
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanisms of crystallisation in polysorbates and sorbitan esters†
Emelie J.
Nilsson‡
 *ab,
Tania K.
Lind‡
ab,
Dieter
Scherer
c,
Tatyana
Skansberger
ab,
Kell
Mortensen
d,
Johan
Engblom
ab and
Vitaly
Kocherbitov
*ab
*ab,
Tania K.
Lind‡
ab,
Dieter
Scherer
c,
Tatyana
Skansberger
ab,
Kell
Mortensen
d,
Johan
Engblom
ab and
Vitaly
Kocherbitov
*ab
aBiomedical Sciences, Faculty of Health and Society, Malmö University, SE-205 06 Malmö, Sweden. E-mail: emelie.nilsson@mau.se; vitaly.kocherbitov@mau.se
bBiofilms – Research Center for Biointerfaces, Malmö University, SE-205 06 Malmö, Sweden
cLONZA AG, Basel, CH-4002, Switzerland
dNiels Bohr Institute, University of Copenhagen, DK-2100 Copenhagen, Denmark
First published on 21st May 2020
Abstract
Polysorbates (PS), commonly known as Tween™, are some of the most extensively used excipients and protein stabilisers in biopharmaceutical products worldwide. It is stipulated in the pharmacopoeia specifications that these ethoxylated surfactants are complex mixtures comprised of a wealth of molecular species. While little is known about the propensity of PSs to crystallise, they are used in applications ranging from food products, cosmetics, different types of drug dosage forms like creams and oral products to parenteral applications. However, in recent years a range of issues and safety concerns have appeared when using them for stabilising biopharmaceutical products including precipitation, particle formation, and adverse biological effects. Therefore, the aim of this study was to thoroughly characterise the thermotropic behaviour and mechanism of crystallisation of polysorbates with different hydrocarbon tails and their non-ethoxylated sorbitan ester equivalents for comparison. A systematic and comprehensive product characterisation was carried out, taking advantage of a combination of complementary techniques such as differential scanning calorimetry, matrix assisted laser desorption ionisation time-of-flight and small- and wide-angle X-ray diffraction. We show that polysorbate 80, having an unsaturated hydrocarbon tail, crystallises by the ethylene oxide chains in the headgroup. Polysorbate 20, 40, and 60, containing saturated hydrocarbon esters tails, crystallise not only by the ethylene oxide chains but also by their hydrocarbon tails. An analogous behaviour was observed for the PS non-ethoxylated equivalents, the sorbitan esters. Sorbitan esters with saturated hydrocarbon tails displayed a crystallisation of the tail upon cooling, whereas the sorbitan ester with unsaturated hydrocarbon tail displayed no crystallisation.
Introduction
Polyoxyethylene (20) sorbitan monoesters, also called polysorbates (PS), are non-ionic surfactants with several brand names such as Canarcel and Tween™. PSs are used in numerous applications within biochemistry, food production, and cosmetics, as well as for pharmaceutical purposes. Examples of the use of PSs are as excipients in vaccines, emulsifiers or stabilization of proteins against unfolding.1 Polysorbates belong to an old class of chemicals. A patent (GB573789) belonging to Atlas Powder Company, describing the synthesis, dates back to 1943 and already in 1947 publications in the life science field appeared.2,3 This was before the days of analytical methods, such as mass spectrometry or NMR, were implemented to elucidate the molecular composition. The composition of PS was an assumption only. While used as surfactants in creams etc. the effects of heterogeneity were not observed or looked for, and were not considered important. According to the US and European pharmacopoeia specifications, polysorbates are mixtures of partial esters of fatty acids with sorbitol and its anhydrides ethoxylated with approximately 20 mol of ethylene oxide (EO) added for each mole of sorbitol and sorbitol anhydrides (sorbitan and isosorbide). PS80, PS60, PS40 and PS20, refer to PS products where the main hydrocarbon tail is oleate, stearate, palmitate and laurate, respectively (C18:1, C18:0, C16:0 and C12:0). Although the pharmacopoeia standards stipulate the primary ester component for each product, within the monograph specifications, they are also all allowed a certain amount of other defined fatty acid ester tails which is product specific, see Table S1 in the ESI† for complete specifications. This means that within these broad specifications, PS60 e.g. can contain 40–60% of stearic acid and 30–50% of palmitic acid whereas PS40 should contain more than 92% palmitic acid. Furthermore, PSs are diverse and complex mixtures containing sorbitol, sorbitan, and isosorbide headgroups, a variety of fatty acid ester species, a broad distribution of EO units, and a varying number of hydrocarbon tails as well as unreacted products from the synthesis (see Fig. 1). In fact, in 2017, as many as 355 different compounds were identified in a commercial PS80 product.4 PSs are furthermore prone to undergo different kinds of chemical degradation such as oxidation, cleavage at the ethylene oxide units, and hydrolysis, which can lead to the formation of several by-products.5–7 The performance of formulations containing PSs is however largely taken for granted despite the fact that it is a complex mixture and that the raw material batch-to-batch variations can be large, although being within the broad specifications of pharmacopoeias.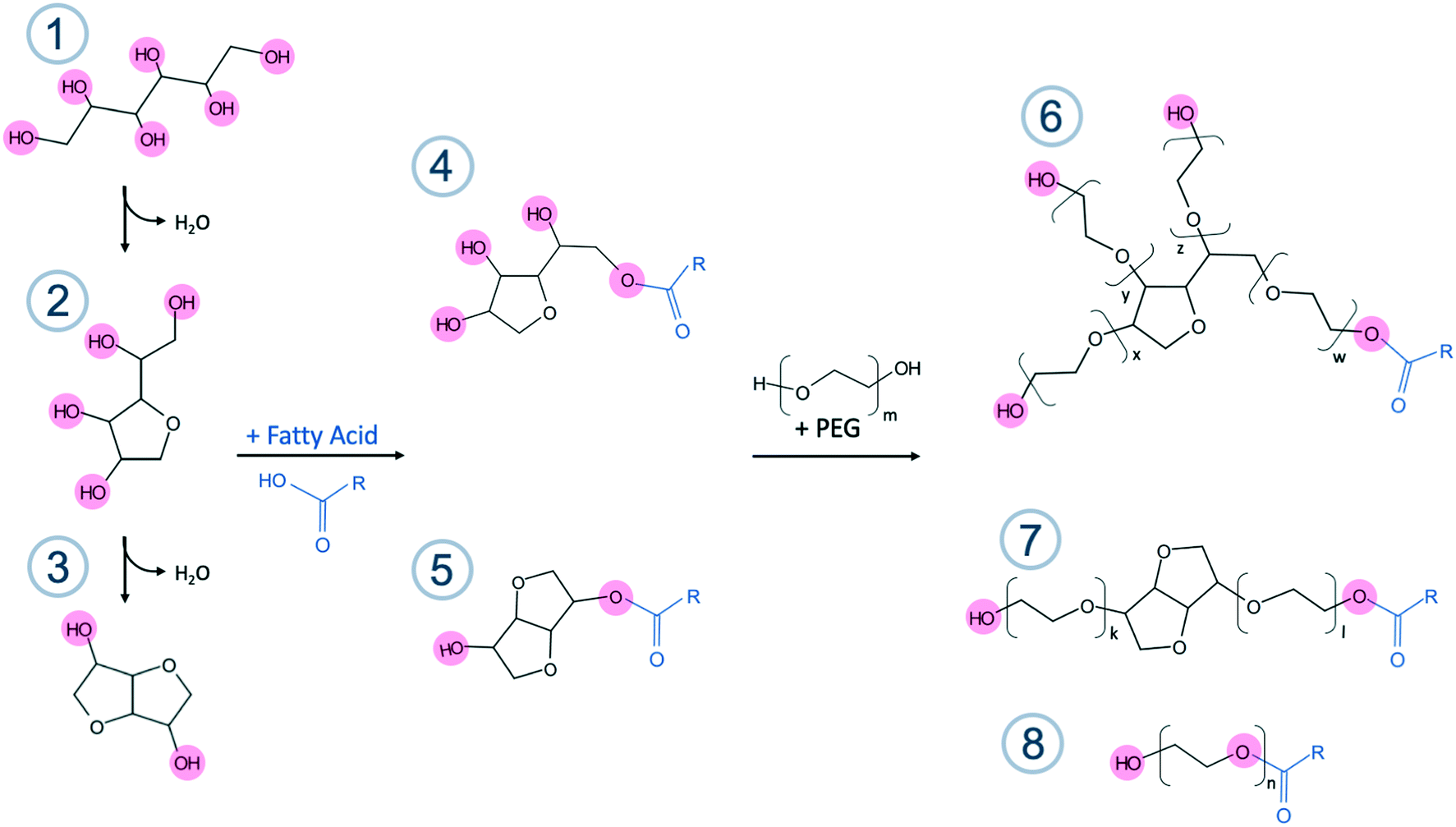 | ||
Fig. 1 Schematic diagram of a common synthesis route of PSs, revealing the origin of some of the more common subspecies present in the products. All active sites, marked in red, are positions where esterification with fatty acids (marked in blue, and R is a linear aliphatic chain) can occur, which explain the tetra-functionality of PS products compared to the di-functionality of isosorbide species. The amount of EO in polyoxyethylene (20) is defined as: x + y + z + w = 20. The numbers refer to 1: sorbitol, 2: sorbitan, 3: isosorbide, 4: sorbitan ester (SPAN), 5: isosorbide ester, 6: PEO sorbitan ester (PS), 7: PEO isosorbide ester and 8: PEO ester. The fatty acid for PS80 is oleic acid (R = (CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7CH3), for PS60 steric acid (R = (CH2)16CH3), for PS40 palmitic acid (R = (CH2)14CH3) and for PS20 it is lauric acid (R = (CH2)10CH3). CH(CH2)7CH3), for PS60 steric acid (R = (CH2)16CH3), for PS40 palmitic acid (R = (CH2)14CH3) and for PS20 it is lauric acid (R = (CH2)10CH3). | ||
Polysorbate 20 and polysorbate 80 are nowadays the most common surfactants used to protect therapeutic proteins against adsorption to interfaces and related instabilities.8 Their high surface activity, even at low concentrations, can be explained by their low critical micelle concentration (CMC) and high hydrophilic–lipophilic balance (HLB). About 80% of the commercial monoclonal antibody formulations contain PS20 or PS80, with typical concentrations ranging from 0.001% to 0.1% (w/v).8 However, particles have been observed in pharmaceutical formulations containing PS20 or PS80, rendering them unusable or possibly harmful.9–12 Moreover particles were observed earlier in formulations containing PS20 as compared to PS80.12 Little is known about the structure, composition and origin of these particles, but they have been suggested to consist of free fatty acids. As PSs are used as excipients in very small amounts in pharmaceutical formulations, and in turn only a fraction of the material forms the particles, the amount of isolated test material has been small, and contradictory conclusions have been reported in previous studies.9–12 Due to the extensive use of these surfactants in food and healthcare it is extremely important to assess their physical properties in order to avoid adverse biological effects. Investigations into how the actual composition, molecular size distribution, purity etc. affect their crystallisation and particle formation behaviour is therefore highly needed. Furthermore, due to the complexity of the formulations, investigations into the behaviour of the individual components, and explicitly the mechanism of crystallisation of neat PS products, is an essential starting point. This may provide new insights into prevention or postponement of particle formation in pharmaceutical formulations containing PS.
