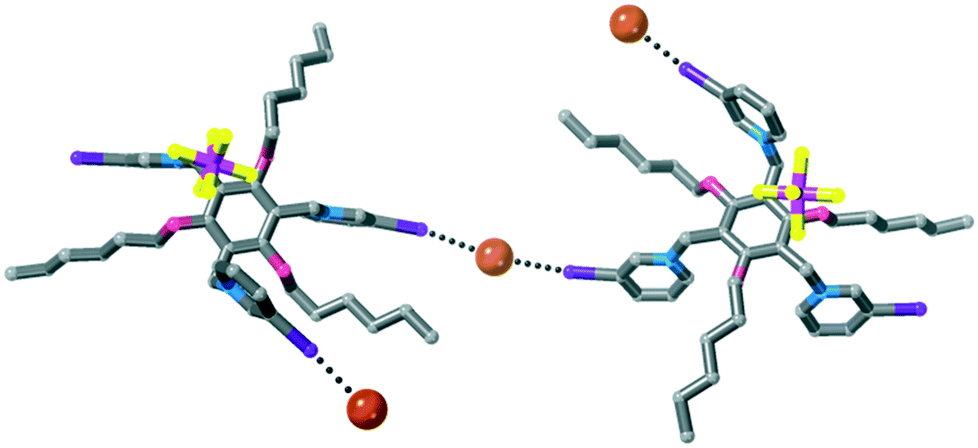 | ||
| Fig. 9 Structure of 4·Br1.5·(PF6)1.5. Hydrogen atoms omitted for clarity. | ||
Survey of the Cambridge Structural Database
We undertook a survey of the Cambridge Structural Database (CSD) to see how typical the interactions that we observed are. The CSD (version 5.40, November 2018 + 1 update) was searched for 2-, 3-, and 4-bromo and iodopyridinium structures containing halogen bonds to either halide or oxoanions (defined as an interaction < ∑rvdW).47,49 These data are shown in Table 2.| Motif | Cl− | Br− | I− | Oxoanions |
|---|---|---|---|---|
| 2-BrPy | — | 87–90% (89%, n = 4) | — | — |
| 3-BrPy | — | 91–92% (92%, n = 2) | 90–93% (92%, n = 2) | — |
| 4-BrPy | — | — | — | — |
| 2-IPy | — | — | 83% (83%, n = 1) | — |
| 3-IPy | 82–87% (84%, n = 2) | 83–87% (85%, n = 7) | 82–88% (85%, n = 7) | 82–99% (88%, n = 11) |
| 4-IPy | — | 84–95% (85%, n = 8) | 82–92% (84%, n = 9) | — |
Perhaps the most notable observation is how few halopyridinium⋯anion halogen bonds have been characterised by X-ray crystallography, particularly given the ease of synthesis of these compounds. From the relatively small number of data available, it is clear that interactions involving the iodopyridinium group tend to be shorter than those involving the bromopyridinium group (mean values 83–88% vs. 89–92%, respectively).
Notably, the shortest halopyridinium⋯anion interactions in the CSD are 82% ∑rvdW, meaning the 80% in 4·Cl·(PF6)2 is the shortest such interaction yet observed. The structure of 3·Cl3 also contains a shorter halogen bond (85% ∑rvdW (ref. 47)) than any bromopyridinium halogen bond previously reported, although given this is the first bromopyridinium⋯chloride interaction this is not hugely surprising (basic chloride often forming quite short halogen bonding interactions). 4·Br1.5·(PF6)1.5 also contains a shorter iodopyridinium⋯bromide interaction (82% ∑rvdW (ref. 47)) than any reported to date. Interestingly, the halogen bonds in 3·Br3, 3·I3, 4·Br3 and 4·I3 are quite a lot longer (bromopyridinium⋯anion bond lengths: 92–93% ∑rvdW, iodopyridinium⋯anion bond lengths: 88–89% ∑rvdW).47
In order to put these results in context, we also searched the CSD for structures involving haloimidazolium⋯anion and halotriazolium⋯anion halogen bonds (Table 3). A similar CSD survey was carried out by Beer in 2014, who noted that iodine halogen bond donors tend to give shorter interactions than bromine, while the choice of anion did not have a significant effect on halogen bond length.50 This appears to still be broadly true, although we note the tendency of chloride to form short interactions.