To assess the thermotropic behaviour of polysorbates and propose a mechanism of crystallisation, differential scanning calorimetry (DSC) was used as it allows for the detection of thermal events such as crystallisation, glass transitions and melting.13 In order to differentiate between the molecular moieties of polysorbates and their individual contribution to the crystallisation process, we focus here on separating the effect of the head from the tails in order to establish which part of the molecules that crystallises. Systematic studies were thus designed using polysorbates with varying tail composition (in terms of hydrocarbon length and level of saturation) and their non-ethoxylated equivalents (SPAN™) – see Table 1.
| Abbreviation | Primary ester | Tail length | EOa | Manufacturer (purity grade) |
|---|---|---|---|---|
| a Average number of EO units as specified by the manufacturer. | ||||
| PS80 | Polyethylene oxide sorbitan monooleate | C18:1 | 20 | KLK OLEO (ST), Croda (ST, HP, SR), NOF (UP) |
| PS60 | Polyethylene oxide sorbitan monostearate | C18:0 | 20 | Croda (ST) |
| PS40 | Polyethylene oxide sorbitan monopalmitate | C16:0 | 20 | Croda (ST) |
| PS20 | Polyethylene oxide sorbitan monolaurate | C12:0 | 20 | KLK OLEO (ST), Croda (ST, HP) |
| SPAN80 | Sorbitan monooleate | C18:1 | 0 | Croda (ST), KLK OLEO (ST), NOF (UP) |
| SPAN60 | Sorbitan monostearate | C18:0 | 0 | Croda (ST) |
| SPAN40 | Sorbitan monopalmitate | C16:0 | 0 | Croda (ST) |
| SPAN20 | Sorbitan monolaurate | C12:0 | 0 | Croda (ST) |
The thermotropic behaviour of the fatty acid equivalents to the hydrocarbon tail, as well as of alkanes, has previously been investigated and revealed a strong correlation between increased carbon chain length and increasing enthalpy of melting (J g−1).14–18 Polyethylene glycol (PEG), the free polymer equivalent, with a molecular weight Mw ranging from 1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (approx. 20–230 EO units), to the EO chains found in the PS headgroup, has also been subjected to similar studies, but revealed less of a trend between the length of the polymer and the melting enthalpy and melting temperature.19,20 These previous publications provide a starting point in understanding the DSC data of the PS and SPAN products studied here. In order to fully understand these materials, the thermal behaviour of the products is interpreted in combination with knowledge gained from both mass spectroscopy, providing insights into the complexity of the products, and X-ray diffraction data, providing detailed information of the structure of the PS products.
000 g mol−1 (approx. 20–230 EO units), to the EO chains found in the PS headgroup, has also been subjected to similar studies, but revealed less of a trend between the length of the polymer and the melting enthalpy and melting temperature.19,20 These previous publications provide a starting point in understanding the DSC data of the PS and SPAN products studied here. In order to fully understand these materials, the thermal behaviour of the products is interpreted in combination with knowledge gained from both mass spectroscopy, providing insights into the complexity of the products, and X-ray diffraction data, providing detailed information of the structure of the PS products.
Materials and methods
Materials
Polyethylene oxide sorbitan monooleate (PS80), polyethylene oxide sorbitan monostearate (PS60), polyethylene oxide sorbitan monopalmitate (PS40), polyethylene oxide sorbitan monolaurate (PS20), sorbitan monooleate (SPAN80), sorbitan monostearate (SPAN60), sorbitan monopalmitate (SPAN40), and sorbitan monolaurate (SPAN20) were obtained from different manufacturers and with different purity grades, as specified in Table 1 and S2 in ESI.† SPAN is a trademark, but for simplicity we refer to all sorbitan esters as SPANs regardless of the manufacturer. PEG 400, polyethylene glycol with an average molecular weight (Mw) of 400 g mol−1, was purchased form Sigma Aldrich. For Karl Fischer titration Coulomat AG and Hydranal Water standard 1.0 were used as received from VWR. Super-DHB matrix (consisting of a 9:1 mixture of 2,5-dihydroxybenzoic acid and 2-hydroxy-5-methoxybenzoic acid) and NaCl were purchased from Sigma Aldrich and dissolved in 99% ethanol purchased from VWR.
Differential scanning calorimetry (DSC)
For the DSC measurements, a DSC1 from Mettler Toledo was used and all samples were sealed in 40 μl aluminium pans. All DSC measurements were carried out with one of the following thermal programs. The standard thermal program consisted of two thermal cycles with cooling from 25 °C to −80 °C (segment 1), equilibration for 5 min at −80 °C (segment 2), heating from −80 °C to 80 °C (segment 3), equilibration for 5 min at +80 °C (segment 4). Segments 1–4 constitute the first thermal cycle. The second thermal cycle was carried out by cooling from 80 °C to −80 °C (segment 5), equilibration at −80 °C (segment 6), heating from −80 °C to 80 °C (segment 7), equilibration at 80 °C (segment 9) and then heating to 25 °C (segment 10). All heating and cooling segments were run with a scan rate of 10 °C min−1 and a nitrogen flow of 80 ml min−1. The second thermal program, designed to correspond to the temperature ramp during SWAXD measurements, consisted of only one thermal cycle. This program consisted of the three first segments from the previous program (cooling from 25 °C to −80 °C, equilibration for 5 min at −80 °C and heating from −80 °C to 80 °C), but this time with a temperature rate of 1 °C min−1 and with the same nitrogen flow of 80 ml min−1. All DSC data was evaluated and integrated using the STARe software (version 16). The melting peak temperature, Tmp, was defined as the peak position in the heating segments in the DSC curves. The enthalpy of melting, ΔHm, was defined as the integrated endothermic peak, see Fig. S1 in the ESI† for schematic explanation of the calculations. All samples were measured once as received, and after drying in vacuum for 12–18 hours to remove any residual water or solvent from the samples. Multiple measurements were carried out for each sample, to confirm the reproducibility of the DSC experiments (see Table S1 in the ESI†).Vacuum drying and sealing
A few hundred mg of each sample, in 1.5 ml vials, were placed in a vacuum pistol, containing 3 Å molecular sieves, connected to a vacuum pump. The samples were afterwards pipetted into aluminium DSC pans and sealed under nitrogen in a glove bag at relative humidity below 10%. The vials containing the samples for SWAXD were filled with nitrogen and sealed with a gas tight lid.Karl Fischer titration (KF)
The moisture content in the samples was determined using coulometric Karl Fischer titration with a 737 KF coulometer from Metrohm. The KF apparatus was calibrated using a standard sample (Hydranal Water standard 1.0), with a known water content of 0.1 wt%, and the averaged result of 3 calibration measurements was 0.1001 ± 0.0006 wt%. For the measurements, a sample was added (by weight) to the anode solution and after subtraction of the drift, the moisture content was provided in wt% (mg g−1) of the added sample. No sample preparation was done before the measurement. See Table 2 for the determined moisture content in the products.| Sample | Average moisture content [wt% ± std] | No. of measurements |
|---|---|---|
| PS80 | ||
| KLK OLEO ST | 2.498 ± 0.068 | 3 |
| Croda ST | 2.629 ± 0.015 | 10 |
| Croda HP | 0.144 ± 0.043 | 7 |
| Croda SR | 0.133 ± 0.045 | 5 |
| NOF UP | 0.124 ± 0.033 | 6 |
| PS20 | ||
| KLK OLEO ST | 2.808 ± 0.032 | 3 |
| Croda ST | 3.098 ± 0.021 | 3 |
| Croda ST | 3.675 ± 0.086 | 3 |
| Croda ST | 2.536 ± 0.402 | 3 |
| Croda HP | 0.185 ± 0.011 | 3 |
| SPAN80 | ||
| KLK OLEO ST | 0.213 ± 0.006 | 3 |
| Croda ST | 0.176 ± 0.010 | 3 |
| NOF UP | 0.122 ± 0.020 | 3 |
| SPAN20 | ||
| KLK OLEO ST | 0.214 ± 0.023 | 3 |
| Croda ST | 0.431 ± 0.010 | 3 |
| Croda ST | 0.146 ± 0.059 | 3 |
Matrix assisted laser desorption/ionisation-time of flight (MALDI-TOF)
2,5-Dihydroxybenzoic acid (super-DHB >99.0, Sigma Aldrich) was used as the matrix for MALDI-TOF experiments and prepared as a 5 mg ml−1 solution in ethanol with 10 mM NaCl added in order to exclusively detect sodiated adducts. Prior to use, the matrix was bath sonicated for 10 min in order to properly dissolve. All PS samples were prepared as 1 mg ml−1 solutions in ethanol and mixed 1:1 (vol:vol) with the matrix. 1 μl of each sample was spotted onto a target plate (MPT 384 polished steel, Bruker) in triplicates. All sample spots were allowed to air dry and crystallise on the target plate before MALDI-TOF measurements were performed. Positive ion MALDI-TOF mass spectrometry was carried out on an Ultraflex TOF/TOF, Bruker Daltonics instrument equipped with a 337 nm N2 laser operated at a frequency of 50 Hz in reflection mode. Spectra were recorded at an accelerating voltage of 25 kV and with matrix suppression until 480 Da with 1000 summed acquisitions per measurement. The laser power was kept slightly above the threshold for detection in order to get optimal peak resolution. All mass spectra were acquired with FlexControl 3.4 and analysed with the FlexAnalysis 3.4 software with basic baseline correction and smoothening. The mass spectra of all PS samples showed multiple distributions all with a specific peak spacing of 44 Da (C2H4O = 1 EO unit), as expected for ethoxylated polymers. As MALDI-TOF generates a mass spectrum with individual peak intensities based purely on the sum of all possible ions of a specific mass, distinguishing between molecules of identical mass is impossible. In the case of PS80, one oleate tail (264 Da) is isobaric with six EO units, meaning that unreacted ethoxylated sorbitan (free ethoxylated headgroups) and the esterified PEO sorbitan mono-, di-, and polyesters will overlap in the mass spectrum. As an example, the non-esterified PEO (26) sorbitan headgroup is identical in mass to PEO (20) sorbitan monooleate, PEO (14) sorbitan dioelate, and to PEO (8) sorbitan trioleate etc. This generates, what would appear to be, a simpler mass spectrum but it prevents elaborate mass data analysis and deconvolution of peaks related to PEO sorbitan and its various esters in PS80. This is in contrast to PS60, PS40, and PS20, where the mass of the main hydrocarbon tail is unique.
Small and wide-angle X-ray diffraction (SWAXD)
All SWAXD experiments were carried out on a Ganesha SAXD/WAXD instrument (SAXSLAB/Xenocs, at UCPH, Denmark) using a CuKα-radiation (λ = 1.541 Å) source. The samples were dried overnight under vacuum and pipetted into a copper sandwich sample cell (under ambient relative humidity), with an O-ring as the spacer between two mica windows. Measurements were carried out in vacuum with temperature control (Linkam LNP95, Linkam Scientific Instruments Ltd., Tadworth, United Kingdom) using liquid nitrogen to achieve temperatures below room temperature. The actual sample temperature was calculated using a calibration curve. Data analysis, including transmission correction, background subtraction, removal of mica peaks and azimuthal averaging, was done using the SAXSGui from SAXSLab. Samples were measured every 1 min with an exposure time of 1 min during both cooling and heating at 10 °C min−1, similar to the standard DSC measurements, from ambient temperatures to below crystallisation (as determined by DSC, see Table 4 for each sample's specific temperature) and back to ambient temperatures or temperatures above melting (as determined by DSC, see Table 4 for each sample's specific temperature). However, for some samples where DSC curves had shown complex crystallisation and melting behaviour (multiple peaks) the sample was equilibrated at certain temperatures, as described in the text. Three selected PS samples were measured with 1 min time frames using a slow cooling/heating program (stepwise temperature change of 2 °C with two or three frames measured at each temperature) to achieve detailed temperature scans, ranging from 23 to −56 °C for PS20 and PS80 and 29 to −56 °C for PS60.Results and discussion
The thermal behaviour of PS and SPAN
To understand the mechanism of crystallisation of PS products, their thermotropic behaviour was studied and compared to that of their non-ethoxylated sorbitan ester equivalents. This approach allowed us to differentiate between the impact of the hydrocarbon tails versus the headgroup upon crystallisation. DSC revealed different thermal behaviour for the investigated products (Fig. 2), however, a more or less distinct exothermic crystallisation peak and an endothermic melting peak were observed in all samples. Fig. 2 shows DSC curves for four PS and four SPAN products, with varying hydrocarbon tail length and saturation level. The first and second thermal cycle were similar for the PSs products, but displayed a change for the SPANs, especially for the products with longer saturated tails.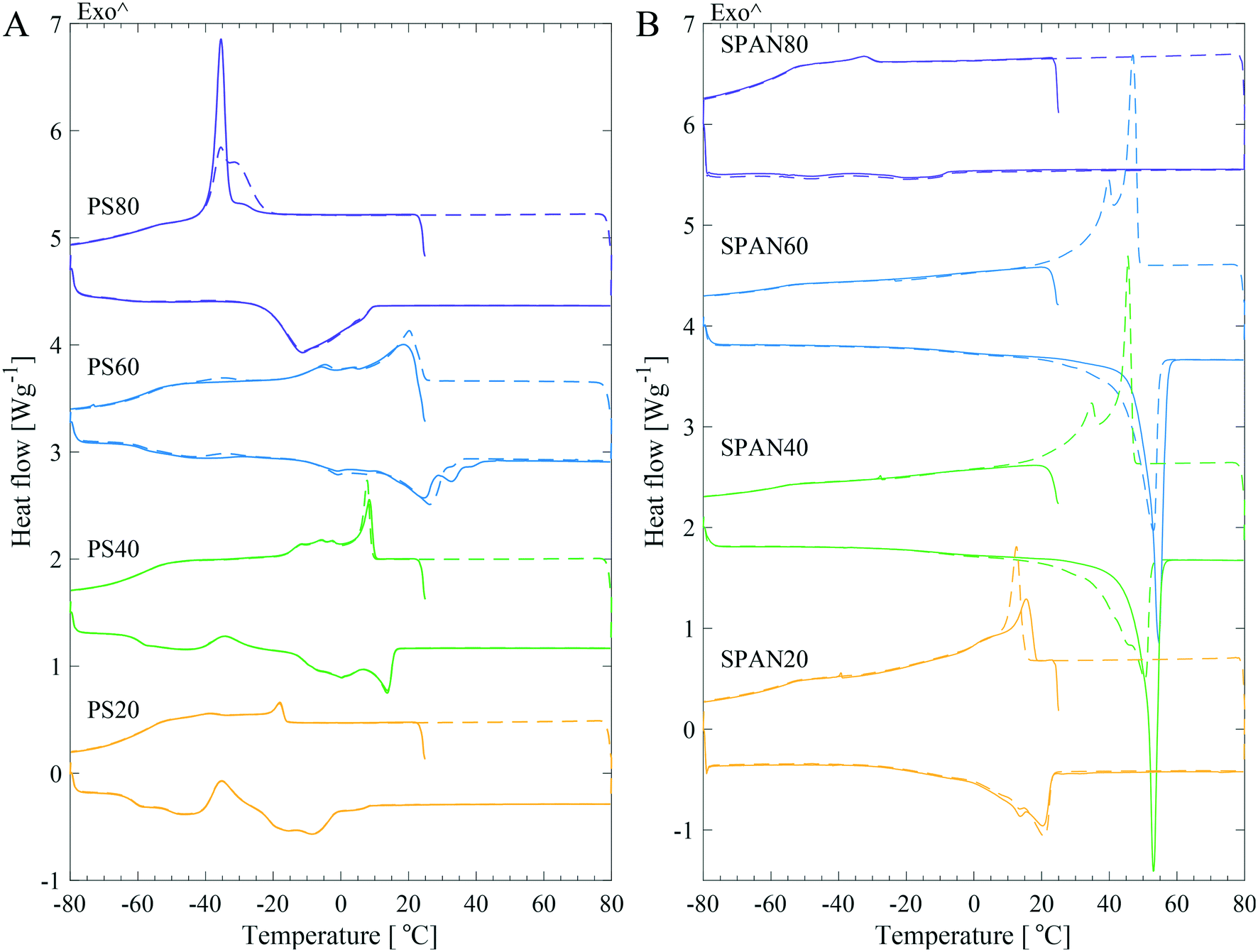 | ||
| Fig. 2 Representative DSC thermograms displaying the first (solid lines) and second (dashed lines) thermal cycle of four different PS (A) and SPAN (B) samples with different hydrocarbon chain length and saturation. The data have been offset along the ordinate for clarity. | ||
Moisture is known to affect the structure of neat surfactants. In the n-octyl α-D-glucoside/water system it was shown that even trace amounts of water in the system had a great impact on the melting behaviour.21 Therefore, Karl Fischer titration measurements were carried out for all samples to assess the moisture content. For the high purity grade products, the manufacturers explicitly stated a low moisture content. The results (see Table 2 for the determined moisture content) corroborated the specifications from the manufacturer by showing lower moisture content for PS samples of high purity (HP), super refined (SR) and ultra-purity (UP) grade but also for all SPANs. Higher moisture contents (approximately tenfold) were found for all standard (ST) grade products compared to higher purity products. The effect of moisture content on the thermal behaviour was tested by measuring the samples with DSC before and after vacuum drying. This data showed that the SPANs were less affected by vacuum drying as compared to the PS products, which for many PS samples showed noticeable changes in the endothermic melting peak shape and temperature (Fig. S2 in the ESI†). Additionally, the shift in peak position and the change in peak shape were significantly larger for standard grade (ST) PS samples, from both Croda and KLK OLEO, in comparison to the higher purity grade samples. Hence, in order to minimise the effect of water on the crystallisation behaviour and to fully understand the underlying mechanism, it was important to control the moisture content by vacuum drying all samples prior to the DSC measurements.
The shape of the endothermic peaks provided information about the PS products. The melting peaks in all studied systems were broad and, in some cases, multiple peaks were detected. Croda HP and Croda ST products had broader endothermic melting peaks, ending at higher temperatures as compared to NOF UP, Croda SR, and KLK OLEO ST, see Fig. 3. This could indicate that Croda HP and ST contained a broader distribution of subspecies. A range of PS20 products were also tested (Fig. S3 in the ESI†), which showed a similar behaviour as for the different PS80 products. Large variations were observed in the endothermic melting peak between samples of different purity grades and samples from different manufacturers, whereas the batch-to-batch variation within the same product was much less pronounced.