| Motif | Cl− | Br− | I− | Oxoanions |
|---|---|---|---|---|
| 2-BrIm | — | 82–89% (86%, n = 10) | 87–91% (90%, n = 5) | 81–99% (87%, n = 4) |
| 4-BrIm | — | 86–93% (88%, n = 7) | 91% (91%, n = 1) | 88–96% (91%, n = 3) |
| 2-IIm | 74–81% (78%, n = 13) | 78–83% (80%, n = 20) | 77–83% (81%, n = 21) | 74–100% (80%, n = 8) |
| 4-IIm | — | 82% (82%, n = 1) | — | 79–96% (84%, n = 4) |
| BrTrz | — | 84–88% (86%, n = 4) | 86% (86%, n = 1) | 85–90% (88%, n = 2) |
| ITrz | 76–78% (77%, n = 7) | 79–81% (80%, n = 2) | 80–80% (80%, n = 2) | 73–99% (80%, n = 10) |
When comparing haloimidazolium and halotriazolium groups with halopyridinium (i.e.Tables 2 and 3), it is clear that there are far more structurally characterised examples involving the five-membered heterocycles than pyridinium groups. Interactions are noticeably shorter for the imidazolium and triazolium groups than halopyridiniums, presumably as the extra nitrogen atoms in these heterocycles further polarise the halogen atom.
Discussion of crystal engineering implications
Solvent appears to play a significant role in the outcome of crystallisations of these systems. The structures of 3·Br3, 4·Br3 and 4·I3 are isostructural: these all crystallise from acetonitrile and contain an acetonitrile solvent located in the electron deficient cleft of the tripodal host. In the case of 4·Br3, even when a mixture of DMF and acetonitrile was used for crystallisation, only the acetonitrile was incorporated into the structure. The structure of 3·I3 was crystallised from acetone instead of acetonitrile and in this case, iodide sits in the tripod's cleft instead of acetone. It is notable that acetone is a better hydrogen bond acceptor (although a worse hydrogen bond donor) than acetonitrile,33 so the incorporation of solvent into the receptor's cleft is not purely due to hydrogen bond accepting ability. Interestingly when 3·Cl3 was crystallised from acetonitrile, a structure different to 3·Br3, 4·Br3 and 4·I3 was formed, with a chloride anion sitting in the centre of the receptor.Our attempts to prepare halogen bonded capsules (Fig. 1) from these tripodal receptors were unsuccessful. We attribute this to a range of factors: firstly it would appear that in solution, binding an anion in the C–H rimmed cleft of the receptors is preferred to halogen bonding interactions (explaining the 1:1 binding stoichiometry observed). Despite the incorporation of solubilising groups, it was not possible to selectively crystallise given ratios of coordinating (i.e. halide) to non-coordinating (i.e. PF6−) anions, which would presumably be necessary to isolate the putative cages in the solid state. To favour the formation of the targeted architectures it may be necessary to add substituents to the pyridinium rings that block the central cavity of the host; alternatively other methods to force the halogen atoms to point upwards (as shown in Fig. 1) rather than outwards may also aid this. Initial attempts to prepare tripodal receptors from 3,5-dibromopyridine, which would have halogen atoms pointing both upwards and outwards, were unsuccessful.51
Conclusions
In this work new tripodal tris(halopyridinium) receptors were synthesised. The ability of these receptors to bind anions in competitive 9:1 CD3CN:d6-DMSO solvent media was investigated using 1H NMR titration experiments. These experiments revealed that the receptors bound anions in a 1:1 binding mode with chloride binding more strongly than bromide, iodide and acetate. The hydrogen and halogen bonded solid state self-assembly of the tripodal hosts with anions was investigated, and it was found that solvent had a surprisingly large effect on the nature of the product. A range of crystal structures were obtained with either solvent or anions located in the receptors' cleft and varying number of halogen bonds forming. A comparison with other halopyridinium structures in the CSD revealed that the structure of 4·Cl·(PF6)2 contains the shortest known halopyridinium⋯anion halogen bond. Generally, halopyridinium⋯anion halogen bonds are longer than haloimidazolium or halotriazolium⋯anion interactions. However, given the ease with which halopyridinium receptors can be synthesised, we suggest they are worthy of further investigations for anion templated crystal engineering applications.