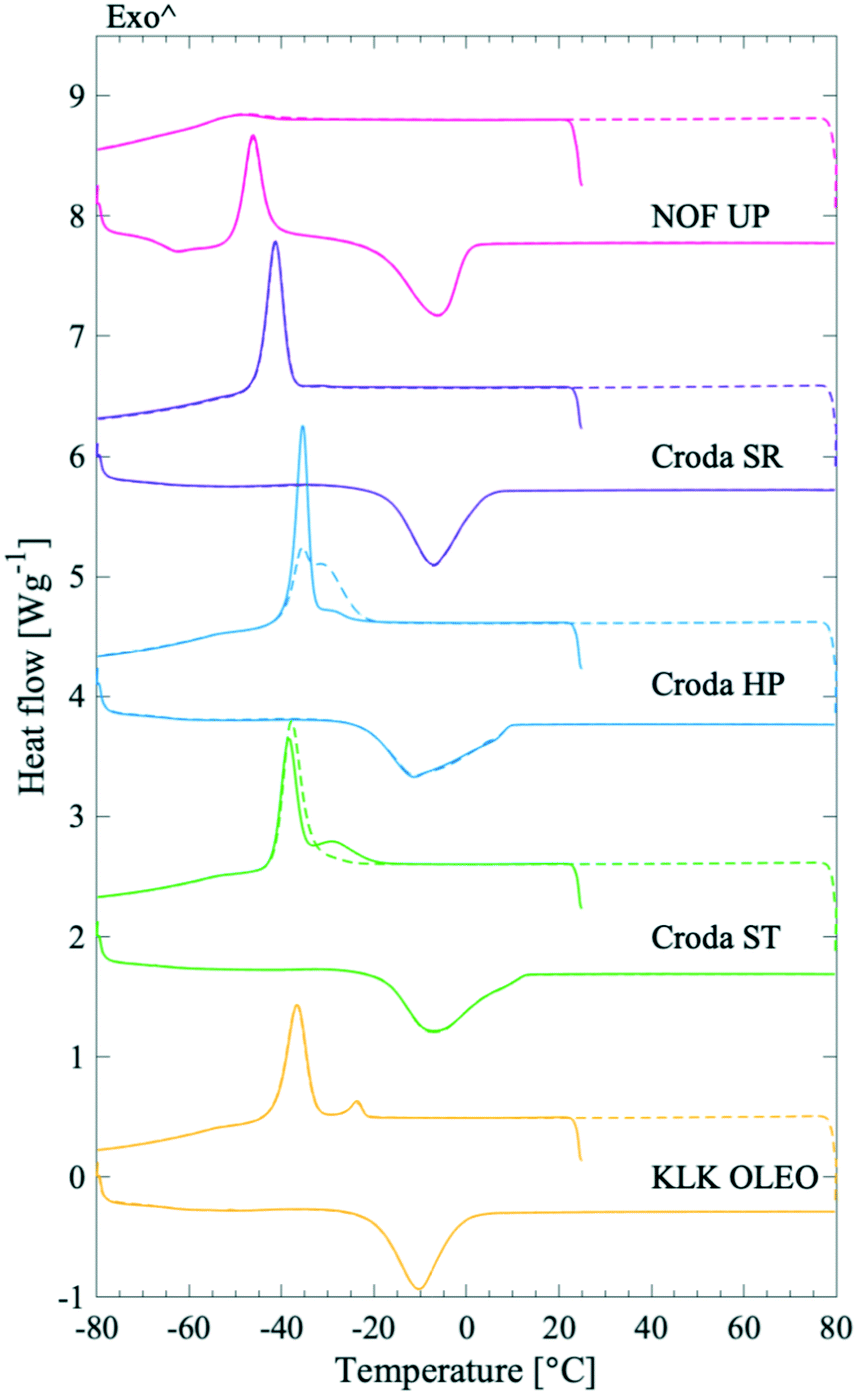 | ||
| Fig. 3 Representative DSC thermograms for PS80 products of varying purity. The graphs show the first (solid lines) and second (dashed lines) thermal cycle of PS80 NOF UP (top, pink), PS80 Croda SR (2nd from top, purple), PS80 Croda HP (middle, blue), PS80 Croda ST (2nd from bottom, green) and PS80 KLK OLEO ST (bottom, yellow). The data have been offset along the ordinate for clarity. | ||
The carbon chain length and saturation level of PS had an overall impact on the melting peak temperature. Hence, these products followed the same trend as fatty acids and alkanes, i.e. the longer the saturated chain, the higher the melting temperature.14–18,22 However, the melting peak temperature (Tmp) values for the PS products were lower as compared to the values for the SPANs. This provided an initial indication that there was a difference in the mechanism of crystallisation for the two types of products. Averaged values of the Tmp and enthalpy of melting (ΔHm) are listed in Table 3. The ΔHm for all the PS samples were similar in magnitude, ranging between approximately 25–50 J g−1, with a maximum difference of a factor of two.
| Sample | No. samples | 1st cycle | 2nd cycle | ||
|---|---|---|---|---|---|
| T mp [°C] | ΔHm [J g−1] | T mp [°C] | ΔHm [J g−1] | ||
| PS 80 KLK OLEO ST | 4 | −10.43 ± 0.07 | 43.10 ± 0.80 | −10.43 ± 0.07 | 43.08 ± 1.03 |
| PS 80 Croda ST | 4 | −7.11 ± 0.25 | 49.75 ± 0.78 | −6.90 ± 0.20 | 49.55 ± 0.80 |
| PS 80 Croda HP | 4 | −11.72 ± 0.16 | 46.61 ± 2.01 | −11.60 ± 0.22 | 46.80 ± 1.98 |
| PS 80 Croda SR | 4 | −7.70 ± 0.33 | 45.44 ± 1.05 | −7.70 ± 0.33 | 45.84 ± 1.15 |
| PS 80 NOF UP | 4 | −6.87 ± 0.29 | 41.36 ± 0.70 | −6.83 ± 0.35 | 41.29 ± 0.92 |
| PS 60 Croda ST | 4 | 24.05 ± 0.23 | 45.65 ± 1.47 | 25.21 ± 0.63 | 49.91 ± 0.76 |
| PS 40 Croda ST | 4 | 13.85 ± 0.17 | 40.04 ± 0.58 | 13.77 ± 0.24 | 40.07 ± 0.59 |
| PS 20 OLEO ST | 5 | −16.33 ± 0.16 | 26.30 ± 1.18 | −16.33 ± 0.16 | 26.40 ± 1.23 |
| PS 20 Croda ST batch 1 | 4 | −8.26 ± 0.22 | 36.42 ± 1.19 | −8.21 ± 0.21 | 36.36 ± 1.04 |
| PS 20 Croda ST batch 2 | 4 | −8.26 ± 0.02 | 35.86 ± 0.81 | −8.30 ± 0.18 | 36.10 ± 0.83 |
| PS 20 Croda HP | 4 | −8.56 ± 0.06 | 26.65 ± 2.16 | −8.56 ± 0.06 | 26.86 ± 1.60 |
| SPAN 80 OLEO ST | 3 | 2.02 ± 0.14 | 15.48 ± 0.33 | 2.08 ± 0.14 | 15.32 ± 0.47 |
| SPAN 80 Croda ST batch 2 | 3 | −19.43 ± 0.42 | 8.96 ± 0.34 | −19.65 ± 0.21 | 9.20 ± 0.79 |
| SPAN 80 NOF UP | 3 | −45.54 ± 0.50 | 2.36 ± 0.15 | −45.70 ± 0.56 | 2.34 ± 0.06 |
| SPAN 60 Croda ST | 3 | 53.53 ± 0.33 | 100.15 ± 2.26 | 52.27 ± 0.19 | 96.63 ± 1.87 |
| SPAN 40 Croda ST | 3 | 52.88 ± 0.54 | 102.11 ± 0.96 | 50.26 ± 0.11 | 95.91 ± 0.53 |
| SPAN 20 OLEO ST | 3 | 12.93 ± 0.13 | 45.76 ± 0.26 | 12.82 ± 0.07 | 44.93 ± 0.81 |
| SPAN 20 Croda ST batch 1 | 3 | 20.41 ± 0.11 | 54.72 ± 1.54 | 20.67 ± 0.10 | 55.60 ± 2.04 |
Data from the first and second thermal cycle of the DSC measurements of the non-ethoxylated material with varying hydrocarbon tail length (all ST grade from Croda), ranging from laurate (C12:0), palmitate (C16:0), stearate (C18:0) to oleate (C18:1), are presented in Fig. 2B. For a quick overview of the relation between the enthalpy of melting, (ΔHm) and the melting peak temperature (Tmp) with the nature of the tail, ΔHm and Tmp were plotted as a function of the primary hydrocarbon tail length for all commercial products (see Fig. 4). From this data it was apparent that the length and saturation level of the SPAN hydrocarbon tails have a larger impact on both the ΔHm and Tmp as compared to the PS products. For the saturated chains the ΔHm and Tmp both increased noticeably with increasing chain length (see Fig. 4 and Table 3). Large variations in the values of ΔHm and Tmp were seen for SPAN80 and SPAN20 products from different manufacturers and with various purity grades (Fig. S4 and Table S3 in the ESI†). Samples of SPAN80 varied substantially between manufacturers (Croda, KLK OLEO and NOF, compared, Fig. S4 in the ESI†), whereas the batch-to-batch variation was insignificant (data not shown). Although the DSC measurements were reproducible, the endothermic melting peak for SPAN80 showed large variations from product to product, giving rise to a large variation in ΔHm, however, the values were much lower in magnitude as compared to SPAN60. These low values were indicative of materials that would form poorly ordered solid phases. Furthermore, the SPANs with saturated hydrocarbon tails displayed ΔHm values close to two times higher than their ethoxylated equivalents. These findings indicate that the crystallising moieties in SPANs and PS products are of different origin.
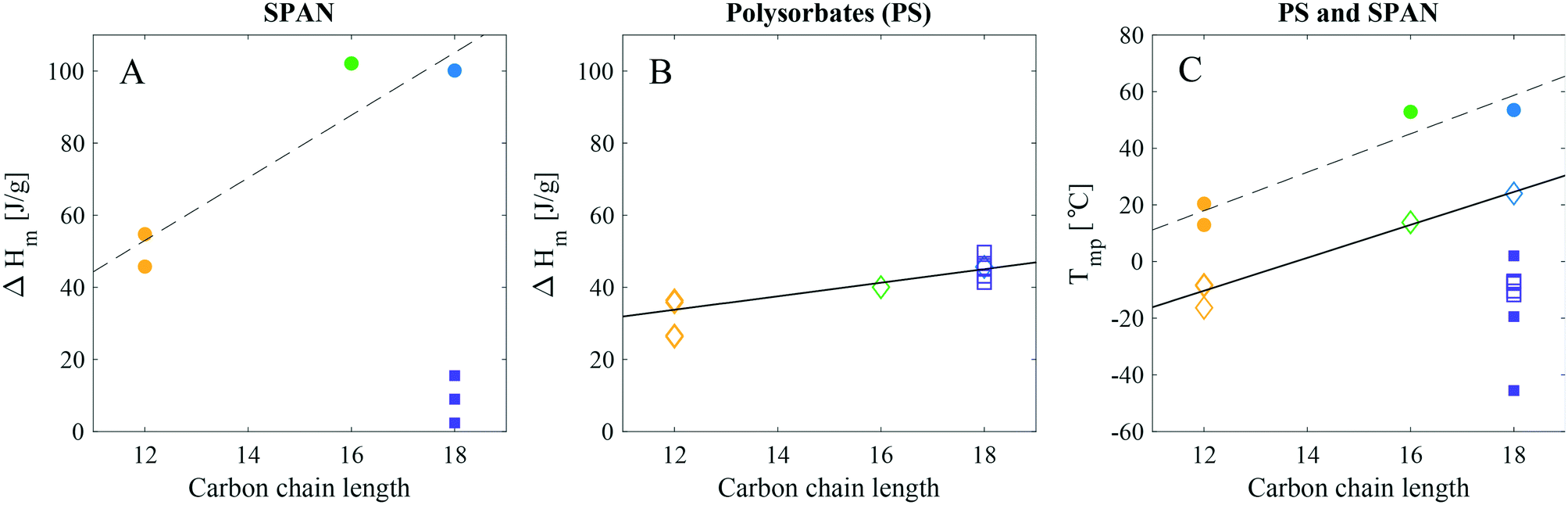 | ||
| Fig. 4 Enthalpy of melting, ΔHm, for A) SPANs (yellow filled circle: SPAN20, green filled circle: SPAN40, blue filled circle: SPAN60 and purple filled square: SPAN80) and B) PSs (yellow diamond: PS20, green diamond: PS40, blue diamond: PS60 and purple square: PS80) as well as C) Tmp for both as a function of carbon chain length. Note that the carbon chain length is the primary hydrocarbon tail length found in each product. The linear regressions, to the saturated tail products, reveal a linear dependence upon both ΔHm and Tmp to the hydrocarbon tail length. | ||
In particular, the plot indicates that saturated SPANs (Fig. 4A) crystallise by the hydrocarbon tail, as they follow a linear trend similar to fatty acids and alkanes.14–18 More energy is required to melt a stearate than a palmitate tail and even less for a laurate tail. The enthalpy of melting for fatty acids is reported to be approximately 170–210 J g−1,14,16–18 (lauric acid: 172–179 J g−1, palmitic acid: 199–206 J g−1, stearic acid: 203–208 J g−1 and oleic acid: 76 J g−1). This increment of approx. 6 J g−1 per CH2 is in good agreement with our findings (9 J g−1 per CH2, as determined by the slope of the linear regression in Fig. 4A), supporting the theory that SPANs (with saturated hydrocarbon chains) crystallise by the hydrocarbon tail. SPAN80 did not fit into the linear trend, which is however not surprising as the degree of saturation in hydrocarbons is known to have significant impact on their melting behaviour. Consequently, the ΔHm value for SPAN80 is very low and could possibly be affected by the presence of impurities in the form of saturated hydrocarbon tails. The ΔHm value for SPAN60 is slightly lower compared to the value for SPAN40. As literature values for ΔHm for palmitic and stearic acid are very similar, the assumption could be made that the difference in values for the SPAN products are comparable. A small variation in hydrocarbon tail distribution could then easily cause such an effect observed for the SPAN40 and 60.