Experimental
General remarks
Triethers 7 (ref. 37) and 8 (ref. 38) were prepared by alkylation of phloroglucinol as previously described. All other materials were bought from commercial suppliers and used as received. Details of instrumentation and characterisation data are provided in the ESI.†Bromomethyl scaffold 9
Triether 7 (1.00 g, 2.64 mmol, 1.00 equiv.) and paraformaldehyde (454 mg, 15.1 mmol, 5.72 equiv.) were suspended in 33% HBr in AcOH (8 mL) in a heavy-walled vial. The reaction mixture was heated at 85 °C for 3 hours and then cooled to room temperature. DCM (20 mL) was added and the organic phase was washed with water (3 × 10 mL) and brine (10 mL) and then dried (MgSO4). The organic layer was concentrated under vacuum, to give the crude product as an orange oil. The crude product was dissolved in DCM and filtered through a short plug of silica, then and concentrated and dried under vacuum to give the pure product as a yellow oil. Yield: 1.02 g, (1.55 mmol, 59%).1H-NMR (400 MHz, CDCl3): 4.58 (s, 6H), 4.25 (t, J = 6.7 Hz, 6H), 1.93 (p, J = 6.7 Hz, 6H), 1.53–1.60 (m, 6H), 1.38–1.41 (m, 12H), 0.93 (t, J = 7.0 Hz, 9H) ppm. 13C-NMR (101 MHz, CDCl3): 159.7, 123.3, 75.2, 31.9, 30.5, 25.7, 23.3, 22.8, 14.2 ppm. HRESI-MS (pos.) 679.0790, calc. for [C27H45Br3O3·Na]+ = 679.0791 Da.
3·Br 3
Compound 9 (150 mg, 0.232 mmol, 1.0 equiv.) and 3-bromopyridine (0.064 mL, 100 mg, 0.66 mmol, 3.0 equiv.) were dissolved in chloroform (5 mL) and heated at reflux under N2 for 4 hours. The resultant yellow solution was concentrated under vacuum to give the crude product as a yellow tacky solid. The crude product was then stirred in boiling toluene for 1 hour. The resulting solid was then isolated by filtration, washed with toluene (3 × 5 mL) and diethyl ether (5 mL) and dried under vacuum to give the product as a pale yellow crystalline solid. Yield: 0.11 g (0.099 mmol, 45%).1H-NMR (400 MHz, DMSO-d6): 9.59 (s, 3H) 9.15 (d, J = 6.5 Hz, 3H), 8.89 (d, J = 8.4 Hz, 3H), 8.03 (dd, J = 8.4, 6.5 Hz, 3H), 5.92 (s, 6H), 3.91 (t, J = 6.8 Hz, 6H), 1.59 (p, J = 6.8 Hz, 6H), 1.10–1.27 (m, 18H), 0.84 (t, J = 6.9 Hz, 9H) ppm.13C-NMR (101 MHz, DMSO-d6): 160.3, 148.1, 145.8, 143.5, 129.0, 121.7, 118.0, 76.0, 54.4, 31.1, 29.3, 24.6, 22.0, 13.9 ppm. HRESI-MS (pos.) 485.5535 [C42H57Br4N3O3]2+ = 485.5541 Da.
3·(PF 6 ) 3
3·3Br (0.10 g, 0.088 mmol, 1.0 equiv.) and NH4PF6 (0.086 g, 0.53 mmol, 6.0 equiv.) were suspended in methanol (10 mL). The suspension was heated at 60 °C and stirred until all the solid was dissolved. Water (3 ml) was then added dropwise until the solution stayed cloudy and the solution was left at room temperature for two hours. The resulting precipitate was isolated by filtration, washed with water (3 × 5 mL) and diethyl ether (5 mL), and dried under vacuum to give a white powder. Yield: 0.072 g (0.054 mmol, 61%).1H-NMR (400 MHz, DMSO-d6): 9.42 (s, 3H), 8.92 (d, J = 8.1 Hz 3), 8.78 (d, J = 6.1 Hz, 3H), 8.08 (m, 3H), 5.86 (s, 6H), 3.85 (t, J = 6.6 Hz, 6H), 1.57–1.64 (m, J = 6.6 Hz, 6H), 1.12–1.28 (m, 18H), 0.85 (t, J = 6.8 Hz, 9H) ppm. 19F-NMR (376 MHz, DMSO-d6): −70.2 (d, J = 711.3 Hz) ppm. 31P-NMR (161 MHz, DMSO-d6): −144.2 (hept, J = 711.3 Hz) ppm. ESI-MS (pos): 518.7, calc. for [C42H57Br3F6N3O3P]2+: 518.6 Da; 1180.5 calc. for [C42H57Br3F12N3P2]+: calc. 1180.1 Da.
4·Br 3
Compound 9 (100 mg, 0.15 mmol, 1.0 equiv.) and 3-iodopyridine (92 mg, 0.45 mmol, 3.0 equiv.) were dissolved in chloroform (5 mL) and the solution was heated to reflux for 4 hours under N2. After this time the solution had gone cloudy and a white solid had formed. The solution was cooled to room temperature, then the solid was isolated by filtration, washed with chloroform (3 × 5 mL) and diethyl ether (5 mL) and dried to give a white powder. Yield: 0.073 g (0.057 mmol, 38%).1H-NMR (400 MHz, DMSO-d6): 9.58 (s, 3H), 9.06 (d, J = 6.3 Hz, 3H), 8.95 (d, J = 8.1 Hz, 3H), 7.84 (dd, J = 8.1, 6.3 Hz, 3H), 5.87 (s, 6H), 3.88 (t, J = 6.8 Hz, 6H), 1.58 (p, J = 6.8 Hz, 6H), 1.06–1.33 (m, 18H), 0.85 (t, J = 6.9 Hz, 9H) ppm. 13C-NMR (101 MHz, DMSO-d6): 160.2, 153.2, 149.9, 143.1, 128.8, 118.1, 95.9, 75.9, 54.0, 31.2, 29.3, 24.6, 22.0, 14.0 ppm. HRESI-MS (pos.): 556.5334 calc. for [C42H57I3N3O3Br]2+, 556.5343 Da.
4·(PF 6 ) 3
4·Br 3 (0.040 g, 0.031 mmol, 1.0 equiv.) and NH4PF6 (0.030 mg, 0.19 mmol, 6.1 equiv) were suspended in methanol (7 mL) and heated to 60 °C until all material dissolved. Water (2 mL) was added dropwise until the solution stayed cloudy, and then the suspension was left to stand at room temperature for 3 days. The solid was isolated by filtration, washed with water (3 × 5 mL) and diethyl ether (5 mL) and dried under vacuum to give the product as a white powder. Yield: 0.034 g (0.023 mmol, 73%).1H-NMR (400 MHz, DMSO-d6): 9.48 (s, 3H), 8.99 (d, J = 8.1 Hz, 3H), 8.69 (d, J = 6.1 Hz, 3H), 7.86 (dd, J = 8.1, 6.1 Hz, 3H), 5.81 (s, 6H), 3.83 (t, J = 6.8 Hz, 6H), 1.61 (p, J = 6.8 Hz, 6H), 1.10–1.36 (m, 18H), 0.86 (t, J = 6.9 Hz, 9H) ppm. 19F-NMR (376 MHz, DMSO-d6): −70.2 (d, J = 711.3 Hz) ppm. 31P-NMR (161 MHz, DMSO-d6): −144.2 (hept, J = 711.3 Hz) ppm. ESI-MS (pos.): 1322.1, calc. for [C42H57F12I3N3O3P2]+ = 1322.5 Da.
Bromomethyl scaffold 10
Triether 8 (0.750 g, 1.20 mmol 1.00 equiv.), paraformaldehyde (0.219 g, 7.14 mmol, 5.95 equiv.) and 33% HBr in glacial acetic acid (6 mL) were suspended in a heavy-walled glass vial. The mixture was then heated at 85 °C for 3 hours. The reaction was cooled to room temperature and DCM (15 mL) was added. The solution was washed with water (3 × 10 mL), brine (10 mL), dried (MgSO4) and then concentrated under vacuum to give an orange oil. The crude product was dissolved in DCM and filtered through a short plug of silica, then concentrated under vacuum to give a yellow oil. The resultant product was then purified using column chromatography (3:2 DCM:petroleum spirits), to give the product as a pale yellow oil. Yield: 0.249 g (0.273 mmol, 23%).