The ΔHm values for the various PS products displayed considerably higher similarities (Fig. 4B) and showed a less pronounced linear increase as compared to the SPANs (only 2 J g−1 per CH2, from slope of linear regression). In particular, and in stark contrast to the non-ethoxylated material, the values for PS80 and PS60 (C18:1 and C18:0) were in fact overlapping. Therefore, in the case of PSs the nature of the carbon chain appeared to have much less of an effect on the crystallisation as compared to the SPANs. This indicates that the PS products crystallise in a way different to the non-ethoxylated SPAN products. However, it does not necessarily mean that PSs crystallise by the head group only as there is still a positive trend in the ΔHm with increasing hydrocarbon tail length. As for the SPANs, the Tmp for the PS products revealed a linear dependence for the saturated tails and a lower value for PS80 (Fig. 4C). This could be explained by the presence of the double bond, if the product crystallises by the hydrocarbon tail. On the other hand, if the products crystallise by the headgroup, their Tmp would be expected to be similar which is not the case. It is possible that a difference in the degree of ethoxylation could give rise to such incoherent thermotropic behaviour of the PS products. Additional information on the EO distribution, the presence of different subspecies, and the hydrocarbon tail composition is thus required to further interpret the DSC data.
Identification of subspecies in the samples
Fig. 5 shows MALDI-TOF mass spectra of PS80 (C18:1), PS60 (C16:0), PS40 (C14:0), and PS20 (C12:0). The spectra revealed substantial differences in the ethoxylation level and hydrocarbon tail distribution. All PS samples contained a low mass distribution at approximately 700–1500 Da (marked with (i) in Fig. 5) consistent with ethoxylated isosorbide esters and ethoxylated esters. Due to isobaric molecules and overlapping distributions in PS80 (as discussed in the methods section) the main mass distribution in the PS80 spectrum corresponded to a sum of ethoxylated sorbitan headgroups (h) and ethoxylated sorbitan oleates including monoesters (m), diesters (d), and triesters (t). The minor distribution imbedded in the main distribution of PS80 corresponded to PEO sorbitan monopalmitate and traces of linoleate and stearate could also be detected. In the case of PS60, PS40, and PS20 the primary esters were not isobaric with a particular number of EO units and thus, although complex, the various distributions could be analysed in more detail. For these three samples the free ethoxylated headgroups (h) constituted a major distribution, which was clearly separated from the esterified species. The presence of substantial amounts of such non-esterified species in PS products is affirmed by previous studies and theoretical calculations.23,24 The distribution marked (m) corresponded to the ethoxylated sorbitan monoester species in all products and the remaining distributions can be assigned to diesters (d) and triesters (t) with various tail compositions. In PS60 the sorbitan monoester distribution (m) consisted of two main distributions (shifted by 28 Da (C2H4)), which were assigned to ethoxylated sorbitan monostearate and sorbitan monopalmitate (as expected from the specifications, Table S1 in the ESI†). The specifications for PS40 require more than 92.0% palmitic acid, and the MALDI-TOF analysis confirmed that it contained no significant amounts of stearate but predominantly sorbitan palmitate. Due to the simpler hydrocarbon tail composition and the lack of overlap for PS40 the sorbitan dipalmitate (d) and the tripalmitate distributions (t) were more clearly distinguished. PS20, showed a main distribution containing laurate but also contributions from myristate and palmitate. All products apart from PS40 had complex peak distributions of diesters and triesters due to the many possible combinations of hydrocarbon tails. Further details and a zoom of the peaks with assignment can be found in the ESI† Fig. S5 and Table S4.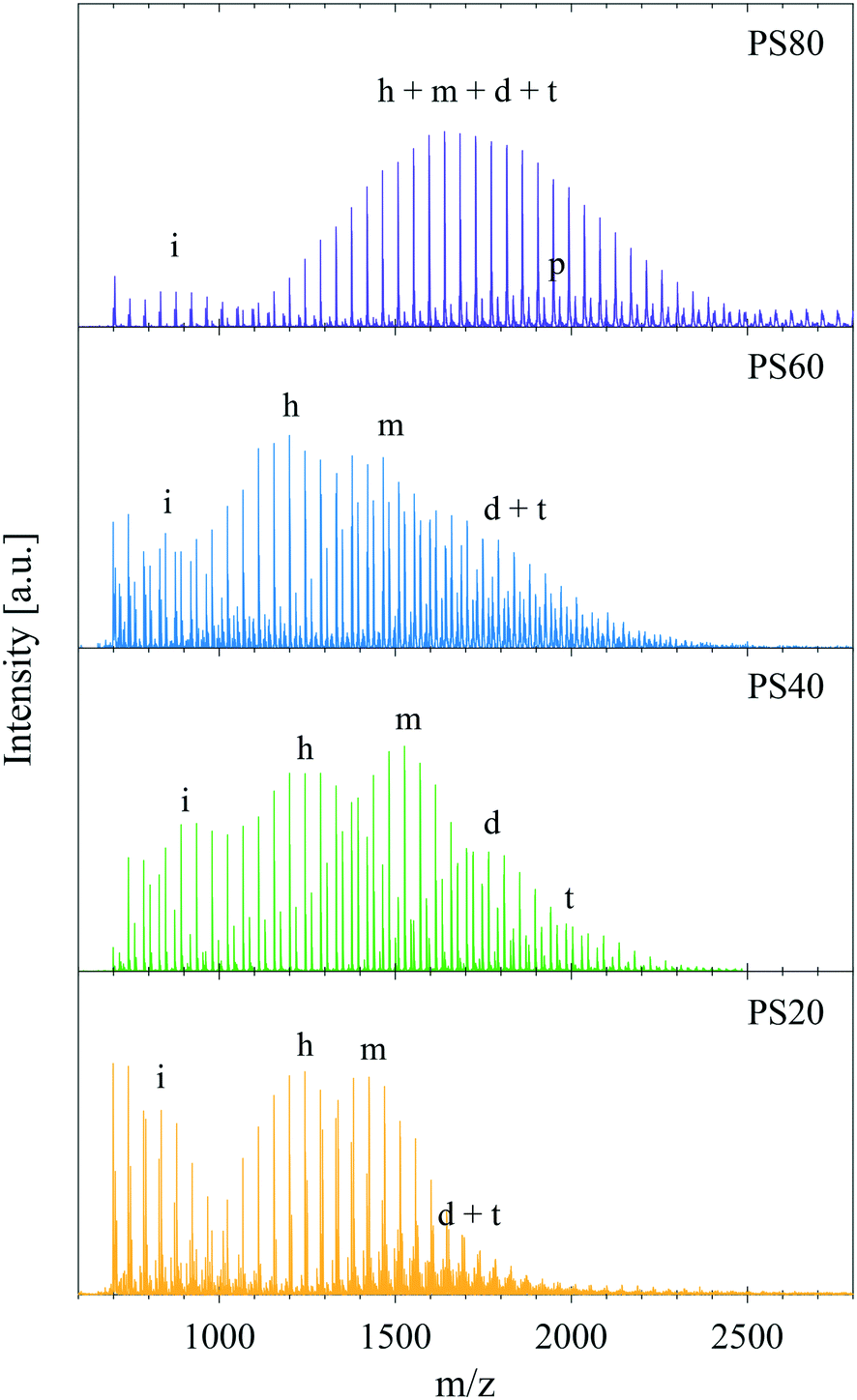 | ||
| Fig. 5 MALDI-TOF of polysorbate samples of different main hydrocarbon tail composition: PS80, PS60, PS40, and PS20. For PS80 the spectrum shows less peaks because of overlapping distributions of isobaric molecules. The letters denote, i) PEO isosorbide and PEO ester species, h) non-esterified headgroups, m) the main monoester distribution, d) PEO sorbitan diester species, t) PEO sorbitan triester species, p) PEO sorbitan palmitates in PS80. A zoom of the peak distributions and peak assignments can be found in ESI† Fig. S5 and Table S4. | ||
The MALDI-TOF data showed that more than one species and various hydrocarbon tails were present in the samples thus explaining the complex endothermic peaks revealed by the DSC data (Fig. 2). The presence of PEO sorbitan, PEO isosorbide esters and PEO esters as well as polyesters, in combination with a hydrocarbon tail and EO distribution, provided a plausible explanation to the complex shape of the endothermic melting (solid to liquid transition) occurring when the sample was heated from −80 °C to 80 °C.