1H-NMR (400 MHz, CDCl3): 4.58 (s, 6H), 4.26 (t, J = 6.7 Hz, 6H), 1.94 (p, J = 6.7 Hz, 6H), 1.50–1.61 (m, 6H), 1.21–1.46 (m, 48H), 0.89 (t, J = 6.8 Hz, 9H). 13C NMR (101 MHz, CDCl3): 159.7, 123.3, 75.2, 32.1, 30.5, 29.9, 29.84, 29.80, 29.7, 29.6, 26.0, 23.2, 22.9, 14.3 ppm. Due to overlap in alkyl peaks in the 13C NMR spectrum there is one fewer carbon environment than expected. HRESI-MS (pos.) 829.4511, calc. for [C45H81Br2O3, i.e. loss of Br−]+ = 829.4527 Da.
5·Br 3
Bromomethyl compound 10 (0.104 g, 0.114 mmol, 1.00 equiv.) and 3-bromopyridine (0.032 mL, 52 mg, 0.36 mmol, 3.2 equiv.) were dissolved in chloroform (6 mL) and heated to reflux for 4 hours under N2. The reaction mixture was then taken to dryness to leave a tacky yellow solid. This was then dissolved in boiling toluene (10 mL) and then cooled in the freezer. The toluene was then decanted off and the resultant waxy solid was dried under vacuum to give the product as a yellow crystalline powder. Yield: 0.052 g (0.037 mmol, 33%).1H NMR (400 MHz, CDCl3): 10.10 (s, 3H), 9.72 (d, J = 6.2 Hz, 3H), 8.44 (d, J = 8.7 Hz, 3H), 7.99 (dd, J = 8.7, 6.2 Hz, 3H), 6.36 (s, 6H), 4.20 (t, J = 6.5 Hz, 6H), 1.60–1.72 (m, 6H), 1.15–1.35 (m, 54H), 0.88 (t, J = 6.8 Hz, 9H) ppm. 13C NMR (101 MHz, CDCl3): 161.2, 147.9, 146.3, 144.9, 129.7, 122.9, 118.2, 78.4, 55.6, 32.1, 30.9, 30.0, 29.94, 29.89, 29.9, 26.2, 22.9, 14.3 ppm. Due to overlap in alkyl carbon environments there are 2 fewer peaks than expected. HRESI-MS (pos.) 1304.3077, calc. for [C60H93Br5O3N3]+ = 1304.3091 Da.
5·(PF 6 ) 3
The bromide salt 5·Br3 (0.100 g, 0.0726 mmol, 1.00 equiv.) and NH4PF6 (0.071 mg, 0.43 mmol, 5.9 equiv.) were suspended in methanol (10 mL). The suspension was then heated to 60 °C until all the solid dissolved. Water (1.5 mL) was then added dropwise until the solution turned cloudy. The solution was left to stand overnight and then the precipitate was isolated by filtration, washed with methanol/water (10:1.5, 3 × 10 mL), and the solid dried under vacuum to leave the product as a white powder. Yield: 0.047 g (0.036 mmol, 50%).
1H NMR (400 MHz, CDCl3): 8.90 (s, 3H), 8.49 (d, J = 6.2 Hz, 3H), 8.44 (d, J = 8.5 Hz, 3H), 7.72–7.81 (m, 3H), 5.69 (s, 6H), 4.07 (t, J = 6.2 Hz, 6H), 1.80–1.93 (m, 6H), 1.18–1.46 (m, 54H), 0.88 (t, J = 6.8 Hz, 9H) ppm. 19F-NMR (376 MHz, CDCl3): −72.1 (d, J = 713 Hz) ppm. 31P-NMR (161 MHz, CDCl3): −142.8 (hept, 713 Hz) ppm. ESI-MS (pos.) 644.7 calc. for [C60H93Br3F6N3O3P3]2+ = 644.7 Da.
6·Br 3
Compound 10 (0.11 g, 0.12 mmol, 1.0 equiv.) and 3-iodopyridine (0.75 g, 0.36 mmol, 3.0 equiv.) were dissolved in chloroform (7 mL) and heated to reflux for 48 hours under N2. Over this time a white solid precipitated out of the reaction mixture. This solid was isolated by filtration, washed with chloroform (3 × 5 mL) and diethyl ether (5 mL) and air dried to give the product as a white powder. Yield: 0.64 g (0.042 mmol, 35%).1H-NMR (400 NMR, DMSO-d6): 9.54 (s, 3H), 8.99 (d, J = 5.7 Hz, 3H), 8.94 (d, J = 8.0 Hz, 3H), 7.85 (t, J = 7.6 Hz, 3H), 5.84 (s, 6H), 3.86 (t, J = 5.5 Hz, 2H), 1.58 (p, J = 6.9 Hz, 6H), 1.11–1.24 (m, 54H), 0.86 (t, J = 6.5 Hz, 9H) ppm. 13C-NMR (101 MHz, DMSO-d6): 160.2, 153.2, 149.8, 143.1, 128.7, 118.1, 95.8, 75.9, 54.0, 31.3, 29.4, 29.0, 28.7, 25.0, 22.1, 14.0 ppm. Due to overlap in alkyl carbon environments there are 4 fewer peaks than expected. HRESI-MS (pos.) 682.6763, calc. for [C60H93I3N3O3Br]2+ = 682.6752 Da.
6·(PF 6 ) 3
The bromide salt 6·Br3 (0.030 g, 0.020 mmol, 1.0 equiv.) and NH4PF6 (0.019 g, 0.12 mmol, 6.0 equiv.) were suspended in methanol (5 mL) and heated to 60 °C until all solid had dissolved. Water (2 mL) was then added dropwise until the solution stayed cloudy. The solution was then left to stand overnight. The resultant solid was filtered, washed with water (3 × 5 mL), and dried under vacuum to give the product as a white powder. Yield: 0.020 g (0.012 mmol, 60%).1H-NMR (400 MHz, CDCl3): 9.06 (s, 3H), 8.61 (d, J = 8.1 Hz, 3H), 8.51 (d, J = 6.2 Hz, 3H), 7.66 (dd, J = 8.1, 6.2 Hz, 3H), 5.69 (s, 6H), 4.07 (t, J = 6.5 Hz, 6H), 1.80–1.90 (m, 6H), 1.20–1.47 (m, 54H), 0.88 (t, J = 6.8 Hz, 9H) ppm. 19F-NMR (376 MHz, CDCl3): −72.0 (d, J = 714 Hz) ppm. 31P-NMR (161 MHz, CDCl3): −144.9 (hept, J = 714 Hz) ppm. ESI-MS (pos.) 715.1, calc. for [C60H93F6I3N3O3P]2+ = 715.2 Da.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Samuel Walker for initial experiments towards the synthesis of compounds 7–10, Drs Stephanie Boer and Michael Gardiner for assistance with X-ray crystallography, and the Australian Research Council for providing an Australian Government RTP scholarship to EMF and a Discovery Early Career Research Award (DE170100200) to NGW. Part of this work was carried out on the MX1 Macromolecular Crystallography beam line at the Australian Synchrotron, Victoria, Australia.52Notes and references
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed.
- A. Caballero, N. G. White and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 1845–1848 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039 CrossRef CAS PubMed.
- M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Chem. – Eur. J., 2016, 22, 185–192 CrossRef CAS PubMed.
- S. P. Cornes, M. R. Sambrook and P. D. Beer, Chem. Commun., 2017, 53, 3866–3869 RSC.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187–7191 CrossRef CAS PubMed.
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- A. Casnati, R. Liantonio, P. Metrangolo, G. Resnati, R. Ungaro and F. Ugozzoli, Angew. Chem., Int. Ed., 2006, 45, 1915–1918 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- C.-F. Ng, H.-F. Chow and T. C. W. Mak, Angew. Chem., Int. Ed., 2018, 57, 4986–4990 CrossRef CAS PubMed.
- C. J. Massena, D. A. Decato and O. B. Berryman, Angew. Chem., Int. Ed., 2018, 57, 16109–16113 CrossRef CAS PubMed.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499–507 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Eur. J. Org. Chem., 2019, 3433–3441 CrossRef CAS.
- M. C. Pfrunder, A. J. Brock, J. J. Brown, A. Grosjean, J. Ward, J. C. McMurtrie and J. K. Clegg, Chem. Commun., 2018, 54, 3974–3976 RSC.
- V. I. Nikolayenko, D. C. Castell, D. P. van Heerden and L. J. Barbour, Angew. Chem., Int. Ed., 2018, 57, 12086–12091 CrossRef CAS PubMed.