MALDI-TOF spectra of PS80 of different purity grades are shown in Fig. S6A in the ESI.† From this data it was clear that all previously stated by-products were found, as well as a tail combination of linoleate, stearate, and palmitate additional to the oleate in all products except the NOF UP and Croda SR, which mainly contained oleate tails (99.3% for NOF UP and 87.2% for Croda SR according to the certificate of analysis). A range of PS20 products were also tested with MALDI-TOF (Fig. S6B in the ESI†) and DSC (Fig. S3 in the ESI†), which showed a similar behaviour as for PS80. Large variations were observed in the endothermic melting peak between samples of different purity grades and samples from different manufacturers, whereas the batch-to-batch variation within the same product was much less pronounced.
The unesterified sorbitans (headgroups) in PS60, PS40, and PS20 were on average ethoxylated by 23–24 EO units (approx. 5–6 EO per chain), and the main monoester species in each product were ethoxylated with a similar number of EO units as determined by the MALDI data. Although it was not possible to deconvolute the monoester peak distribution from the non-esterified PEO sorbitan or the polyesters in PS80, it was evident that the molecular species had a noticeably higher ethoxylation level than the other products. This was corroborated by earlier findings that suggest approximately 7–8 EO units per chain, corresponding to 28–32 EO units per sorbitan ester.23–25 Addition of 20 mol of EO to 1 mol of sorbitol during synthesis (assuming full conversion of sorbitol to sorbitan only) should theoretically give a sorbitan monoester with four EO chains having 5 EO units each on average. However, the higher degree of ethoxylation originates from the synthesis route, where sorbitol is first dehydrated to sorbitan. Due to the harsh conditions during synthesis, sorbitan molecules can be further dehydrated into isosorbide molecules. In the traditional synthesis route these species are then esterified resulting in formation of both sorbitan and isosorbide esters. The isosorbide esters only have two active sites (hydroxyl groups), where esterification and ethoxylation can occur, i.e. bi-functional, as compared to the sorbitan esters that have four active sites (Fig. 1). This entails that, post esterification, the 20 mol of EO added will distribute over less than four chains on average per mol of sorbitan/isosorbide species, leaving the chains to grow longer and the finished product to have a higher degree of ethoxylation compared to the expected 5 EO units per chain on average. In effect, this means that the higher the amount of isosorbide by-products produced during synthesis the higher the overall ethoxylation level of the sorbitan species. PEG formed during the ethoxylation step in the synthesis can, however, also be esterified and form linear ethoxylated esters, thus counteracting the increase in EO chain length. The higher degree of ethoxylation could to some extent explain the differences between the ΔHm for the different PS products, if the products would crystallise by the headgroups, or in particular by the EO chains. The slightly higher values found for the PS80 would then correspond to a longer EO chain. PEG 400, corresponding to an average of 9–10 EO units per chain, was measured with DSC (Fig. S7 in ESI†) and provided an ΔHm of 110 J g−1. This EO chain length is only marginally longer than for the PS80 products, but the ΔHm is still a factor of two larger. Additionally, if the product would only crystallise by the EO chains why would the ΔHm values be different for PS20, PS40 and PS60? Is it an effect of the complexity of the PS products affecting the melting of EO chains or is it a more intricate structure that melts?
What crystallises? – Structure determination by SWAXD
In order to resolve which parts of the molecule that crystallise, small- and wide-angle diffraction (SWAXD) experiments were performed at different temperatures to obtain structural data on the order on different length scales of the products, both in their liquid and solid states. Fig. 6A and B show the SWAXD data for PEG 400 and various PS products of different hydrocarbon tail composition in the solid (Fig. 6A) and in the liquid state (Fig. 6B), respectively. Fig. 6C and D show the corresponding data for SPAN products (SPAN80, SPAN60 and SPAN20).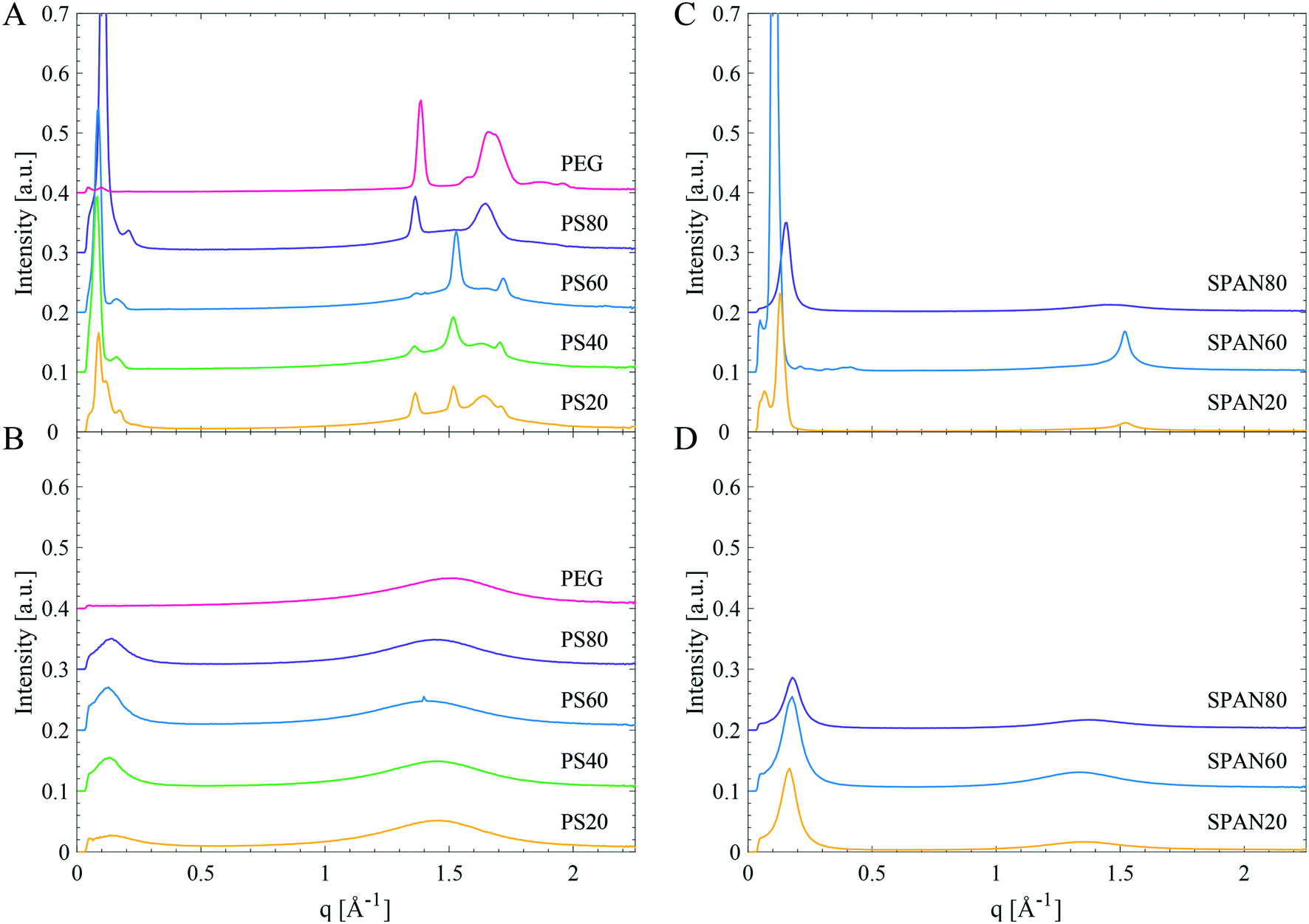 | ||
| Fig. 6 SWAXD data for PEG 400, PS and SPAN products. Left: PEG 400, PS80, PS40, PS60, and PS20 in the solid (A) and liquid (B) state. Right: SPAN80, SPAN60, and SPAN20 in the solid (C) and liquid (D) state. Specific temperatures at which the samples were measured are given in Table 4. | ||
Low temperature structure of the samples
All PS products revealed distinct Bragg peaks in the WAXD region in the solid state (Fig. 6A and Table 4), demonstrating long-range order (on the sub-nanometre length scale) in the material. The PS products all displayed two Bragg peaks (at q-values around 1.4 and 1.6 Å−1, corresponding to d-spacings of 4.8 Å and 3.8 Å), coinciding with the Bragg peaks found in PEG 400 (Fig. 6A and Table 4). These peaks were attributed to crystallisation of the EO chains in PS products, as it coincides with the main reflections arising from a PEG crystal as established by Takahashi et al.27 However, these two Bragg peaks differed in intensities for different PS products. They were most intense in PS80, whereas only just detectable for PS60. They were also the only Bragg peaks found in the WAXD region for PS80, while the products containing saturated tails, PS60, PS40 and PS20, showed two additional Bragg peaks in the WAXD region (at q-values of 1.5 and 1.7 Å−1, corresponding to d-spacings of 4.1 and 3.7 Å). These two latter Bragg peaks were assigned to an orthorhombic hydrocarbon chain packing.26 The relative intensity of these two different sets of Bragg peaks shows a correlation to the hydrocarbon tail length. The hydrocarbon tail Bragg peaks for PS60 were more pronounced than for PS40 and the peaks were found even less intense for PS20, see Fig. 6A. In order to gain deeper insight into the solidification and crystallisation mechanism of the PS products, PS80, PS60, and PS20 were subjected to a slow cooling cycle with SWAXD data acquired every minute (see methods section for more detailed information on the temperature scans). The SWAXD curves were plotted as a function of temperature (Fig. 7) and showed intricate features of the process of crystallisation not revealed by the diffractogram (Fig. 6) obtained before and after the main crystallisation event. For PS80 (Fig. 7A), the two Bragg peaks in the WAXD region, associated with PEG crystallisation, appeared just below 0 °C and the intensity of the peaks stabilised at −30 °C, as corroborated by the broad exothermic crystallisation peak in the corresponding DSC thermogram in Fig. 7D. The data obtained while slowly cooling PS60 revealed how the hydrocarbon tails initially packed in a hexagonal phase (one Bragg peak at a q value of 1.5 Å−1), starting at approx. 20 °C. The hydrocarbon chains gradually rearranged into the denser orthorhombic packing as noted by the presence of two Bragg peaks (at q 1.5 and 1.7 Å−1, a small shift is noted in the Bragg peak at q 1.5 Å−1, which is expected when rearranging from a hexagonal to orthorhombic phase). The two Bragg peaks stabilised in intensity at approximately −20 °C, coinciding with the end of the exothermic peak in the corresponding DSC thermogram in Fig. 7D. The two Bragg peaks originating from EO chain crystallisation (at q values 1.36–1.37 and 1.64–1.65 Å−1) were visible at all temperatures during the slow cooling measurement (from 29 to −56 °C) of PS60 (Fig. 7B). For PS20 the temperature scans revealed a similar behaviour (Fig. 7C). For PS20 the hydrocarbon tails crystallised in an orthorhombic packing, as noticed by the two Bragg peaks that stabilised in intensity at −35 °C. The two Bragg peaks associated to the EO chain crystallisation appeared around −24 °C and their intensity stabilised at −32 °C. This was supported by a smaller exothermic plateau in the DSC thermogram in Fig. 7D starting at around −10 °C, consistent with the hydrocarbon tail crystallisation, and a larger exothermic peak at approx. −35 °C connected to crystallisation of the EO chains.
| Sample | State | Temp [°C] | SAXD | WAXD | |||||
|---|---|---|---|---|---|---|---|---|---|
| Low q-spacing [Å−1] | d-Spacing [Å] | Structure | Large q-spacing [Å−1] | d-Spacing [Å] | PEG crystallisation | Hydrocarbon tail packing | |||
| a q-Spacing data from shoulder peaks rather than well-defined Bragg peaks, see Fig. 6. b This spacing might correspond to a metastable isotropic liquid since no phase transition was observed in the SWAXD experiments. | |||||||||
| PEG400 | Solid | −65 | 0.048a | 131 | Lamellar | 1.387 | 4.53 | Yes | n.a. |
| 1.580a | 3.98 | ||||||||
| 1.659 | 3.79 | ||||||||
| 0.099a | 63.5 | 1.687a | 3.72 | ||||||
| 1.875a | 3.35 | ||||||||
| 1.960 | 3.21 | ||||||||
| Liquid | 20 | — | — | None | 1.517 | 4.14 | No | n.a. | |
| PS80 | Solid | −56 | 0.105 | 59.8 | Lamellar | 1.364 | 4.61 | Yes | None |
| 0.210 | 29.9 | ||||||||
| 0.060a | 105 | 1.648 | 3.81 | ||||||
| 0.162a | 38.8 | ||||||||
| Liquid | 20 | 0.139 | 45.2 | None | 1.449 | 4.34 | No | None | |
| PS60 | Solid | −56 | 0.085 | 73.9 | Lamellar | 1.370 | 4.59 | Yes | Orthorhombic |
| 0.162 | 38.8 | ||||||||
| 1.529 | 4.11 | ||||||||
| 0.054a | 116 | ||||||||
| 1.653 | 3.80 | ||||||||
| 0.190a | 54.2 | ||||||||
| 1.721 | 3.65 | ||||||||
| 0.116a | 33.1 | ||||||||
| Liquid | 53 | 0.128 | 49.1 | None | 1.421 | 4.42 | No | None | |
| PS40 | Solid | −22 | 0.080 | 78.5 | Lamellar | 1.364 | 4.61 | Yes | Orthorhombic |
| 0.162 | 38.8 | ||||||||
| 1.517 | 4.14 | ||||||||
| 0.054a | 116 | ||||||||
| 1.631 | 3.85 | ||||||||
| 0.178a | 52.4 | ||||||||
| 0.120a | 35.3 | 1.708 | 3.68 | ||||||
| Liquid | 20 | 0.128 | 49.1 | None | 1.455 | 4.32 | No | None | |
| PS20 | Solid | −65 | 0.088 | 71.4 | Lamellar | 1.364 | 4.61 | Yes | Orthorhombic |
| 0.173 | 36.3 | 1.517 | 4.14 | ||||||
| 0.054a | 116 | 1.636 | 3.84 | ||||||
| 0.120a | 52.4 | 1.710 | 3.67 | ||||||
| Liquid | 20 | 0.140 | 44.9 | None | 1.461 | 4.30 | No | None | |
| SPAN80 | Solid | −65 | 0.153b | 41.1b | Lamellar | 1.466 | 4.29 | n.a. | None |
| Liquid | 20 | 0.180 | 34.9 | None | 1.375 | 4.57 | n.a. | None | |
| SPAN60 | Solid | 20 | 0.105 | 59.8 | Lamellar | 1.520 | 4.13 | n.a. | Hexagonal |
| 0.210 | 29.9 | ||||||||
| 0.247a | 25.4 | ||||||||
| 0.318 | 19.8 | ||||||||
| 0.352a | 17.8 | ||||||||
| 0.389a | 16.2 | ||||||||
| 0.414 | 15.2 | ||||||||
| Liquid | 64 | 0.179 | 35.1 | None | 1.341 | 4.69 | n.a. | None | |
| SPAN20 | Solid | −65 | 0.131 | 48.0 | Lamellar | 1.523 | 4.13 | n.a. | Hexagonal |
| Liquid | 28 | 0.167 | 37.6 | None | 1.364 | 4.61 | n.a. | None | |
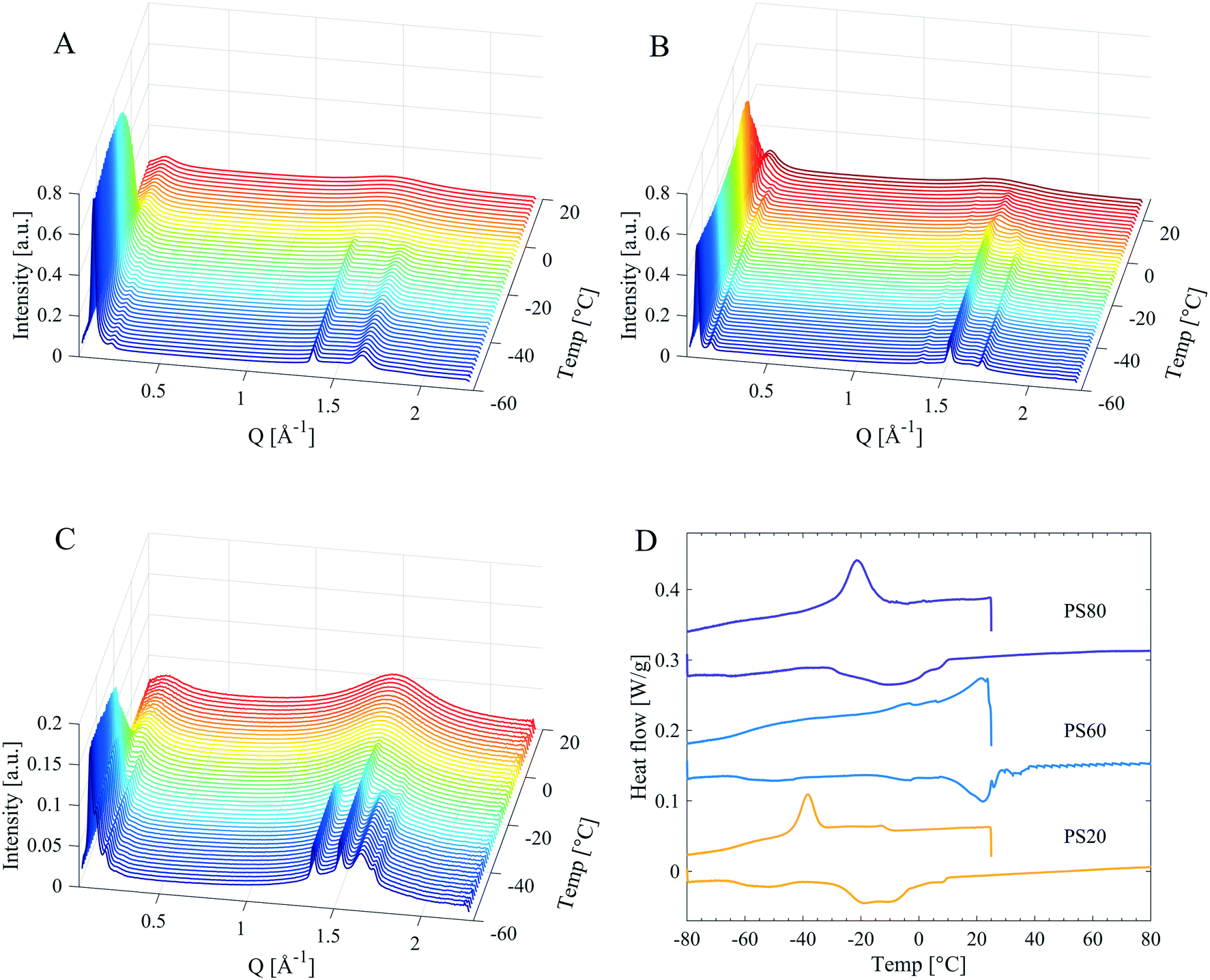 | ||
| Fig. 7 SWAXD of A) PS80, B) PS60 and C) PS20 from the slow cooling measurements, with D) corresponding DSC data measured at 1 °C min−1. One SWAXD measurement curve is displayed every 2 °C, which is the last data set at each temperature. | ||
The data clearly suggest that in the unsaturated PS80 only the headgroup crystallised, whereas saturated PS products crystallised in part by both the EO chains and the hydrocarbon tails. The contribution to crystallinity from the tails increased as the length of the saturated hydrocarbon chains increased. The enthalpies of melting for PEG and fatty acids (ranging from lauric acid to stearic and oleic acid) are in the same order of magnitude and hence it was difficult to completely differentiate between their contributions to the melting enthalpy based on only DSC data. However, from the SWAXD data, the Tmp dependence for the PSs on saturated hydrocarbon tail length can be explained to be a consequence of tail crystallisation.
The length of a hydrocarbon chain, l, can be estimated using the following equation: l = 1.5 + 1.27nc Å, where nc is the number of carbon atoms in the hydrocarbon chain,29 and the length of a sorbitan is approximated to be 8–9 Å in its longest conformation. SPAN60 would according to this approximation form a bilayer, with two extended hydrocarbon tails, with a thickness of 65 Å (2 × 24.4 + 2 × 8 Å), corresponding well to the repeat distance of 60 Å found in the diffraction data (Fig. 6C). This structure is also consistent with SPAN20, where the equivalent length of two fully extended hydrocarbon tails and two heads would be 49 Å, matching well with the experimentally determined repeat distance of 48 Å. The estimated repeat distance of 41 Å for SPAN80 is consistent with a bilayer of fluid hydrocarbon tails. The length of an oleate tail in a glycerol monooleate lamellar liquid crystal has previously been determined to be 17 Å.30 A bilayer in the SPAN80 system would thus be 17 × 2 + 2 × 8 = 50 Å, corresponding reasonably to the observed 41 Å.