- C. B. Aakeröy, A. Rajbanshi, P. Metrangolo, G. Resnati, M. F. Parisi, J. Desper and T. Pilati, CrystEngComm, 2012, 14, 6366–6368 RSC.
- O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339–12344 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343–8351 RSC.
- N. K. Beyeh, F. Pan and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 7303–7307 CrossRef CAS PubMed.
- V. Amendola, G. Bergamaschi, M. Boiocchi, N. Fusco, M. V. La Rocca, L. Linati, E. L. Presti, M. Mella, P. Metrangolo and A. Miljkovic, RSC Adv., 2016, 6, 67540–67549 RSC.
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8, 5392–5398 RSC.
- D. E. Barry, C. S. Hawes, S. Blasco and T. Gunnlaugsson, Cryst. Growth Des., 2016, 16, 5194–5205 CrossRef CAS.
- C.-F. Ng, H.-F. Chow and T. C. W. Mak, CrystEngComm, 2019, 21, 1130–1136 RSC.
- T. Ueda and T. Mochida, Organometallics, 2015, 34, 1279–1286 CrossRef CAS.
- M. L. Bushey, A. Hwang, P. W. Stephens and C. Nuckolls, J. Am. Chem. Soc., 2001, 123, 8157–8158 CrossRef CAS PubMed.
- B. E. Nielsen, H. Gotfredsen, B. Rasmussen, C. G. Tortzen and M. Pittelkow, Synlett, 2013, 24, 2437–2442 CrossRef CAS.
- We have used the same assignment of the pyridinium ring protons as Amendola, Mella and Metrangolo.31 This suggests that the peaks that move significantly are H1, next to the halogen bonding group, and H4 - the interior proton that can point into the interior of the tripodal host allowing C–H⋯anion hydrogen bonding (see solid state structures). An alternative assignment could be possible, where H4 is assigned to be the proton para to the pyridinium nitrogen atom, as this is also near to the halogen bonding group. However, for the reasons discussed later in the section on the discussion of crystal engineering implications, we think this is less likely and Amendola, Mella and Metrangolo's assignment is the most probable.
- Bindfit, accessed at http://supramolecular.org Search PubMed.
- A 1:1 binding stoichiometry may appear somewhat surprising given that anions could bind either within the cleft of the tripodal receptor, or to any of the three halopyridinium groups. However, the binding data appear quite clear, with 1:1 binding isotherms fitting
well (Fig. 3, and Fig. S26, S28, S30, S32 and S34†) and attempts to fit the data to other binding stoichiometries giving entirely non-sensible results. We note that Amendola, Mella and Metrangolo observed 1:1 binding isotherms for the binding of anions to related receptors.31.
- M. Cametti, K. Raatikainen, P. Metrangolo, T. Pilati, G. Terraneo and G. Resnati, Org. Biomol. Chem., 2012, 10, 1329–1333 RSC.
- L. C. Gilday and P. D. Beer, Chem. – Eur. J., 2014, 20, 8379–8385 CrossRef CAS PubMed.
- Given this surprising result, we repeated the titration and obtained the same result (within experimental error). We also added AgI to 6·(PF6)3 to rule out any residual halide being present from incomplete anion exchange, but observed no precipitation.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- Wang and Hof have studied the effect of incorporating ethyl substituents in the 1, 3 and 5 positions on the conformation of other substituents in the 2, 4 and 6 positions. Their work showed that the alternating up–down arrangement is generally favoured according to gas phase calculations, but this effect is not overwhelmingly. A study of crystal structures in the Cambridge Structural Database49 revealed that only about two thirds of 2,4,6-substituted-1,3,5-triethylbenzene structures had the alternating up–down conformation: X. Wang and F. Hof, Beilstein J. Org. Chem., 2012, 8, 1–10 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- N. G. White, A. Caballero and P. D. Beer, CrystEngComm, 2014, 14, 3722–3729 RSC.
- We heated 9 with an excess of 3,5-dibromopyridine in refluxing chloroform, 1,4-dioxane or toluene for several days, but there was no evidence of any reaction occurring.
- T. M. McPhillips, S. E. McPhillips, H.-J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter and R. P. Phizackerley, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data, details of anion binding titrations, and crystallographic data. CCDC 1984628–1984635. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00241k |
| This journal is © The Royal Society of Chemistry 2020 |