All PS products displayed at least two distinct Bragg peaks in the SAXD region (q-values in Table 4), in the solid state, corresponding to lamellar phases with repeat distances of 60 Å for PS80, 74 Å for PS60, 77 Å for PS40 and 72 Å for PS20. The Bragg peaks for PS80, from the lamellar phase, appeared simultaneously as the Bragg peaks corresponding to the EO chain crystallisation in the WAXD region (Fig. 7A). For PS60 and PS20 the Bragg peaks corresponding to the lamellar phase appeared together with the Bragg peaks corresponding to the hydrocarbon tail crystallisation in the WAXD region (Fig. 7B and C). Additionally, in all PS products, and especially in PS20, a second lamellar phase with a repeat distance of approx. 105 and 116 Å exists for the products with unsaturated and saturated hydrocarbon tails respectively. This likely originates from domains with a different organisation of the molecules or domains of different subspecies.
Liquid state structure of the samples
In their fluid state the SPAN products displayed one single broad peak in the WAXD region (d-spacing of 4.6–4.7 Å) originating from melted, disordered hydrocarbon chains.26 For the investigated SPANs, the sharper peak in the SAXD region could be from remnant domains of lamellar structures, with repeat distances of 35 and 38 Å, similar to what have been observed for liquid triglycerides.22 However, all interaction peaks are of low intensity suggesting disordered isotropic liquids, with only a small degree of order.The PS products in the liquid state behaved similar to the SPANs (see Fig. 6B and temperatures in Table 4). There were two different broad peaks, one in the SAXD region and one in the WAXD region, but no obvious Bragg peaks were detected. The first peak corresponded to repeat distances of 45, 49, 49, and 45 Å for PS80, PS60, PS40, and PS20 respectively. The peak in the WAXD region, at a d-spacing of 4.3–4.4 Å, was at slightly smaller repeat distances as compared to the SPANs. This most likely arises from fluid disordered hydrocarbon tails (d-spacing of 4.6–4.7 Å for the SPANs) and fluid EO chains (d-spacing of 4.1 Å in the fluid PEG 400), resulting in a broad peak with at d-spacing in between the two. However, all peaks were weak in intensity, indicating a large degree of disorder in the liquid state of the PS products.
Agreement between SWAXD and DSC data
The PS products crystallised by the EO chains in the headgroup, as well as by the hydrocarbon tails for the saturated hydrocarbon tail PS products. Values in literature show a clear and increasing relation between ΔHm (J g−1) and increasing hydrocarbon tail length of free fatty acids,26 however only a small positive trend in ΔHm was revealed for PS products. The experimentally determined ΔHm values were between 25–50 J g−1 for the PS products, but around 110 J g−1 for PEG 400 and 170–210 J g−1 (ref. 14 and 16–18) for free fatty acids, thus it is clear that only parts of the material crystallise. This was supported by a broad background in the SWAXD data (Fig. 6), indicating a certain degree of amorphous material. The similarity between the ΔHm values for PEG and free fatty acids, in combination with the overall lower ΔHm values for the PS products made it difficult to conclude which part was crystallising. Therefore, a combination of techniques, i.e. both DSC and SWAXD, was required to establish a mechanism of crystallisation for PS products.Conclusions
We have characterised the thermal behaviour and mechanism of crystallisation of commonly used PS products and their non-ethoxylated equivalents (SPAN). The investigated PS products included commercially available PS80, PS60, PS40, and PS20 differing mainly by the nature of their hydrocarbon tail i.e. length and saturation.Extensive calorimetric analysis revealed that the enthalpy of melting (ΔHm) of saturated SPANs increased linearly by approx. 9 J g−1 per CH2, clearly indicating that these products crystallise by the hydrocarbon tail. However, unsaturated SPAN80 had a much lower ΔHm and thus did not follow the same trend. WAXD data confirmed the hydrocarbon tail crystallisation in saturated SPANs, revealing hexagonal packing of the tails, while no hydrocarbon tail crystallisation was observed for SPAN80. The SAXS data showed that on the nanometre length scale, the structures obtained upon crystallisation of SPANs were lamellar.
The ΔHm of both saturated and unsaturated polysorbates were much more comparable in magnitude and did not show a significant trend, suggesting that the hydrocarbon tail was not the only contributor to the crystallisation process for the ethoxylated products, if even contributing at all. In fact, WAXD diffractograms revealed that unsaturated PS80 only crystallised by the EO chains in the headgroup, and not by the hydrocarbon tail. All polysorbates with saturated hydrocarbon tails crystallised by the EO chains, but also in part by their tails. The extent of hydrocarbon tail crystallisation increased with length, ranging from C12:0 to C18:0. Upon cooling, the hydrocarbon tails first arranged in a hexagonal structure before rearranging into orthorhombic packing. Upon crystallisation of the PS the SAXD data showed that the obtained structures, on the nanometre length scale, were lamellar.
Using MALDI-TOF we showed that ethoxylated sorbitan ester molecules in commercial PS products had a higher degree of ethoxylation than 20 EO units. As polysorbates crystallise by the EO chains in the headgroup the degree of ethoxylation could have a profound impact on the propensity to crystallise. This correlation is a topic of interest in pending studies as it is of major importance for PS80 in particular, since it only crystallises by the EO chains.
In conclusion, the thermotropic behaviour and solid structure of a range of well-known and commonly used PS and SPAN products have been determined. We show a clear effect of hydrocarbon tail saturation and ethoxylation on the mechanism of crystallisation. Due to the complexity of the commercial products, further analysis and separation of the respective products into their individual components is needed in order to propose a more comprehensive description of the different constituents.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge Lonza AG (Switzerland) for funding the project, and Dr Benjamin Wyler (Lonza AG, Switzerland) for the fruitful discussions. Yana Znamenskaya Falk is recognised for all of her help with the DSC measurements. Johan Engblom and Vitaly Kocherbitov also acknowledge the Gustaf Th Olsson foundation for financial support.References
- T. A. Khan, H.-C. Mahler and R. S. K. Kishore, Eur. J. Pharm. Biopharm., 2015, 97, 60–67 CrossRef CAS PubMed.
- B. D. Davis, Arch. Biochem., 1947, 15, 359–364 CAS.
- B. D. Davis, Arch. Biochem., 1947, 15, 351–358 CAS.
- H. Sun, R. Yang, J. Wang, X. Yang, J. Tu, L. Xie, C. Li, Q. Lao and C. Sun, RSC Adv., 2017, 7, 15127–15138 RSC.
- D. Hewitt, M. Alvarez, K. Robinson, J. Ji, Y. J. Wang, Y.-H. Kao and T. Zhang, J. Chromatogr. A, 2011, 1218, 2138–2145 CrossRef CAS PubMed.
- E. Hvattum, W. L. Yip, D. Grace and K. Dyrstad, J. Pharm. Biomed. Anal., 2012, 62, 7–16 CrossRef CAS PubMed.
- R. S. K. Kishore, A. Pappenberger, I. B. Dauphin, A. Ross, B. Buergi, A. Staempfli and H.-C. Mahler, J. Pharm. Sci., 2011, 100, 721–731 CrossRef CAS PubMed.
- A. Martos, W. Koch, W. Jiskoot, K. Wuchner, G. Winter, W. Friess and A. Hawe, J. Pharm. Sci., 2017, 106, 1722–1735 CrossRef CAS PubMed.
- X. Cao, R. Matthew Fesinmeyer, C. J. Pierini, C. C. Siska, J. R. Litowski, S. Brych, Z.-Q. Wen and G. R. Kleemann, J. Pharm. Sci., 2015, 104, 433–446 CrossRef CAS PubMed.
- V. Hampl, X. Guo, C. Ehrenstrasser, M. Viertler, L. Rayner, G. Campanelli, R. Schipflinger, K. Thewes, A. Cerreti, S. Boehm and C. Sonderegger, J. Pharm. Sci., 2018, 107, 1552–1561 CrossRef CAS PubMed.
- M. Saggu, J. Liu and A. Patel, Pharm. Res., 2015, 32, 2877–2888 CrossRef CAS PubMed.
- C. C. Siska, C. J. Pierini, H. R. Lau, R. F. Latypov, R. Matthew Fesinmeyer and J. R. Litowsk, J. Pharm. Sci., 2015, 104, 447–456 CrossRef CAS PubMed.
- M. Wagner, Thermal Analyiss in Practice, Mettler Toledo, 2009 Search PubMed.
- F. O. Cedeno, M. M. Prieto, A. Espina and J. R. Garcia, Thermochim. Acta, 2001, 369, 39–50 CrossRef CAS.
- M. Dirand, M. Bouroukba, A.-J. Briard, V. Chevallier, D. Petitjean and J.-P. Corriou, J. Chem. Thermodyn., 2002, 34, 1255–1277 CrossRef CAS.
- W. B. Kong, X. W. Fu, Y. Yuan, Z. M. Liu and J. X. Lei, RSC Adv., 2017, 7, 29554–29562 RSC.
- J. Sohns, B. Seifert and E. Hahne, Int. J. Thermophys., 1981, 2, 71–87 CrossRef CAS.
- Z. L. Zhang, Y. P. Yuan, N. Zhang and X. L. Cao, J. Chem. Eng. Data, 2015, 60, 2495–2501 CrossRef CAS.
- C. P. Buckley and A. J. Kovacs, Colloid Polym. Sci., 1976, 254, 695–715 CrossRef CAS.
- K. Pielichowski and K. Flejtuch, Polym. Adv. Technol., 2002, 13, 690–696 CrossRef CAS.
- V. Kocherbitov and O. Söderman, Phys. Chem. Chem. Phys., 2003, 5, 5262–5270 RSC.
- K. Larsson and P. J. Quinn, in The Lipid handbook, ed. F. D. Gunstone, J. L. Harwood and F. B. Padley, Chapman & Hall, 2nd edn, 1995, pp. 401–485 Search PubMed.
- J. D. Brandner, Drug Dev. Ind. Pharm., 1998, 24, 1049–1054 CrossRef CAS PubMed.
- S. Frison-Norrie and P. Sporns, J. Agric. Food Chem., 2001, 49, 3335–3340 CrossRef CAS PubMed.
- Y. Wang, R. Zhang, L. Tan, H. Y. Zhang and M. Yang, J. Chromatogr. Sci., 2012, 50, 598–607 Search PubMed.
- D. M. Small, The Physical Chemistry of Lipids, Plenum Press, 1988 Search PubMed.
- Y. Takahashi and H. Tadokoro, Macromolecules, 1973, 6, 672–675 CrossRef CAS.
- J. M. Seddon and R. H. Templer, in Handbook of Biological Physics, ed. R. Lipowsky and E. Sackmann, North-Holland, 1995, vol. 1, pp. 97–160 Search PubMed.
- D. F. Evans and H. K. Wennerström, The colloidal domain : where physics, chemistry, biology, and technology meet, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- J. Engblom and S. T. Hyde, J. Phys. II, 1995, 5, 171–190 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce00236d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |